

Abstract
Bombesin receptor subtype (BRS)-3 is a G protein coupled receptor (GPCR) for the bombesin (BB)-family of peptides. BRS-3 is an orphan GPCR and little is known of its physiological role due to the lack of specific agonists and antagonists. PD168368 is a nonpeptide antagonist for the neuromedin B (NMB) receptor (R) whereas PD176252 is a nonpeptide antagonist for the gastrin releasing peptide (GRP) R and NMBR but not BRS-3. Here nonpeptide analogs of PD176252 e.g. the S-enantiomer ML-18, and the R-enantiomer, EMY-98, were investigated as BRS-3 antagonists using lung cancer cells. ML-18 and EMY-98 inhibited specific 125I-BA1 (DTyr-Gln-Trp-Ala-Val-βAla-His-Phe-Nle-NH2)BB6-14 binding to NCI-H1299 lung cancer cells stably transfected with BRS-3 with IC50 values of 4.8 and > 100 μM, respectively. In contrast, ML-18 bound with lower affinity to the GRPR and NMBR with IC50 values of 16 and >100 μM, respectively. ML-18 (16 μM), but not its enantiomer EMY-98, inhibited the ability of 10 nM BA1 to elevate cytosolic Ca2+ in a reversible manner using lung cancer cells loaded with FURA2-AM. ML-18 (16 μM), but not EMY-98, inhibited the ability of 100 nM BA1 to cause tyrosine phosphorylation of the EGFR and ERK in lung cancer cells. ML-18 but not EMY-98 inhibited the proliferation of lung cancer cells. The results indicate that ML-18 is a nonpeptide BRS-3 antagonist that should serve as a template to improve potency and selectivity.
Keywords: bombesin receptor subtype 3, nonpeptide antagonist, lung cancer, proliferation
1. Introduction
Bombesin receptor subtype (BRS)-3 is an orphan G-protein coupled receptor (GPCR) whose role in normal physiology is unknown due to a lack of specific agonists and antagonists [14]. In BRS-3 knockout mice, however, obesity develops associated with hypertension and impairment of glucose metabolism [27]. The knockout mice have increased serum leptin levels, increased feeding behavior and hyperphagia. BRS-3 knockout mice have altered taste perception as a result of reduced receptor densities in the amygdala and hypothalamus [40]. In the periphery, BRS-3 is present in pancreatic islets and BRS-3 deficient mice have increased plasma insulin [10]. Also, BRS-3 is overexpressed in a number of cancer tumors [15]. The results indicate that BRS-3 may be important in the normal and malignant CNS as well as peripheral tissues.
Human BRS-3 contains 399 amino acids [9]. It has 51% sequence homology with the gastrin releasing peptide (GRP) receptor (R) which contains 384 amino acids [3,34] and 47% sequence homology with the neuromedin B (NMB) R which contains 390 amino acids [37]. The BB family of receptors interact with a guanine nucleotide binding protein (Gq) activating phospholipase C resulting in phosphatidylinositol (PI) turnover [31, 32]. The IP3 released causes elevation of cytosolic calcium (Ca2+) whereas the diacylglycerol released causes activation of protein kinase (PK) C [5, 23]. BRS-3 binds synthetic, [D-Tyr6, β-Ala11, Phe13, Nle14]BB(6-14), abbreviated BA1, with high affinity [36]. Also, BA1 binds with high affinity to the GRPR and NMBR causing their activation. BRS-3 does not bind BB, NMB or GRP with high affinity whereas the NMBR prefers NMB relative to GRP or BB and the GRPR prefers GRP or BB relative to NMB [29]. Thus BA1 represents a universal agonist for the BB family of receptors.
Non-peptide antagonists for the BBR family have been identified. PD168368 is a nonpeptide antagonist, which binds with high affinity to the NMBR and inhibits the growth of lung cancer cells [20]. PD176252 is a non-peptide antagonist for the NMBR and GRPR which inhibits the growth of lung cancer cells [2, 22]. Both PD168368 and PD176252, which are S-3-(1H-indol-3-yl)-2-[3-(4-nitrophenyl)ureido]propanamide analogs, do not bind with high affinity to BRS-3 [11].
In this communication, two related PD176252 analogs were screened as BRS-3 antagonists. The (S) PD176252 analog ML-18, but not EMY-98, (R) analog, inhibited specific 125I-BA1 binding to lung cancer cells transfected with BRS-3 or GRPR but not NMBR. ML-18, but not EMY-98 antagonized the ability of BA1 to elevate cytosolic Ca2+ in lung cancer cells. BA1 addition to lung cancer cells caused the tyrosine phosphorylation of EGFR and ERK which was blocked by ML-18 but not EMY-98. Finally, ML-18 but not EMY-98 inhibited the growth of lung cancer cells. These results indicate that ML-18 is a nonpeptide BRS-3 antagonist.
2. Materials and Methods
2.1 Chemical synthesis
ML-18 and EMY-98 were synthesized as pure enantiomers using N-BOC-R-tryptophan or N-BOC-S-tryptophan as described [33]. Enantiomeric purity was assessed by chiral high-performance liquid chromatography (HPLC) analysis on a Perkin-Elmer series 200LC instrument using a Daicel Chiral Cell OD column (250 mm × 4.6 mm, 5 μm particle size). The compounds were eluted with n-hexane/ethanol, 4/1 v/v at a flow rate of 0.8 ml/min. The absorbance was determined at 230 nm using a Perkin-Elmer 785A UV/VIS detector. All compounds had >95% enantiomeric excesses. The molecular weight of (S)-3-(1H-indol-3-yl)-N-[[1-(4-methoxyphenyl)cyclohexyl]methyl]-2-[(4-nitrophenyl)carbamoylamino]propanamide, ML-18, and (R)- 3-(1H-indol-3-yl)-N-[[1-(4-methoxyphenyl)cyclohexyl]methyl]-2-[(4-nitrophenyl)carbamoylamino]propanamide, EMY-98, was 569.9 Daltons. Routinely ML-18 and EMY-98 were dissolved in DMSO at a concentration of 10 mM prior to use.
2.2 Cell culture
Human NCI-H727 lung cancer cells, which are known to contain BRS-3 and wild type EGFR [6, 21] were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium containing 10% heat-inactivated fetal bovine serum (FBS; Invitrogen, Grand Island, NY). Human NCI-H1299 lung cancer cells with increased stable expression of the BRS-3 were used [4] and were grown in RPMI 1640 containing 10% FBS supplemented with 300 mg/l G418 sulfate (Sigma-Aldrich, St. Louis, MO). The cells were split weekly 1/20 with trypsin-ethylenediaminotetraacetic acid (EDTA). The cells were mycoplasma free and were used when they were in exponential growth phase after incubation at 37°C in 5% CO2/95% air.
2.3. Receptor binding
BA1, which binds with high affinity to all human BB receptors, was radiolabeled using iodogen and HPLC purified as reported previously [18]. The ability of BA1, EMY-98, GRP, ML-18 and NMB to inhibit specific 125I-BA1 binding to lung cancer cells was investigated. GRP and NMB were purchased from Bachem Inc. (Torrance, CA) and BA1 was provided by Dr. D. Coy (Tulane Univ.). The lung cancer cells were washed 3 times in SIT medium (RPMI-1640 containing 3 × 10-8 M sodium selenite, 5 μg/ml bovine insulin and 10 μg/ml transferrin (Sigma-Aldrich, St. Louis, MO)). The cells were incubated in SIT buffer containing 0.25% bovine serum albumin and 250 μg/ml bacitracin (Sigma-Aldrich, St. Louis, MO) and 125I-BA1 (100,000 cpm) added, as well as various concentrations of unlabelled competitor. After incubation at 37°C for 30 min, free 125I-BA1 was removed by washing 3 times in buffer and the cells which contained bound 125I-BA1 dissolved in 0.2 N NaOH and counted in a gamma counter. The half maximal inhibitory concentration (IC50) was calculated for each unlabeled competitor.
2.4. Western Blot
The ability of BA1 to stimulate tyrosine phosphorylation of EGFR or ERK (p42/p44 MAP kinase) was investigated by Western blot [25]. Lung cancer cells were cultured in 10 cm dishes. When a monolayer of cells formed they were placed in SIT media for 3 hr. Routinely, lung cancer cells were treated with 16 μM EMY-98 or ML-18 for 30 min prior to stimulation with BA1. Then cells were treated with 0.1 μM BA1 for 2 min, washed twice with PBS and lysed in buffer containing 50 mM Tris.HCl (pH 7.5), 150 mM sodium chloride, 1% Triton X-100, 1% deoxycholate, 1% sodium azide, 1 mM ethyleneglycoltetraacetic acid, 0.4 M EDTA, 1.5 μg/ml aprotinin, 1.5 μg/ml leupeptin, 1 mM phenylmethylsulfonylfluoride and 0.2 mM sodium vanadate (Sigma-Aldrich, St. Louis, MO). The lysate was sonicated for 5 s at 4°C and centrifuged at 10,000 × g for 15 min. Protein concentration was measured using the BCA reagent (Pierce Chemical Co., Rockford, IL), and 400 μg of protein was incubated with 4 μg of anti-phosphotyrosine (PY) monoclonal antibody, 4 μg of goat anti-mouse immunoglobulin IgG and 15 μl of immobilized protein G overnight at 4°C. The immunoprecipitates were washed 3 times with phosphate buffered saline and analyzed by sodium dodecyl sulfate/polyacrylamide gel electrophoresis and Western blotting. Immunoprecipitates were fractionated using 4-20% polyacrylamide gels (Novex, San Diego, CA). Proteins were transferred to nitrocellulose membranes and the membranes were blocked overnight at 4°C using blotto (5% non-fat dried milk in solution containing 50 mM Tris/HCl (pH 8.0), 2 mM CaCl2, 80 mM sodium chloride, 0.05% Tween 20 and 0.02% sodium azide) and incubated for 16 h at 4°C with 1 μg/ml anti-EGFR antibody (Cell Signaling Technologies, Danvers, MA) followed by anti-rabbit immunoglobulin G-horseradish peroxidase conjugate (Upstate Biotechnologies, Lake Placid, NY). The membrane was washed for 10 min with blotto and twice for 10 min with washing solution (50 mM Tris/HCl (pH 8.0), 2 mM CaCl2, 80 mM sodium chloride, 0.05% Tween 20 and 0.02% sodium azide). The blot was incubated with enhanced chemiluminescence detection reagent for 5 min and exposed to Kodak XAR film. The intensity of the bands was determined using a densitometer.
Alternatively, 20 μg of cellular extract was loaded onto a 15 well 4-20% polyacrylamide gels. After transfer to nitrocellulose, the blot was probed with anti PY1068-EGFR, anti-EGFR, anti-FAK, anti-PY397FAK, anti-PY204ERK, anti-ERK or anti-tubulin (Cell Signaling Technologies, Danvers, MA).
2.5 Cytosolic Ca2+
NCI-H727 or BRS-3 transfected NCI-H1299 cells were treated with trypsin-EDTA and harvested. After centrifugation the cells were resuspended in SIT medium (2.5 × 106 cells/ml) containing Fura-2AM (Calbiochem, La Jolla, CA) at 37°C for 30 min [23]. The cells were centrifuged at 1000 × g for 5 min and resuspended at a concentration of 2.5 × 106/ml and 2 ml placed in a Quartz cuvette containing a stirbar. The excitation ratio was determined at 340 and 380 nm and the emission at 510 nm using a spectrofluorometer equipped with a magnetic stirring mechanism and temperature (37°C) regulated cuvette holder before and after addition of drug.
2.6 Proliferation
Growth studies in vitro were conducted using the 3-(4,5-dimethylthiazol-2-yl)-2.5-diphenyl-2H-tetrazolium bromide (MTT) and clonogenic assays [17]. In the MTT assay, NCI-H727 or NCI-H1299 cells transfected with BRS-3 (104/well) were placed in SIT medium and various concentrations of ML-18 or gefitinib added. After 2 days, 15 μl of 0.1 % MTT solution added. After 4 h, 150 μl of dimethylsulfoxide was added. After 16 h, the optical density at 570 nm was determined. In the clonogenic assay, the effects of ML-18 and EMY-98 were investigated on NCI-H727 cells. The bottom layer contained 0.5% agarose in SIT medium containing 5% FBS in 6 well plates. The top layer consisted of 3 ml of SIT medium in 0.3% agarose, BA1, ML-18, EMY-98 and/or gefitinib using 5 × 104 lung cancer cells. Triplicate wells were plated and after 2 weeks, 1 ml of 0.1% p-iodonitrotetrazolium violet was added and after 16 hours at 37°C, the plates were screened for colony formation; the number of colonies larger than 50 μm in diameter were counted using an Omnicon image analysis system.
3. Results
3.1 Receptor binding
The ability of ML-18 and EMY-98 to bind to the BBR was investigated using lung cancer cells. Figure 1A shows that specific 125I-BA1 binding to NCI-H1299 cells transfected with BRS-3 is inhibited moderately by ML-18 at 3, 10 and 30 μM but not 0.1 μM. In contrast, specific 125I-BA1 binding to BRS-3 tranfected cells is inhibited weakly by 30 μM EMY-98 but not 1 μM. Table I shows that the IC50 values for BRS-3 binding BA1, ML-18, EMY-98, GRP and NMB are 0.006, 4.8, >100, >10 and >10 μM, respectively using NCI-H1299 cells transfected with BRS-3. Similar results were obtained using NCI-H727 lung cancer cells (Table I). Figure 1B indicates that ML-18 and EMY-98 bind with moderate and low affinity to GRPR, respectively. Table I shows that the IC50 values for GRPR binding for BA1, ML-18, EMY-98, GRP and NMB are 0.0004, 16, >100, 0.0003 and 0.06 μM, respectively. Figure 1C shows that ML-18 and EMY-98 had little effect on specific 125I-BA1 binding to the NMBR. Table I shows that the IC50 values for NMBR binding for BA1, ML-18, EMY-98, GRP and NMB are 0.0025, >100, >100, 0.15 and 0.0007 μM, respectively. The results indicate that ML-18 but not EMY-98 binds with moderate affinity to BRS-3 and the GRPR but not the NMBR.
Figure 1.

Receptor binding. The ability of ML-18 (•) and EMY-98 (■) to inhibit specific 125I-BA1 binding to (A) BRS-3 transfected NCI-H1299, (B) GRPR transfected Balb/3T3 and (C) NMBR transfected NCI-H1299 cells is shown as a function of ligand concentration. The mean value ± S.D. of 3 determinations each repeated in duplicate is shown; p < 0.05. *; p < 0.01, ** relative to control using the Student's t-test.
Table I. Binding to BBR.
| IC50, μM | ||||
|---|---|---|---|---|
| Ligand | BRS3 | GRPR | NMBR | NCI-H727 |
| BA1 | 0.006 ± .001 | 0.0004 ± .0001 | 0.0025 ± .0003 | 0.004 ± .001 |
| EMY-98 | >100 | >100 | >100 | >100 |
| GRP | >10 | 0.0003 ± 0.0001 | 0.150 ± 23 | >10 |
| ML-18 | 4.8 ± 0.6 | 16 ± 2 | >100 | 6.4 ± 1.1 |
| NMB | >10 | 0.060 ± .008 | 0.0007 ± .0001 | >10 |
The mean IC50 ± S.D. of 3 determinations is indicated to inhibit specific 125I-BA1 binding to NCI-H727 lung cancer cells or cells transfected with BRS-3, GRPR or NMBR.
3.2 Calcium-Effect on peptide agonist, BA1
The ability of ML-18 to function as a BRS-3 antagonist was investigated using a calcium assay. Figure 2A shows the cytosolic Ca2+ increased within seconds after addition of 10 nM BA1 to lung cancer cells from 0.18 to 0.20 μM and then the Ca2+ returned to baseline over a 2 min period. Addition of 16 μM ML-18 but not EMY-98 antagonized the ability of 10 nM BA1 to increase cytosolic Ca2+ in lung cancer cells (Figs.2B, 2C) whereas ML-18 and EMY-98 had little effect on basal cytosolic Ca2+. Figure 2D shows that 16 μM ML-18 blocked the ability of 10 nM BA1 to increase cytosolic Ca2+ in NCI-H727 cells. Subsequent addition of 0.1 μM BA1 and 1 μM BA1 increased the cytosolic Ca2+ weakly and strongly, respectively. The structure of ML-18 is shown (Fig. 2E). The results indicate that ML-18 but not EMY-98 is a reversible nonpeptide BRS-3 antagonist.
Figure 2.

Cytosolic Ca2+. BRS-3 transfected NCI-H1299 cells were loaded with Fura-2AM and the cytosolic Ca2+ determined for 4 min after the addition of (A) 10 nM BA1, (B) 16 μM EMY-98 followed by 10 nM BA1, (C) 16 μM ML-18 followed by 10 nM BA1, (D) NCI-H727 cells are treated with 16 μM ML-18 followed by 10 nM BA1, 0.1 μM BA1 and 1 μM BA1. This experiment is representative of 4 others. (E) The structure of ML-18 is shown.
Tyrosine phosphorylation
BRS-3 regulates EGFR and ERK tyrosine phosphorylation in lung cancer cells [25]. Fig. 3A shows that addition of 0.1 μM BA1 addition to NCI-H1299 cells transfected with BRS-3 increases FAK (125 kDa) tyrosine phosphorylation after 2 min. BA1 significantly increased FAK tyrosine phosphorylation 10.9-fold (Fig. 3B). The increase in FAK tyrosine phosphorylation caused by BA1 was significantly inhibited 16 but not 1.6 or 0.16 μM ML-18 (Fig. 3B). As a control, total FAK was not altered by ML-18 (Fig. 3A). Addition of BA1 to NCI-H1299 cells transfected with BRS-3 increased ERK (44 and 42 kDa) tyrosine phosphorylation (Fig. 3C). BA1 significantly increased ERK tyrosine phosphorylation 2.9-fold (Fig. 3D). The increased in ERK tyrosine phosphorylation caused by BA1 was significantly inhibited by 16 but not 1.6 or 0.16 μM ML-18 (Fig. 3D). Addition of BA1 to NCI-H1299 cells transfected with BRS-3 increased EGFR (170 kDa) tyrosine phosphorylation (Fig. 3E). BA1 increased significantly EGFR tyrosine phosphorylation 5.9-fold (Fig. 3F). The increase in EGFR tyrosine phosphorylation caused by BA1 was significantly inhibited by 16 but not 1.6 or 0.16 μM ML-18 (Fig. 3F). Similar data were obtained using NCI-H727 cells (T. Moody, unpublished). The results indicate that ML-18 in a dose-dependent manner decreases lung cancer FAK, ERK and EGFR tyrosine phosphorylation that is regulated by BRS-3.
Figure 3.

Western blot. BRS-3 transfected NCI-H1299 cells were treated with 0.1 μM BA1 for 2 min in the presence or absence of varying doses of ML-18 and the P-FAK and FAK (A), P-ERK and ERK (C) as well as P-EGFR and EGFR (E) is indicated. By densitometry analysis, the mean % ± S.D. of 3 determinations is indicated for P-FAK (B), P-ERK (D) and P-EGFR (F); p < 0.05, *; p < 0.01, ** relative to the no additions control; p < 0.05, a; p < 0.01, aa relative to BA1 using the Student's t-test. This experiment is representative of 3 others.
3.4 Proliferation
The ability of ML-18 to inhibit lung cancer cellular proliferation was investigated. ML-18 (4.8 μM) had little effect on the proliferation of NCI-H1299 cells transfected with BRS-3 whereas proliferation was moderately and strongly inhibited by 16 and 48 μM ML-18, respectively (Fig. 4). Gefitinib weakly inhibited the proliferation of NCI-H1299 cells transfected with BRS-3 in the absence of ML-18 (IC50 > 30 μM). In the presence of ML-18 the gefitinib dose-response curve shifted to the left. In the presence of 48 μM ML-18 the gefitinib IC50 was approximately 4.5 μM. Thus ML-18 increases the gefitinib sensitivity by approximately 1-order of magnitude.
Figure 4.

MTT assay. The proliferation of BRS-3 transfected NCI-H1299 cells is indicated as a function of gefitinib concentration in the presence of (○) 0, (■) 4.8, (Δ) 16 and (◆) 48 μM ML-18. The mean value ± S.D. of 8 determinations is indicated. This experiment is representative of 3 others.
Using the clonogenic assay, 1.6 μM ML-18 but not EMY-98 significantly reduced the NCI-H727 colony number (Table II). Gefitinib (1 μM) or gefitinib plus ML-18 moderately and strongly reduced NCI-H727 colony number. In contrast, 0.01 μM BA1 significantly increased lung cancer colony number. The increase in colony number caused by BA1 addition to lung cancer cells was inhibited by ML-18 and/or gefitinib but not EMY-98. The results indicate that BA1 stimulates lung cancer proliferation, EMY-98 has no effect and ML-18 as well as gefitinib inhibit lung cancer growth.
Table II. Clonogenic assay.
| Addition | Colony number |
|---|---|
| None | 32 ± 4 |
| ML-18, 1.6 uM | 22 ± 3* |
| EMY-98, 1.6 uM | 34 ± 3 |
| Gefitinib, 1 uM | 20 ± 4* |
| Gefitinib + ML-18 | 13 ± 2** |
| Gefitinib + EMY-98 | 21 ± 3* |
| BA1, 0.01 uM | 46 ± 7* |
| BA1 + ML-18 | 35 ± 2 |
| BA1 + EMY-98 | 44 ± 6* |
| BA1 + Gefitinib | 33 ± 5 |
| BA1 + ML-18 + Gef | 24 ± 3* |
The mean value ± S.D. of 3 determinations is indicated using NCI-H727 cells;
p < 0.05,
p < 0.01,
relative to no additions using the Student's t-test. This experiment is representative of 3 others.
Calcium-Effect on specific BRS-3 agonists, MK-5046
MK5046 stimulated and increase in cytosolic calcium of NCI-H1299 cells transfected with BRS-3 (Fig. 5) and this increase was inhibited by ML-18 or EMY-18, however neither antagonist had an effect on calcium when present alone (Fig. 5).
Figure 5.
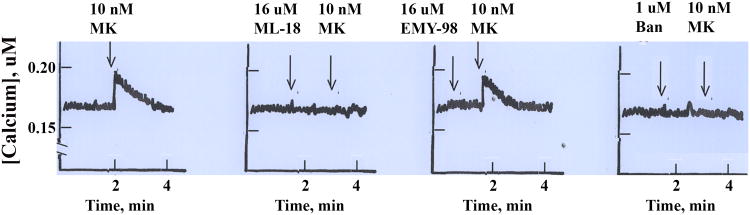
Cytosolic Ca2+. NCI-H727 cells were loaded with Fura-2AM and the cytosolic Ca2+ determined for 4 min after the addition of (A) 10 nM MK 5046 (MK), (B) 16 μM ML-18 followed by 10 nM MK, (C) 16 μM EMY-98 followed by 10 nM MK and (D) 1 μM Bantag (Ban) by 10 nM MK. This experiment is representative of 3 others.
Discussion
Lung cancer is a serious public health problem which causes approximately 160,000 deaths in the United States annually. Traditionally lung cancer is treated with chemotherapy but the 5 year survival rate is only 16%. Thus new therapeutic agents are needed to treat lung cancer patients. The tyrosine kinase inhibitors (gefitinib or erlotinib) for the EGFR were approved to treat lung cancer patients who fail chemotherapy and have EGFR mutations [16, 28]. Because only 13% of the lung cancer patients have EGFR mutations new therapeutic approaches are needed to treat lung cancer patients with wild type EGFR. Recently we found that GPCR antagonists improve the potency of gefitinib in lung cancer cells which have wild type EGFR [20]. BBRs have been detected in lung cancer tumors using in vitro autoradiographic and immunocytochemical techniques [19,30]. Here the effects of nonpeptide BRS-3 antagonists were investigated in lung cancer cells.
ML-18 binds with higher affinity to BRS-3 than the GRPR or NMBR. Previously PD165929 and PD168368 were discovered to bind with high affinity to the NMBR but not the GRPR or BRS-3 [8]. The structure of PD168368 is characterized by three moieties: the scaffold of (S)-α-methyltryptophan, the 4-nitrophenylurea group and the 1-(2-pyridyl)cyclohexyl ring system. Site-directed mutagenesis indicates that Tyr220 in transmembrane domain 5 of the NMBR is essential for high affinity PD168368 binding [35]. The 4-nitrourea group may interact with Tyr220 of the NMBR via hydrogen bonding. Subsequently, PD176252 was discovered to bind with high affinity to the NMBR and GRPR but not BRS-3 [2, 11]. PD176252 differs from PD168368 in that it has a 1-(2-pyridyl-5-methoxy)cyclohexyl ring. The methoxy group may interact with basic amino acids such as Arg287 of the GRPR via electrostatic interactions [1]. In contrast, ML-18 has a similar structure to PD176252 but differs from PD168368 and PD176252 in that it has a phenyl group instead of the 2-pyridyl. Because ML-18 will have less positive charge than PD176252, due to the substitution of a phenyl ring for the pyridine, it may interact with Phe 222 of BRS-3 by hydrophobic interactions. Previously, it was determined that the IC50 for PD176252 was >10 μM for BRS-3, 0.2 μM for GRPR and 0.0002 μM for the NMBR, respectively [11]. Thus ML-18 binds to BRS-3 with approximately an order of magnitude greater affinity than does PD176252, whereas ML-18 binds with 2-orders of magnitude lower affinity to the GRPR and 5-orders of magnitude lower affinity for the NMBR than it does PD176252.
ML-18 is a steroselective BRS-3 antagonist. Within seconds after addition of BA1 to lung cancer cells the cytosolic Ca2+ transiently increased. The increase in cytosolic Ca2+ caused by BA1 was blocked by the (S) isomer ML-18 but not the (R) isomer EMY-98. Addition of 16 μM ML-18 blocked the increase in cytosolic Ca2+ caused by 10 nM BA1, however, if 100 nM and 1000 nM BA1 was subsequently added, there was a moderate and strong Ca2+ response, respectively. These results indicate that ML-18 is a reversible BRS-3 antagonist. In contrast, ML-18 is an agonist for N-formyl peptide receptors (FPR) which are GPCR involved in inflammatory processes [33]. ML-18 interaction with FPR1 and FPR2 increases cytosolic Ca2+ in HL-60 cells with ED50 values of 15 and 10 μM. ML-18 does not increase Ca2+ in NCI-H727 or NCI-H1299 lung cancer cells transfected with BRS-3.
Recently MK-5046 was identified as a selective nonpeptide BRS-3 agonist [12, 13]. MK-5046 binds with high affinity to BRS-3 (IC50 = 0.018 μM) but not the GRPR or NMBR [26]. Addition of 10 nM MK-5046 increased the cytosolic Ca2+ in NCI-H1299 cells transfected with BRS-3 (Fig. 1S-A). The increase in cytosolic Ca2+ caused by MK-5046 was antagonized by 16 μM ML-18 but not EMY-98 (Fig. 1S B,C). In addition the increase in cytosolic Ca2+ caused by MK-5046 was antagonized by Bantag-1, a peptide BRS-3 antagonist (Fig. 1S-D). Bantag-1 binds with high affinity to BRS-3 (IC50 = 0.001 μM) but not the GRPR or NMBR [26]. Thus ML-18 represents a useful nonpeptide BRS-3 antagonist for in vitro experiments, however, for in vivo experiments new ML-18 analogs are needed which bind with higher affinity and specificity to BRS-3.
BRS-3 regulates EGFR and ERK tyrosine phosphorylation within minutes after addition to lung cancer cells [25]. In NCI-H1299 cells, transforming growth factor (TGF)α is released from the cells in a Src- and matrix metalloprotease (MMP) dependent manner. The TGFα binds with high affinity to the EGFR causing its tyrosine phosphorylation. BA1 addition to NCI-H1299 cells transfected with BRS-3 caused FAK tyrosine phosphorylation which was inhibited by ML-18. Preliminary data (T. Moody, unpublished) indicate that the Src inhibitor PP2 inhibits the ability of BA1 to cause tyrosine phosphorylation of FAK, EGFR and ERK. BRS-3 regulates EGFR and ERK tyrosine phosphorylation which is blocked by gefitinib [25]. ML-18 inhibits tyrosine phosphorylation of EGFR and ERK caused by BA1 addition to lung cancer cells. Phosphorylated ERK can enter the nucleus and alter gene expression. BRS-3 regulates c-fos mRNA in a MEK-dependent manner [38].
Previously, immunoreactive GRP was detected in high concentrations in lung cancer cells [24, 39]. Because monoclonal antibodies to GRP inhibited the growth of lung cancer cell lines, GRP was hypothesized to be an autocrine growth factor for lung cancer cells [7]. Antagonists such as PD176252 inhibited the growth of lung cancer in vitro and in vivo [22]. ML-18 inhibited the proliferation of lung cancer cells in a dose-dependent manner. Using the MTT assay, which takes 2 days, the IC50 for ML-18 was 5 μM. Gefitinib inhibited the growth of NCI-H1299 cells transfected with BRS-3, however, its potency was increased in the presence of ML-18. These results indicate that BRS-3 antagonists increase the potency of gefitinib in lung cancer cells with wild type EGFR. Previously PD168368 and PD176252 increased the potency of gefitinib in cancer cells [20, 41]. It remains to be determined if BBR antagonists increase the potency of gefitinib in vivo. Using the clonogenic assay which takes 2 weeks, BA1 increased NCI-H727 colony number. The increase in colony number caused by BA1 is inhibited by ML-18 and/or gefitinib but not EMY-98. These results suggest that BA1 may increase colony number in an EGFR-dependent manner.
Summary
ML-18 is a new non-peptide BRS-3 antagonist. ML-18 inhibits binding to BRS-3, and the ability of BA1 to increase cytosolic Ca2+, increase EGFR tyrosine phosphorylation and increase the proliferation of lung cancer cells, whereas EMY-98 is inactive. ML-18 potentiates the cytotoxicity of gefitinib on lung cancer cells. This represents the first report of a nonpeptide BRS-3 antagonist and ML-18 will serve as a good lead compound to modify and increase the affinity/specificity for BRS-3.
Highlights.
Nonpeptide antagonists for BRS-3 were synthesized
ML-18 but not EMY-98 bound with moderate affinity to cells containing BRS-3 and GRPR but not NMBR
ML-18 antagonized the ability of BRS-3 agonists to increase EGFR and ERK tyrosine phosphorylation, elevate cytosolic Ca2+ and increase proliferation of lung cancer cells.
Acknowledgments
The authors thank Dr. D. Coy (Tulane Univ.) for the BA1 and Drs. M. Nicklaus and M. Peach for helpful discussions. This research is supported by the intramural programs of NCI and NIDDK of the NIH.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest.
References
- 1.Akeson M, Sainz E, Mantey SA, Jensen RT, Battey JF. Identification of four amino acids in the gastrin-releasing peptide receptor that are required for high affinity agonist binding. J Biol Chem. 1997;272:17405–9. doi: 10.1074/jbc.272.28.17405. [DOI] [PubMed] [Google Scholar]
- 2.Ashwood V, Brownhill V, Higginbottom M, Horwell DC, Hughes J, Lewthwaite RA, et al. PD176252- The first high affinity non-peptide gastrin releasing peptide (BB2) receptor antagonist. Bioorg Med Cem. 1998;8:2589–94. doi: 10.1016/s0960-894x(98)00462-4. [DOI] [PubMed] [Google Scholar]
- 3.Battey JF, Way JM, Corjay MH, Shapira H, Kusano K, Harkins RW, et al. Molecular cloning of the bombesin/gastrin releasing peptide receptor from Swiss 3T3 cells. Proc Natl Acad Sci USA. 1991;88:395–9. doi: 10.1073/pnas.88.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benya RV, Kusui, Pradhan T, Battey JF, Jensen RT. Expression and characterization of cloned human bombesin receptors. Mol Pharmacol. 1995;47:10–20. [PubMed] [Google Scholar]
- 5.Bunn PA, Dienhart DB, Chan D, Puck TT, Tagawa M, Jewett PB, Braunschweiger E. Neuropeptide stimulation of calcium flux in human lung cancer cells: Delineation of alternative pathways. Proc Natl Acad Sci USA. 1990;87:2162–6. doi: 10.1073/pnas.87.6.2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corjay MH, Dobrzanski DJ, Way JM, Viallet J, Shapira H, Worland P, et al. Two distinct bombesin receptor subtypes are expressed and functional in human lung carcinoma cell lines. J Biol Chem. 1991;266:18771–9. [PubMed] [Google Scholar]
- 7.Cuttitta F, Carney DN, Mulshine J, Moody TW, Fedorko J, Fischler A, Minna JD. Bombesin-like peptides can function as autocrine growth factors in human small cell lung cancer. Nature. 1985;316:823–6. doi: 10.1038/316823a0. [DOI] [PubMed] [Google Scholar]
- 8.Eden JM, Hall MD, Higginbottom H, Horwell DC, Howson W, Hughes J, et al. PD165929-The first high affinity non-peptide neuromedin B (NMB) receptor selective antagonist. Bioorg Med Chem Lett. 1996;6:2617–23. [Google Scholar]
- 9.Fathi Z, Corjay MH, Shapira H, Wada E, Benya R, Jensen R, et al. BRS-3: A novel bombesin receptor subtype selectivity expressed in testis and lung carcinoma cells. J Biol Chem. 1993;268:5979–84. [PubMed] [Google Scholar]
- 10.Fleischmann A, Laderach U, Friess H, Buechler NWO, Reubi JC. Bombesin receptors in distinct tissue compartments of human pancreatic diseases. Lab Inves. 2000;80:1807–17. doi: 10.1038/labinvest.3780192. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez N, Mantey SA, Pradhan T, Sancho V, Moody TW, Coy DH, et al. Characterization of putative GRP- and NMB-receptor antagonist's interaction with human receptors. Peptides. 2009;30:1473–86. doi: 10.1016/j.peptides.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guan XM, Chen H, Dobbelaar PH, Dong Y, Fong TM, Gagen K, et al. Regulation of energy homeostasis by bombesin receptor subtype-3: Selective receptor agonists for the treatment of obesity. Cell Metab. 2010;11:101–12. doi: 10.1016/j.cmet.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 13.Guan XM, Metzger JM, Yang L, Raustad KA, Wang SP, Spann SK, et al. Antiobesity effect of MK-5046, a novel bombesin receptor subtype-3 agonist. J Pharmacol Exp Ther. 2011;336:356–64. doi: 10.1124/jpet.110.174763. [DOI] [PubMed] [Google Scholar]
- 14.Jensen RT, Battey JF, Spindel ER, Benya RV. Mammalian bombesin receptors: Nomenclature, distribution, pharmacology, signaling, and functions in normal and disease states. Pharmacological reviews. 2008;60:1–42. doi: 10.1124/pr.107.07108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jensen RT, Moody TW. Bombesin Peptides. In: Kastin A, editor. Handbook of biologically active peptides. Elsevier Press; Amsterdam: 2013. pp. 506–11. [Google Scholar]
- 16.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 17.Mahmoud S, Staley J, Taylor J, Bogden A, Moreau JP, Coy D, et al. (Psi13,14)bombesin analogs inhibit the growth of small cell lung cancer in vitro and in vivo. Cancer Res. 1991;51:1798–1802. [PubMed] [Google Scholar]
- 18.Mantey SA, Weber HC, Sainz E, Akeson M, Ryan RR, Pradhan TK, Searles RP, et al. Discovery of a high affinity radioligand for the human orphan receptor, bombesin receptor subtype 3, which demonstrates it has a unique pharmacology compared to other mammalian bombesin receptors. J Biol Chem. 1997;272:26062–71. doi: 10.1074/jbc.272.41.26062. [DOI] [PubMed] [Google Scholar]
- 19.Mattei J, Achcar RD, Cano CH, Macedo BR, Meurer L, Battle BS, et al. Gastrin-releasing peptide receptor expression in lung cancer. Arch Pathol Lab Med. 2014;138:98–104. doi: 10.5858/arpa.2012-0679-OA. [DOI] [PubMed] [Google Scholar]
- 20.Moody TW, Berna MJ, Mantey S, Sancho V, Ridnour L, Wink DA, et al. Inhibition of Neuromedin B mediated transactivation of EGF receptors in lung cancer cells enhances gefitinib sensitivity. Eur J Pharm. 2010;637:38–45. doi: 10.1016/j.ejphar.2010.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moody TW, Lee M, Kris RM, Bellot F, Bepler G, Oie H, et al. Lung carcinoid cell lines have bombesin-like peptides and EGF receptors. J Cellular Biochem. 1990;43:139–47. doi: 10.1002/jcb.240430205. [DOI] [PubMed] [Google Scholar]
- 22.Moody TW, Leyton J, Garcia-Marin L, Jensen RT. Nonpeptide gastrin releasing peptide receptor antagonists inhibit the proliferation of lung cancer cells. Eur J Pharm. 2003;474:21–9. doi: 10.1016/s0014-2999(03)01996-4. [DOI] [PubMed] [Google Scholar]
- 23.Moody TW, Murphy A, Mahmoud S, Fiskum GF. Bombesin-like peptides elevate cytosolic calcium in small cell lung cancer cells. Biochem Biophys Res Commun. 1987;147:189–95. doi: 10.1016/s0006-291x(87)80105-5. [DOI] [PubMed] [Google Scholar]
- 24.Moody TW, Pert CB, Gazdar AF, Carney DN, Minna JD. High levels of intracellular bombesin characterize human small-cell lung carcinoma. Science. 1981;214:1246–8. doi: 10.1126/science.6272398. [DOI] [PubMed] [Google Scholar]
- 25.Moody TW, Sancho V, Di Florio A, Nuche-Berenguer B, Mantey S, Jensen RT. Bombesin-receptor subtype-3 agonists stimulate the growth of lung cancer cells and increase EGF receptor tyrosine phosphorylation. Peptides. 2011;32:1677–84. doi: 10.1016/j.peptides.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moreno P, Mantey SA, Nuche-Berenguer B, Reitman ML, Gonzalez N, Coy DH, Jensen RT. Comparative pharmacology of bombesin receptor subtype-3 nonpeptide agonist MK-5046, a universal peptide agonist, and peptide antagonist Bantag-1 for human bombesin receptors. J Pharmacol Expt Ther. 2013;347:110–6. doi: 10.1124/jpet.113.206896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohki-Hamazaki H, Watese K, Yamamoto K, Ogura H, Yamano M, et al. Mice lacking bombesin receptor subtype 3 develop metabolic defects and obesity. Nature. 1997;390:165–9. doi: 10.1038/36568. [DOI] [PubMed] [Google Scholar]
- 28.Paez JG, Janne PA, Lee JC, Tracey S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 29.Pradhan TK, Katsuno T, Taylor JE, Kim SH, Ryan RR, Mantey SA, et al. Identification of a unique ligand which has high affinity for all four bombesin receptor subtypes. Eut J Pharmacol. 1998;343:275–87. doi: 10.1016/s0014-2999(97)01527-6. [DOI] [PubMed] [Google Scholar]
- 30.Reubi JC, Wenger S, Schmuckli-Mauer J, Schaer JC, Gugger M. Bombesin receptor subtypes in human cancers: Detection with the universal radioligand [125I-D-Tyr6, B-Ala11, Phe13, Nle14]bombesin(6-14) Clin Cancer Res. 2002;8:1139–46. [PubMed] [Google Scholar]
- 31.Rozengurt E. Signal transduction pathways in the mitogenic response to G-protein coupled neuropeptide receptor agonists. J Cell Physiol. 1998;177:507–17. doi: 10.1002/(SICI)1097-4652(199812)177:4<507::AID-JCP2>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 32.Ryan RR, Weber HC, Mantey SA, Hou W, Hilburger ME, Pradhan TK, et al. Pharmacology and intracellular signaling mechanisms of the native human orphan receptor BRS-3 in lung cancer cells. J Pharm Exp Ther. 1998;273:13613–24. [PubMed] [Google Scholar]
- 33.Schepetkin AI, Kirpotina LN, Khlebnikov Ai, Leopoldo M, Lucente E, Lacivita E, et al. 3-(1H-indol-3-yl)-2-[3-(4-nitrophenyl)ureido]propanamide enantiomers with human formyl-peptide receptor agonist activity: Molecular modeling of chiral recognition by FRP2. Biochemical Pharmacology. 2013;85:404–16. doi: 10.1016/j.bcp.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spindel ER, Giladi E, Brehm P, Goodman RH, Segerson TP. Cloning and functional characterization of a complementary DNA encoding the murine fibroblast bombesin/gastrin-releasing peptide receptor. Proc Natl Acad Sci USA. 1990;87:9813–7. doi: 10.1210/mend-4-12-1956. [DOI] [PubMed] [Google Scholar]
- 35.Tokita K, Hocart SJ, Katsuo R, Mantey SA, Coy DH, Jensen RT. Tyrosine 220 in the 5th transmembrane domain of the neuromedin B receptor is critical for the high selectivity of the peptoid antagonist PD168368. J Biol Chem. 2001;276:495–504. doi: 10.1074/jbc.M006059200. [DOI] [PubMed] [Google Scholar]
- 36.Uehara H, Gonzalez N, Sancho V, Mantey SA, Nuche-Berenguer B, Pradhan T, et al. Pharmacology and selectivity of various natural and synthetic bombesin related peptide agonists for human and rat bombesin receptors differs. Peptides. 2011;32:1685–99. doi: 10.1016/j.peptides.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wada E, Way J, Shapira H, Kusano K, Lebacq-Verheyden AM, Coy DH, et al. cDNA cloning, characterization and brain region specific expression of a neuromedin-B preferring receptor. Neuron. 1991;6:421–30. doi: 10.1016/0896-6273(91)90250-4. [DOI] [PubMed] [Google Scholar]
- 38.Weber HC, Walters J, Leyton J, Casibang M, Purdom S, Jensen RT, et al. A bombesin receptor subtype-3 peptide increases nuclear oncogene expression in a MEK-1 dependent manner in human lung cancer cells. Eur J Pharm. 2001;412:13–20. doi: 10.1016/s0014-2999(00)00941-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wood SM, Wood JR, Ghatei MA, Lee YC, O'Shaughnessy D, Bloom SR. Bombesin, somatostatin and neurotensin-like immunoreactivity in bronchial carcinoma. J Clin Endocrinol Metab. 1981;53:1310–2. doi: 10.1210/jcem-53-6-1310. [DOI] [PubMed] [Google Scholar]
- 40.Yamada K, Wada E, Imaki J, Ohki-Hamazaki H, Wada K. Hyperresponsiveness to palatable and adversive taste stimuli in genetically obese (bombesin receptor subtype-3 deficient) mice. Physiol Behavior. 1999;66:863–867. doi: 10.1016/s0031-9384(99)00032-3. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Q, Bhola NE, Lui VW, Siwak DR, Thomas SM, Gubish CT, et al. Antitumor mechanisms of combined gastrin-releasing peptide receptor and epidermal growth factor receptor targeting in head and neck cancer. Mol Cancer Ther. 2007;6:1414–24. doi: 10.1158/1535-7163.MCT-06-0678. [DOI] [PubMed] [Google Scholar]