

Abstract
The proliferation of vascular smooth muscle cells (VSMCs) is one of the main features of atherosclerosis induced by high glucose. Mevalonate pathway is an important metabolic pathway that plays a key role in multiple cellular processes. The aim of this study was to define whether the enzyme expression in mevalonate pathway is changed in proliferated VSMCs during atherogenic process in diabetic mice. Diabetes was induced in BALB/c mice with streptozotocin (STZ, 50 mg/kg/day for 5 days). Induction of diabetes with STZ was associated with an increase of lesion area and media thickness after 8 and 16 weeks of diabetes. In aorta, there were overexpressions of some enzymes, including 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGR), farnesyl pyrophosphate synthase (FPPS), geranylgeranyl pyrophosphate synthase (GGPPS), farnesyltransferase (FNT), and geranylgeranyltransferase-1 (GGT-1), and unchanged expression of squalene synthase (SQS) and phosphor-3-hydroxy-3-methylglutaryl-coenzyme A reductase (P-HMGR) in 8 and 16 weeks of diabetes. In vitro, VSMCs were cultured and treated with different glucose concentrations for 48 h. High glucose (22.2 mM) induced VSMC proliferation and upregulation of HMGR, FPPS, GGPPS, FNT, and GGT-1 but did not change the expressions of SQS and P-HMGR. In conclusion, altered expression of several key enzymes in the mevalonate pathway may play a potential pathophysiological role in atherogenic process of diabetes macrovascular complication.
1. Introduction
Diabetes mellitus is a rapidly growing global problem, which is associated with increased morbidity and mortality mainly due to chronic macrovascular complications including cardiovascular disease and stroke [1, 2]. Improving macrovascular outcomes through glucose-lowering interventions has remained a difficult, complicated, and, to date, largely unsuccessful enterprise. There is an ongoing need for new therapeutic targets which would slow the development and progression of diabetic macrovascular diseases [3].
Atherosclerosis (AS) is the cause of a majority of cardio- and cerebrovascular events. Despite familiarity with this disease, some of its fundamental characteristics remain poorly recognized and understood. The proliferation of vascular smooth muscle cells (VSMCs) is one of the main features of atherosclerosis that are induced by high-glucose conditions [4]. In both type 1 and type 2 diabetes, atherosclerosis is accelerated [5]. So what is unique about proliferated vascular smooth muscle from diabetes? The answer may probably provide the next therapeutic target for macrovascular complication.
Mevalonate pathway is an important metabolic pathway which coverts mevalonate into sterol isoprenoids, such as cholesterol, and nonsterol isoprenoids, including isoprenoid intermediates-farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP) [6, 7]. These nonsterol isoprenoid intermediates are essential for the posttranslational prenylation of a multitude of proteins involved in intracellular signaling (especially the small GTP-binding proteins) and are essential in cell growth/differentiation, gene expression, protein glycosylation, and cytoskeletal assembly [6, 7].
In our previous studies [8–10], we found that VSMCs from spontaneously hypertensive rats (SHR) were remarkably proliferated since 3 weeks of age; meanwhile, the expressions of key enzymes in mevalonate pathway were significantly upregulated, including 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGR), farnesyl pyrophosphate synthase (FPPS), farnesyltransferase (FNT), and geranylgeranyltransferase-1 (GGT-1). Moreover, our team revealed that inhibition of FPPS prevented VSMCs remodeling in SHR [11]. All these data were obtained from hypertensive proliferated VSMCs. Our question then is, in diabetic proliferated VSMCs, which of the key enzymes of mevalonate pathway are altered and whether these changes are responsible for the diabetic proliferation. An earlier paper has reported that expression of GGT was significantly upregulated in brain, fat, and skeletal muscle from ob/ob diabetic mice [12]. However, detailed data about the expression of mevalonate pathway in diabetic proliferated VSMCs remain to be determined.
Therefore, the present study was designed to investigate the expression of key enzymes of mevalonate pathway in (1) aortas from streptozotocin- (STZ-) induced 0-, 8-, and 16-week diabetic mice in vivo and (2) high-glucose-induced proliferated VSMCs in vitro, including HMGR, FPPS, FNT-β, GGT-1β, squalene synthase (SQS), phosphor-3-hydroxy-3-methylglutaryl-coenzyme A reductase (P-HMGR), and geranylgeranyl pyrophosphate synthase (GGPPS) (Figure 1).
Figure 1.

Schematic diagram of the mevalonate pathway. HMGR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; FPPS, farnesyl pyrophosphate synthase; SQS, squalene synthase; FNT, farnesyltransferase; GGT-1, geranylgeranyltransferase-1; GGPPS, geranylgeranyl pyrophosphate synthase.
2. Materials and Methods
2.1. Animals
Female BALB/c mice (20 ± 2.5 g) were purchased from the Shanghai Laboratory Animal Center (Chinese Academy of Sciences) and housed in a pathogen-free laboratory at The First Affiliated Hospital of Zhejiang University. The procedures and protocols of the study conformed to the Guide for the Care and Use of Laboratory Animal published by the US National Institutes of Health (NIH Publication number 85-23, revised 1996) and the guidelines of the Animal Care and Use Committee of Zhejiang University.
Diabetic mice were induced by daily injection of STZ (Sigma-Aldrich, St. Louis, MO, USA) at a dose of 50 mg/kg for 5 days after a 4 h fast; control animals received the vehicle (citrate buffer; 0.05 mol/L, pH: 4.5). At the time of 0, 8, and 16 weeks after the fifth STZ injection, fasting blood glucose levels were measured in venous blood drawn from the tail by using a CONTOUR glucose meter (Bayer, Mishawaka, IN, USA). Mice with fasting plasma glucose over 13.9 mM were considered diabetic. After measurement of glucose level, diabetic mice were sacrificed after, respectively, 0, 8, and 16 weeks of diabetic duration, and their blood samples were collected. The diabetic duration was confirmed according to the process of atherosclerosis (pre-AS, labile-AS, and moderate-AS) as previously reported [13–15].
2.2. Glucose and Lipid Analysis
Fasting blood glucose (FBG) levels were evaluated as described above. Serum total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), and triglyceride (TG) concentrations were determined by commercial enzymatic methods (test kits from Shanghai Rongsheng Biotech, Inc., Shanghai, China).
2.3. Histological Analysis
The aorta was dissected in situ from the ascending aorta to the iliac bifurcation, cleaned of peripheral fat under a dissecting microscope, and then fixed in 10% neutral formalin, embedded in paraffin, and sequentially stained with hematoxylin and eosin. Lesion areas (LA) per section were counted by taking the average of 6 sections spaced 30 μm apart, beginning at the base of the aortic root. Media thickness (MT) at 10 different points of the thoracic aorta was measured and calculated. Morphometric analysis above was performed with Image-Pro Plus 6.0.
2.4. Cell Culture
Mouse VSMCs were isolated from thoracic aortic explants of female BALB/c mice as previously described [16, 17]. In brief, aortic explants were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; Gibco) and maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air. After 2 weeks, cells that had migrated onto the tissue culture dish were collected by trypsinization and subcultured successively. The identity of the VSMC was determined by the positive immunocytochemistry reactivity to smooth muscle specific α-actin. To ensure the consistency of results, passages 5–12 of VSMCs were used for experiment.
2.5. Proliferation Assay
VSMC proliferation was measured by the 3-[4,4-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay. Cells were cultured in 10% FBS-DMEM on 96-well plates (5 × 103 cells/well). When the VSMCs reached a 60% confluent state, the cells were rendered quiescent by incubation in DMEM containing NG and 0.1% FBS for 24 h. Next, the VSMCs were treated with different glucose concentrations (5.6, 11.1, 16.7, and 22.2 mM) for 48 h. The same concentrations of mannitol were used as an osmotic control. The cells of 96 wells were incubated with 100 μL of 0.5 mg/mL MTT at 37°C for 4 h, washed with cold PBS, and lysed with 100 μL of DMSO (Sigma, St. Louis, MO, USA). After the insoluble crystals were completely dissolved, the optical density of each well was immediately measured at 570 nm using an automatic microplate reader (Molecular Devices, Sunnyvale, CA).
2.6. Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction (QRT-PCR)
Total RNA of the aortic media (VSMCs were the only cell type in this layer) or cultured VSMCs was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Complementary DNA was subsequently synthesized from RNA using the PrimeScript 1st Strand cDNA Synthesis Kit (TAKARA Biotechnology Co. Ltd., Dalian, China) following the manufacturer's instructions. cDNA was amplified afterward using the SYBR Premix Ex Taq II for Perfect Real Time (TAKARA Biotechnology Co. Ltd., Dalian, China). A housekeeping gene, β-actin, was used as internal control for normalizing the QRT-PCR data. Primers employed for β-actin and key enzymes were listed in Table 1.
Table 1.
Glucose and lipid analysis at 0-, 8-, and 16-week diabetic duration in BALB/c mice.
| Week | FBG (mM) | TC (mM) | HDL-C (mM) | LDL-C (mM) | TG (mM) |
|---|---|---|---|---|---|
| Control mice | |||||
| 0 (n = 6) | 7.82 ± 0.70 | 1.82 ± 0.07 | 0.93 ± 0.06 | 0.48 ± 0.06 | 1.31 ± 0.05 |
| 8 (n = 6) | 7.66 ± 0.62 | 1.88 ± 0.07 | 0.86 ± 0.07 | 0.44 ± 0.06 | 1.30 ± 0.05 |
| 16 (n = 6) | 8.40 ± 0.61 | 1.90 ± 0.08 | 0.93 ± 0.06 | 0.45 ± 0.06 | 1.25 ± 0.05 |
| Diabetic mice | |||||
| 0 (n = 6) | 8.20 ± 0.63 | 1.93 ± 0.09 | 0.85 ± 0.06 | 0.47 ± 0.05 | 1.26 ± 0.06 |
| 8 (n = 6) | 19.54 ± 1.39# | 1.91 ± 0.08 | 0.84 ± 0.05 | 0.48 ± 0.07 | 1.35 ± 0.06 |
| 16 (n = 6) | 21.04 ± 1.42# | 1.96 ± 0.08 | 0.77 ± 0.06 | 0.53 ± 0.07 | 1.40 ± 0.05 |
Data expressed as mean ± SEM.
# P < 0.01 versus age-matched control mice.
FBG, fasting blood glucose; TC, total cholesterol; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; TG, triglyceride.
2.7. Western Blot Analysis
Total proteins were isolated from the aortic media (VSMCs were the only cell type in this layer) or cultured VSMCs, and the procedure of western blot analysis was performed as described in our previous reports [8–10]. The expressions of key enzymes in mevalonate pathway were detected using their specific antibodies: HMGR (ab174830, Abcam, UK), P-HMGR (09-356, Millipore, USA), FPPS (ab38854, Abcam, UK), SQS (sc-136372, Santa Cruz, Japan), GGPPS (ab167168, Abcam, UK), FNT-β (ab74206, Abcam, UK), or GGT-1β (sc-18996, Santa Cruz, Japan). To ensure equal protein loading, β-actin was used as an endogenous control (ab8226, Abcam, UK).
2.8. Statistical Analysis
Values are reported as mean ± SEM. All analyses were performed with SPSS (ver. 13.0; Chicago, IL, USA). One-way analysis of variance (ANOVA) followed by Bonferroni post hoc test was used to determine significant differences between groups. Differences were considered statistically significant at a value of P < 0.05.
3. Results
3.1. Glucose and Lipid Content
As expected the STZ-induced mice had extremely higher levels of glucose than age-matched control groups. At 8- and 16-week diabetic groups, the levels of fasting blood glucose were separately 19.54 ± 1.39 mM and 21.04 ± 1.42 mM (both P < 0.01 versus age-matched control) (Table 1). However, in all groups, serum lipid levels (TC, LDL-C, HDL-C, and TG) were comparable (Table 2).
Table 2.
Sequences of forward and reverse primer.
| cDNA | Forward primer | Reverse primer |
|---|---|---|
| HMGR | 5-GAATGCAGAGAAAGGTGCAA-3 | 5-CACCACGTTCATGAGTTTCC-3 |
| FPPS | 5-GCACTGACATCCAGGACAAC-3 | 5-TAAGATCTGGCGCTGTTGAG-3 |
| SQS | 5-CAAGCAGGTCATCTCCAAGA-3 | 5-GCAGCCAAGAGCATGATAAA-3 |
| GGPPS | 5-TGGAACTTCATCAGGGACAA-3 | 5-CCTGTCTTCTGCAACACCAT-3 |
| FNT-β | 5-TGGCAGTAATCCTGGAACTG-3 | 5-GAGCTCGCTGTGACTGTAGC-3 |
| GGT-1β | 5-GCTAAATGGCCAGACAGTCA-3 | 5-CAAATCCCACTCTCCTCCAT-3 |
| β-actin | 5-CTCCATCCTGGCCTCGCTGT-3 | 5-GCTGTCACCTTCACCGTT-3 |
HMGR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; FPPS, farnesyl pyrophosphate synthase; SQS, squalene synthase; GGPPS, geranylgeranyl pyrophosphate synthase; FNT-β, farnesyltransferase-β; GGT-1β, geranylgeranyltransferase-1β.
3.2. Atherosclerosis Development
The morphologic data of lesion area (LA) and media thickness (MT) are summarized in Figure 2. In our study, STZ-induced diabetic mice for 8 weeks developed mild aortic atherosclerotic plaques, with a mean LA of (6.82 ± 0.69) × 103 μm2 (P < 0.01 versus age-matched control). Moreover, in 16-week diabetic mice, aortic atherosclerotic plaques were moderate, with a mean LA of (10.32 ± 0.77) × 103 μm2 (P < 0.01 versus age-matched control). In contrast, no lesions were seen for control mice regardless of time duration. On the other hand, the MT of the thoracic aorta was increased in both 8- and 16-week diabetic mice when compared with age-matched controls.
Figure 2.

Histological analysis of aortas. (a) Lesion areas (LA) of aorta from BALB/c mice at 0-, 8-, and 16-week diabetic duration. (b) Media thickness (MT) of the thoracic aorta from BALB/c mice at 0-, 8-, and 16-week diabetic duration. Data were obtained from 6 mice for each group. Data expressed as mean ± SEM. ∗ P < 0.05 versus age-matched control mice; # P < 0.01 versus age-matched control mice.
3.3. Proliferation of VSMCs
Incubation of VSMCs with various concentrations of glucose (5.6, 11.1, 16.7, and 22.2 mM) resulted in an increase of VSMC proliferation in a dose-dependent manner after 48 h of treatment. This increase was not seen in the osmotic control cells treated with mannitol to maintain the same osmolarity as cell grown in a high concentration of glucose (data not shown). We found that 22.2 mM glucose significantly increased VSMC proliferation (Figure 3) compared to control cells exposed to 5.6 mM glucose (P < 0.01).
Figure 3.
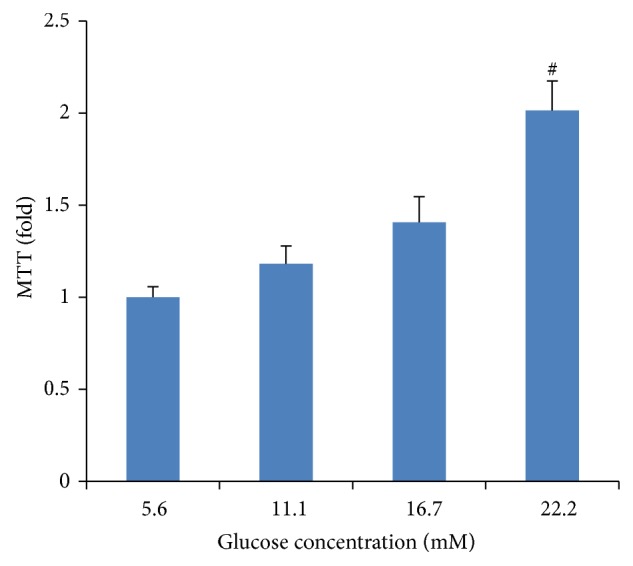
High-glucose-induced proliferation of vascular smooth muscle cells (VSMCs). VSMCs were incubated in various concentrations of glucose (5.6, 11.1, 16.7, and 22.2 mM) for 48 h. Cell proliferation was measured by an MTT assay. Mannitol was used as an osmotic control. Data expressed as mean ± SEM. ∗ P < 0.05 versus normal glucose (5.6 mM) group; # P < 0.01 versus normal glucose (5.6 mM) group.
3.4. HMGR Expression Levels
HMGR is the rate-controlling enzyme of the mevalonate pathway and is modulated through negative feedback regulation by sterols and nonsterol metabolites derived from mevalonate (Figure 1). The mRNA expressions of HMGR were significantly higher in both 8-week (mild-AS phase) and 16-week (moderate-AS phase) diabetic mice than those in age-matched control mice (Figure 4(a)). In vitro, high glucose concentrations (16.7 and 22.2 mM) significantly upregulated mRNA expression of HMGR compared to control VSMCs exposed to 5.6 mM glucose (Figure 6(a)).
Figure 4.

Aortic mRNA expressions of key enzymes using QRT-PCR. The relative expression level of each sample was calibrated by the comparative threshold cycle method, using β-actin as an endogenous control. Data are expressed as fold changes (mean ± SEM), normalized to β-actin mRNA expression. ∗ P < 0.05 versus age-matched control mice; # P < 0.01 versus age-matched control mice.
Figure 6.

Changes of key enzymes in cultured VSMCs treated with different glucose concentrations for 48 h. (a) Gene expressions from QRT-PCR. The relative expression level of each sample was calibrated by the comparative threshold cycle method, using β-actin as an endogenous control. (b) Protein expressions from western blot analysis. β-actin was used as a loading control. Data expressed as mean ± SEM. ∗ P < 0.05 and # P < 0.01 versus normal glucose group (5.6 mmol/L). HMGR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; P-HMGR, phosphor-3-hydroxy-3-methylglutaryl-coenzyme A reductase; FPPS, farnesyl pyrophosphate synthase; SQS, squalene synthase; GGPPS, geranylgeranyl pyrophosphate synthase; FNT-β, farnesyltransferase-β; GGT-1β, geranylgeranyltransferase-1β.
Then, short-term regulation of HMGR is mainly ensured by phosphorylation and dephosphorylation process [18]. The phosphorylated HMGR is inactive, while the dephosphorylated one is active. In our experiment, the protein expression of HMGR in the aorta was significantly higher in both diabetic groups than the control group (Figures 5(a) and 5(b)). In contrast, the expression of P-HMGR, inactivated enzyme reversibly by phosphorylation [18], was similar in diabetes and nondiabetes mice (Figure 5(a); the densitometric average of P-HMGR was not shown). In vitro, VSMCs treated with 22.2 mM glucose remarkably induced protein expression of HMGR but not the P-HMGR (Figures 6(b) and 6(c); the densitometric average of P-HMGR was not shown).
Figure 5.

Aortic protein expressions of key enzymes by western blot. (a) Representative blots are shown. β-actin was used as a loading control. ((b)–(g)) The densitometric average was normalized to the values obtained from the analysis of β-actin as an internal control. Data expressed as mean ± SEM. ∗ P < 0.05 versus age-matched control mice; # P < 0.01 versus age-matched control mice. HMGR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; P-HMGR, phosphor-3-hydroxy-3-methylglutaryl-coenzyme A reductase; FPPS, farnesyl pyrophosphate synthase; SQS, squalene synthase; GGPPS, geranylgeranyl pyrophosphate synthase; FNT-β, farnesyltransferase-β; GGT-1β, geranylgeranyltransferase-1β.
3.5. FPPS Expression Levels
FPPS is a key enzyme in mevalonate pathway and directly catalyzes the formation of FPP. As seen in Figure 1, FPP is a major branching point intermediate of the mevalonate pathway that leads to several classes of essential metabolites including sterols, such as cholesterol, and nonsterols substrates for farnesylation and geranylgeranylation of proteins [19]. In both 8-week and 16-week diabetic mice, mRNA expressions of FPPS were remarkably higher than those in age-matched control groups (Figure 4(b)). Similarly, the protein expressions were also significantly higher in diabetic mice than those in control (Figure 5(c)). In cultured VSMCs, we found that 22.2 mM glucose significantly increased FPPS mRNA and protein expressions compared to control cells exposed to 5.6 mM glucose (Figure 6).
3.6. SQS Expression Levels
SQS represents one of the branches in mevalonate pathway, locating downstream of FPP (Figure 1). It catalyzes the synthesis of squalene from FPP, acting as the first and solely committed step towards the biosynthesis of the cholesterol, and, hence, is considered to be a potential regulatory point for sterol metabolism [20]. In our study, neither mRNA nor protein expression of SQS was different among control and two diabetic groups (Figures 4(c) and 5(d)). These findings could explain the comparable lipid levels in both control and diabetic groups. Also, after treatment with various concentrations (5.6, 11.1, 16.7, and 22.2 mM) of glucose for 48 h, VSMCs exhibited no remarkable change of SQS expression (Figure 6).
3.7. GGPPS, FNT-β, and GGT-1β Expression Levels
As seen in Figure 1, another branch locating downstream of FPP is nonsterol isoprenoid biosynthetic pathway, including several key enzymes of GGPPS, FNT, and GGT-1. GGPPS mediates the condensation of FPP with isopentenyl diphosphate to yield GGPP, a precursor of geranylgeranylated proteins [21]. FNT and GGT-1 are enzymes that carry out the process of prenylation in the cell [22]. They are heterodimers that share the same α subunit but have different β subunits. Thus, to assess differences of FNT and GGT-1 between diabetic and control mice, mRNA and protein expression were detected for FNT-β and GGT-1β. In our study, aortas from both 8- and 16-week diabetic duration exhibited greater mRNA and protein levels of GGPPS, FNT-β, and GGT-1β than those from age-matched controls (Figures 4(d)–4(f) and 5(e)–5(g)). In cultured VSMCs, high concentration of glucose (22.2 mM) significantly stimulated mRNA and protein expressions of GGPPS, FNT-β, and GGT-1β compared to 5.6 mM glucose treatment (Figure 6).
4. Discussion
Elevated levels of glucose are related to most of the complications secondary to diabetes, especially the macrovascular complication [23, 24]. The role of glucose in this complication is not completely understood and probably has a multifactorial origin. The present results show, in an experimental mouse model of atherosclerosis secondary to hyperglycemia, that there is a complicated system of enzyme expression between the sterol branch and the nonsterol isoprenoid branch signaling pathway in STZ-induced diabetic aortas.
A clear relationship between diabetes and cardiovascular disease has been established for decades. Despite this, the mechanisms by which diabetes contributes to plaque formation remain in question. Pathophysiologically, the lesions of atherosclerosis represent a series of highly specific cellular and molecular responses. Atherosclerosis can develop in response to endothelial cell (EC) injury caused by hyperglycemia. After initial injury, different cell types including EC, platelets, and inflammatory cells release mediators, such as growth factors and cytokines that induce multiple effects. These growth factors and cytokines will promote the changes of VSMC from the quiescent contractile state to the synthetic proliferative state, exaggerating VSMC proliferation and migration [4, 5, 25]. It has been widely accepted that VSMC proliferation is an important event in the pathophysiological course of atherosclerosis and attracts more attention of the researchers. In our study, BALB/c mice made diabetic by STZ treatment exhibited different levels of plaque formation, from mild to moderate, due to different diabetic duration, which is in agreement with most of the previous studies [26, 27].
The signaling pathways by which changes in glucose metabolism modulate VSMC proliferation are poorly understood. Numerous molecular and cellular studies have demonstrated that small GTP-binding proteins, consisting of the Ras and Rho family (RhoA and Rac1), participate in the VSMC mitogenic machinery triggered by hyperglycemia. Indeed, extracellular signal-regulated protein kinases (ERKs) (also known as the 42 and 44 kDa isoforms (ERK1/2) pathway) are typically initiated by Ras, which can be stimulated by inflammatory cytokines from high-glucose injured EC [28–30]. The activated ERK1/2 proteins eventually lead to VSMC proliferation and intimal thickening [28, 29, 31]. Moreover, RhoA has been reported to be abundantly expressed in cardiovascular tissues. RhoA and Rho kinase may also be involved in hyperglycemia-induced cell growth and serum response element- (SRE-) dependent c-fos gene expression in rat aortic smooth muscle cells [32]. Furthermore, several studies have identified the key role for Rac1 GTPase in the regulation of high-glucose- and diabetes-induced VSMC proliferation [33, 34]. Zhu and coworkers reported that high-glucose treatment resulted in phosphorylation and membrane translocation of Rac1, p47phox, and p67phox subunits, leading to NADPH oxidase activation and reactive oxygen species (ROS) production, subsequently promoting VSMC proliferation and vascular dysfunction [34].
As stated above, small GTPases (including Ras, RhoA, and Rac1) appear to exert multiple effects on high-glucose-induced VSMC proliferation. For their part, the membrane localization and activation of Ras, RhoA, and Rac1 depend on the process of isoprenylation [35–37]. Ras translocation from the cytoplasm to the plasma membrane is dependent on farnesylation by FNT with FPP, whereas translocation of RhoA and Rac1 is dependent on geranylgeranylation by GGT-1 with GGPP [22]. In our study, we found increased expression of HMGR and FPPS and similar SQS in the aorta from STZ-induced diabetic mice. As shown in Figure 1, HMGR and FPPS are two key enzymes, locating upstream of mevalonate pathway, and directly or indirectly catalyze the synthesis of isoprenoid intermediates (FPP and GGPP). SQS represents one of the branches of mevalonate pathway, locating downstream of FPP. Our findings support the viewpoint that pathologic VSMC proliferation in diabetic aorta may be caused by toxic accumulation of isoprenoid intermediates, including FPP and GGPP, which leads to excessive small GTPase activation.
Theoretically, both HMGR inhibitors (statins) and FPPS inhibitors (nitrogen-containing bisphosphonate, N-BP) may inhibit the synthesis of isoprenoid intermediates, suppress the activation of small GTPase, attenuate the VSMC proliferation, and finally slow the progression of atherosclerosis induced by diabetes. Indeed, the reality is more complex. A wealth of both experimental and clinical evidence reveals the beneficial effects of statins on atherosclerotic progression with diabetes [38, 39], including inhibition of VSMC proliferation and migration [40, 41]. However, large trials suggested an increased incidence of new onset diabetes with long-term use of statins [42–44]. No exact mechanism has been established. Some investigators have proposed that statins may cause an immune response that interferes with insulin signaling [45]. On the other hand, N-BPs, which are extensively used in bone-related disorders [46], have been reported to inhibit the development of experimental atherosclerosis without altering serum lipid profile [47, 48]. In vitro study showed that the antiatherosclerotic effect of N-BPs might be due to inhibition of VSMCs, at least for zoledronate [49]. However, current evidence is not conclusive and further research is necessary to confirm these actions in the clinical setting.
In our study, beside HMGR and FPPS, the expressions of GGPPS, FNT, and GGT-1 were also remarkably upregulated in diabetic aortas. In cultured VSMCs, high glucose induced cell proliferation, accompanying upregulated HMGR, FPPS, GGPPS, FNT, and GGT-1. All these results showed that these enzymes of mevalonate pathway were upregulated in atherosclerotic aortas from diabetes, and the changes may mainly focus on the vascular media-VSMCs. Therefore, we inferred that these enzymes may play important roles in diabetic atherosclerosis, especially in VSMC proliferation. Then, GGPPS, FNT, and GGT-1 locating downstream of FPP to synthesize nonsterol isoprenoid might be regarded as candidate targets for antiatherosclerosis intervention. Further study is necessary.
In conclusion, our work provided the evidence that alteration of the expression of key enzymes in the mevalonate pathway in aorta from diabetes mice is implicated in the atherogenic process, probably through activation of small GTPases and inhibition of VSMC proliferation. Although there are many essential differences between experimental and clinical studies, our study indicates that interfering with the upregulation of key enzymes and biosynthesis of isoprenoids in the mevalonate pathway may open the field for novel therapeutic indications and give many possible therapeutic targets for diabetic macrovascular diseases in the future.
Acknowledgments
The authors express their sincere gratitude to Dr. Iain C. Bruce for revising the paper. This work was supported by the Zhejiang Provincial Natural Science Foundation of China (Project for Young Scientists, no. LQ13H070001), Medical Science and Technology Project of Zhejiang Province (no. 2014KYA025), and the Scientific Research Fund of Zhejiang Provincial Education Department (no. Y201223930).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Guo-Ping Chen and Xiao-Qin Zhang contributed equally to this work.
References
- 1.Nickerson H. D., Dutta S. Diabetic complications: current challenges and opportunities. Journal of Cardiovascular Translational Research. 2012;5(4):375–379. doi: 10.1007/s12265-012-9388-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmidt A. M. Recent highlights of ATVB: diabetes mellitus. Arteriosclerosis, Thrombosis, and Vascular Biology. 2014;34(5):954–958. doi: 10.1161/atvbaha.114.303522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porter K. E., Riches K. The vascular smooth muscle cell: a therapeutic target in type 2 diabetes? Clinical Science. 2013;125(4):167–182. doi: 10.1042/cs20120413. [DOI] [PubMed] [Google Scholar]
- 4.Rudijanto A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Medica Indonesiana. 2007;39(2):86–93. [PubMed] [Google Scholar]
- 5.Chait A., Bornfeldt K. E. Diabetes and atherosclerosis: is there a role for hyperglycemia? Journal of Lipid Research. 2009;50(supplement):S335–S339. doi: 10.1194/jlr.r800059-jlr200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldstein J. L., Brown M. S. Regulation of the mevalonate pathway. Nature. 1990;343(6257):425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 7.Buhaescu I., Izzedine H. Mevalonate pathway: a review of clinical and therapeutical implications. Clinical Biochemistry. 2007;40(9-10):575–584. doi: 10.1016/j.clinbiochem.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 8.Li L., Hu S.-J., Dong H.-T., Kang L., Chen N.-Y., Fang Y.-Q. Alterations in gene expression of series key enzymes in mevalonic acid pathway detected by RNA array in spontaneously hypertensive rats. Chinese Journal of Pathophysiology. 2008;24(1):54–59. [Google Scholar]
- 9.Chen G.-P., Yao L., Lu X., Li L., Hu S.-J. Tissue-specific effects of atorvastatin on 3-hydroxy-3-methylglutaryl- coenzyme A reductase expression and activity in spontaneously hypertensive rats. Acta Pharmacologica Sinica. 2008;29(10):1181–1186. doi: 10.1111/j.1745-7254.2008.00855.x. [DOI] [PubMed] [Google Scholar]
- 10.Han J., Jiang D.-M., Du C.-Q., Hu S.-J. Alteration of enzyme expressions in mevalonate pathway—possible role for cardiovascular remodeling in spontaneously hypertensive rats. Circulation Journal. 2011;75(6):1409–1417. doi: 10.1253/circj.cj-10-1101. [DOI] [PubMed] [Google Scholar]
- 11.Du C.-Q., Yang L., Yang J., et al. Inhibition of farnesyl pyrophosphate synthase prevents norepinephrine-induced fibrotic responses in vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension Research. 2014;37(1):26–34. doi: 10.1038/hr.2013.96. [DOI] [PubMed] [Google Scholar]
- 12.Vicent D., Maratos-Flier E., Kahn C. R. The branch point enzyme of the mevalonate pathway for protein prenylation is overexpressed in the ob/ob mouse and induced by adipogenesis. Molecular and Cellular Biology. 2000;20(6):2158–2166. doi: 10.1128/mcb.20.6.2158-2166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu K.-K., Huan Y. Diabetic atherosclerosis mouse models. Atherosclerosis. 2007;191(2):241–249. doi: 10.1016/j.atherosclerosis.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 14.Shen X., Bornfeldt K. E. Mouse models for studies of cardiovascular complications of type 1 diabetes. Annals of the New York Academy of Sciences. 2007;1103:202–217. doi: 10.1196/annals.1394.004. [DOI] [PubMed] [Google Scholar]
- 15.Müller O. J., Katus H. A., Backs J. Macrovascular disease in diabetes: is the mouse a suitable model? Experimental and Clinical Endocrinology and Diabetes. 2012;120(4):194–196. doi: 10.1055/s-0032-1304580. [DOI] [PubMed] [Google Scholar]
- 16.Mehrhof F. B., Schmidt-Ullrich R., Dietz R., Scheidereit C. Regulation of vascular smooth muscle cell proliferation: role of NF-kappaB revisted. Circulation Research. 2005;96(9):958–964. doi: 10.1161/01.res.0000166924.31219.49. [DOI] [PubMed] [Google Scholar]
- 17.Chen G.-P., Ye Y., Li L., Yang Y., Qian A.-B., Hu S.-J. Endothelium-independent vasorelaxant effect of sodium ferulate on rat thoracic aorta. Life Sciences. 2009;84(3-4):81–88. doi: 10.1016/j.lfs.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 18.Pallottini V., Martini C., Cavallini G., et al. Age-related HMG-CoA reductase deregulation depends on ROS-induced p38 activation. Mechanisms of Ageing and Development. 2007;128(11-12):688–695. doi: 10.1016/j.mad.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Dhar M. K., Koul A., Kaul S. Farnesyl pyrophosphate synthase: a key enzyme in isoprenoid biosynthetic pathway and potential molecular target for drug development. New Biotechnology. 2013;30(2):114–123. doi: 10.1016/j.nbt.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 20.Kourounakis A. P., Katselou M. G., Matralis A. N., Ladopoulou E. M., Bavavea E. Squalene synthase inhibitors: an update on the search for new antihyperlipidemic and antiatherosclerotic agents. Current Medicinal Chemistry. 2011;18(29):4418–4439. doi: 10.2174/092986711797287557. [DOI] [PubMed] [Google Scholar]
- 21.Wiemer A. J., Wiemer D. F., Hohl R. J. Geranylgeranyl diphosphate synthase: an emerging therapeutic target. Clinical Pharmacology and Therapeutics. 2011;90(6):804–812. doi: 10.1038/clpt.2011.215. [DOI] [PubMed] [Google Scholar]
- 22.Park J., Matralis A. N., Berghuis A. M., Tsantrizos Y. S. Human isoprenoid synthase enzymes as therapeutic targets. Frontiers in Chemistry. 2014;2:p. 50. doi: 10.3389/fchem.2014.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gray S. P., Jandeleit-Dahm K. The pathobiology of diabetic vascular complications—cardiovascular and kidney disease. Journal of Molecular Medicine. 2014;92(5):441–452. doi: 10.1007/s00109-014-1146-1. [DOI] [PubMed] [Google Scholar]
- 24.Potenza M. A., Nacci C., Gagliardi S., Montagnani M. Cardiovascular complications in diabetes: lessons from animal models. Current Medicinal Chemistry. 2011;18(12):1806–1819. doi: 10.2174/092986711795496755. [DOI] [PubMed] [Google Scholar]
- 25.Orasanu G., Plutzky J. The pathologic continuum of diabetic vascular disease. Journal of the American College of Cardiology. 2009;53(5) supplement:S35–S42. doi: 10.1016/j.jacc.2008.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kunjathoor V. V., Wilson D. L., LeBoeuf R. C. Increased atherosclerosis in streptozotocin-induced diabetic mice. The Journal of Clinical Investigation. 1996;97(7):1767–1773. doi: 10.1172/jci118604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otero P., Bonet B., Herrera E., Rabano A. Development of atherosclerosis in the diabetic BALB/c mice: prevention with vitamin E administration. Atherosclerosis. 2005;182(2):259–265. doi: 10.1016/j.atherosclerosis.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 28.Chen J., Dai M., Wang Y. Paeonol inhibits proliferation of vascular smooth muscle cells stimulated by high glucose via Ras-Raf-ERK1/2 signaling pathway in coculture model. Evidence-Based Complementary and Alternative Medicine. 2014;2014:9. doi: 10.1155/2014/484269.484269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mebratu Y., Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death: is subcellular localization the answer? Cell Cycle. 2009;8(8):1168–1175. doi: 10.4161/cc.8.8.8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Y., Liu J., Li L., Liu L., Wu L. Role of Ras/PKCζ/MEK/ERK1/2 signaling pathway in angiotensin II-induced vascular smooth muscle cell proliferation. Regulatory Peptides. 2005;128(1):43–50. doi: 10.1016/j.regpep.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 31.Popov D., Nemecz M., Dumitrescu M., Georgescu A., Böhmer F. D. Long-term high glucose concentration influences Akt, ERK1/2, and PTP1B protein expression in human aortic smooth muscle cells. Biochemical and Biophysical Research Communications. 2009;388(1):51–55. doi: 10.1016/j.bbrc.2009.07.141. [DOI] [PubMed] [Google Scholar]
- 32.Ishiko K., Sakoda T., Akagami T., et al. Hyperglycemia induced cell growth and gene expression via the serum response element through rhoa and rho-kinase in vascular smooth muscle cells. Preparative Biochemistry & Biotechnology. 2010;40(2):139–151. doi: 10.1080/10826060903558927. [DOI] [PubMed] [Google Scholar]
- 33.Sawada N., Li Y., Liao J. K. Novel aspects of the roles of Rac1 GTPase in the cardiovascular system. Current Opinion in Pharmacology. 2010;10(2):116–121. doi: 10.1016/j.coph.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu L.-H., Wang L., Wang D., et al. Puerarin attenuates high-glucose and diabetes-induced vascular smooth muscle cell proliferation by blocking PKCbeta2/Rac1-dependent signaling. Free Radical Biology & Medicine. 2010;48(4):471–482. doi: 10.1016/j.freeradbiomed.2009.10.040. [DOI] [PubMed] [Google Scholar]
- 35.McTaggart S. J. Isoprenylated proteins. Cellular and Molecular Life Sciences. 2006;63(3):255–267. doi: 10.1007/s00018-005-5298-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roskoski R., Jr. Protein prenylation: a pivotal posttranslational process. Biochemical and Biophysical Research Communications. 2003;303(1):1–7. doi: 10.1016/s0006-291x(03)00323-1. [DOI] [PubMed] [Google Scholar]
- 37.Casey P. J. Protein lipidation in cell signaling. Science. 1995;268(5208):221–225. doi: 10.1126/science.7716512. [DOI] [PubMed] [Google Scholar]
- 38.Nicholls S. J., Ballantyne C. M., Barter P. J., et al. Effect of two intensive statin regimens on progression of coronary disease. The New England Journal of Medicine. 2011;365(22):2078–2087. doi: 10.1056/nejmoa1110874. [DOI] [PubMed] [Google Scholar]
- 39.Stegman B., Puri R., Cho L., et al. High-intensity statin therapy alters the natural history of diabetic coronary atherosclerosis: insights from SATURN. Diabetes Care. 2014;37(11):3114–3120. doi: 10.2337/dc14-1121. [DOI] [PubMed] [Google Scholar]
- 40.Liao J. K., Laufs U. Pleiotropic effects of statins. Annual Review of Pharmacology and Toxicology. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laufs U., Liao J. K. Direct vascular effects of HMG-CoA reductase inhibitors. Trends in Cardiovascular Medicine. 2000;10(4):143–148. doi: 10.1016/S1050-1738(00)00044-X. [DOI] [PubMed] [Google Scholar]
- 42.Sattar N., Preiss D., Murray H. M., et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. The Lancet. 2010;375(9716):734–742. doi: 10.1016/S0140-6736(09)61965-6. [DOI] [PubMed] [Google Scholar]
- 43.Preiss D., Seshasai S. R. K., Welsh P., et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: a meta-analysis. The Journal of the American Medical Association. 2011;305(24):2556–2564. doi: 10.1001/jama.2011.860. [DOI] [PubMed] [Google Scholar]
- 44.Swerdlow D. I., Preiss D., Kuchenbaecker K. B., et al. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. The Lancet. 2015;385(9965):351–361. doi: 10.1016/S0140-6736(14)61183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henriksbo B. D., Lau T. C., Cavallari J. F., et al. luvastatin causes NLRP3 inflammasome-mediated adipose insulin resistance. Diabetes. 2014;63(11):3742–3747. doi: 10.2337/db13-1398. [DOI] [PubMed] [Google Scholar]
- 46.Russell R. G. G. Bisphosphonates: the first 40 years. Bone. 2011;49(1):2–19. doi: 10.1016/j.bone.2011.04.022. [DOI] [PubMed] [Google Scholar]
- 47.Bevilacqua M., Dominguez L. J., Rosini S., Barbagallo M. Bisphosphonates and atherosclerosis: why? Lupus. 2005;14(9):773–779. doi: 10.1191/0961203305lu2219oa. [DOI] [PubMed] [Google Scholar]
- 48.Ylitalo R. Bisphosphonates and atherosclerosis. General Pharmacology: The Vascular System. 2000;35(6):287–296. doi: 10.1016/s0306-3623(01)00121-5. [DOI] [PubMed] [Google Scholar]
- 49.Wu L., Zhu L., Shi W.-H., Zhang J., Ma D., Yu B. Zoledronate inhibits the proliferation, adhesion and migration of vascular smooth muscle cells. European Journal of Pharmacology. 2009;602(1):124–131. doi: 10.1016/j.ejphar.2008.10.043. [DOI] [PubMed] [Google Scholar]