

Abstract
Effective immunotherapeutic strategies require the ability to generate a systemic antigen-specific response capable of impacting both primary and metastatic disease. We have built on our oncolytic vaccinia GM-CSF strategy by adding recombinant tumor antigen to increase the response in the tumor microenvironment and systemically. In the present study, orthotopic growth of a syngeneic HER2/neu-overexpressing mammary carcinoma in FVB/N mice (NBT1) was associated with increased Gr1+CD11b+ myeloid derived suppressor cells (MDSCs) both systemically and in the tumor microenvironment. This MDSC population had inhibitory effects on the HER2/neu specific Th1 immune response. VVneu and VVGMCSF are recombinant oncolytic vaccinia viruses that encode HER2/neu and GM-CSF, respectively. Naïve FVB mice vaccinated with combined VVneu and VVGMCSF given systemically developed systemic HER2/neu-specific immunity. NBT1 bearing mice became anergic to systemic immunization with combined VVneu and VVGMCSF. Intratumoral VVGMCSF failed to result in systemic antitumor immunity until combined with intratumoral VVneu. Infection/transfection of the tumor microenvironment with combined VVGMCSF and VVneu resulted in development of systemic tumor-specific immunity, reduction in splenic and tumor MDSC, and therapeutic efficacy against tumor. These studies demonstrate the enhanced efficacy of oncolytic vaccinia virus recombinants encoding combined tumor antigen and GM-CSF in modulating the microenvironment of MDSC-rich tumors.
Keywords: cancer vaccine, myeloid derived suppressor cells, HER2, oncolytic virus, vaccinia vector
Introduction
The goal of tumor immunotherapy is the development of modalities given alone or in combination that result in the generation of an effective systemic tumor-specific immune response by the host that can overcome immune escape mechanisms. The identification of immunogenic targets unique to or overexpressed by tumor cells is critical to achieving this goal. The HER2/neu oncogene encodes Human Epidermal growth factor Receptor 2 (HER2/neu), a member of the Epidermal Growth Factor Receptor (EGFR) family of transmembrane tyrosine kinase receptors, which participates in processes including physiology, proliferation, and differentiation of various human tissues 1, 2. Overexpression of HER2/neu is found in approximately 20% of invasive breast cancers, and is associated with a more invasive phenotype and a poorer prognosis 3. Development of an active immune response using a vaccine targeting HER2/neu represents an attractive immunotherapeutic strategy for overcoming immune escape mechanisms induced by the tumor microenvironment.
Myeloid derived suppressor cells (MDSCs), a population of immature myeloid cells that are increased systemically and in the tumor microenvironment of both murine cancer models and human malignancies, are prominent contributors to tumor immune escape 4, 5. This heterogeneous population is characterized phenotypically in mice by the cell surface antigens CD11b and Gr-1 5. Gr-1 encompasses two subtypes, Ly-6C and Ly-6G, which have been used to further differentiate MDSCs into CD11b+Ly-6Chigh Ly-6G− monocytic (mMDSC) and CD11b+Ly-6ClowLy-6G+ granulocytic (gMDSC) subpopulations, respectively 6, 7. Consistent with their heterogeneous phenotype, MDSCs suppress the anti-tumor immune response through multiple mechanisms 8. MDSCs interfere with lymphocyte proliferation via deprivation of essential amino acids such as arginine and cysteine 7, 9, 10. They also mediate oxidative stress via production of reactive oxygen species (ROS) and peroxynitrate, which leads to nitration of tyrosine in CD8 and the T cell receptor (TCR) and, thus, changes in the rigidity the TCR 11. Furthermore, MDSCs support induction of other immune inhibitory populations such as regulatory T cells (Tregs) through the production of Transforming Growth Factor-β (TGF-β) and IL-10 12–15. Given these immune suppressive effects, therapies that can overcome systemic anergy induced by MDSCs have generated great interest.
Studies from our group were the first to develop and test recombinant Vaccinia vectors encoding the immune-enhancing GM-CSF for the localized treatment of solid tumors. In preclinical studies we demonstrated that vaccinia and vaccinia recombinants were effective in infecting/transfecting tumors and importantly that despite the immunogenicity of the vaccinia vector, high levels of transfection could be obtained following repeated injections of tumor in mice 16 and subsequently in patients with recurrent superficial melanoma 17. We developed and took clinical VVGMCSF into Phase I trials in melanoma 18. Subsequent to our studies, this recombinant (JX-594) was shown to have antitumor activity in preclinical models and clinical trials in a number of diseases 19, 20.
In the present study using orthotopic growth of an aggressive HER2/neu expressing murine tumor characterized by high levels of CD11b+Gr-1+ MDSCs in the tumor microenvironment and systemically that suppressed HER2/neu-specific Th1, we show that intratumoral treatment with the oncolytic VVGMCSF is ineffective at reducing tumor growth nor does it lead to the development of a systemic tumor specific immune response. However, when combined with a neu-encoding vaccinia VVneu and administered into the tumor microenvironment, mice develop systemic anti-neu immunity, significant reduction in tumoral and systemic MDSC, and manifest a major antitumor response. The same virus combination (vaccine) fails to generate a similar response when given systemically in NBT1 bearing mice. We characterize the presence and function of the MDSC response and the resultant CTL and interferon responses to HER2/neu. These results point to the ability of the tumor microenvironment to both promote immune escape and act as an effective vaccination site for tumor antigen-encoding oncolytic viruses that can result in a systemic immune response capable of mediating tumor regression.
Materials and Methods
Cell Culture
The HER2/neu expressing NBT1 mammary tumor cell line was derived by serial cell culture of tumor cells obtained from a spontaneous breast tumor that arose in a FVB/neuT mouse transgenic for a HER2/neu receptor that is constitutively activated by a point mutation in the transmembrane region. NBT1 was maintained in media composed of CMRL-1066 (Invitrogen, Carlsbad, CA) supplemented with 10% FBS, 2mM L-glutamine, 50 IU/mL penicillin/streptomycin, and 4μM Dexamethasone. Splenic cell cultures used in immune analyses were carried out using a supplemented RPMI-1640 media as described 21. The Sf9 and Sf21 cell lines (Invitrogen) were maintained in Grace’s Supplemented Medium (Invitrogen) supplemented with 10% FBS and 50 IU/ml penicillin/streptomycin. HER2/neu Sf21 cell lysates used for in vitro restimulation of CTL assay described below were prepared by resuspending cells in RPMI medium with Protease Inhibitor Cocktail (Sigma-Aldrich), lysing via 4 freeze/thaw cycles, sonicating for 2 minutes, and centrifugation for 15 minutes at 13,500 rcf.
Construction of recombinant vaccinia virus expressing HER2/neu (VVneu)
The rat neu cDNA was cloned from pSV2-neuNT (provided by R.A. Weinberg), ligated into pBluescript (Stratagene, La Jolla, CA) using SalI and Hind III restriction sites, and then removed from pBluscript using SalI and NotI. It was cloned into vaccinia recombination plasmid pSC11/9. VVGMCSF was prepared as previously described 21–23. VVβGal was provided as a negative control by Dr. Laurence Eisenlohr (Thomas Jefferson University, Philadelphia, PA).
Construction of Recombinant Baculovirus Expressing rat HER2/neu
The rat neu cDNA coding sequence was assembled in the pFASTBac/CT-TOPO recombination vector (Invitrogen). Rat neu transforming cDNA was excised from a vaccinia recombination plasmid containing the full length rat neuNT cDNA sequence 24 in the vector pSC11/9 using NotI and SalI restriction enzymes. The 4727bp rat neu DNA was gel purified, then ligated to pBluescript II SK(+) (Stratagene) at Not/Sal to produce plasmid BSratneuNT. This neuNT-containing plasmid was used to generate 3 DNA fragments which, when ligated together, prepared the rat neuNT coding sequences for ligation into pFASTBac/CT-TOPO. pBSratneuNT was first digested with HincII which cut at two sites: a HincII site at residue 1599 and the SalI site at the 3′-end of the neuNT cDNA, residue 4622. HincII digestion released a 3023bp fragment, residues 1599-4622, which contained two problematic NcoI sites. The HincII-cut vector was gel purified, religated, and cloned then digested with NcoI. Filling in the NcoI site, located at position -19, with T4 DNA polymerase followed by ligation transformed the NcoI site into a NsiI site, creating plasmid BSNeuNsi. Plasmid BSNeuNsi was digested with both SbfI and SalI releasing a 1366bp fragment and leaving 338bp of cDNA containing 233bp of 5′ coding sequence. Plasmid pBSratneuNT was next digested with both SbfI and SalI releasing a 4389bp fragment, residues 234-4622, which, after gel purification, was cut with NcoI to generate a 2775bp middle fragment comprising residues 234-3008. Lastly pBSratneuNT was PCR-amplified using primers NTNco (5′-gcatagcggccgccatggacagtaccttctaccgtt-3′) and NTSal (5′-ctacgcgtcgacacaggtacatccaggcctaggtac-3′) to produce a 768bp fragment of neu coding sequence comprising residues 3009-3777. At its 3′ end this amplicon contained a SalI site for cloning and a final residue codon mutation of GTA to GTG. The NTNco/NTSal PCR amplicon--residues 3009-3777--was used without subcloning in a three-way ligation with the SbfI/NcoI middle fragment—residues 234-3008—and the plasmid BSNeuNsi--residues -105-233—cut with SbfI/SalI. Following transformation, clone selection, and DNA sequencing, a correct full-length neuNT cDNA clone was identified, pBSNeuNsiSal. This plasmid was cut with both NsiI and SalI to release the neuNT cDNA and treated with mung bean nuclease to remove 3′ and 5′ overhangs generating a blunt-end cDNA. Treatment with Antarctic alkaline phosphatase removed terminal phosphates allowing neuNT cDNA to be TOPO cloned into pFASTBac/CT-TOPO vector. The recombinant bacmid was transfected into the Sf9 cell line to produce P1baculovirus stock expressing HER2/neu (bac HER2/neu), which was further amplified with several rounds of infection of Sf21 cells. Bac HER2/neu was used to infect Sf21 cells. Infected HER2/neu Sf21 cells were pelleted and used to generate Sf21neu lysate for restimulation and ELISA as described below.
Animal Experiments
Four to 6-week old FVB/N mice (Jackson Labs, Bar Harbor, ME) were maintained in a HEPA-filtered cage system for at least 1 week prior to use. For in vivo experiments involving tumor-bearing mice, anesthetized FVB/N mice were injected using a 27 Gauge tuberculin syringe (Becton Dickinson, Franklin Lakes, NJ) into the right second mammary fat pad with 2×106 NBT1 cells suspended in Hanks Balanced Salt Solution (Sigma-Aldrich) as previously described 25. For in vivo vaccination studies in naïve FVB/N mice, mice received 2 s.c. or intra-mammary fat pad (i.m.f.) injections, 2 weeks apart, using a cocktail of 1×106 pfu VVGMCSF plus either 1×106 pfu VVneu or VVβGal using a 27 Gauge tuberculin syringe. In in vivo vaccination studies of NBT1 tumor-bearing mice, mice were treated with 2 s.c. or i.t. injections, 2 weeks apart, of 1×106 pfu VVGMCSF plus 7.5μg KLH (Sigma-Aldrich) with either 1×106 pfu VVneu or VVβGal. For vaccinations, i.m.f. injections were made directly into the number 2 fat pad. All intratumoral (i.t.) injections were made directly into the mammary fat pad tumors. S.c. injections were placed in the contralateral (left) groin. All viral injections used the same total of 2×106 vaccinia PFU. Five animals per group were used for each independent in vivo experiment. Animal experiments were conducted in accordance with protocols approved by the Rutgers Institutional Animal Care and Use Committee.
CTL assays
CTL assays were performed as previously described 21, 26, with several modifications. Briefly, effector cells were prepared from splenocytes, vaccination site draining lymph nodes (VDN), or tumor draining lymph nodes (TDN) of treated/vaccinated FVB/N mice. Spleens or lymph nodes were homogenized and red blood cells were lysed using ammonium chloride buffer (ACK buffer, 0.15M NH4Cl, 1.0mM KHCO3, 0.1mM EDTA), washed with TCM and filtered through a 70μm nylon mesh (BD Biosciences, San Jose, CA). Effector cells were resuspended at 7×106 cells/ml and cultured with 3×106 cells/ml of irradiated (25 Gray) splenocytes from naïve FVB/N mice that had been incubated overnight in 50ml conicals at 37°C at 4×106 cells/ml in TCM + 2-ME with 300ug/ml of the RNEU420-429 (PDSLRDLSVF) (Genscript, Piscataway, NJ) immunodominant epitope of the rat neu protein 27, lymphocytic choriomeningitis virus nucleoprotein NP118-126 (RPQASGVYM) control peptide (Genscript), or indicated cell lysates. Restimulation cultures were in a total of 2mL of TCM + 2-ME in 24-well plates for 5 days at 37°C, 5% CO2. On day 3 of restimulation, 50μL of supernatant was assayed for IFN-γ using ELISA as described previously 28. On day 5, stimulated effector cells were harvested and cultured for 4 hours with 51Cr -labeled NBT1 cells as targets. 100μL of supernatant were removed and 51Cr release of target cells was measured with a gamma counter (Packard Bioscience, Meriden, CT). Percent specific lysis was calculated from the formula [(experimental release − spontaneous release) × 100/(maximal release in 1% SDS − spontaneous release)].
Magnetic Bead Depletion of MDSCs
MDSCs were depleted from the splenocyte population of NBT1 tumor-bearing mice using a mouse MDSC Isolation Kit (Miltenyi Biotec, Auburn, CA) as per manufacturer’s instructions. Briefly, 1×108 splenocytes were resuspended in 350μl MACS buffer (PBS, 0.5% BSA, 2mM EDTA) and blocked for 10 minutes with 50μl FcR Blocking Reagent at 4°C. 100μl of anti-Ly6G-Biotin was added and incubated for 10 minutes at 4°C. Cells were washed in 10ml MACS buffer and resuspended in 100ul MACS buffer. 200μl anti-biotin microbeads were added and incubated at 4°C for 15 minutes. Magnetic separation was performed using LS disposable column and MACS Magnetic separator (Miltenyi Biotec). The negative fraction was collected and restimulated as described above. MDSC depletion was validated using flow cytometry.
Flow Cytometry
Flow cytometry for RNEU420-429-tetramer (provided by the NIAID Tetramer Facility, Atlanta, GA) was conducted as described previously 21. Briefly, effectors from treated mice were restimulated for 5 days as described above. Effectors were harvested, washed, and resuspended in PBS/5% FBS with 0.1% w/v sodium azide at 1×106 cells/100μL. Cells were stained with anti-CD8α-FITC at manufacturer’s recommended concentration (BD Biosciences, San Jose, CA) and the RNEU420-429 -specific tetramer labeled with phycoerythrin (1:100 dilution). Flow cytometry for MDSC was conducted on splenocytes, LN cells, and tumor suspensions straight ex vivo. Tumor suspensions were prepared as described previously 29, with modification. Tumors were cut into small pieces and incubated at 37°C with gentle shaking for 20 minutes in 5ml RPMI with collagenase D (1mg/ml, Roche). Tumor pieces were dissociated with a metal strainer, washed in TCM + 2-ME, red blood cells were lysed using ACK buffer, washed again with TCM and filtered through a 70μm nylon mesh (BD Biosciences, San Jose, CA). Cells were stained using anti-Gr-1 PE (eBioscience, San Diego, CA), CD11b FITC (eBioscience), anti-Ly6G APC (BD Biosciences), and anti-Ly6C PE-Cy7 (eBioscience). Flow cytometry data was acquired using a FC-500 flow cytometer (Beckman Coulter, Miami, FL) and analyzed using CXP (Beckman Coulter) provided by the CINJ flow cytometry shared resource.
Statistical Analysis
Results were expressed as mean +/− Standard Error. Significance for experiments with >2 conditions (P < 0.05) was determined by Analysis of Variance (ANOVA) with post-hoc Tukey multiple comparisons test using the InStat software package (GraphPad Software, La Jolla, CA). Significance for experiments with 2 conditions was determined using Student’s T-test, two-tailed with unequal variances in Microsoft Excel (Microsoft, Redmond, WA). Kaplan-meier survival curves were generated using Prism (GraphPad Software), with significance calculated using Log-Rank (Mantel-Cox) Test and correction for multiple comparisons using Bonferroni method.
Results
Vaccination of naïve FVB/N mice results in a systemic HER2 MHC Class I epitope-specific CTL response
We hypothesized that vaccination of a naïve host with a vaccinia construct expressing HER2/neu would induce an antigen-specific Th1 immune response, regardless of the site of vaccination. We utilized intra-mammary fat pad (i.m.f.) injection or contralateral subcutaneous (s.c.) injection of VVneu + VVGMCSF in naïve FVB/N mice. Two weeks after the final vaccination, splenocytes and draining lymph nodes of the vaccination site (VDN) were restimulated with irradiated splenocytes from naïve female FVB/N mice that had been pulsed with immunodominant RNEU420-429 peptide (Fig. 1a). Both s.c. and i.m.f. vaccination with VVneu and VVGMCSF resulted in increased percent specific lysis of 51Cr-labeled NBT1 tumor cells by splenic CTLs in vitro, when compared to controls (Fig. 1b).
Fig. 1. Vaccination of naïve FVB/N mice with VVneu induces a systemic HER2-specific Th1 response.
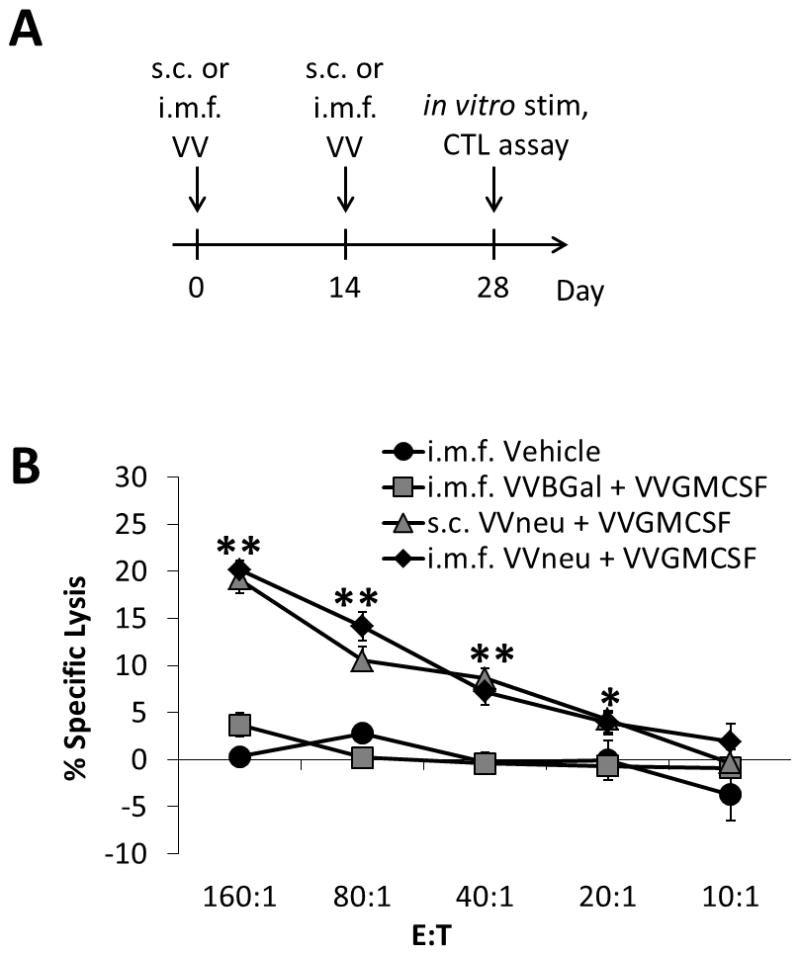
(a) Female FVB/N mice were injected twice, two weeks apart, with VVneu + VVGMCSF (either s.c. or i.m.f.), VVBGal + VVGMCSF (i.m.f.), or vehicle (i.m.f.). Two weeks after the second and final vaccination, spleens were restimulated with irradiated splenocytes from naïve female FVB/N mice that had been pulsed with immunodominant RNEU420-429 peptide. (b) NBT1 tumor-specific systemic (spleen) CTL activity against NBT1 target cells after restimulation. Differences of both s.c. and i.m.f. VVneu + VVGMCSF groups compared to controls were significant (** P < 0.01, * P < 0.05). Results are representative of 3 independent experiments.
Myeloid Derived Suppressor Cells (MDSCs) suppress the systemic anti-tumor immune response
MDSCs are a heterogeneous population of immature granulocytic and monocytic cells that have inhibitory effects on tumor-specific T cell activation and function 5, 6, 30. Given previous studies demonstrating the importance of a Type 1 anti-tumor response in effective immune therapies 21, 28, we hypothesized that MDSCs inhibited a systemic anti-tumor immune response in the NBT1 model. To determine whether the presence of orthotopic NBT1 tumor affected levels of MDSC in FVB/N mice, we injected FVB/N mice with NBT1 cells in the right number 2 mammary fat pad. After 4–5 weeks of tumor growth, we used flow cytometry to measure levels of Gr-1, Ly-6G, Ly-6C and CD11b on cells from spleen and tumor. The CD11b+Gr1+ population was significantly increased in the systemic population of tumor-bearing mice compared to naïve mice (Fig. 2a). A large population of CD11b+Ly-6CintLy6G+ cells was detected, a phenotype consistent with granulocytic MDSCs (gMDSCs, Fig. 2b).
Fig. 2. NBT1 growth in female FVB/N mice leads to increased levels of Ly6G+ gMDSC, both systemically and in the tumor microenvironment.

(a) Flow cytometry was performed on spleen, TDN, and tumor samples stained for Gr-1, CD11b, Ly-6C, and Ly-6G. Levels of CD11b+Gr1+ cells in spleen, TDN, and tumor were compared to levels in spleen and axillary LN of naïve female FVB/N mice. Cumulative results are presented from 5 independent experiments (*P < 0.05). (b) Cells from spleen and tumor were gated to include monocytic and granulocytic populations for evaluation of Gr-1 and CD11b. This population was then further gated for CD11b+ cells, which were evaluated for Ly-6G and Ly-6C. Results are representative of 3 independent experiments.
To demonstrate the immune suppressive nature of the MDSCs induced by NBT1, we used a MDSC magnetic bead isolation kit to deplete Ly6G+ cells from the splenocyte population from NBT1 tumor-bearing mice. We validated depletion of MDSCs from the negative fraction using flow cytometry staining for CD11b, Gr-1, Ly6G, and Ly6C (Fig. 3a). We then restimulated this MDSC-depleted splenocyte population with irradiated splenocytes from naïve female FVB/N mice that had been pulsed with immunodominant MHC Class I RNEU420-429 peptide or HER2/neu protein in Sf21 neu baculovirus-infected lysate, with NP118-126 peptide and unmodified Sf21 cells used as restimulation controls. IFN-γ production by MDSC-depleted splenocytes was increased in most conditions compared to pre-depletion controls, with HER2/neu restimulated effectors significantly increased compared to all other conditions (Fig. 3b).
Fig. 3. MDSCs suppress a HER2/neu-specific Th1 response in NBT1 tumor-bearing mice.

MDSCs from spleen of NBT1 tumor-bearing mice were depleted using a MDSC (Ly6G+) magnetic bead isolation kit. (a) Flow cytometry was performed on pre and post-depletion samples stained for Gr-1, CD11b, Ly-6C, and Ly-6G. (b) The MDSC-depleted negative fraction was restimulated with irradiated naïve splenocytes pulsed with control NP118-126 peptide, RNEU420-429 peptide, lysate from Sf21 insect cells that had been infected with baculovirus expressing HER2/neu, and control Sf21 cell lysate. IFN-γ ELISA was performed in triplicate on supernatant from MDSC-depleted effectors. Results representative of 3 independent experiments. (†<0.001 compared to all other stimulation conditions.)
Vaccination with VVneu + VVGMCSF into the tumor microenvironment, but not VVGMCSF alone, results in a systemic HER2-specific CTL response, decreased MDSCs, and tumor regression
We asked whether vaccination with HER2/neu antigen-encoding VVneu and VVGMCSF could overcome MDSC-associated anergy against HER2/neu induced by the NBT1 model. After two weeks of orthotopic NBT1 tumor growth, two doses of VVneu + VGMCSF + KLH, two weeks apart, were administered subcutaneously (s.c.) or by injection into the tumor microenvironment (i.t., Fig 4a). Keyhole Limpet Hemocyanin (KLH) was added to all treatment groups and controls as previous studies by our group and others had demonstrated its effectiveness as a vaccine adjuvant21, 31, 32. Two weeks after the final vaccination, splenocytes and vaccination-site draining lymph nodes (VDN) were restimulated with irradiated splenocytes from naïve female FVB/N mice that had been pulsed with immunodominant RNEU420-429 peptide. Splenocytes from mice vaccinated with i.t. VVneu + VVGMCSF + KLH showed significantly increased ability to lyse target NBT1 cells after in vitro restimulation when compared to both equivalent s.c. vaccination and i.t. VVBGal + VVGMCSF + KLH (Fig. 4b). Similarly, cytolytic activity of the VDN population was highest in mice given i.t. VVneu + VVGMCSF + KLH (Fig. 4c). Increased CTL activity was specific to the RNEU420-429 MHC Class I epitope, as restimulation with control NP118-126 did not result in increased cytolytic activity (not shown). The percentage of restimulated CD8+ lymphocytes that were RNEU tetramer-positive from both spleen and VDN was significantly higher in mice that were treated with i.t. VVneu + VVGMCSF + KLH than in all other groups (Fig. 4d).
Fig. 4. Vaccination with VVneu into the tumor microenvironment generates a systemic antitumor CTL response.

(a) Female FVB/N mice were injected with 2×106 NBT1 tumor cells into the right second mammary fat pad. Mice were treated twice (days 14 and 28) with VVneu + VVGMCSF + KLH (s.c. or i.t.), VVBGal + VVGMCSF + KLH (i.t.), or vehicle control (i.t.). On day 42, spleens and VDN were restimulated with irradiated naïve splenocytes that had been pulsed with RNEU420-429 peptide. (b) Ability of splenocyte effectors to lyse target NBT1cells was measured by 51Cr release assay. Difference between i.t. VVneu + VVGMCSF + KLH and all other conditions was significant (** P < 0.01). (c) Tumor-specific CTL activity of VDN effector cells was measured by 51Cr lysis. Differences between i.t. VVneu + VVGMCSF + KLH and all other groups were statistically significant (** P < 0.01, * P < 0.05). (d) Restimulated effectors were evaluated for RNEU tetramer-positive CD8+ cells using flow cytometry. The percent of CD8+ cells that are RNEU tetramer positive was measured, with representative dot plots and cumulative results of 3 independent experiments presented (**P < 0.01).
MDSC levels were evaluated using flow cytometry. In mice treated with i.t. VVneu + VVGMCSF + KLH, MDSC levels in spleen on day 28 were significantly decreased compared to i.t. Vehicle (Fig. 5). Systemic MDSC levels were not significantly decreased in mice treated with s.c. VVneu + VVGMCSF + KLH. Notably, tumor-bearing mice treated with i.t. VVBGal + VVGMCSF + KLH exhibited a significant increase in intratumoral, though not systemic, MDSC levels (Fig. 5). A potential explanation may be that, in addition to its enhancing effects on antigen presentation, GM-CSF is one of multiple cytokines that support development of MDSCs 30.
Fig. 5. Vaccination with VVneu into the tumor microenvironment results in decreased levels of systemic and tumor microenvironment MDSCs.
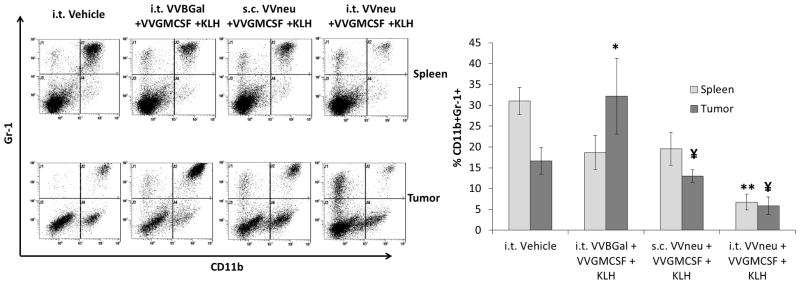
Splenocytes and tumor were stained ex vivo for Gr-1 and CD11b using flow cytometry on day 28. Representative dot plots and combined mean %CD11b+Gr-1+ cells from 3 independent experiments are shown (*P < 0.05 compared to i.t. Vehicle tumor, **P < 0.01 compared to i.t. Vehicle spleen, ¥P < 0.05 compared to i.t. VVBGal + VVGMCSF + KLH tumor; all other comparisons not significant).
Primary tumors in mice vaccinated with i.t. VVneu + VVGMCSF + KLH regressed from peak size and were significantly smaller than all other treatment conditions on day 42, including equivalent s.c. VVneu and i.t. VVGMCSF without antigen (Fig. 6a, b, c). Subcutaneous VVneu + VVGMCSF + KLH vaccination or i.t. VVBGal + VVGMCSF + KLH did not inhibit NBT1 growth.
Fig. 6. Vaccination with VVneu into the tumor microenvironment results in regression of NBT1 tumor.

(a) Mean tumor size and standard deviation from 5 combined independent experiments are shown. (b) Day 42 tumor size from 5 combined experiments, each point representing one mouse from indicated treatment condition († P < 0.001). (c) Kaplan-meier survival curve from 5 combined experiments († P < 0.001).
Discussion
The studies presented here demonstrate using an aggressive orthotopic model of HER2/neu driven mammary tumor (NBT1) that growth of the tumor results in a significant infiltration of intratumor and systemic MDSC resulting in the lack of development of systemic immunity and anergy to peripheral s.c. immunization using a neu encoding vaccinia vaccine that is effective in naïve (non tumor bearing) mice. Treatment of tumor bearing mice using intratumoral oncolytic VVGMCSF fails to induce systemic immunity nor does it lead to tumor regression. In fact, intratumoral VVGMCSF and resulting GM-CSF expression in tumor leads to a significant expansion of tumor resident MDSC as suggested by the Vonderheide group 33. However, when tumor antigen encoding VVneu is combined with VVGMCSF, thus providing virally expressed tumor antigen to the tumor microenvironment, the combination results in the generation of systemic neu-specific immunity, a significant reduction in tumor and systemic MDSC, and a significant antitumor response. We confirmed the contribution of MDSCs to immune escape in our model by using flow cytometry to identify a population of CD11b+ Ly-6CintLy-6G+ cells. We also demonstrated that these cells have suppressive effects by finding, despite an overall increased background of IFN-γ in MDSC-depleted cultures, increased HER2/neu-specific IFN-γ production of the systemic splenocyte population after depletion of Ly-6G+ cells.
The goal of tumor immunotherapy is the development of modalities given alone or in combination that result in the generation of an effective systemic tumor-specific immune response by the host that can overcome immune escape mechanisms. Original studies from our laboratory using a murine bladder cancer model demonstrated the unexpected expansion of antigen-specific (tetramer positive) CD8+ T cells in the tumor microenvironment but not systemically 21. This led us to immunize intratumorally using a vaccinia encoding vaccine with resultant induction of systemic T cell immunity 21. Coincidently, studies from the Schlom group using a colon cancer model demonstrated the induction of significant antitumor immunity when priming or boosting immunizations were given intratumorally 34. These initial responses to intratumoral poxvirus based vaccines have been translated to Phase I trials in prostate cancer 35 and by us in pancreatic cancer 36 using antigen encoding non-replicating fowlpox vectors.
The strategy of delivering oncolytic virus to tumor has long been studied with the overall hypothesis being that virus-mediated oncolysis could have primary antitumor effectiveness and as a sequellae of tumor lysis, antigen released into the tumor microenvironment could lead to a consolidating systemic antitumor response active against metastases. As recently reviewed by Lichty et al, preclinical and clinical studies are ongoing that use a variety of oncolytic vectors and encoded immune regulatory molecules 37. Our initial studies focused on the potential of generating systemic immunity using the oncolytic VVGMCSF which would have the potential of lysing tumor, eliciting antigen, and enhancing antigen presentation via the GM-CSF. This agent studied by others as JX-594 was subsequently characterized as to its bioavailability, and antitumor and immune responses 19, 20, 38. Of particular note in these studies, JX-594 was shown to preferentially persist and replicate in the tumor microenvironment following i.v. administration in preclinical models and in patients 19, 20. Given that accessibility of tumor for local administration is a limitation to the use of these approaches, this finding suggests that the uniqueness of the vaccinia platform may allow systemic delivery of virus, immune modulating encoded cytokines as, well as antigen as we present here. Clinical studies using another DNA viral vector HSV-1-GMCSF construct (OncoVEX GM-CSF ) have also demonstrated local infectivity, antitumor activity, and local and systemic immune responses 39, 40 further supporting the potential of this approach. Ongoing preclinical and clinical studies combining oncolytic virus with other modalities such as chemotherapy and immune checkpoint inhibitors are also under development and ongoing by a number of investigators 41, 42.
In conclusion, the studies presented here provide support for delivering to the tumor microenvironment a combination of virally encoded tumor antigen and immunomodulating GM-CSF, both encoded by the oncolytic vaccinia vector. Our studies demonstrate the requirement for antigen delivered to the microenvironment in overcoming MDSC-based immune escape and subsequent development of a systemic anti-tumor immune response in our model. Our and others’ studies have proven the feasibility of using DNA viral vectors to deliver multiple gene constructs to the tumor microenvironment via direct injection 18–20, 38–40 and in some cases following intravenous administration 19, 20. These combined studies support the conclusion that this approach has the potential of impacting tumor alone and supports future approaches combining this with added immune targeted and antitumor approaches.
Acknowledgments
Studies were supported by NCI R01 CA42908 and the CINJ shared resources supported by NCI P30 CA72720.
Footnotes
Conflict of Interest
Dr. Lattime is an inventor of the patented recombinant Vaccinia-GMCSF which has been licensed to Sillajen and is being studied as JX-594. As such, he derives royalties and licensing fees from Thomas Jefferson University where the patent is held. Dr. de Vries and Dr. Monken have no conflicts.
References
- 1.Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW, Jr, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421(6924):756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 2.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nature Reviews Molecular Cell Biology. 2001;2(2):127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 3.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 4.Ostrand-Rosenberg S. Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunology, Immunotherapy : CII. 2010;59(10):1593–1600. doi: 10.1007/s00262-010-0855-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. Journal of immunology. 2009;182(8):4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Youn J-I, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. Journal of immunology. 2008;181(8):5791–802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Youn J-I, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. Journal of Leukocyte Biology. 2012;91 (1):167–81. doi: 10.1189/jlb.0311177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nature Reviews Immunology. 2012;12(4):253–68. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Z, Pang Y, Gara SK, Achyut BR, Heger C, Goldsmith PK, et al. Gr-1+CD11b+ cells are responsible for tumor promoting effect of TGF-beta in breast cancer progression. International Journal of Cancer. 2012;131(11):2584–95. doi: 10.1002/ijc.27572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-Derived Suppressor Cells Inhibit T-Cell Activation by Depleting Cystine and Cysteine. Cancer Research. 2009;70(1):68–77. doi: 10.1158/0008-5472.CAN-09-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nature Medicine. 2007;13(7):828–35. doi: 10.1038/nm1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takaku S, Terabe M, Ambrosino E, Peng J, Lonning S, McPherson JM, et al. Blockade of TGF-[beta] enhances tumor vaccine efficacy mediated by CD8+ T cells. International Journal of Cancer. 2009 doi: 10.1002/ijc.24961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goulart MR, Pluhar GE, Ohlfest JR. Identification of myeloid derived suppressor cells in dogs with naturally occurring cancer. PloS one. 2012;7(3):e33274–e33274. doi: 10.1371/journal.pone.0033274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mundy-Bosse BL, Young GS, Bauer T, Binkley E, Bloomston M, Bill Ma, et al. Distinct myeloid suppressor cell subsets correlate with plasma IL-6 and IL-10 and reduced interferon-alpha signaling in CD4+ T cells from patients with GI malignancy. Cancer Immunology, Immunotherapy : CII. 2011;60(9):1269–79. doi: 10.1007/s00262-011-1029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terabe M, Matsui S, Park JM, Mamura M, Noben-Trauth N, Donaldson DD, et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. Journal of Experimental Medicine. 2003;198(11):1741–52. doi: 10.1084/jem.20022227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Intravesical gene therapy: Vaccinia virus recombinants transfect murine bladder tumors and urothelium. Proceedings of the American Association for Cancer Research; March 1993; 1993. [Google Scholar]
- 17.Mastrangelo MJ, Maguire HC, McCue PA, Lee SS, AA, LNN A pilot study demonstrating the feasibility of using intratumoral vaccinia injections as a vector for gene transfer. Vaccine Res. 1995;4(2):55–69. [Google Scholar]
- 18.Mastrangelo MJ, Maguire HC, Jr, Eisenlohr LC, Laughlin CE, Monken CE, McCue PA, et al. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Therapy. 1999;6(5):409–22. doi: 10.1038/sj.cgt.7700066. [DOI] [PubMed] [Google Scholar]
- 19.Parato KA, Breitbach CJ, Le Boeuf F, Wang J, Storbeck C, Ilkow C, et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther. 2012;20(4):749–58. doi: 10.1038/mt.2011.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQM, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477(7362):7102–7362. doi: 10.1038/nature10358. [DOI] [PubMed] [Google Scholar]
- 21.Yang AS, Monken CE, Lattime EC. Intratumoral vaccination with vaccinia-expressed tumor antigen and granulocyte macrophage colony-stimulating factor overcomes immunological ignorance to tumor antigen. Cancer Research. 2003;63(20):6956–6961. [PubMed] [Google Scholar]
- 22.Chakrabarti S, Sisler JR, Moss B. Compact, synthetic, vaccinia virus early/late promoter for protein expression. BioTechniques. 1997;23(6):1094–1097. doi: 10.2144/97236st07. [DOI] [PubMed] [Google Scholar]
- 23.Eisenlohr LC, Yewdell JW, Bennink JR. Flanking sequences influence the presentation of an endogenously synthesized peptide to cytotoxic T lymphocytes. The Journal of Experimental Medicine. 1992;175(2):481–487. doi: 10.1084/jem.175.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bargmann CI, Hung M-C, Weinberg RA. Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell. 1986;45(5):649–657. doi: 10.1016/0092-8674(86)90779-8. [DOI] [PubMed] [Google Scholar]
- 25.Rusciano D, Burger MM. Cancer metastasis: experimental approaches (laboratory techniques in biochemistry and molecular biology) In: Welch DR, editor. Cancer Metastasis: Experimental Approaches. Elsevier Science Publ. Co; Amsterdam, Netherlands: 2000. [Google Scholar]
- 26.McAveney KM, Gomella LG, Lattime EC. Induction of TH1- and TH2-associated cytokine mRNA in mouse bladder following intravesical growth of the murine bladder tumor MB49 and BCG immunotherapy. Cancer Immunology, Immunotherapy : CII. 1994;39(6):401–406. doi: 10.1007/BF01534428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ercolini AM, Machiels JP, Chen YC, Slansky JE, Giedlen M, Reilly RT, et al. Identification and characterization of the immunodominant rat HER-2/neu MHC class I epitope presented by spontaneous mammary tumors from HER-2/neu-transgenic mice. Journal of immunology. 2003;170(8):4273–4280. doi: 10.4049/jimmunol.170.8.4273. [DOI] [PubMed] [Google Scholar]
- 28.Nikitczuk KP. PLGA-Polymer Encapsulating Tumor Antigen and CpG DNA Administered into the Tumor Microenvironment Elicits a Systemic Antigen-Specific IFN-γ Response and Enhances Survival. Journal of Cancer Therapy. 2013;04(01):280–290. doi: 10.4236/jct.2013.41035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sinha P, Chornoguz O, Clements VK, Artemenko KA, Zubarev RA, Ostrand-Rosenberg S. Myeloid-derived suppressor cells express the death receptor Fas and apoptose in response to T cell-expressed FasL. Blood. 2011;117(20):5381–90. doi: 10.1182/blood-2010-11-321752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abe F, Dafferner AJ, Donkor M, Westphal SN, Scholar EM, Solheim JC, et al. Myeloid-derived suppressor cells in mammary tumor progression in FVB Neu transgenic mice. Cancer Immunology, Immunotherapy : CII. 2010;59(1):47–62. doi: 10.1007/s00262-009-0719-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riggs DR, Jackson B, Vona-Davis L, McFadden D. In vitro anticancer effects of a novel immunostimulant: keyhole limpet hemocyanin. The Journal of surgical research. 2002;108(2):279–84. doi: 10.1006/jsre.2002.6548. [DOI] [PubMed] [Google Scholar]
- 32.Sabbatini PJ, Ragupathi G, Hood C, Aghajanian CA, Juretzka M, Iasonos A, et al. Pilot study of a heptavalent vaccine-keyhole limpet hemocyanin conjugate plus QS21 in patients with epithelial ovarian, fallopian tube, or peritoneal cancer. Clin Cancer Res. 2007;13(14):4170–7. doi: 10.1158/1078-0432.CCR-06-2949. [DOI] [PubMed] [Google Scholar]
- 33.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822–35. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kudo-Saito C, Schlom J, Hodge JW. Intratumoral vaccination and diversified subcutaneous/intratumoral vaccination with recombinant poxviruses encoding a tumor antigen and multiple costimulatory molecules. Clinical Cancer Research. 2004;10(3):1090–1099. doi: 10.1158/1078-0432.ccr-03-0145. [DOI] [PubMed] [Google Scholar]
- 35.Gulley JL, Heery CR, Madan RA, Walter BA, Merino MJ, Dahut WL, et al. Phase I study of intraprostatic vaccine administration in men with locally recurrent or progressive prostate cancer. Cancer Immunol Immunother. 2013;62(9):1521–31. doi: 10.1007/s00262-013-1448-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Repaka A, Poplin Ea, August Da, Ben-Menachem T, Michael H, Artymyshyn R, et al. 350 Phase I Trial of Endoscopic Ultrasound (EUS) Guided Intratumoral Vaccination With Recombinant Panvac-F and Systemic Panvac-V in Patients With Locally Advanced Pancreatic Cancer. Gastrointestinal Endoscopy. 2013;77(5, Supplement):AB143. [Google Scholar]
- 37.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14(8):559–67. doi: 10.1038/nrc3770. [DOI] [PubMed] [Google Scholar]
- 38.Park BH, Hwang T, Liu TC, Sze DY, Kim JS, Kwon HC, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. The Lancet Oncology. 2008;9(6):533–42. doi: 10.1016/S1470-2045(08)70107-4. [DOI] [PubMed] [Google Scholar]
- 39.Hu JC, Coffin RS, Davis CJ, Graham NJ, Groves N, Guest PJ, et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 2006;12(22):6737–47. doi: 10.1158/1078-0432.CCR-06-0759. [DOI] [PubMed] [Google Scholar]
- 40.Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Annals of surgical oncology. 2010;17(3):718–30. doi: 10.1245/s10434-009-0809-6. [DOI] [PubMed] [Google Scholar]
- 41.Lee SY, Kang TH, Knoff J, Huang Z, Soong RS, Alvarez RD, et al. Intratumoral injection of therapeutic HPV vaccinia vaccine following cisplatin enhances HPV-specific antitumor effects. Cancer Immunol Immunother. 2013;62(7):1175–85. doi: 10.1007/s00262-013-1421-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Science translational medicine. 2014;6(226):226ra32. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]