

Abstract
Co- and post-transcriptional regulation of gene expression is complex and multi-faceted, spanning the complete RNA lifecycle from genesis to decay. High-throughput profiling of the constituent events and processes is achieved through a range of technologies that continue to expand and evolve. Fully leveraging the resulting data is non-trivial, and requires the use of computational methods and tools carefully crafted for specific data sources and often intended to probe particular biological processes. Drawing upon databases of information pre-compiled by other researchers can further elevate analyses. Within this review, we describe the major co- and post-transcriptional events in the RNA lifecycle that are amenable to high-throughput profiling. We place specific emphasis on the analysis of the resulting data, in particular the computational tools and resources available, as well as looking towards future challenges that remain to be addressed.
Co- and post-transcriptional regulation encompasses a multifaceted and interconnected group of events including RNA processing, translation and decay. Each stage involves multiple regulatory steps and interactions with complexes containing RNA-binding proteins (RBPs) and non-coding RNAs 1. The list of regulators, which often participate in multiple processes, is long, with a possible >1,000 RBPs and thousands of non-coding RNAs in human 2, 3. Dissecting co- and post-transcriptional regulatory events at the genomic level poses numerous challenges in terms of methods and computational analyses.
RNA biology reached genome-wide scale only recently, when RIP-chip (ribonucleoprotein immuno-precipitation followed by microarray analysis), the first approach for en masse identification of RBP targets, gained popularity in the early 2000’s 4. Other methods are still under development. For instance, ribosomal profiling (RP), which is now the method of choice for the study of translation regulation, was developed just a few years ago and continues to evolve 5, 6 As a result, computational methods to support these technologies have yet to reach the level of maturity seen, for example, in the transcriptomic field. Also in contrast to transcriptomics, where some consensus has been reached in terms of methods and analysis pipelines 7–10, RNA biologists continue to use a range of different experimental and analysis approaches. For example, although still used, RIP-chip and RIP-seq have been mostly replaced by a plethora of different cross-linking methods such as cross-linking and analysis of cDNAs (CRAC)11 and CLIP (Cross-linking and Immuno-Precipitation) approaches, i.e. HITS-CLIP, PAR-CLIP and iCLIP 12–15. All methods have their pros and cons and, due to their technical differences and biases, deliver slightly different datasets 16. When comparing datasets, it is hard to say why one method but not the others captured a particular binding site. We clearly need to conduct more extensive comparative analyses coupled with functional assays to better understand what each method is producing. An understanding of the idiosyncrasies of each technology used in the lab and how they relate to analysis methods is essential. They will give us the means to improve computational tools and include filters that at the end will deliver the highest number of functional RBP sites with a minimum of false positives.
At a higher level, the need for effective integration of disparate data sources in the study of co- and post-transcriptional regulation is particularly pronounced. Assigning function to RBP binding can be a complex task due to the polyvalent nature of these proteins. For example, binding of a given RBP to 3′UTRs (untranslated regions) could affect mRNA decay, translation or interfere with poly(A) site selection; multiple angles of analysis are necessary, but data integration is nontrivial. There is need to centralize all co- and post-transcriptional datasets and develop tools to allow cross-platform comparisons.
Figure 1 summarizes the relation between the major experimental high-throughput assays with both the stages and regulators of the RNA lifecycle they inform on. In the next sections, we cover different high-throughput approaches used in RNA biology, tailoring the discussion to the computational methods available and challenges in terms of development and data integration.
Figure 1.

Summary of post-transcriptional regulation processes and corresponding computational methods.
Profiling RNA-binding protein activities
Experimental methods
RNA binding proteins are, next to non-coding RNAs, the central drivers of co- and post-transcriptional regulation, and can have hundreds to thousands of target mRNAs thanks to flexibility in their binding specificity. En masse identification of in vivo binding has become possible only within the last decade, first with RIP and then with CLIP. They were developed by the Keene and Darnell labs respectively 4, 12. They both consist of immuno-precipitation approaches where RNPs containing the RBP of choice are isolated and associated mRNAs are subsequently purified and identified. Quantification of the resultant RNA, was originally carried out using micro-arrays or Sanger sequencing, but is now more commonly performed using next- and second-generation deep sequencing. When RIP was established, there were some concerns regarding the possibility of re-assortment of RNPs during the IP process. This issue was essentially raised by a study from the Steitz lab 17, in which a very simplistic analysis was conducted. To the best of our knowledge, similar claims have not been reported by other scientists using RIP. In fact, RIP was used successfully in cell systems and organisms to generate cell type specific gene expression profiles and no problems of cross-contamination between cell types have been reported 18–20.
We focus on the analysis of data from these high-throughput assays, termed RIP-seq and CLIP-seq. While CLIP-seq is more frequently used, RIP-seq continues to be used, especially if there are limitations in terms of antibodies, or the amount and type of tissue. Recently, ‘reversed CLIP’ assays have been developed in which mRNAs are extracted; the binding sites and identities of bound proteins are determined by RNA-seq and proteomics, respectively 21, 22. These studies have revealed the enormous extent of the protein-RNA interaction landscape. In a more recent study Tombe et al.23 developed a high-throughput sequencing–RNA affinity profiling (HiTS-RAP) assay that employs high-throughput sequencing to measure RNA aptamer affinities in large scale by quantifying the binding of fluorescently labeled protein to millions of RNAs anchored to sequenced cDNA templates. This is an extension of high-throughput sequencing–fluorescent ligand interaction profiling (HiTS-FLIP) protocol24 that was previously developed to image and analyze the binding of fluorescently labeled proteins to DNA clusters for direct quantitative measurement of protein-DNA binding affinity.
Finding targets and binding sites of RNA-binding proteins
RIP and CLIP aim to answer two closely related questions: which transcripts are bound by an RBP, and where. The key distinction lies in resolution. Generally, RIP-seq does not involve digestion of bound RNA fragments, and provides transcript-level resolution, enriching reads in bound RNAs but not necessarily with positional information. In contrast, CLIP-seq allows for much higher resolution. From a technical perspective though, identifying targets at the full transcript level and finding binding sites at the resolution of ten or twenty nucleotides are essentially the same problem: we search for genomic regions which are enriched for reads. This process is referred to as peak-calling, and forms the basis for any downstream analyses. Peak-calling follows read-mapping (alignment of short sequenced reads to the reference genome), which we will not address as it has been covered fully elsewhere 25. Peak-calling assumes that some loci will receive reads, but not all of these represent true binding sites. There are a number of possible reasons for this, including transient or non-specific interactions26–29, cross-linking biases (modest uridine preference caused by UV cross-linking in HITS-CLIP and iCLIP)30, re-association after cell lysis17 (the artifactual RNA-protein complexes formed in cell lysate, depending on lysis conditions, generally only a problem with RIP-seq), and background cross-linking (background caused by random UV cross-linking of RNAs to proteins that are not the RBP of interest)31. However, it is expected that such false-positive loci will generally accumulate few reads. There is generally no specific way of defining such binding activities and different groups use different measure. For instance Friedersdorf et al.31 performed an experimental method to define background cross-linking in PAR-CLIP data. Freeberg et al. 32 calculated the cross-link score (CLS) for each T in the genome, where CLS is the ratio of CLIP reads containing one or two T-to-C conversion events to the number of mRNA-seq reads and associate low CLS values to transient binding 32. Similar methods can be used to define cross-lining biases and background cross-linking. Peak-calling aims to differentiate these loci from those that represent targeted binding of the RBP, i.e. are true-positives. This differentiation is particularly important in RIP, where the lack of cross-linking and RNAse treatment results in much higher background signal. Although CLIP has a high-degree of accuracy that cannot be achieved by RIP, it exhibits both cross-link biases and background cross-linking. In addition, due to inefficiency of UV cross-linking33 it is not clear what proportion of binding activities is really captured by cross-linking. Nonetheless, even with these problems CLIP has proven to be useful for identifying mRNA targets of RBPs. However, due to the above-mentioned problems rendering careful separation of signal from noise essential 17, 30, 31.
The simplest peak-calling scheme considers only the number of reads mapped to a locus. The exact read-count threshold to use must be calibrated for each dataset, since sequencing depth varies. A major challenge is selecting an appropriate resolution. Reads are counted into bins tiled along the genome. If bin size is too small, it is difficult to distinguish the underlying distribution of the read counts in peaks from the background. If the bin size is too large, resolution suffers. Most methods defer the decision to the analyst, although there are some attempts to automize selection of resolution, such as RIP-Seeker 34.
Further, one must consider the statistical distribution of the read counts. In previous work, we demonstrated that read-counts are Poisson over-dispersed in CLIP-seq datasets 16. An appropriate model to capture their distribution is thus the negative binomial. When only a single sample is analyzed, loci with zero-counts are not considered, and in this case it is better to use a zero-truncated negative binomial, which appropriately adjusts for the missing zero counts. These distributions were used as the basis for the Piranha peak-caller 16. In addition, other methods proposed Hidden Markov Model (HMM) for modeling and analyzing CLIP-seq data, such as dCLIP35 and MiCLIP36. At the first step, dCLIP normalizes CLIP-seq data across datasets and subsequently employs an HMM to detect common or different RBP-binding regions across conditions35. MiCLIP uses two rounds of HMM to first infer enriched vs. non-enriched regions and then to distinguish binding sites of RBPs vs. non-binding sites within those enriched regions36.
Additional information beyond read-counts can be used to improve peak calling. One example is transcript abundance. The number of reads mapping to a given genomic locus will be proportional to the binding strength of the RBP to that site, but also the abundance of the RNA. Abundant RNAs will take a greater slice of the sequencing pie, leaving less abundant RNAs, even if strongly bound, starving for coverage. Piranha was developed to account for this sequencing inequality, allowing the significance threshold, at which a locus is considered a true interaction, to vary as a function of RNA abundance, measured by RNA-seq 16. AS-peak 37 is another peak caller, tailored specifically to RIP-seq data, that considers transcript abundance.
Other markers of true RBP-RNA interactions are modifications in nucleotide reads as a result of UV cross-linking, coined cross-link induced mutation sites (CIMS). In HITS-CLIP, CIMS are ‘deletions’ at the cross-linked nucleotide 38, while in PAR-CLIP, reads exhibit T-to-C nucleotide conversions due to incorporation of 4SU photoactivatable-ribonucleoside into transcripts 15. Not only are these changes useful in distinguishing true from false interactions, but they have also been used to improve localization. Without considering CIMS, only iCLIP achieves single-nucleotide resolution. Zhang & Darnell proposed a systematic method based on CIMS for the analysis of HITS-CLIP, elevating HITS-CLIP to single nucleotide resolution, and allowing exact localization of the cross-link location 38. They applied their genome-wide analysis to Nova and Ago HITS-CLIP data, identifying CIMS deletions in ~8% of mRNA tags mapped to Nova targets. Corcoran et al. 39 proposed a method for PAR-CLIP data, based on the characteristic conversion. They allow a read to contain up to two mismatches restricted to T-to-C conversions during the mapping. At each genomic locus, they calculate the likelihood of T to C conversion and use this to predict interaction sites.
To date, most analyses employing CLIP- and RIP-seq have been restricted to identifying targets and binding sites under single conditions. Moving forward, comparative analyses will become more important, and a few studies have already taken steps in this direction 35, 40–42. Firstly Tenenbaum et al. 4 used RIP-chip to determine dynamic changes in mRNA targets during neuronal differentiation. Moreover, Mukherjee et al. 43 employed Gaussian Mixture Modeling to RIP-seq data with probabilistic LOD scores and background quantification of each mRNA target to quantify dynamic changes in mRNA targets during T cell activation. However, computational tools to facilitate comparative peak-calling are few. To date, only Piranha and dCLIP provide support for identifying differential binding 16, 35.
Most tools for identifying interaction sites are stand-alone programs intended to run on a local machine. There are some online tools that can be used for CLIP data analysis, for example PIPE-CLIP 44 and pyCRAC 45, both of which run on the web-based Galaxy 46 platform.
Characterizing and understanding RBP specificity
Nucleic acid binding proteins interact with their substrate (DNA or RNA) and participate in biochemical reactions that lead to specific cellular functions 47. In the case of RNA, these interactions happen between a subset of residues in the protein (the RNA binding domains, or RBDs) and a subset of nucleotides within the RNA (the binding sites). Certain nucleotide sequences present high affinity for the protein’s RBDs, causing the protein to bind to these locations with high frequency. These patterns are called motifs, and observing these patterns in a genomic location is called a motif occurrence. Motifs can be characterized by both sequence and structural elements and show tremendous variation amongst RBPs, even between members of the same RBP family 48.
Until the early 2000s, characterization of binding sites was mostly restricted to individual studies involving a particular RBP and one target gene/binding motif. Such studies include a variety of assays from mutagenesis and binding shifts to more elaborate analyses involving 3D structures of RBP bound to RNA1, 49, 50. One exception, SELEX experiments, combined a recombinant RBP and large pools of short random RNA sequences. After several rounds of selection, a consensus motif is defined based on the sequence of RNA fragments preferentially bound 51, 52. RNAcompete is another in vitro method that is much less expensive than SELEX due to a smaller designed pool of RNA oligo-nucleotides 53.
Finding statistically enriched motifs in biological sequences is one of the most well studied problems in computational biology. The inherent variability in the motif sequence for RBPs renders methods based on exact matches of little use54. More flexible models have been proposed, the most well established being the position weight matrix, constructed by counting occurrences of each type of nucleotide at each position in the motif 47, 55–57. Methods employing this representation can generally be divided into two groups, 1) exhaustive enumeration methods, which are based on enumerating possible motifs then progressively narrowing the search to the neighborhood of highest scoring motifs and 2) probabilistic models, which construct the motif model and find the occurrences of the motif simultaneously in an iterative manner 58. Much of the extensive body of work on motif discovery is due to the attention paid to transcription factors and the need to understand transcriptional regulation through protein-DNA interactions. MEME 59, MDScan 60, AlignACE 61 and DME 62 are just a handful of the highly successful methods. The interested reader is encouraged to pursue one or more of the extensive reviews written on the details of these methods 63–67. In comparison, motif finding in RNA brings its own unique set of challenges that must be considered. Early applications of motif-finding algorithms optimized for transcription factor binding sites to finding regulatory regions in RNA, especially RBP binding sites, encountered a number of challenges, chief amongst which are the shorter length of RBP motifs68, 69 and the role of RNA secondary structure in binding site recognition 70.
An early approach for modeling RNA structure involves covariance models (CMs) 71, 72. CMs deliver both a sequence alignment and a consensus structure for a set of RBP-bound RNA sequences. Training a CM constructs a model from a set of sequences, which in turn can be used for aligning new sequences in an integrative approach. Other methods, such as Dynalign, a software for simultaneous sequence and structural alignment of RNA molecules using dynamic programing73, evolutionary methods 74, and text indexing approaches 75 have been used for sequence and structural motif discovery for RNAs. However, evolutionary and text indexing methods are very limited in terms of the range of RNA secondary structures that they can discover, while CM and Dynalign are computationally expensive.
MEMERIS 76 was proposed for RNA binding site characterization, and it takes both sequence and structure into account. MEMERIS calculates the probability of RNA regions to be single-stranded, and uses these values as prior knowledge to guide the search for the motif. RNAcontext 77 is another approach for RNA binding site characterization and motif discovery that takes both sequence and structure of the RNA into account. The model developed in this program has a much simpler representation than MEMERIS: a position weight matrix for describing the motif sequence and an additional vector to describe the structural context of each nucleotide in the motif. RNAcontext performs well, both in vitro and in vivo, in terms of recovering experimentally validated motifs. However, both MEMERIS and RNAcontext suffer from the assumption that RNA sequence and structure are independent. In addition, MEMERIS takes only single stranded regions into account, which is a limiting factor for RBPs that bind double-stranded RNA. More recently, a new method called GraphProt was proposed as a machine-learning framework for learning models of RBP binding preferences from different types of high-throughput experimental data. GraphProt in essence is a supervised learning algorithm that builds a model using positive and negative sets of binding sites and then scans the genome to find instances of binding sites based on sequence and structure profiles 78. For Identification of miRNA-RISC complex target sites, handful of studies has done CLIP experiment for transcriptome-wide mapping of miRNA targets, which have proven to be quite useful15, 39, 79, 80. In addition, the computational methods take advantage of predictive features of the binding regions, most notably sequence characteristics of the seed region, phylogenic conservation of binding sites and secondary structure accessibility of the target81–83.
Several databases of RNA-protein interaction sites have been developed. RBPDB 84 contains a collection of experimental motifs of RNA-binding sites from human, mouse, fly and worm. This database includes RBP binding sites derived from in vitro methods, motifs in position weight matrix format, and sets of sequences of binding sites obtained from immunoprecipitation experiments in vivo. CLIPZ 85 is a database of binding sites that are constructed from CLIP data for a limited number of proteins. However, users can upload their short read sequences from CLIP, small RNA sequencing, and mRNA sequencing experiments for analysis 85. RBPmap is a webserver for prediction of RBP binding sites. Users can input their sequences and motif in the form of a consensus sequence or position weight matrix or select from a large database of experimentally validated motifs. The algorithm then searches sequences for the motif, compares matches to the embedded background model, calculates a weighted rank for all the positions, and outputs a summary of all predicted binding sites 86.
Regulators and function
Binding of a given RBP to a target transcript can produce a variety of outcomes, both promoting and repressing events – for instance increasing or decreasing translation or mRNA decay, promoting or repressing exon skipping or the usage of a distal poly A site. A variety of genomics methods are necessary to link binding to function. For instance proteomics studies have been combined with RIP-chip and CLIP experiments to identify functional RBP binding sites, e.g. to characterize the translation regulators such as HuR 27, Msi1 87, IGF2BP1-3, QKI and PUM2 88, or the splicing regulator RBM20 89. Other methods combine the analysis of miRNAs with proteome, transcriptome or translatome profiling, e.g. for miR-124 90, mir-223 and others 91–93. The analysis of this data (and integration with data from binding assays) brings a new set of computational challenges that we discuss in the remaining sections.
Regulating transcript abundance
Quantifying gene expression is a well-studied problem in computational genomics. Expression profiling is now largely performed by RNA-seq. Read counts are the main source of information to calculate a gene’s expression profile, though they must be correctly normalized to obtain meaningful information. There are primarily two concerns during normalization, which arise from transcript length and sequencing depth. The former is the result of RNA fragmentation during library construction in which longer transcripts naturally generate more reads than shorter transcripts even if they have similar abundance. Sequencing depth refers to the variability in the total number of reads sequenced and mapped in each run, which causes variations across samples 8. To account for these issues, the reads per kilobase of transcript per million mapped reads (RPKM) metric was introduced by Mortazavi et al. 94 to normalize a transcript’s read count by both its length and the total number of mapped reads in the sample 8. With paired-end data, to avoid counting reads that fall into mapped fragments twice, a similar measure called reads per kilobase of transcript per million mapped fragments (FPKM) was developed 95. However, Wagner et al.96 showed evidence that RPKM is not suitable for comparison between samples and proposed a new measure called transcript per million (TPM) for this purpose96. For a comprehensive review on normalization methods for transcript abundance, refer to Dillies et al. 97.
Often the goal of analyses is to compare expression between conditions and identify transcripts whose concentration changed. Methods such as Cuffdiff 95, edgeR 98 and DESeq 99 are frequently used. Cuffdiff 95 is based on beta negative binomial model and estimates the variance of RNA-seq data by t-like statistics from FPKM values. edgeR98 is based on an over-dispersed Poisson model in order to explain the variation in the read count data. The evaluation of differences across transcripts, are estimated using Empirical Bayes method. DESeq99 uses a negative binomial for estimation of variability in read count data. Differential expression analysis for RNA-seq is a widely explored area; for a comprehensive survey refer to 8, 100.
RNA-binding proteins have the capacity to directly regulate mRNA levels. However, many studies observe substantial changes in transcript abundance upon knockdown or knockout of RBPs, but find a surprisingly small overlap with the set of RBP targets identified by binding assays 101. This discrepancy is most likely due to a large number of indirect effects. As a result, the question of whether data from binding assays can be effectively married with mRNA expression data remains open.
Alternative splicing
The “one gene, one enzyme” hypothesis postulated by Beadle and Tatum 102 is no longer valid; we know that the number of human genes is much smaller than the number of expressed proteins 103. This discrepancy can be explained by several levels of gene regulation, co- and post-transcriptional modifications, especially alternative splicing 104.
More than 90% of human genes are alternatively spliced, with a role in many physiological functions 105, 106. Alternative splicing, coupled to nonsense-mediated decay (NMD), can also directly regulate gene expression by producing unstable transcripts that contain premature stop codons 107, 108. Splicing-related changes in gene expression can be triggered in response to stress and other environmental signals 109, and are increasingly recognized as a participant in many diseases 110–113. Cancer-related studies have revealed specific changes in alternative splicing patterns that can be used for diagnosis 65 and therapy 114.
Many mathematical models, algorithms and statistical methods have been developed and employed to explore alternative splicing. The goal of these methods is generally to identify and quantify the abundance of individual transcripts 115, 116, or more commonly, to profile changes in splicing either at the full transcript level or at the level of individual splice sites and exons 117–121. The latter task is called differential splicing analysis. An example of such an analysis would be to calculate exon inclusion from exon-junction arrays, microarrays or RNA-seq data, and then compare the values between samples or conditions to infer occurrences of different alternative splicing events. Although some approaches to either problem may employ a reference dataset of exons or splice junctions and only considers splicing events with known splice junctions, a frequent goal is to identify novel splicing events with previously unknown donor and acceptor sites. Addressing this challenge relies heavily on split-read mappers, which are able to map reads containing previously unknown splice junctions – a task that regular short-read mappers generally fail with, as the read is not derived from a single contiguous region of genomic sequence, nor one that can easily be constructed in silico 122–132.
Several excellent reviews of computational methods for splicing and alternative splicing analysis already exist; for a detailed review of methods and databases refer to Hooper et al. 133, and the EURASNET website 134, respectively. Despite much work in this field, it remains challenging to link the observed changes in splicing regulation with their regulators, such as RNA-binding proteins. In the case of RBPs, one approach is to profile cells with a regulator of interest either silenced or deleted, and compare against the wild-type. RIP and CLIP have been used to match observed changes in splicing to the putative binding sites identified, as has been done for example for Nova 13, hnRNP proteins (namely, hnRNP C 14, H1 116, L 135, A1, A2, A2B1, F, M, U 136), TDP43 137, Fox 138, 139, PTB 140, 141, Mbnl1 142, TIA1, and TIAL1 143. However, the analysis is generally ad-hoc; no effective computational tools yet exist for linking functional assays such as RNA-seq with binding assays such as RIP- or CLIP-seq. One main reason for this problem is that observing binding activities of an RBPs according to RIP or CLIP experiments is not an evidence of direct binding.
Alternative poly-adenylation
Poly-adenylation is the addition of a stretch of adenosine nucleotides to the end of RNA molecules. This polyA tail aids nuclear export and translation, and protects the transcript from degradation. The point at which the RNA is cleaved and the tail is added can vary – a mechanism known as alternative polyadenylation (APA). APA can result in mRNAs with differences in coding sequence and 3′UTR, contributing to altered regulation, function, stability, localization, and translational efficiency 144. Although alternative polyA sites, that are situated between coding exons, can lead to isoforms encoding different proteins 145, more often APA events result in shorter 3′UTRs which lack sequences that are targets of microRNAs and RNA-binding proteins 146. The earliest examples of APA were described in the mRNAs of IgM and DHFR 147, 148. Subsequently, EST databases and microarray analyses allowed the identification of several other APA sites 149, 150. Recent RNAseq methods have enormously improved our understanding of APA 151.
Genomic studies have shown that APA is a widespread phenomenon in metazoan genomes. For example, about 70% of mammalian genes and about 50% of the genes in flies and worms are subjected to APA 146, 152, 153. This mechanism is known to regulate a range of biological processes, often associated with development, cellular differentiation and proliferation. Shortened 3′ UTRs due to alternative poly-adenylation are associated with increased pluripotency and cell proliferation 154, 155, and relaxation of microRNA repression of oncogenes 156.
Computational methods for the prediction of alternative polyadenylation are mainly based on the Direct RNA Sequencing (DRS) technology 157, in which RNA molecules are sequenced without prior conversion to cDNA or the need for biasing ligation or amplification steps 157. This method was employed to develop a map of over 1 million polyA sites in major cancers and tumor cell lines 158, 159. An alternative method, PolyA-seq, allows for the high-throughput sequencing of the 3′ ends of polyadenylated transcripts, and has been used to obtain a global map of polyadenylation sites in human, rhesus, dog, mouse, and rat 153. Purely computational methods for predicting the locations of polyA signals also exist, such as the classification-based method polyA-predict, which was used to construct a database of predicted sites 160. Other databases of polyA sites include PACdb 161 and PolyA_DB 162.
Stability and decay
Regulation of mRNA stability and decay
Another major contributor to expression regulation is mRNA degradation which has also been linked to several diseases163. Two major regulatory routes control mRNA decay: quality control mechanisms eliminate the production of aberrant protein products while another group of mechanisms influence mRNA life time with the main purpose of controlling protein abundance.
A prevalent example of degradation for quality control is Nonsense Mediated Decay (NMD), which eliminates mRNAs that prematurely terminate translation 107. It can be regulated in multiple ways, such as relative concentration and phosphorylation of NMD factors and miRNAs – a detailed review is provided by Kervestin et al. 164.
Another important mechanism is the ARE-mediated mRNA decay. It is predicted that 9% of the human transcriptome contains ARE elements in the 3′UTR; these are characteristic short AU rich or U-rich sequences 165. ARE-containing mRNAs have been implicated in important physiological functions as well as diseases and tumorigenesis 166. Several RBPs like TTP, BRF1, KSRP and AUF1 interact with ARE-sequences and help recruit degradative enzymes. Another group of RBPs, which include the highly studied HuR, binds ARE elements and increase their stability167. These ARE binding proteins have their activities modulated by cell signaling, phosphorylation and cellular localization 168, 169. For a comprehensive review on mRNA decay see 170.
Transcriptome-wide profiling and computational tools
Transcriptome-wide analysis of mRNA decay generally relies on time-series data in which mRNA levels are measured at different time points 171. For example, data from genomic run-on experiments is used by the computational tool mRNAStab to determine mRNA stability by calculating mRNA half-lives 172. Dölken et al. 173 developed a pioneering approach to separate total cellular RNA into newly transcribed and preexisting RNA upon metabolic labeling. Other methods are based on Dynamic Transcriptome Analysis (DTA) 174 to calculate mRNA half-lives 175. From a functional perspective, the influence of RNA sequence and structural elements on mRNA stability and other post-transcriptional regulatory mechanisms has been the subject of recent studies 176. For instance, TEISER 177 is a computational framework to calculate the correlation between the presence or absence of sequence and structural motifs with experimentally determined mRNA stability. MIST-Seq (Measurement of Isoform-Specific Turnover using Sequencing) is another recently introduced method designed to estimate the decay rate of a population of RNAs accurately 178. Its application revealed that even minor differences in sequence composition could lead to large changes in decay rates between isoforms, highlighting the functional effect of particular 3′ UTR elements on mRNA stability. Similar studies have been carried out in yeast, comparing mRNA isoform half-lives across different isoforms of particular genes and inferring biological functions for particular sequence elements 179.
Micro-RNA biogenesis and function in mRNA decay
Over the last decade though, probably the most heavily studied mechanism for regulating mRNA levels has been through micro-RNAs (miRNAs). Micro-RNAs regulate gene expression by base-pairing with complementary sequences in mRNAs 180. To accomplish this, miRNAs rely on an Argonaute protein to form a complex, called the RNA-induced silencing complex (RISC) that facilitates the binding of miRNAs to mRNAs, and their gene silencing function. However, the actual mediators of gene silencing are members of the GW182 protein family, which regulate all downstream steps in gene silencing 181–186. Watson-Crick base-pairing between the miRNA and target mRNA determines the specificity of the complex, while the Argonaute protein exerts the gene regulatory function 187. A given miRNA can have hundreds of targets and a given gene can be regulated by multiple miRNAs. A more comprehensive review on the mechanisms of miRNA gene regulation is presented elsewhere 188. The end result of miRNA-mediated gene regulation is reduced protein output from the cognate mRNA92.
The most successful methods to date for computational identification of miRNA binding sites have been miRanda 189, TargetScan 190, and PicTar 191. miRanda uses a dynamic programming algorithm to search for complementarity matches between miRNAs and 3′ UTRs. For each match, it estimates the stability of interaction using thermodynamic calculation of the complex free energy and calculates a conservation score with closely related species 189. Validations have shown this approach to be highly successful. TargetScan 190 takes a similar approach based on the thermodynamics of RNA-RNA interactions and comparative sequence analysis to predict miRNA targets conserved between species. The algorithm in PicTar is based on Ahab 192, 193, which is a probabilistic algorithm for the identification of combinations of transcription factor binding sites 190 and identifies common targets of microRNAs in eight vertebrate genomes.
Several research groups have developed databases of miRNA target sites. ExprTargetDB 194 is a database obtained using an integrative approach combining the results form TargetScan, miRanda, and PicTar. Other databases include miRBase 195, the repository for miRNA gene set annotations and TarBase 196, which is a collection of miRNA gene interactions coupled with experimental observations for any listed interaction. STarMir 197 is a web-server that predicts miRNA binding sites and computes several other features of the targets such as consensus sequence, thermodynamic and target structure to calculate a measure of confidence for each predicted site.
Micro-RNAs act in concert with RBPs. Some databases leverage this for greater accuracy. For instance, Starbase 198, which uses CLIP experiments to compile a set of computationally predicted miRNA target sites for several species. They also filter false positive miRNA target sites, which can be used for the detection of false negative binding sites absent from current prediction sets. Another database employing CLIP-seq data is doRiNA 199, which uses PicTar 191, and offers the advantage of easy visualization via the UCSC genome browser. Target prediction algorithms for miRNAs that rely on a trusted set of miRNA target sites can greatly benefit from such a feature 176.
Translation
Translation and its role in biological processes
Translation regulation plays an important role in many biological processes 200–202. It accounts for up to 30% of variation in protein expression in both yeast 203 and mammalian cells 204. Certain cell types are even more reliant on post-transcriptional regulation than others. Examples include blood platelets, which lack nuclei, so their cellular responses must be modulated post-transcriptionally, and the final stages of sperm development, where transcription is silenced 205, 206. Translation regulation is also essential in development. During early embryogenesis it controls embryonic axis, body patterning and cell fate, as transcription is largely quiescent at this stage 207. Since translation reacts faster than transcription, it often forms the basis for rapid responses to environmental changes 202.
Due to its important role in cellular biology, translation is also recognized as a nexus susceptible to disruption in diseases. For example, abnormal translation is now a recognized characteristic of tumor cells and a potential target for therapy 208. Elevated levels of the translation initiation factor elF4E have been found in many cancer cell lines and tumors, and over-expression in rodent cells results in malignancies 209. Close to 60% of the mRNAs classified as proto-oncogenes have atypical 5′UTRs with complex structure and high GC content, hindering ribosome binding 210. There are implications for understanding cancer treatment as well. Radiotherapy is the preferred approach for many tumor types. Genome-wide analyses of irradiated cells revealed that the number of genes with translation affected by radiation is close to 10-fold greater than those with altered transcription 203.
Methods and challenges
Genome-scale knowledge of translation regulation has lagged behind that of transcription, despite its central role. Integrative analysis of RNA-seq and shotgun proteomics and comparison of protein to mRNA concentrations is one approach to estimate translation efficiency 211. However, this approach is limited, for example by the number of genes covered by proteomics analysis and ignorance of protein degradation. More direct approaches use ribosome binding to mRNAs as a proxy of translation efficiency. For decades polysome profiling has been used to study translation regulation. This method is based on separation of mRNAs that are heavily loaded with ribosome from free mRNAs using ultracentrifugation on sucrose gradients. Coupling polysomal profiling and microarrays or RNA-seq enable translation studies to enter the world of genomics 212, 213. In recent years, the field has experienced a dramatic boost with the advent of ribosome profiling 6.
Ribosome profiling
Ribosome profiling (RP) is a relatively new method that promises to provide researchers with quantitative information about the relative number and locations of ribosomes bound to RNA 6. In the RP method, ribosome-protected mRNA fragments are sequenced deeply. Figure 2 demonstrates the detailed steps in this protocol. RP can be used for examining translational control in a range of settings, from basic mechanistic investigations to studies of disease and drug treatments 214, 215. It provides an excellent tool to investigate, discover and catalog translational products present in a cell type at single-nucleotide resolution. Despite its challenging protocol, the RP technology is now more and more used, and computational analysis tools are under development. Currently, the number and position of reads is used to estimate ribosome binding.
Figure 2. (A) Overview of ribosomal profiling (RP) experiments. (B) Detailed steps in the ArtSeq protocol for ribosomal profiling.
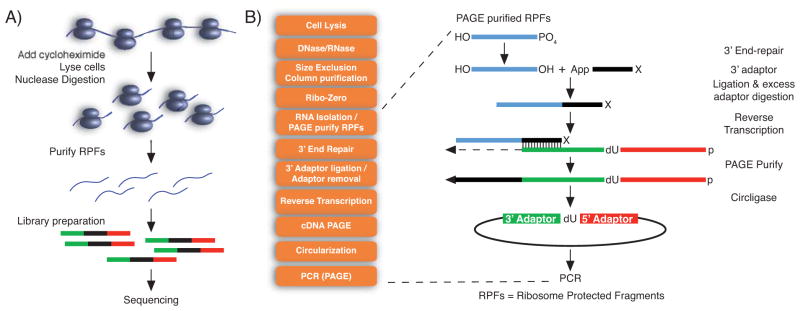
The protocol starts with cell fragmentation; the resulting cell extract is submitted to nuclease digestion, which will generate ribosome-protected RNA fragments. Ribosome-RNA complexes are purified using gel filtration columns (SV400 samples) or sucrose cushion (sucrose samples), followed by RNA extraction and elimination of ribosomal RNAs (rRNA). rRNA-depleted samples are submitted to electrophoresis, and ribosome-protected fragments (about 35 nt long) are eluted from gel. These RNAs are used as templates for library preparation and sequencing 236. Figure adapted from 237.
A fundamental contribution of RP has been the identification of open reading frames (ORFs). An ORF is a segment of an mRNA, bounded by a translation initiation site (TIS) and translation termination site (TTS), which causes formation of the elongation-competent 80S ribosome complex 216. Identifying ORFs is one of the classical analysis problems of computational genomics 217. HMMs have been used to identify ORFs for more than 20 years 218, and have done exceptionally well due to their flexibility and the natural sequential dependence within ORFs. The most sophisticated ORF-predicting HMMs were developed in the context of determining the complete gene structure (promoter, exon, intron, etc.) 219.
However, factors such as transcripts with multiple ORFs, internal ribosome entry sites, leaky translation, ribosome shunting, and near-cognate start codons make the purely computational identification of ORFs problematic, as evidenced by the discovery of many novel ORFs by RP studies 220–223. Despite the success of RP, no public tools are available to date. Many studies simply assume known ORFs224. Those that predict them rely on read patterns in ribosome profiling data from samples treated with elongation inhibitors, which cause ribosome arrest at the TIS (Figure 3). Ingolia et al. 222 employed a classification approach to provide genome-wide maps of protein synthesis. Lee et al. 223 defined a measure based on the number of reads at each position and the total number of reads on the same transcript in their data to identify peaks of ribosome activities and therefore obtain a global map of translation initiation sites in mammalian cells. Fritsch et al. 221 employed a neural network method for genome-wide identification of novel upstream ORFs in human. Stern-Ginossar et al. 225 used a method similar to Ingolia et al. 222 to discover diverse short reading frames in human Cytomegalovirus. Clear read patterns denote both TIS and stop codons in untreated samples too, but have not so far been leveraged to improve our definition of ORFs.
Figure 3. Read profiles of untreated and harringtonine-treated RP data.
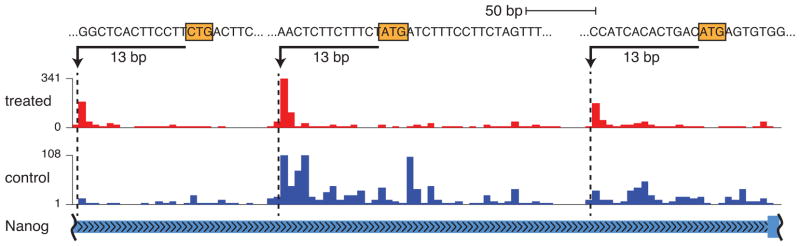
The genic ORF and two uORFs in the Nanog transcript are shown. Start codons are highlighted, and the offset of the 5′ end of reads is indicated.
Another complication is that elongation-inhibited samples only approximately identify the TIS, since the start of the reads marking the protected fragment is offset from the A-site – by about 12 nucleotides generally 223, 226, 227. The TIS is then determined by searching for a sequence (codon) nearby, which requires an existing model and precludes unbiased TIS characterization. Existing methods for detecting the read-pattern indicative of TIS in RP data have been trained on known exemplars 221, 222, which may not always be available and biases towards sites similar to those already known.
Identification of ORFs also opens up the possibility of finding and characterizing regulatory reading frames. Many mRNAs contain ORFs upstream of the genic ORF, called uORFs, which also engage ribosomes 216, 228. Whether uORFs produce viable proteins with any function remains open, though the fact that they regulate translation of their downstream genic counterparts is now well established through several recent studies 216, 228–231.
RP analysis provides several measures of translation regulation. It reports the number of mRNAs bound by ribosomes compared to unbound mRNAs (occupancy), it reports the total number of ribosomes per mRNA (density), and the ribosome position at nucleotide resolution. While these data are insufficient to calculate actual rates of translation, they serve as a detailed proxy of translation efficiency per gene. Mass-spectrometry based approaches have recently provided methods to measure actual translation rates 211, 232, but in contrast to RP, these methods only cover a fraction of the human genome. To the best of our knowledge no comparison of RP and actual protein expression levels exists to-date.
Via the clever use of time-series data and drug treatments that inhibit translation initiation, RP can also provide insights into translation elongation speed using so-called “run-off” experiments 222. Following treatment, ribosomes inside active ORFs will move away from the TIS leaving a “depleted” region, where RP reads are only observed at the noise level. In addition, we can also define the unaffected region, where ribosomes still exist, and the “depleting” region, where some intermediate fraction of messages have been depleted of ribosomes (i.e. stochastic variation in speed between molecules with the same ORF). Analysis of the position and lengths of these regions after specific treatment times provides estimates of elongation speed.
Despite the successes of RP, there are a number of outstanding computational challenges. One major challenge is correctly adjusting for ribosome pausing. Protein synthesis by ribosomes takes place at non-uniform speeds between ORFs, and also with varying speeds within an ORF; one extreme is pausing 227, 233, 234. Metrics aimed at measuring translation levels must therefore be adjusted to remove the influence of stalled ribosomes. These might be stalled preinitiation complexes, ribosomes paused during elongation or awaiting release upon termination. Because these ribosomes are not actively translating, they do not contribute to protein levels. Previous studies either ignore the pausing phenomenon, or assume important pausing happens near TIS and stop sites, discarding all reads falling within a fixed distance to these. This discards information, alters the effective size of the region when normalizing, and cannot be done for short coding sequences.
Conclusion
Controlled and coordinated binding of one or more RNA binding proteins or miRNAs is the key mechanism that drives co- and post-transcriptional regulation of gene expression. These processes are often complex, inter-related, and dynamic in terms of their timing. Efforts to understand them at global scale therefore require multiple lines of investigation, and necessitate a range of computational methods to interpret the resultant data. Transcriptome-wide profiling of co- and post-transcriptional regulation is still a young field, and the development of computational tools to complement the emerging biological assays is pending. Some fundamental problems still exist. For example it remains unclear what proportion of sites identified in CLIP or RIP are actual binding sites. Moreover, our understanding of what makes a functional RBP binding site, as opposed to one that has little or no functional impact is still thin. As a result, there are no effective computational tools for determining whether a given RIP- or CLIP-seq site represents functional binding or not. Nevertheless, substantial progress has been made and a range of methods aimed both at fundamental processing of data, and the more high-level goal of understanding specific biological processes are now available. These are supplemented by a growing collection of databases and online resources.
Moving forward, new biological questions will be asked. Questions aimed at expanding our understanding of the interactions between regulators, regulatory networks, the timing of events, and how perturbation of the cellular state affects them. These questions will drive the next generation of computational methods. One key issue will be the development of tools that effectively handle multi-factorial experimental designs, with multiple replicates, and are able to leverage the additional statistical information they bring. First studies exist which combine several of these large-scale approaches. For example, to distinguish functional from non-functional RBP binding sites, proteomics studies have been combined with RIP-chip and CLIP experiments to characterize the translation regulators. Other efforts combine the analysis of miRNAs with proteome, transcriptome or translatome profiling. As more and more studies on multi-dimensional approaches arise, we need computational methods to integrate and analyze these data. In recent years there has been some studies to gain insight into functions of RBPs by studying mRNA targets of particular RBPs obtained by RIP or CLIP together with changes in mRNA stability or splicing and before and after knockdown of that specific RBP235. These approaches will help to drive the consolidation of information about co- and post-transcriptional gene regulation into more holistic and comprehensive models.
Acknowledgments
Work related to the topics covered in this review article was supported by NIH grant R01HG006015 to ADS and LOFP. CV acknowledges funding by the NYU University Research Challenge Fund.
Contributor Information
Emad Bahrami-Samani, Molecular and Computational Biology, Department of Biological Sciences, University of Southern California, Los Angeles, CA.
Dat T. Vo, Children’s Cancer Research Institute and Department of Cellular and Structural Biology, University of Texas Health Science Center, San Antonio, TX
Patricia Rosa de Araujo, Children’s Cancer Research Institute and Department of Cellular and Structural Biology, University of Texas Health Science Center, San Antonio, TX.
Christine Vogel, Center for Genomics and Systems Biology, Department of Biology, New York University, New York, NY.
Andrew D. Smith, Molecular and Computational Biology, Department of Biological Sciences, University of Southern California, Los Angeles, CA
Luiz O. F. Penalva, Children’s Cancer Research Institute and Department of Cellular and Structural Biology, University of Texas Health Science Center, San Antonio, TX
Philip J. Uren, Molecular and Computational Biology, Department of Biological Sciences, University of Southern California, Los Angeles, CA.
References
- 1.Glisovic T, Bachorik JL, Yong J, Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582:1977–1986. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galante PA, Sandhu D, de Sousa Abreu R, Gradassi M, Slager N, Vogel C, de Souza SJ, Penalva LO. A comprehensive in silico expression analysis of RNA binding proteins in normal and tumor tissue: Identification of potential players in tumor formation. RNA Biol. 2009;6:426–433. doi: 10.4161/rna.6.4.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 4.Tenenbaum SA, Carson CC, Lager PJ, Keene JD. Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc Natl Acad Sci U S A. 2000;97:14085–14090. doi: 10.1073/pnas.97.26.14085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arava Y, Wang Y, Storey JD, Liu CL, Brown PO, Herschlag D. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences. 2003;100:3889–3894. doi: 10.1073/pnas.0635171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. Genome-Wide Analysis in Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011;12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods. 2011;8:469–477. doi: 10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- 9.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oshlack A, Robinson MD, Young MD. From RNA-seq reads to differential expression results. Genome Biol. 2010;11:220. doi: 10.1186/gb-2010-11-12-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Granneman S, Kudla G, Petfalski E, Tollervey D. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proceedings of the National Academy of Sciences. 2009;106:9613–9618. doi: 10.1073/pnas.0901997106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 13.Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr, Jungkamp A-C, Munschauer M. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uren PJ, Bahrami-Samani E, Burns SC, Qiao M, Karginov FV, Hodges E, Hannon GJ, Sanford JR, Penalva LOF, Smith AD. Site identification in high-throughput RNA-protein interaction data. Bioinformatics. 2012;28:3013–3020. doi: 10.1093/bioinformatics/bts569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mili S, Steitz JA. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. Rna. 2004;10:1692–1694. doi: 10.1261/rna.7151404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roy PJ, Stuart JM, Lund J, Kim SK. Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature. 2002;418:975–979. doi: 10.1038/nature01012. [DOI] [PubMed] [Google Scholar]
- 19.Penalva LO, Burdick MD, Lin SM, Sutterluety H, Keene JD. RNA-binding proteins to assess gene expression states of co-cultivated cells in response to tumor cells. Molecular cancer. 2004;3:24. doi: 10.1186/1476-4598-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, et al. Application of a Translational Profiling Approach for the Comparative Analysis of CNS Cell Types. Cell. 2008;135:749–762. doi: 10.1016/j.cell.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Molecular cell. 2012;46:674–690. doi: 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 22.Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 23.Tome JM, Ozer A, Pagano JM, Gheba D, Schroth GP, Lis JT. Comprehensive analysis of RNA-protein interactions by high-throughput sequencing-RNA affinity profiling. Nature methods. 2014 doi: 10.1038/nmeth.2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nutiu R, Friedman RC, Luo S, Khrebtukova I, Silva D, Li R, Zhang L, Schroth GP, Burge CB. Direct measurement of DNA affinity landscapes on a high-throughput sequencing instrument. Nature biotechnology. 2011;29:659–664. doi: 10.1038/nbt.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fonseca NA, Rung J, Brazma A, Marioni JC. Tools for mapping high-throughput sequencing data. Bioinformatics. 2012;28:3169–3177. doi: 10.1093/bioinformatics/bts605. [DOI] [PubMed] [Google Scholar]
- 26.Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano M, Jr, Tuschl T, Ohler U, Keene JD. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Molecular cell. 2011;43:327–339. doi: 10.1016/j.molcel.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lebedeva S, Jens M, Theil K, Schwanhäusser B, Selbach M, Landthaler M, Rajewsky N. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Molecular cell. 2011;43:340–352. doi: 10.1016/j.molcel.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Ascano M, Mukherjee N, Bandaru P, Miller JB, Nusbaum JD, Corcoran DL, Langlois C, Munschauer M, Dewell S, Hafner M. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012 doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Erhard F, Dölken L, Zimmer R. RIP-chip enrichment analysis. Bioinformatics. 2013;29:77–83. doi: 10.1093/bioinformatics/bts631. [DOI] [PubMed] [Google Scholar]
- 30.Sugimoto Y, König J, Hussain S, Zupan B, Curk T, Frye M, Ule J. Analysis of CLIP and iCLIP methods for nucleotide-resolution studies of protein-RNA interactions. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friedersdorf MB, Keene JD. Advancing the functional utility of PAR-CLIP by quantifying background binding to mRNAs and lncRNAs. Genome Biology. 2014;15:16. doi: 10.1186/gb-2014-15-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freeberg MA, Han T, Moresco JJ, Kong A, Yang Y-C, Lu ZJ, Yates JR, Kim JK. Pervasive and dynamic protein binding sites of the mRNA transcriptome in Saccharomyces cerevisiae. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klass DM, Scheibe M, Butter F, Hogan GJ, Mann M, Brown PO. Quantitative proteomic analysis reveals concurrent RNA–protein interactions and identifies new RNA-binding proteins in Saccharomyces cerevisiae. Genome research. 2013;23:1028–1038. doi: 10.1101/gr.153031.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y, Zhao DY, Greenblatt JF, Zhang ZL. RIPSeeker: a statistical package for identifying protein-associated transcripts from RIP-seq experiments. Nucleic Acids Research. 2013;41:18. doi: 10.1093/nar/gkt142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang T, Xie Y, Xiao GH. dCLIP: a computational approach for comparative CLIP-seq analyses. Genome Biology. 2014;15:13. doi: 10.1186/gb-2014-15-1-r11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang T, Chen B, Kim M, Xie Y, Xiao G. A Model-Based Approach to Identify Binding Sites in CLIP-Seq Data. PLoS ONE. 2014;9:e93248. doi: 10.1371/journal.pone.0093248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kucukural A, Ozadam H, Singh G, Moore MJ, Cenik C. ASPeak: an abundance sensitive peak detection algorithm for RIP-Seq. Bioinformatics. 2013;29:2485–2486. doi: 10.1093/bioinformatics/btt428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang C, Darnell RB. Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nature biotechnology. 2011;29:607–614. doi: 10.1038/nbt.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Corcoran DL, Georgiev S, Mukherjee N, Gottwein E, Skalsky RL, Keene JD, Ohler U. PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biology. 2011;12:16. doi: 10.1186/gb-2011-12-8-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loeb GB, Khan AA, Canner D, Hiatt JB, Shendure J, Darnell RB, Leslie CS, Rudensky AY. Transcriptome-wide miR-155 binding map reveals widespread noncanonical microRNA targeting. Molecular cell. 2012;48:760–770. doi: 10.1016/j.molcel.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zarnack K, König J, Tajnik M, Martincorena I, Eustermann S, Stévant I, Reyes A, Anders S, Luscombe NM, Ule J. Direct competition between hnrnp c and u2af65 protects the transcriptome from the exonization of alu elements. Cell. 2013;152:453–466. doi: 10.1016/j.cell.2012.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xue Y, Ouyang K, Huang J, Zhou Y, Ouyang H, Li H, Wang G, Wu Q, Wei C, Bi Y. Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell. 2013;152:82–96. doi: 10.1016/j.cell.2012.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mukherjee N, Lager PJ, Friedersdorf MB, Thompson MA, Keene JD. Coordinated posttranscriptional mRNA population dynamics during T cell activation. Molecular systems biology. 2009:5. doi: 10.1038/msb.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen BB, Yun J, Kim MS, Mendell JT, Xie Y. PIPE-CLIP: a comprehensive online tool for CLIP-seq data analysis. Genome Biology. 2014;15:10. doi: 10.1186/gb-2014-15-1-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Webb S, Hector RD, Kudla G, Granneman S. PAR-CLIP data indicate that Nrd1-Nab3-dependent transcription termination regulates expression of hundreds of protein coding genes in yeast. Genome Biology. 2014:15. doi: 10.1186/gb-2014-15-1-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu JS, Neuwald AF, Lawrence CE. Bayesian models for multiple local sequence alignment and Gibbs sampling strategies. Journal of the American Statistical Association. 1995;90:1156–1170. [Google Scholar]
- 48.Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perez-Canadillas JM, Varani G. Recent advances in RNA-protein recognition. Current Opinion in Structural Biology. 2001;11:53–58. doi: 10.1016/s0959-440x(00)00164-0. [DOI] [PubMed] [Google Scholar]
- 50.Sanchez-Diaz P, Penalva LO. Review Post-Transcription Meets Post-Genomic. RNA biology. 2006;3:101–109. doi: 10.4161/rna.3.3.3373. [DOI] [PubMed] [Google Scholar]
- 51.Klug SJ, Famulok M. ALL YOU WANTED TO KNOW ABOUT SELEX. Molecular Biology Reports. 1994;20:97–107. doi: 10.1007/BF00996358. [DOI] [PubMed] [Google Scholar]
- 52.Tuerk C. Using the SELEX combinatorial chemistry process to find high affinity nucleic acid ligands to target molecules. Methods in Molecular Biology; PCR cloning protocols: From molecular cloning to genetic engineering. 1997;67:219–230. doi: 10.1385/0-89603-483-6:219. [DOI] [PubMed] [Google Scholar]
- 53.Ray D, Kazan H, Chan ET, Castillo LP, Chaudhry S, Talukder S, Blencowe BJ, Morris Q, Hughes TR. Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nature biotechnology. 2009;27:667–670. doi: 10.1038/nbt.1550. [DOI] [PubMed] [Google Scholar]
- 54.Stormo GD, Schneider TD, Gold L, Ehrenfeucht A. Use of the ‘Perceptron’ algorithm to distinguish translational initiation sites in E. coli. Nucleic Acids Research. 1982;10:2997–3011. doi: 10.1093/nar/10.9.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cardon LR, Stormo GD. Expectation maximization algorithm for identifying protein-binding sites with variable lengths from unaligned DNA fragments. Journal of Molecular Biology. 1992;223:159–170. doi: 10.1016/0022-2836(92)90723-w. [DOI] [PubMed] [Google Scholar]
- 56.Lawrence CE, Reilly AA. An expectation maximization (EM) algorithm for the identification and characterization of common sites in unaligned biopolymer sequences. Proteins: Structure, Function, and Bioinformatics. 1990;7:41–51. doi: 10.1002/prot.340070105. [DOI] [PubMed] [Google Scholar]
- 57.Liu JS. The collapsed Gibbs sampler in Bayesian computations with applications to a gene regulation problem. Journal of the American Statistical Association. 1994;89:958–966. [Google Scholar]
- 58.Abdullah SLS, Hussin NM, Harun H, Khalid NEA. Comparative study of random-PSO and Linear-PSO algorithms. Computer & Information Science (ICCIS), 2012 International Conference on; IEEE; 2012. [Google Scholar]
- 59.Bailey TL, Elkan C. Unsupervised learning of multiple motifs in biopolymers using expectation maximization. Machine learning. 1995;21:51–80. [Google Scholar]
- 60.Liu XS, Brutlag DL, Liu JS. An algorithm for finding protein–DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nature biotechnology. 2002;20:835–839. doi: 10.1038/nbt717. [DOI] [PubMed] [Google Scholar]
- 61.Roth FP, Hughes JD, Estep PW, Church GM. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nature biotechnology. 1998;16:939–945. doi: 10.1038/nbt1098-939. [DOI] [PubMed] [Google Scholar]
- 62.Smith AD, Sumazin P, Zhang MQ. Identifying tissue-selective transcription factor binding sites in vertebrate promoters. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1560–1565. doi: 10.1073/pnas.0406123102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Das MK, Dai H-K. A survey of DNA motif finding algorithms. BMC bioinformatics. 2007;8:S21. doi: 10.1186/1471-2105-8-S7-S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hu J, Li B, Kihara D. Limitations and potentials of current motif discovery algorithms. Nucleic acids research. 2005;33:4899–4913. doi: 10.1093/nar/gki791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gardina PJ, Clark TA, Shimada B, Staples MK, Yang Q, Veitch J, Schweitzer A, Awad T, Sugnet C, Dee S, et al. Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array. Bmc Genomics. 2006:7. doi: 10.1186/1471-2164-7-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qiu P. Recent advances in computational promoter analysis in understanding the transcriptional regulatory network. Biochemical and Biophysical Research Communications. 2003;309:495–501. doi: 10.1016/j.bbrc.2003.08.052. [DOI] [PubMed] [Google Scholar]
- 67.Stormo GD. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- 68.Akerman M, David-Eden H, Pinter RY, Mandel-Gutfreund Y. A computational approach for genome-wide mapping of splicing factor binding sites. Genome Biol. 2009;10:R30. doi: 10.1186/gb-2009-10-3-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang C, Lee K-Y, Swanson MS, Darnell RB. Prediction of clustered RNA-binding protein motif sites in the mammalian genome. Nucleic acids research. 2013;41:6793–6807. doi: 10.1093/nar/gkt421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li X, Kazan H, Lipshitz HD, Morris QD. Finding the target sites of RNA binding proteins. Wiley Interdisciplinary Reviews: RNA. 2014;5:111–130. doi: 10.1002/wrna.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Eddy SR, Durbin R. RNA sequence analysis using covariance models. Nucleic acids research. 1994;22:2079–2088. doi: 10.1093/nar/22.11.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yao Z, Weinberg Z, Ruzzo WL. CMfinder—a covariance model based RNA motif finding algorithm. Bioinformatics. 2006;22:445–452. doi: 10.1093/bioinformatics/btk008. [DOI] [PubMed] [Google Scholar]
- 73.Mathews DH, Turner DH. Dynalign: an algorithm for finding the secondary structure common to two RNA sequences. Journal of molecular biology. 2002;317:191–203. doi: 10.1006/jmbi.2001.5351. [DOI] [PubMed] [Google Scholar]
- 74.Fogel GB, Porto VW, Weekes DG, Fogel DB, Griffey RH, McNeil JA, Lesnik E, Ecker DJ, Sampath R. Discovery of RNA structural elements using evolutionary computation. Nucleic Acids Research. 2002;30:5310–5317. doi: 10.1093/nar/gkf653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mauri G, Pavesi G. Combinatorial Pattern Matching. Springer; 2003. Pattern discovery in RNA secondary structure using affix trees. [Google Scholar]
- 76.Hiller M, Pudimat R, Busch A, Backofen R. Using RNA secondary structures to guide sequence motif finding towards single-stranded regions. Nucleic acids research. 2006;34:e117–e117. doi: 10.1093/nar/gkl544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kazan H, Ray D, Chan ET, Hughes TR, Morris Q. RNAcontext: a new method for learning the sequence and structure binding preferences of RNA-binding proteins. PLoS computational biology. 2010;6:e1000832. doi: 10.1371/journal.pcbi.1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maticzka D, Lange SJ, Costa F, Backofen R. GraphProt: modeling binding preferences of RNA-binding proteins. Genome Biol. 2014;15:R17. doi: 10.1186/gb-2014-15-1-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chou CH, Lin FM, Chou MT, Hsu SD, Chang TH, Weng SL, Shrestha S, Hsiao CC, Hung JH, Huang HD. A computational approach for identifying microRNA-target interactions using high-throughput CLIP and PAR-CLIP sequencing. Bmc Genomics. 2013;14:11. doi: 10.1186/1471-2164-14-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moore MJ, Zhang C, Gantman EC, Mele A, Darnell JC, Darnell RB. Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nature protocols. 2014;9:263–293. doi: 10.1038/nprot.2014.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zheng H, Fu R, Wang J-T, Liu Q, Chen H, Jiang S-W. Advances in the techniques for the prediction of microRNA targets. International journal of molecular sciences. 2013;14:8179–8187. doi: 10.3390/ijms14048179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grimson A, Farh KK-H, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Molecular cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nature genetics. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 84.Cook KB, Kazan H, Zuberi K, Morris Q, Hughes TR. RBPDB: a database of RNA-binding specificities. Nucleic acids research. 2011;39:D301–D308. doi: 10.1093/nar/gkq1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Khorshid M, Rodak C, Zavolan M. CLIPZ: a database and analysis environment for experimentally determined binding sites of RNA-binding proteins. Nucleic Acids Research. 2011;39:D245–D252. doi: 10.1093/nar/gkq940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Paz I, Kosti I, Ares M, Cline M, Mandel-Gutfreund Y. RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic acids research. 2014:gku406. doi: 10.1093/nar/gku406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vo DT, Subramaniam D, Remke M, Burton TL, Uren PJ, Gelfond JA, de Sousa Abreu R, Burns SC, Qiao M, Suresh U. The RNA-binding protein Musashi1 affects medulloblastoma growth via a network of cancer-related genes and is an indicator of poor prognosis. The American journal of pathology. 2012;181:1762–1772. doi: 10.1016/j.ajpath.2012.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Scheibe M, Butter F, Hafner M, Tuschl T, Mann M. Quantitative mass spectrometry and PAR-CLIP to identify RNA-protein interactions. Nucleic acids research. 2012;40:9897–9902. doi: 10.1093/nar/gks746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Maatz H, Jens M, Liss M, Schafer S, Heinig M, Kirchner M, Adami E, Rintisch C, Dauksaite V, Radke MH. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. The Journal of clinical investigation. 2014;124:3419. doi: 10.1172/JCI74523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hendrickson DG, Hogan DJ, McCullough HL, Myers JW, Herschlag D, Ferrell JE, Brown PO. Concordant regulation of translation and mRNA abundance for hundreds of targets of a human microRNA. PLoS biology. 2009;7:e1000238. doi: 10.1371/journal.pbio.1000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 92.Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tamim S, Vo DT, Uren PJ, Qiao M, Bindewald E, Kasprzak WK, Shapiro BA, Nakaya HI, Burns SC, Araujo PR. Genomic Analyses Reveal Broad Impact of miR-137 on Genes Associated with Malignant Transformation and Neuronal Differentiation in Glioblastoma Cells. PloS one. 2014;9:e85591. doi: 10.1371/journal.pone.0085591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 95.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology. 2010;28:511–U174. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory in Biosciences. 2012;131:281–285. doi: 10.1007/s12064-012-0162-3. [DOI] [PubMed] [Google Scholar]
- 97.Dillies M-A, Rau A, Aubert J, Hennequet-Antier C, Jeanmougin M, Servant N, Keime C, Marot G, Castel D, Estelle J. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Briefings in bioinformatics. 2013;14:671–683. doi: 10.1093/bib/bbs046. [DOI] [PubMed] [Google Scholar]
- 98.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biology. 2010;11:12. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nookaew I, Papini M, Pornputtapong N, Scalcinati G, Fagerberg L, Uhlen M, Nielsen J. A comprehensive comparison of RNA-Seq-based transcriptome analysis from reads to differential gene expression and cross-comparison with microarrays: a case study in Saccharomyces cerevisiae. Nucleic Acids Research. 2012;40:10084–10097. doi: 10.1093/nar/gks804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nakaya T, Alexiou P, Maragkakis M, Chang A, Mourelatos Z. FUS regulates genes coding for RNA-binding proteins in neurons by binding to their highly conserved introns. Rna. 2013;19:498–509. doi: 10.1261/rna.037804.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Beadle GW, Tatum EL. Genetic control of biochemical reactions in Neurospora. Proceedings of the National Academy of Sciences of the United States of America. 1941;27:499. doi: 10.1073/pnas.27.11.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Valdivia HH. One gene, many proteins - Alternative splicing of the ryanodine receptor gene adds novel functions to an already complex channel protein. Circulation Research. 2007;100:761–763. doi: 10.1161/01.RES.0000263400.64391.37. [DOI] [PubMed] [Google Scholar]
- 104.Brett D, Pospisil H, Valcarcel J, Reich J, Bork P. Alternative splicing and genome complexity. Nature Genetics. 2002;30:29–30. doi: 10.1038/ng803. [DOI] [PubMed] [Google Scholar]
- 105.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing (vol 40, pg 1413, 2008) Nature Genetics. 2009;41:762–762. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 106.Wang ET, Sandberg R, Luo SJ, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schweingruber C, Rufener SC, Zünd D, Yamashita A, Mühlemann O. Nonsense-mediated mRNA decay—mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms. 2013;1829:612–623. doi: 10.1016/j.bbagrm.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 108.Kalyna M, Simpson CG, Syed NH, Lewandowska D, Marquez Y, Kusenda B, Marshall J, Fuller J, Cardle L, McNicol J, et al. Alternative splicing and nonsense-mediated decay modulate expression of important regulatory genes in Arabidopsis. Nucleic Acids Research. 2012;40:2454–2469. doi: 10.1093/nar/gkr932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ali GS, Reddy ASN. Regulation of Alternative Splicing of Pre-mRNAs by Stresses. Nuclear Pre-Mrna Processing in Plants. 2008;326:257–275. doi: 10.1007/978-3-540-76776-3_14. [DOI] [PubMed] [Google Scholar]
- 110.Yu P, Zhou L, Ke W, Li K. Clinical significance of pAKT and CD44v6 overexpression with breast cancer. Journal of cancer research and clinical oncology. 2010;136:1283–1292. doi: 10.1007/s00432-010-0779-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Todaro M, Gaggianesi M, Catalano V, Benfante A, Iovino F, Biffoni M, Apuzzo T, Sperduti I, Volpe S, Cocorullo G. CD44v6 Is a Marker of Constitutive and Reprogrammed Cancer Stem Cells Driving Colon Cancer Metastasis. Cell stem cell. 2014;14:342–356. doi: 10.1016/j.stem.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 112.Shi J, Zhou Z, Di W, Li N. Correlation of CD44v6 expression with ovarian cancer progression and recurrence. BMC cancer. 2013;13:182. doi: 10.1186/1471-2407-13-182. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 113.Liang Y, Fang T, Xu H, Zhuo Z. Expression of CD44v6 and Livin in gastric cancer tissue. Chinese medical journal. 2012;125:3161–3165. [PubMed] [Google Scholar]
- 114.Hagiwara M. Alternative splicing: A new drug target of the post-genome era. Biochimica Et Biophysica Acta-Proteins and Proteomics. 2005;1754:324–331. doi: 10.1016/j.bbapap.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 115.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nature Biotechnology. 2013;31:46-+. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nature Methods. 2010;7:1009–U1101. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Research. 2012;22:2008–2017. doi: 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang WC, Qin ZY, Feng ZX, Wang X, Zhang XG. Identifying differentially spliced genes from two groups of RNA-seq samples. Gene. 2013;518:164–170. doi: 10.1016/j.gene.2012.11.045. [DOI] [PubMed] [Google Scholar]
- 119.Shen SH, Park JW, Huang J, Dittmar KA, Lu ZX, Zhou Q, Carstens RP, Xing Y. MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Research. 2012;40:13. doi: 10.1093/nar/gkr1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hu Y, Huang Y, Du Y, Orellana CF, Singh D, Johnson AR, Monroy A, Kuan PF, Hammond SM, Makowski L, et al. DiffSplice: the genome-wide detection of differential splicing events with RNA-seq. Nucleic Acids Research. 2013;41:18. doi: 10.1093/nar/gks1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Aschoff M, Hotz-Wagenblatt A, Glatting KH, Fischer M, Eils R, Konig R. SplicingCompass: differential splicing detection using RNA-Seq data. Bioinformatics. 2013;29:1141–1148. doi: 10.1093/bioinformatics/btt101. [DOI] [PubMed] [Google Scholar]
- 122.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10:10. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Huang S, Zhang J, Li R, Zhang W, He Z, Lam T-W, Peng Z, Yiu S-M. SOAPsplice: genome-wide ab initio detection of splice junctions from RNA-Seq data. Frontiers in genetics. 2011;2:46. doi: 10.3389/fgene.2011.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hoffmann S, Otto C, Doose G, Tanzer A, Langenberger D, Christ S, Kunz M, Holdt L, Teupser D, Hackermüller J. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing, and fusion detection. Genome biology. 2014;15:R34. doi: 10.1186/gb-2014-15-2-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He XP, Mieczkowski P, Grimm SA, Perou CM, et al. MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Research. 2010;38:14. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ameur A, Wetterbom A, Feuk L, Gyllensten U. Global and unbiased detection of splice junctions from RNA-seq data. Genome Biology. 2010;11:9. doi: 10.1186/gb-2010-11-3-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hoffmann S, Otto C, Kurtz S, Sharma CM, Khaitovich P, Vogel J, Stadler PF, Hackermuller J. Fast Mapping of Short Sequences with Mismatches, Insertions and Deletions Using Index Structures. Plos Computational Biology. 2009;5:10. doi: 10.1371/journal.pcbi.1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Au KF, Jiang H, Lin L, Xing Y, Wong WH. Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Research. 2010;38:4570–4578. doi: 10.1093/nar/gkq211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jean G, Kahles A, Sreedharan VT, Bona FD, Rätsch G. RNA Seq Read Alignments with PALMapper. Current protocols in bioinformatics. :11.16.11–11.16.37. doi: 10.1002/0471250953.bi1106s32. [DOI] [PubMed] [Google Scholar]
- 133.Hooper JE. A survey of software for genome-wide discovery of differential splicing in RNA-Seq data. Human Genomics. 2014:8. doi: 10.1186/1479-7364-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.EURASNET. Alternative Splicing Databases. Available at: http://www.eurasnet.info/tools/asdatabases.
- 135.Hung LH, Heiner M, Hui JY, Schreiner S, Benes V, Bindereif A. Diverse roles of hnRNP L in mammalian mRNA processing: A combined microarray and RNAi analysis. Rna-a Publication of the Rna Society. 2008;14:284–296. doi: 10.1261/rna.725208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Huelga SC, Vu AQ, Arnold JD, Liang TY, Donohue JP, Shiue L, Hoon S, Brenner S, Ares M, Yeo GW. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Faseb Journal. 2012;26:1. doi: 10.1016/j.celrep.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Konig J, Hortobagyi T, Nishimura AL, Zupunski V, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nature Neuroscience. 2011;14:452–U180. doi: 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nature Structural & Molecular Biology. 2009;16:130–137. doi: 10.1038/nsmb.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang CL, Zhang Z, Castle J, Sun SY, Johnson J, Krainer AR, Zhang MQ. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes & Development. 2008;22:2550–2563. doi: 10.1101/gad.1703108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Llorian M, Schwartz S, Clark TA, Hollander D, Tan LY, Spellman R, Gordon A, Schweitzer AC, la Grange P, Ast G, et al. Position-dependent alternative splicing activity revealed by global profiling of alternative splicing events regulated by PTB. Nature Structural & Molecular Biology. 2010;17:1114–U1112. doi: 10.1038/nsmb.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Xue YC, Zhou Y, Wu TB, Zhu T, Ji X, Kwon YS, Zhang C, Yeo G, Black DL, Sun H, et al. Genome-wide Analysis of PTB-RNA Interactions Reveals a Strategy Used by the General Splicing Repressor to Modulate Exon Inclusion or Skipping. Molecular Cell. 2009;36:996–1006. doi: 10.1016/j.molcel.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Du HQ, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, Hall MP, Shiue L, Swanson MS, Thornton CA, et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nature Structural & Molecular Biology. 2010;17:187–U188. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang Z, Kayikci M, Briese M, Zarnack K, Luscombe NM, Rot G, Zupan B, Curk T, Ule J. iCLIP Predicts the Dual Splicing Effects of TIA-RNA Interactions. Plos Biology. 2010;8:16. doi: 10.1371/journal.pbio.1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nature Reviews Genetics. 2013;14:496–506. doi: 10.1038/nrg3482. [DOI] [PubMed] [Google Scholar]
- 145.Hoque M, Ji Z, Zheng D, Luo W, Li W, You B, Park JY, Yehia G, Tian B. Analysis of alternative cleavage and polyadenylation by 3 [prime] region extraction and deep sequencing. Nature methods. 2013;10:133–139. doi: 10.1038/nmeth.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Smibert P, Miura P, Westholm JO, Shenker S, May G, Duff MO, Zhang DY, Eads BD, Carlson J, Brown JB, et al. Global Patterns of Tissue-Specific Alternative Polyadenylation in Drosophila. Cell Reports. 2012;1:277–289. doi: 10.1016/j.celrep.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Alt FW, Bothwell AL, Knapp M, Siden E, Mather E, Koshland M, Baltimore D. Synthesis of secreted and membrane-bound immunoglobulin mu heavy chains is directed by mRNAs that differ at their 3′ ends. Cell. 1980;20:293–301. doi: 10.1016/0092-8674(80)90615-7. [DOI] [PubMed] [Google Scholar]
- 148.Setzer DR, McGrogan M, Nunberg JH, Schimke RT. Size heterogeneity in the 3′ end of dihydrofolate reductase messenger RNAs in mouse cells. Cell. 1980;22:361–370. doi: 10.1016/0092-8674(80)90346-3. [DOI] [PubMed] [Google Scholar]
- 149.Beaudoing E, Freier S, Wyatt JR, Claverie JM, Gautheret D. Patterns of variant polyadenylation signal usage in human genes. Genome Research. 2000;10:1001–1010. doi: 10.1101/gr.10.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gautheret D, Poirot O, Lopez F, Audic S, Claverie JM. Alternate polyadenylation in human mRNAs: A large-scale analysis by EST clustering. Genome Research. 1998;8:524–530. doi: 10.1101/gr.8.5.524. [DOI] [PubMed] [Google Scholar]
- 151.Gruber AR, Martin G, Keller W, Zavolan M. Means to an end: mechanisms of alternative polyadenylation of messenger RNA precursors. Wiley Interdisciplinary Reviews-Rna. 2014;5:183–196. doi: 10.1002/wrna.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Haenni S, Ji Z, Hoque M, Rust N, Sharpe H, Eberhard R, Browne C, Hengartner MO, Mellor J, Tian B, et al. Analysis of C. elegans intestinal gene expression and polyadenylation by fluorescence-activated nuclei sorting and 3′-end-seq. Nucleic Acids Research. 2012;40:6304–6318. doi: 10.1093/nar/gks282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Derti A, Garrett-Engele P, MacIsaac KD, Stevens RC, Sriram S, Chen R, Rohl CA, Johnson JM, Babak T. A quantitative atlas of polyadenylation in five mammals. Genome Research. 2012;22:1173–1183. doi: 10.1101/gr.132563.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mueller AA, Cheung TH, Rando TA. All’s well that ends well: alternative polyadenylation and its implications for stem cell biology. Current Opinion in Cell Biology. 2013;25:222–232. doi: 10.1016/j.ceb.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–1647. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mayr C, Bartel DP. Widespread Shortening of 3′ UTRs by Alternative Cleavage and Polyadenylation Activates Oncogenes in Cancer Cells. Cell. 2009;138:673–684. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ozsolak F, Kapranov P, Foissac S, Kim SW, Fishilevich E, Monaghan AP, John B, Milos PM. Comprehensive Polyadenylation Site Maps in Yeast and Human Reveal Pervasive Alternative Polyadenylation. Cell. 2010;143:1018–1029. doi: 10.1016/j.cell.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lembo A, Di Cunto F, Provero P. Shortening of 3′ UTRs Correlates with Poor Prognosis in Breast and Lung Cancer. Plos One. 2012:7. doi: 10.1371/journal.pone.0031129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lin Y, Li Z, Ozsolak F, Kim SW, Arango-Argoty G, Liu TT, Tenenbaum SA, Bailey T, Monaghan AP, Milos PM, et al. An in-depth map of polyadenylation sites in cancer. Nucleic Acids Research. 2012;40:8460–8471. doi: 10.1093/nar/gks637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cheng YM, Miura RM, Tian B. Prediction of mRNA polyadenylation sites by support vector machine. Bioinformatics. 2006;22:2320–2325. doi: 10.1093/bioinformatics/btl394. [DOI] [PubMed] [Google Scholar]
- 161.Brockman JM, Singh P, Liu DL, Quinlan S, Salisbury J, Graber JH. PACdb: PolyA cleavage site and 3′-UTR database. Bioinformatics. 2005;21:3691–3693. doi: 10.1093/bioinformatics/bti589. [DOI] [PubMed] [Google Scholar]
- 162.Lee JY, Yeh I, Park JY, Tian B. PolyA_DB 2: mRNA polyadenylation sites in vertebrate genes. Nucleic Acids Research. 2007;35:D165–D168. doi: 10.1093/nar/gkl870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Eberhardt R, Anantham D, Herth F, Feller-Kopman D, Ernst A. Electromagnetic navigation diagnostic bronchoscopy in peripheral lung lesions. CHEST Journal. 2007;131:1800–1805. doi: 10.1378/chest.06-3016. [DOI] [PubMed] [Google Scholar]
- 164.Kervestin S, Jacobson A. NMD: a multifaceted response to premature translational termination. Nature Reviews Molecular Cell Biology. 2012;13:700–712. doi: 10.1038/nrm3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bakheet T, Williams BR, Khabar KS. ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic acids research. 2006;34:D111–D114. doi: 10.1093/nar/gkj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Brennan SE, Kuwano Y, Alkharouf N, Blackshear PJ, Gorospe M, Wilson GM. The mRNA-destabilizing protein tristetraprolin is suppressed in many cancers, altering tumorigenic phenotypes and patient prognosis. Cancer research. 2009;69:5168–5176. doi: 10.1158/0008-5472.CAN-08-4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Uren PJ, Burns SC, Ruan J, Singh KK, Smith AD, Penalva LO. Genomic analyses of the RNA-binding protein Hu antigen R (HuR) identify a complex network of target genes and novel characteristics of its binding sites. Journal of Biological Chemistry. 2011;286:37063–37066. doi: 10.1074/jbc.C111.266882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Barreau C, Paillard L, Méreau A, Osborne HB. Mammalian CELF/Bruno-like RNA-binding proteins: molecular characteristics and biological functions. Biochimie. 2006;88:515–525. doi: 10.1016/j.biochi.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 169.Gruber AR, Fallmann J, Kratochvill F, Kovarik P, Hofacker IL. AREsite: a database for the comprehensive investigation of AU-rich elements. Nucleic acids research. 2011;39:D66–D69. doi: 10.1093/nar/gkq990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Schoenberg DR, Maquat LE. Regulation of cytoplasmic mRNA decay. Nature Reviews Genetics. 2012;13:246–259. doi: 10.1038/nrg3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wang Y, Liu CL, Storey JD, Tibshirani RJ, Herschlag D, Brown PO. Precision and functional specificity in mRNA decay. Proceedings of the National Academy of Sciences. 2002;99:5860–5865. doi: 10.1073/pnas.092538799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Alic A, Pérez-Ortín JE, Moreno J, Arnau V. mRNAStab—a web application for mRNA stability analysis. Bioinformatics. 2013;29:813–814. doi: 10.1093/bioinformatics/btt040. [DOI] [PubMed] [Google Scholar]
- 173.Dölken L, Ruzsics Z, Rädle B, Friedel CC, Zimmer R, Mages J, Hoffmann R, Dickinson P, Forster T, Ghazal P. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. Rna. 2008;14:1959–1972. doi: 10.1261/rna.1136108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Miller C, Schwalb B, Maier K, Schulz D, Dümcke S, Zacher B, Mayer A, Sydow J, Marcinowski L, Dölken L. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Molecular systems biology. 2011:7. doi: 10.1038/msb.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Schwalb B, Schulz D, Sun M, Zacher B, Dümcke S, Martin DE, Cramer P, Tresch A. Measurement of genome-wide RNA synthesis and decay rates with Dynamic Transcriptome Analysis (DTA) Bioinformatics. 2012;28:884–885. doi: 10.1093/bioinformatics/bts052. [DOI] [PubMed] [Google Scholar]
- 176.Dieterich C, Stadler PF. Computational biology of RNA interactions. Wiley Interdisciplinary Reviews: RNA. 2013;4:107–120. doi: 10.1002/wrna.1147. [DOI] [PubMed] [Google Scholar]
- 177.Goodarzi H, Najafabadi HS, Oikonomou P, Greco TM, Fish L, Salavati R, Cristea IM, Tavazoie S. Systematic discovery of structural elements governing stability of mammalian messenger RNAs. Nature. 2012;485:264–268. doi: 10.1038/nature11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gupta I, Clauder Münster S, Klaus B, Järvelin AI, Aiyar RS, Benes V, Wilkening S, Huber W, Pelechano V, Steinmetz LM. Alternative polyadenylation diversifies post transcriptional regulation by selective RNA–protein interactions. Molecular systems biology. 2014:10. doi: 10.1002/msb.135068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Geisberg JV, Moqtaderi Z, Fan X, Ozsolak F, Struhl K. Global analysis of mRNA isoform half-lives reveals stabilizing and destabilizing elements in yeast. Cell. 2014;156:812–824. doi: 10.1016/j.cell.2013.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 181.Zhang H, Kolb FA, Brondani V, Billy E, Filipowicz W. Human Dicer preferentially cleaves dsRNAs at their termini without a requirement for ATP. EMBO J. 2002;21:5875–5885. doi: 10.1093/emboj/cdf582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Provost P, Dishart D, Doucet J, Frendewey D, Samuelsson B, Radmark O. Ribonuclease activity and RNA binding of recombinant human Dicer. EMBO J. 2002;21:5864–5874. doi: 10.1093/emboj/cdf578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, Shiekhattar R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740–744. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005;123:631–640. doi: 10.1016/j.cell.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 185.Hammond SM, Boettcher S, Caudy AA, Kobayashi R, Hannon GJ. Argonaute2, a link between genetic and biochemical analyses of RNAi. Science. 2001;293:1146–1150. doi: 10.1126/science.1064023. [DOI] [PubMed] [Google Scholar]
- 186.Pfaff J, Hennig J, Herzog F, Aebersold R, Sattler M, Niessing D, Meister G. Structural features of Argonaute–GW182 protein interactions. Proceedings of the National Academy of Sciences. 2013;110:E3770–E3779. doi: 10.1073/pnas.1308510110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Song JJ, Smith SK, Hannon GJ, Joshua-Tor L. Crystal structure of Argonaute and its implications for RISC slicer activity. Science. 2004;305:1434–1437. doi: 10.1126/science.1102514. [DOI] [PubMed] [Google Scholar]
- 188.Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 189.Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome biology. 5:R1–R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 191.Krek A, Grün D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M. Combinatorial microRNA target predictions. Nature genetics. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 192.Rajewsky N, Vergassola M, Gaul U, Siggia ED. Computational detection of genomic cis-regulatory modules applied to body patterning in the early Drosophila embryo. BMC bioinformatics. 2002;3:30. doi: 10.1186/1471-2105-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Schroeder MD, Pearce M, Fak J, Fan H, Unnerstall U, Emberly E, Rajewsky N, Siggia ED, Gaul U. Transcriptional control in the segmentation gene network of Drosophila. PLoS biology. 2004;2:e271. doi: 10.1371/journal.pbio.0020271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Gamazon ER, Im H-K, Duan S, Lussier YA, Cox NJ, Dolan ME, Zhang W. Exprtarget: an integrative approach to predicting human microRNA targets. PLoS One. 2010;5:e13534. doi: 10.1371/journal.pone.0013534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic acids research. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Vergoulis T, Vlachos IS, Alexiou P, Georgakilas G, Maragkakis M, Reczko M, Gerangelos S, Koziris N, Dalamagas T, Hatzigeorgiou AG. TarBase 6. 0: capturing the exponential growth of miRNA targets with experimental support. Nucleic acids research. 2012;40:D222–D229. doi: 10.1093/nar/gkr1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Rennie W, Liu C, Carmack CS, Wolenc A, Kanoria S, Lu J, Long D, Ding Y. STarMir: a web server for prediction of microRNA binding sites. Nucleic acids research. 2014:gku376. doi: 10.1093/nar/gku376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Yang J-H, Li J-H, Shao P, Zhou H, Chen Y-Q, Qu L-H. starBase: a database for exploring microRNA–mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic acids research. 2011;39:D202–D209. doi: 10.1093/nar/gkq1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Anders G, Mackowiak SD, Jens M, Maaskola J, Kuntzagk A, Rajewsky N, Landthaler M, Dieterich C. doRiNA: a database of RNA interactions in post-transcriptional regulation. Nucleic acids research. 2012;40:D180–D186. doi: 10.1093/nar/gkr1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bazzini AA, Lee MT, Giraldez AJ. Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science. 2012;336:233–237. doi: 10.1126/science.1215704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Loayza-Puch F, Drost J, Rooijers K, Lopes R, Elkon R, Agami R. p53 induces transcriptional and translational programs to suppress cell proliferation and growth. Genome Biology. 2013;14:12. doi: 10.1186/gb-2013-14-4-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Sonenberg N, Hinnebusch AG. New modes of translational control in development, behavior, and disease. Molecular Cell. 2007;28:721–729. doi: 10.1016/j.molcel.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 203.Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nature Biotechnology. 2007;25:117–124. doi: 10.1038/nbt1270. [DOI] [PubMed] [Google Scholar]
- 204.Vogel C, Abreu RD, Ko DJ, Le SY, Shapiro BA, Burns SC, Sandhu D, Boutz DR, Marcotte EM, Penalva LO. Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Molecular Systems Biology. 2010;6:9. doi: 10.1038/msb.2010.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Bettegowda A, Wilkinson MF. Transcription and post-transcriptional regulation of spermatogenesis. Philosophical Transactions of the Royal Society B: Biological Sciences. 2010;365:1637–1651. doi: 10.1098/rstb.2009.0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Brown GT, McIntyre TM. Lipopolysaccharide signaling without a nucleus: Kinase cascades stimulate platelet shedding of proinflammatory IL-1β–rich microparticles. The Journal of Immunology. 2011;186:5489–5496. doi: 10.4049/jimmunol.1001623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Kuersten S, Goodwin EB. The power of the 3′ UTR: Translational control and development. Nature Reviews Genetics. 2003;4:626–637. doi: 10.1038/nrg1125. [DOI] [PubMed] [Google Scholar]
- 208.Bilanges B, Stokoe D. Mechanisms of translational deregulation in human tumors and therapeutic intervention strategies. Oncogene. 2007;26:5973–5990. doi: 10.1038/sj.onc.1210431. [DOI] [PubMed] [Google Scholar]
- 209.Lazaris-Karatzas A, Montine KS, Sonenberg N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′cap. 1990. [DOI] [PubMed] [Google Scholar]
- 210.Kozak M, Evans M, Gardner PD, Flores I, Mariano TM, Pestka S, Phelps A, Wohlrab H, Zushi M, Gomi K. Structural features in eukaryotic mRNAs that mod. Biological Chemistry. 1991:266. [Google Scholar]
- 211.Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 212.Kapeli K, Yeo GW. Genome-wide approaches to dissect the roles of RNA binding proteins in translational control: implications for neurological diseases. Frontiers in neuroscience. 2012:6. doi: 10.3389/fnins.2012.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Zong Q, Schummer M, Hood L, Morris DR. Messenger RNA translation state: the second dimension of high-throughput expression screening. Proceedings of the National Academy of Sciences. 1999;96:10632–10636. doi: 10.1073/pnas.96.19.10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–U196. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kuersten S, Radek A, Vogel C, Penalva LOF. Translation regulation gets its ‘omics’ moment. Wiley Interdisciplinary Reviews-Rna. 2013;4:617–630. doi: 10.1002/wrna.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nature reviews Molecular cell biology. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Staden R. Measurements of the effects that coding for a protein has on a DNA sequence and their use for finding genes. Nucleic acids research. 1984;12:551–567. doi: 10.1093/nar/12.1part2.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Krogh A, Brown M, Mian IS, Sjölander K, Haussler D. Hidden Markov models in computational biology: Applications to protein modeling. Journal of molecular biology. 1994;235:1501–1531. doi: 10.1006/jmbi.1994.1104. [DOI] [PubMed] [Google Scholar]
- 219.Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. Journal of molecular biology. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 220.Crappé J, Van Criekinge W, Trooskens G, Hayakawa E, Luyten W, Baggerman G, Menschaert G. Combining in silico prediction and ribosome profiling in a genome-wide search for novel putatively coding sORFs. BMC genomics. 2013;14:648. doi: 10.1186/1471-2164-14-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Fritsch C, Herrmann A, Nothnagel M, Szafranski K, Huse K, Schumann F, Schreiber S, Platzer M, Krawczak M, Hampe J, et al. Genome-wide search for novel human uORFs and N-terminal protein extensions using ribosomal footprinting. Genome Research. 2012;22:2208–2218. doi: 10.1101/gr.139568.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Ingolia NT, Lareau LF, Weissman JS. Ribosome Profiling of Mouse Embryonic Stem Cells Reveals the Complexity and Dynamics of Mammalian Proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Lee S, Liu BT, Huang SX, Shen B, Qian SB. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E2424–E2432. doi: 10.1073/pnas.1207846109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Stern-Ginossar N, Weisburd B, Michalski A, Vu TKL, Hein MY, Huang SX, Ma M, Shen B, Qian SB, Hengel H, et al. Decoding Human Cytomegalovirus. Science. 2012;338:1088–1093. doi: 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Chew G-L, Pauli A, Rinn JL, Regev A, Schier AF, Valen E. Ribosome profiling reveals resemblance between long non-coding RNAs and 5′ leaders of coding RNAs. Development. 2013;140:2828–2834. doi: 10.1242/dev.098343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Reid DW, Nicchitta CV. Genome-scale ribosome footprinting identifies a primary role for endoplasmic reticulum-bound ribosomes in the translation of the mRNA transcriptome. Journal of Biological Chemistry. 2011 doi: 10.1074/jbc.M111.312280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Barbosa C, Peixeiro I, Romão L. Gene expression regulation by upstream open reading frames and human disease. PLoS genetics. 2013;9:e1003529. doi: 10.1371/journal.pgen.1003529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Calvo SE, Pagliarini DJ, Mootha VK. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proceedings of the National Academy of Sciences. 2009;106:7507–7512. doi: 10.1073/pnas.0810916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Hood HM, Neafsey DE, Galagan J, Sachs MS. Evolutionary roles of upstream open reading frames in mediating gene regulation in fungi. Annual review of microbiology. 2009;63:385–409. doi: 10.1146/annurev.micro.62.081307.162835. [DOI] [PubMed] [Google Scholar]
- 231.Morris DR, Geballe AP. Upstream open reading frames as regulators of mRNA translation. Molecular and Cellular Biology. 2000;20:8635–8642. doi: 10.1128/mcb.20.23.8635-8642.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Howden AJ, Geoghegan V, Katsch K, Efstathiou G, Bhushan B, Boutureira O, Thomas B, Trudgian DC, Kessler BM, Dieterich DC. QuaNCAT: quantitating proteome dynamics in primary cells. Nature methods. 2013;10:343–346. doi: 10.1038/nmeth.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Varenne S, Buc J, Lloubes R, Lazdunski C. Translation is a non-uniform process: effect of tRNA availability on the rate of elongation of nascent polypeptide chains. Journal of molecular biology. 1984;180:549–576. doi: 10.1016/0022-2836(84)90027-5. [DOI] [PubMed] [Google Scholar]
- 234.Shalgi R, Lindquist S, Burge CB. Widespread regulation of translation by elongation pausing in heat shock. Faseb Journal. 2013;27:1. doi: 10.1016/j.molcel.2012.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Mukherjee N, Jacobs NC, Hafner M, Kennington EA, Nusbaum JD, Tuschl T, Blackshear PJ, Ohler U. Global target mRNA specification and regulation by the RNA-binding protein ZFP36. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Freeberg L, Kuersten S, Syed F. Isolate and sequence ribosome-protected mRNA fragments using size-exclusion chromatography. Nature Methods. 2013:10. [Google Scholar]
- 237.ARTSeq Ribosome Profiling Kits from Epicentre. http://www.immunodiagnostic.fi/en/tuoteuutisten-arkisto-english/271-artseq-ribosome-profiling-kits-from-epicentre.