

Abstract
Rheumatoid arthritis (RA) is a prevalent inflammatory joint disease with enigmatic flares, which causes swelling, pain, and irreversible connective tissue damage. Recently, it has been demonstrated in murine models of RA that the popliteal lymph node (PLN) is a biomarker of arthritic flare, as it “expands” in size and contrast enhancement during a prolonged asymptomatic phase, prior to when it “collapses” with accelerated synovitis and joint erosion. This PLN collapse is associated with adjacent knee flare, decreases in PLN volume and contrast enhancement, lymphatic pulse and pumping pressure, and an increase in PLN pressure. Currently, it is known that PLN collapse is accompanied by a translocation of B cells from the follicles to the sinuses, effectively clogging the lymphatic sinuses of the PLN, and that B cell depletion therapy ameliorates arthritic flare by eliminating these B cells and restoring passive lymphatic flow from inflamed joints. Here we review the technological advances that have launched this area of research, describe future directions to help elucidate the potential mechanism of PLN collapse, and speculate on clinical translation towards new diagnostics and therapies for RA.
Keywords: Rheumatoid arthritis, lymph node, flare, lymphatic vessel
1. Introduction1
Rheumatoid arthritis (RA) is a chronic inflammatory joint disease with episodic flares that affects 0.5–1% of the population [1]. While RA is considered an autoimmune disease, due to the prevalence of autoantibodies that are diagnostic for the disease (i.e. rheumatoid factor (RF) and anti-citrillunated protein antibodies (ACPA)) [1], not all RA patients present with autoantibodies (seronegative RA), and these autoantibodies are known to exist in normal healthy people [2]. Based on this discrepancy, and the remarkable success of biologic therapies that target pro-inflammatory cytokines rather than adaptive host responses [3], RA is now considered to be an immune-inflammatory disorder (IMID) caused by multiple etiologies that are not well understood [4], all of which involve lymphatic changes.
It is known that a subset of RA patients have ACPA during the pre-clinical and early stage of the disease [5] and the development of these antibodies is associated with specific Major Histocompatibility Complex (MHC) haplotypes DR1 and/or DR4. It is now established that smoking greatly increased the risk of RA in patients with DR1 or DR4 haplotypes serving as a model of gene-environmental interaction [6, 7]. For the majority of patients, the disease waxes and wanes in arthritic flares, characterized by joint swelling, pain and fatigue along with increased synovial volumes [8]. Current therapeutic strategies include “Treat to Target” [9] and “Window of Opportunity” [10]. These treatments combine biological therapies and disease modifying anti-rheumatic drugs (DMARDs) with the goal of sustained remission. While approximately 30% of patients achieve a remission, a significant proportion of RA patients (70%) are partial responders or prove refractory to all current therapies.
Several clinical observations implicate the lymphatic system in RA pathogenesis. First, it was reported that the lymphangiogenic factor vascular endothelial growth factor C (VEGF-C) and its receptors, VEGFR-2 and VEGFR-3, are expressed more abundantly in RA synovium compared to both healthy and osteoarthritis controls [11, 12]. Second, a greater size and number of popliteal lymph nodes (PLNs) have been found to positively correlate with larger knee synovial volumes (SV) in RA patients, and it has been demonstrated that RA patients have larger and a greater number of PLNs [13]. Third, there is evidence for altered lymph flow in RA patients, as case reports have documented patients with lymphedema that synovitis alone cannot explain, as evidenced by a lack of deep lymphatic vessels and extensive dermal reflux via lymphography [14]. The etiology of the poor lymphatic drainage remains an enigma but blockage in the lymph nodes or lymphatic vessels may be important. It is also known that RA patients have slower lymphatic clearance rate compared to healthy controls, as evidenced by injecting 125I albumin intradermally into the forearm and measuring the half clearance time [15]. Interestingly, cytokine and chemokine levels in lymph are elevated above serum levels, indicating local production by synovial cells [16]. Finally, treatment of RA patients with anti-tumor necrosis factor (anti-TNF) therapy resulted in an increase in lymphatic vessel density in the synovium [17], demonstrating that the lymphatic system responds to anti-inflammatory therapy. This result is complicated as increased lymphatic vessels were found to positively correspond with disease severity and treatment further increased the number of lymphatic vessels. One current hypothesis is that the tissue further primes itself for future inflammation by promoting lymphangiogenesis, possibly through macrophage differentiation into lymphatic endothelial cells [17]. These studies document altered lymphangiogenesis and transport in RA patients combined with increased lymphangiogenesis following anti-inflammatory treatment. Thus, restoration of flow by formation of vessels or improved transport may be part of the response to achieve homeostasis following flare.
Despite major progress in the field, the mechanisms responsible for arthritic flare remain a mystery. Several murine models of inflammatory-erosive arthritis, such as the tumor necrosis factor transgenic (TNF-Tg) mouse model [18] and K/B×N mouse model [19], have been helpful towards elucidating potential mechanisms of flare. Here we discuss our work with these models, and the interactions between joint inflammation and changes in the lymphatic system.
2. TNF stimulates VEGF-C expression by osteoclast precursors
Our first discovery that lymphatics could be playing a major role in murine inflammatory-erosive arthritis was the finding that CD11b+/Gr-1−/lo osteoclast precursors (OCPs), which have been shown to be increased in patients [20] and animals [21] with inflammatory-erosive arthritis, express high levels of VEGF-C via microarray analysis [22]. Next, we found that both TNF-Tg and K/B×N mice have more lymphatic vessels within the synovium compared to WT control mice [23], suggesting that lymphangiogenesis is a common occurrence in this disease. Furthermore, TNF treatment of OCPs induced VEGF-C expression [23], and VEGF-C treatment stimulates osteoclastic bone resorption [22]. Therefore, OCP-derived VEGF-C likely has two roles in RA pathogenesis: 1) stimulating lymphangiogenesis and 2) inducing osteoclastic bone resorption.
3. Establishment of lymph node contrast enhancement and lymph node capacity as biomarkers for knee flare in TNF-Tg mice, a murine model of arthritis
While cross-sectional outcome studies (i.e. histology) of murine models have vastly contributed to our understanding of RA pathogenesis [24], they suffer from limited information on arthritic progression and translational biomarkers. To address this, we developed various longitudinal imaging outcome measures to assess joint inflammation, bone erosions and lymph node parameters, including volume and contrast enhancement [25]. We mainly used TNF-Tg mice (the TNF-Tg line 3647), which were originally obtained from Dr. G. Kollias and carry a 3′-modified human TNF transgene in which the 3′-region of the TNF gene was replaced with that of the human β-globin gene [18]. They develop ankle arthritis starting at 2 months of age, and arthritis progress to knee joints with age. Contrast-enhancement magnetic resonance imaging (CE-MRI) was developed to assess synovial changes in the ankle and knee joints, and draining lymph nodes in TNF-Tg mice. It was found that young (~5 months) TNF-Tg mice had a significantly higher lymph node contrast enhancement (LNCE, intensity of the LN normalized to the combined mean quadriceps and hamstrings muscle intensity) and volume compared to WT mice, which corresponded to an increase in LYVE-1+ PLN sinuses. This is likely due to lymphangiogensis, which increases the ability of contrast to enter the PLN from afferent lymphatic vessels. Lymph node capacity (LNCap), which is the LNCE multiplied by the LN volume, is also increased in TNF-Tg mice. The MRI contrast was injected retro-orbitally and allowed to circulate for 5 minutes. On this time scale, LNCE is likely dependent on both blood and lymphatic transport to the PLN. Blood vessels residing within the PLN may be responsible for a portion of the LNCE, but also, contrast leaking from blood capillaries and taken up by lymphatics in the afferent tissue may also contribute to the LNCE. However, much of the signal resides in the PLN periphery where the sinuses reside. Therefore, afferent lymphatics are understood to be the dominate source of CE (Figure 1). In young TNF-Tg mice, anti-TNF therapy decreases the LNCE and LNCap, demonstrating that these parameters are sensitive to therapy and can be used as disease biomarkers. However, LN volume never returns to WT levels due to the persistence of LYVE-1+ lymphatic sinuses in the LNs, indicating that the tissue becomes primed to respond to chronic inflammation. Moreover, it was found that knee SV significantly correlates with LNCap, demonstrating that larger LNs with greater CE are associated with smaller SV in knee joints: the greater the lymphatic transport the smaller the SV [26].
Figure 1. Routes of transport afferent and efferent to the PLN affect lymph node contrast enhancement (LNCE).
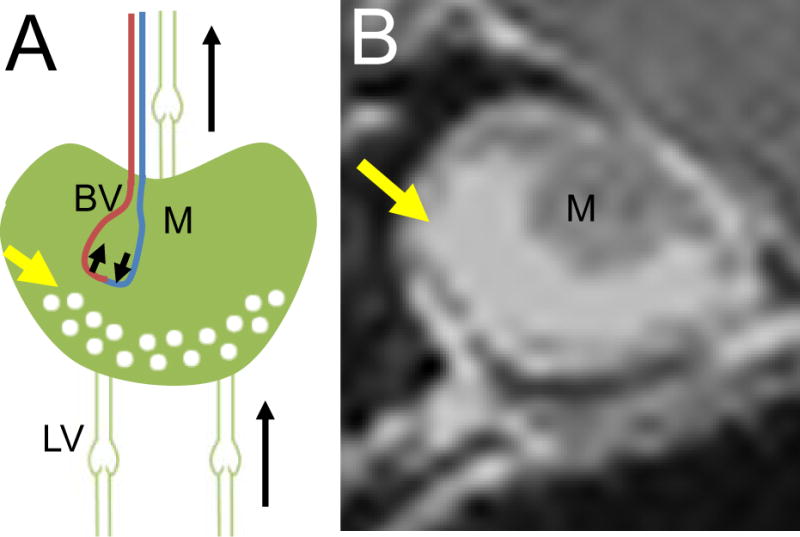
CE-MRI is preformed of the PLN by injecting gadolinium-diethylenetriamine pentaacetic acid (Gd-DTPA) retro-orbitally and allowing it to circulate for five minutes before beginning the MRI scan. This timepoint was chosen because the CE signal of the tissue is stable for the duration of the scan. Following the scan, LNCE is calculated by measuring the intensity of the LN and normalizing it to that of the surrounding muscle. LNCE is likely dependent on the routes of transport to the PLN: sources include arterial blood vessels (BV) from the Gd-DTPA remaining in circulation from the retro-orbital injection, and afferent lymphatic vessels (LV) from the Gd-DTPA leaking from blood capillaries in the distal tissue and being taken up by afferent LV (A). Gd-DTPA arriving from afferent LV will primarily reside within the sinuses (yellow arrow, A), correlating to where the signal is seen in the CEMRI image (yellow arrow, B) which surrounds the dark medulla (M) that excluded the contrast agent. Drainage of Gd-DTPA from the PLN can occur from venous return of the PLN and efferent lymphatic vessels (A). Due to the short time scale in which CE-MRI is preformed, clearance of the Gd-DTPA from the PLN is not observed.
4. The role of lymphatic drainage and lymphangiogenesis in murine inflammatory-erosive arthritis
Given that VEGF-C signaling through VEGFR-3 is the primary pathway for lymphangiogenesis, we performed formal gain and loss of function studies in murine models of arthritis to assess the potential role of lymphatic drainage in RA pathogenesis. In the loss of function studies, TNF-Tg mice were treated with VEGFR-3 neutralizing antibody. VEGFR-3 blockade increased joint inflammation and focal erosions, which was accompanied by decreased joint and PLN lymphangiogenesis and lymphatic clearance. Furthermore, it also decreased the number of CD11b+ myeloid cells in PLNs and joints [27]. These findings indicate that lymphangiogenesis is an important compensatory mechanism for regulating joint inflammation during chronic arthritis [27].
Collecting lymphatic vessels exhibit the ability to contract, which moves fluid against a pressure gradient. Evidence outlined above demonstrated that lymphatics are important in inflammatory-erosive arthritis, thus we developed near infrared indocyanine green (NIR-ICG) imaging to observe whether inflammatory-erosive arthritis affected lymphatic contraction in preclinical models. K/B×N mice underwent NIR-ICG imaging, revealing that during acute arthritis (1 month of age), the lymphatic pulsing frequency was increased [28]. In contrast, during the chronic phase (3 months of age), the lymphatic pulsing frequency returned to WT levels. Congruent with this, it was found that during the acute phase, there is a higher lymphatic clearance versus the chronic phase, which has a clearance similar to WT levels. This study demonstrated that there are two distinct lymphatic phenotypes that occur in inflammatory arthritis: an initial period of increased lymphatic transport with increased clearance, followed by a chronic phase when lymphatic pulsing and clearance is subdued [28]. To formally demonstrate the role of lymphangiogenesis with gain of function studies, recombinant VEGF-C expressing adeno-associated virus was administered into the joints of TNF-Tg mice, which demonstrated significant therapeutic efficacy; as it decreases synovitis, and reduces bone erosion, cartilage loss and osteoclast numbers [29]. This therapeutic effect was accompanied with an increase in lymphatic transport (i.e. lymphatic clearance from the footpad), and suggests that increasing lymphatic transport is a potential treatment for inflammatory-erosive arthritis.
5. Discovery of expanding and collapsed lymph nodes and B-in cells
The aforementioned research inspired a comprehensive longitudinal study of the natural history of inflammatory-erosive arthritis in TNF-Tg mice, in which the animals underwent CE-MRI every 2-weeks to observe their PLNs and SVs, and NIR-ICG imaging to assess changes in lymphatic transport [30]. Interestingly, it was found that knee arthritis in this model is asymmetrical, similar to the human disease, indicating epigenetic factors trigger joint inflammation and arthritic flare in the setting of a systemic IMID. This asymmetry was mirrored in the PLN; as knees with a low SV presented with adjacent PLN that were large in size and CE, while the contralateral knee with severe arthritis presented with a PLN that was smaller and failed to take up contrast [30]. Consistent with previous reports, this study demonstrated the two phases of the PLN phenotype during arthritic progression. In the first phase, the PLN is “expanding” (i.e. presents with increasing volume and CE), which corresponds to knee inflammation that is stable and mild with little bone erosion. Following the expanding phase, the PLN “collapses” (i.e. presents with decreasing volume and CE), which corresponds to a sharp increase in knee synovitis and bone erosions [30]. By NIR-ICG imaging, no lymphatic pulse was detected in legs with collapsed PLN, while legs from WT mice and those with expanding PLN showed a similar lymphatic pulse frequency. This is consistent with the fact that collapsed PLNs also show a decreased LNCE via CE-MRI, indicative of decreased lymphatic transport from ankle to the PLN [31].
To further elucidate the nature of expanding and collapsed PLN, we performed formal molecular and cellular characterization studies via microarray, ex vivo stimulation assays, flow cytometry and immunohistochemistry [30–33]. Remarkably, these studies failed to demonstrate any significant differences in gene expression or cellular composition. However, we found that both forms of TNF-Tg PLN contained a significant 2-fold increase in a B cell subset that was CD21high and CD23+. Moreover, this B cell population was selectively increased in LNs efferent to arthritic joints, and thus were named B cells in inflamed nodes (B-in) [30]. This name was further justified by subsequent studies that identified B-in cell increases in the K/B×N model [30], and in WT murine PLN efferent to sterile inflammation [34]. Importantly, these B-in cells are polyclonal and do not expresses any markers of activation or proliferation [30], strongly suggesting that they do not play an active role in arthritic flare. However, immunohistochemistry demonstrated that in contrast to B-in cells of expanding PLN, which reside in the peripheral follicles, B-in cells within collapsed PLN are translocated into the lymphatic sinuses and appear to be clogging the lymphatic transport [30]. Taken together, these results are consistent with B-in cell clogging of lymphatic sinuses as a novel mode of arthritic flare. The current model of B-in cell clogging of the lymph node sinuses is that macrophages present in lymphatic vessels and sinuses express CXCL13, a B cell chemoattractant (Figure 2). During the collapsed stage, the lymphatic vessels are no longer contracting, which causes an increase in immobile macrophages within these sinuses and vessels, providing a chemotactic gradient that triggers B cells to translocate from the follicles to the sinuses.
Figure 2. CXCL13 secreting macrophages are the prominent cells in TNF-Tg popliteal lymph node (PLN) sinuses.
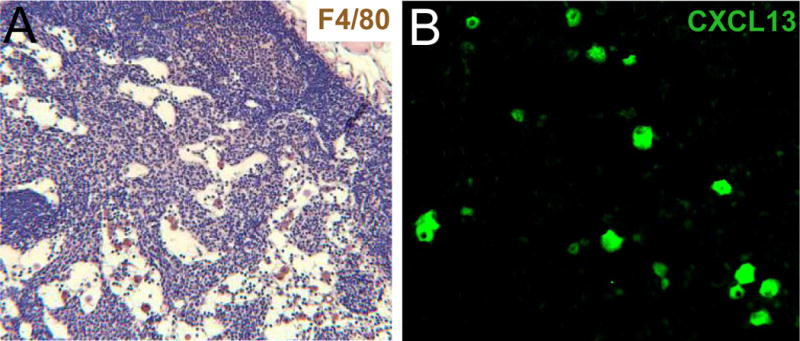
PLN were harvested from TNF-Tg mice and were processed for immunohistochemistry against F4/80 (A, brown) and CXCL13 (B, green). Representative images are shown at 20× and 40×, respectively. Note the presence of the F4/80+ macrophages within the sinus (A) and the sporadic staining of CXCL13 (B), likely due to cells within the sinuses and not within the T/B cell zone.
One apparent contradiction with our findings regarding PLN as a biomarker of inflammation in the adjacent joint is that the knee drains to the iliac lymph node (ILN), while the ankle drains to the PLN. Thus, the PLN cannot be a direct measure of lymphatic egress from the knee. To resolve this we tested the hypothesis that LN collapse during asymmetric knee flare in TNF-Tg mice occurs in series along the ipsilateral axis [35]. Consistently, we found that B cells were located within the LYVE-1+ lymphatic sinuses in both collapsed ILN and PLN ipsilateral to flaring knees, and NIR-ICG imaging demonstrated that a decrease in lymphatic transport to the PLN was accompanied with a decrease in transport from the knee to the ILN. Finally, ICG signal intensity, a marker of lymphatic transport, of both the ILN and PLN significantly inversely correlated with knee synovial volume [35]. Taken together, these data demonstrate that ILN and PLN collapse occur in unison; and therefore PLN collapse is a valid biomarker for knee flare. This is an important point as longitudinal in vivo imaging (PD-US, CE-MRI, NIR-ICG) is possible in the PLN whereas the ILN resides too deep to complete these important measurements. A summary of how the PLN, ILN and lymphatic vessel changes relate to synovitis in the ankle and knee is shown in Figure 3.
Figure 3. Overview of the changes that occur in expanding and collapsed TNF-Tg mice.
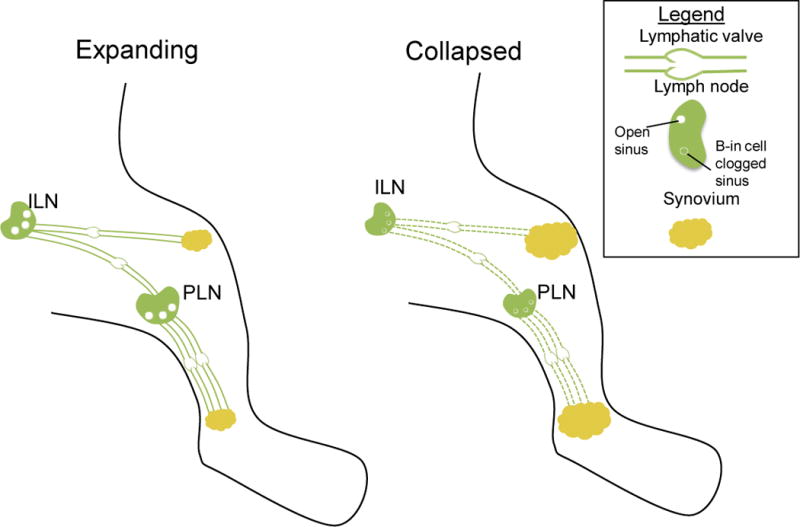
The PLN drains the ankle synovium while the iliac lymph node (ILN) drains the knee synovium. During the expanding phase, both PLN and ILN are large in volume with open sinuses, and afferent lymphatic vessels exhibit contractions. This keeps the synovial volume stable and the small amount of synovitis at this point is due to overwhelming of the capacity of the lymphatic system. This mild disease transitions rapidly to advanced inflammatory-erosive arthritis following the simultaneous collapse of ipsilateral PLN and ILN via poorly understood mechanisms. At this point, there is a lack of lymphatic contraction, the PLN and ILN decrease in size and show B cell clogging of the sinuses. This lack of lymphatic transport results in a very large synovial volume and pannus formation in the knee and ankle, commonly known as arthritic flare.
6. B cell depletion therapy restores lymphatic flow to collapsed PLN
RA is a notoriously refractory disease, particularly for the ~30% of patients who fail multiple treatments including anti-TNF therapy [36, 37]. As many of these patients have high circulating levels of autoantibodies that may be pathogenic, B cell depletion therapy (BCDT) with anti-CD20 (rituximab) was used as a third line agent to treat longstanding RA. Interestingly, results from the definitive phase 3 clinical trial of BCDT in RA showed that effective therapy does not correlate with a decrease in autoantibody levels [38], and that clinical improvement was most strongly correlated with a decrease in synovial macrophages [39], which do not express CD20 and are not direct targets of BCDT. As these results are consistent with our preclinical findings suggesting that B cells play a passive role in arthritic flare, we completed a series of experiments in TNF-Tg mice to formally test this hypothesis. In our prophylactic study, pre-arthritic TNF-Tg mice with expanding PLN were randomized to placebo and anti-CD20 treatment, and the results showed that BCDT significantly prevented the onset of knee arthritis [30]. To assess the effects of BCDT on established arthritic flare, TNF-Tg mice with collapsed PLN were randomized to anti-CD20 vs. placebo, and the therapy was found to significantly increase LNCE and decrease knee SV [31]. The increase in LNCE suggests an “unclogging” of the B cells in the PLN that then allows MRI contrast to fill the sinuses. This conclusion was supported by the fact that lymphatic flow from the footpad increased, demonstrated by a significant increase in footpad clearance. It was also found that ICG clearance had a significant inverse correlation to knee SV, suggesting more than a casual relationship between lymphatic drainage and joint inflammation. Interestingly, BCDT did not restore the lymphatic contraction, but did restore passive transport, indicated by the fact that lymphatic vessels could now take up more ICG and therefore appeared brighter during NIR-ICG imaging. Macrophages are major cellular effectors in the inflammatory-erosive arthritis through the release of pivotal pro-inflammatory cytokines and because they can differentiate into osteoclasts that resorb bone [40]. Macrophages are CD11b+, therefore, the first intravital microscopy of CD11b+ cells moving in lymphatic vessels was performed to examine if this cell subset travels from the distal inflamed ankle joint through lymphatic vessels. It was found that monocytes/macrophages move at great velocity in expanding PLN mice (186 ± 37 μm/s). In contrast, these CD11b+ cells are stationary in lymphatic vessels afferent to collapsed PLN (0 μm/s). Of note is that these immobile CD20− cells were removed by the BCDT therapy, which most likely occurred from recovery of the passive flow. In summary, it has been demonstrated that BCDT, which effectively treats arthritic flare, also correlates with: 1) increased lymphatic flow, 2) removal of B cells from PLN sinuses, and 3) restoration of macrophage (CD11b+ cells) migration via passive lymphatic transport [31].
7. Use of ultrasound for PLN phenotyping
While CE-MRI can be used to phenotype expanding and collapsed PLN, the technique has many limitations including costs, availability of MRI scanners, and the Gd-DTPA contrast that constrains temporal resolution. Therefore, there are benefits to using a more cost-effective technique, such as ultrasound (US). Others have demonstrated that US can be used to monitor changes in the LN of RA patients undergoing treatment [41], and we have demonstrated that US is comparable to CE-MRI for determining relative PLN volume in mice [42]. Power Doppler (PD) is an US technique that can be used to observe blood flow in various tissues. It was found that the normalized power Doppler volume (NPDV) within the PLN is higher in expanding PLN compared to both WT and collapsed PLN [43]. This indicates that there is increased blood flow to the PLN during the expanding phase, and that blood flow decreases during the dramatic changes of the PLN during the collapse phase. Moreover, it demonstrates that PD-US can be used to phenotype PLN. Given the significant differences between expanding and collapsed PLN, one imaging session can phenotype the PLN. It is noteworthy that we also investigated the utility of contrast enhance US in these studies [43]. Interestingly, we found that multiple (≥3) US contrast treatments induce PLN collapsed in younger mice, which corresponds to increased damage to the lymphatic vessels. This lymphatic vessel damage is likely due to the US contrast bubbles inducing mechanical damage to the lymphatic endothelial and smooth muscle cells, but more importantly, demonstrates that PLN collapse can be induced by damaging lymphatic vessels and therefore, lowering lymphatic transport [43].
8. Measurement of lymph node pressure, lymphatic pumping pressure, and lymph speed and viscosity
In an effort to better understand the mechanism of PLN collapse, methods were developed to measure popliteal lymph node pressure (LNP), lymphatic pumping pressure (LPP) and lymph viscosity and speed [44, 45]. Multiphoton fluorescence after photobleaching (MP-FRAP) of injected FITC-BSA was used to calculate the lymph viscosity and speed [46]. It was found that there was no difference in lymph viscosity between WT, expanding or collapsed PLN mice. However lymph speed was found to be significantly lower in mice with collapsed PLN, supporting the hypothesis that lymphatic transport is hindered in this PLN phenotype. LPP was measured by visualizing lymphatic vessels via NIR-ICG imaging, placing a transparent cuff on the mouse leg, increasing the pressure of the cuff to induce occlusion of the lymphatic vessel of interest and then lowering the pressure in the cuff until ICG was able to refill the previously occluded lymphatic vessel. This technique demonstrated that LPP is higher in expanding PLN mice compared to WT, which suggests that the vessels are contracting more forcibly to compensate for the increase fluid load due to extensive inflammation in the expanding TNF-Tg mice. LPP is extremely low after PLN collapse, conceivably due to the lack of lymphatic pulse. Furthermore, we found that LPP was decreased in WT mice that were treated with VEGFR3 neutralizing antibody, which decreased lymphatic transport from footpad (our unpublished observation). LNP was found to be much lower in expanding PLN compared to WT, suggesting increased lymphatic transport leaving the PLN, which then increased after collapse, due to the increase in cell density and lack of egress from the PLN [45].
Table 1 is presented to summarize our quantitative results on murine lymphatics and joints in WT mice and TNF-Tg mice with expanding and collapsed PLN. Taken together, these data provide the bases for a new theory of arthritic flare. In the normal joint, lymphatic egress is achieved by a pressure gradient where the LPP exceeds LNP in addition to active transport. However, the LNP is not “sensed” by the lymph within the lymphatic vessels due to functional intraluminal lymphatic valves. During the early or acute stages of inflammation, LPP increases to compensate for the rapidly increased fluid load. LNP is decreased at this point due to increased lymphangiogenesis that occurs in PLN, further widening the gap between LNP and LPP. Due to extensive vessel dilation during this phase (expanding PLN), intraluminal valves are unlikely to be fully functional as cell egress through lymphatic vessels appears to be independent of the lymphatic pulse [31]. As the PLN collapses during extensive chronic inflammation, the lymphatic pulse is lost, leading to a decrease in LPP, and B-in cells translocate to the sinuses, inducing an increase in LNP as lymphatic egress from the PLN is blocked. In addition, the lymphatic vessels become damaged from the chronic inflammation, leading to leaky lymphatic vessels and a deregulated lymphatic pulse. At this point, the pressure gradient shifts, as the LNP is higher than the LPP, which would impede passive transport. As active transport is already inhibited at this stage due to the loss of the lymphatic pulse, this leads to a dramatic decrease in lymph and inflammatory cell egress from the joint, which is manifested as joint swelling from edema and synovial hyperplasia. This also leads to massive accumulation of catabolic factors (matrix metaloproteases, MMPs) and osteoclasts which mediate irreversible joint damage [45].
Table 1.
Summary of measured and calculated values in TNF-Tg mice, a model of inflammatory erosive arthritis.
| WT | Expanding | Collapsed | |
|---|---|---|---|
| LNCE (a.u.) | 2.22 ± 0.30 [26] | 6.8 ± 0.8 [35] | 3.4 ± 0.7# [35] |
| LNCap (a.u.) | 2.83 ± 0.91 [26] | 52.9 ± 19.1 [35] | 18.6 ± 6.8# [35] |
| Knee Synovial Volume (mm3) | 1.44 ± 0.42 [25] | 3.5 ± 0.7 [35] | 5.5 ± 1.2# [35] |
| B-in cell (%) | 9.9 ± 5 [30] | 35.1 ± 6.9*** [30] | 31.2 ± 5.1*** [30] |
| B-in cell (no. ×106) | 0.08 ± 0.06 [30] | 1.3 ± 0.8*** [30] | 1.2 ± 0.6*** [30] |
| Lymphatic pulsing frequency (pulses/minute) | 1.4 ± 0.40 [31] | 1.4 ± 0.03 [31] | 0 [31] |
| CD11b+ cell velocity within PLV (μm/s) | n.d. [31] | 186 ± 37 [31] | 0 [31] |
| NPDV | 0.0041 ± 0.0017 | 0.5536 ± 0.007* [43] | 0.0086± 0.003# [43] |
| LN volume (mm3) | 0.416 ± 0.02 | 5.64 ± 0.54*** | 7.62 ± 1.26*** |
| Dlymph/DH2O | 0.63 ± 0.02 [45] | 0.63 ± 0.05 [45] | 0.70 ± 0.03 [45] |
| Viscosity (mPa s) | 1.81 ± 0.07 [45] | 1.84 ± 0.16 [45] | 1.61 ± 0.07 [45] |
| Lymph speed (μm/s) | 109.21 ± 3.99 [45] | 104.09 ± 5.71 [45] | 73.61 ± 6.79 **,## |
| LPP (cmH2O) | 11.04 ± 1.47 [45] | 18.76 ± 2.34** [45] | 2.61 ± 0.72 **,### [45] |
| LNP (cmH2O) | 6.86 ± 0.56 [45] | 3.41 ± 0.43* [45] | 9.92 ± 1.79## [45] |
LNCE, lymph node contrast enhancement; LN, lymph node; LNCap, LN capacity; B-in cells, B cells in inflamed nodes; PLV, popliteal lymphatic vessel; n.d., not detectable; NPDV, normalized power Doppler signal within the LN; Dlymph, diffusion coefficient of FITC-BSA in lymph; DH2O, diffusion coefficient of FITC-BSA in water; LPP, lymphatic pumping pressure; LNP, LN pressure; Values are mean ± standard error, n ≥ 4 for each group.
p<0.05 vs. WT;
p<0.01 vs. WT;
p<0.001 vs. WT;
p<0.05 vs. Expanding;
p<0.01 vs. Expanding;
p<0.001 vs. Expanding.
9. Future and therapeutic opportunities
Much is still unknown about what induces PLN collapse, and therefore arthritic flare, in humans and murine models of inflammatory-erosive arthritis. To us, the most burning questions are focused on the mechanism responsible for the loss of the lymphatic pulse and PLN collapse during arthritic flare. Current theories to explain the regulation of the lymphatic pulse are centered on nitric oxide (NO) as an important molecule, as it has been shown to contribute to relaxation of the lymphatic vessel between basal contractions [47–49]. Additionally, activated CD11b+ cells known to express inducible nitric oxide synthase (iNOS) [48] have been shown to adhere to lymphatic vessels in TNF-Tg mice [31]. Based on this information we propose the iNOS squelching hypothesis as illustrated in Figure 4. In this model the homeostatic lymphatic pulse that is driven by endothelial cell release of NO via endothelial nitric oxide synthase (eNOS), is overwhelmed by NO from iNOS expressing CD11b+ macrophages that have adhered to the lymphatic endothelium of lymphatic vessels efferent to chronically inflamed joints. This leads to constitutive smooth muscle cell relaxation and loss of the lymphatic pulse, which leads to PLN collapse due to decreased LPP, B-in cell clogging, and arthritic flare due to the loss of lymph and inflammatory cell egress from the afferent joint.
Figure 4. A schematic of lymphatic pulsing under normal and inflammatory conditions.
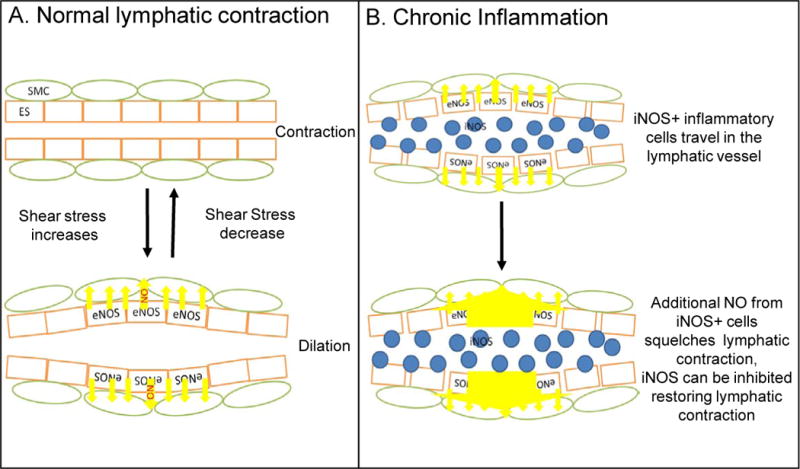
(A) Under normal condition, eNOS is induced by shear stress generated by lymphatic flow in endothelial cells (ES, orange) to maintain a regular lymphatic pulse via nitric oxide (NO, yellow arrows) regulation of lymphatic smooth muscle cell (SMC, green) contractions. (B) During chronic inflammatory conditions, such as inflammatory-erosive arthritis, high levels of NO produced by CD11b+/Gr-1+/iNOS+ cells (blue) that have attached to the lymphatic vessel squelching the eNOS NO gradient such that SMCs are in a constant state of relaxation, therefore causing the loss of lymphatic pulse and reduction of lymphatic clearance.
Taken together, our work demonstrates two distinct mechanisms of arthritic flare. Early flare is accompanied with increased LPP, decreased LNP and increased lymphangiogenesis. This acute flare is likely caused by egressing cells and lymph that overwhelm the lymphatic capacity, which is effectively treated by first line DMARDs. However, this does not treat the underlining pathophysiology. Eventually, chronic inflammation leads to a reduction in lymphatic transport due to increased NO, including the loss of lymphatic contraction, decreased LPP, and increased LNP. At this point, it might be most beneficial to treat with drugs that target the lymphatic system. Roughly 30% of patients are refractory to anti-TNF therapies [36, 37], demonstrating that DMARD therapy is not effective in all patients, and our theory offers a potential way to stratify the RA patient population to determine the most effective treatment. Many of the techniques developed for murine models were developed with clinical translation as the end goal, and can therefore be used in RA patients. Therefore, our hypothesis is that lymphatic research can be used to determine mechanisms of arthritic flare and determine the best treatment for individual patients. Future work includes confirming that PLN collapse occurs in a patient population with chronic RA via techniques we developed in mice, including both CE-MRI and NIR-ICG imaging. This will confirm what patients are undergoing collapse and will give insight as to the state of their lymphatic vessels. Drug studies have begun in mice to determine what drugs are efficient during disease progression and, in addition, to get to the core question that affects so many inflammatory diseases: what causes the loss of lymphatic pulse and can it be rescued? These experiments will be done with lineage tracing of progenitor smooth muscle cells to determine when the lymphatics reach their repair threshold, and determine the origin of a yet-to-be-determined lymphatic muscle stem cell population.
10. Conclusion
As the field of lymphatic research progresses it is evident that the lymphatic system plays a major but under-recognized role in many diseases, including cancer progression [50–52], irritable bowel disease [53], organ rejection [54], atherosclerosis [55, 56] and high blood pressure [57]. The contribution of the lymphatic system the initiation of flare and persistence of inflammation in RA can be inferred by the aforementioned clinical observations, but has not been formally tested in human disease. To elucidate the mechanism of arthritic flare in murine models, we developed several modalities to characterize both lymphatic and joint parameters, many of which are clinically translatable (Table 1). Consistent with the IMID hypothesis of RA, which posits multiple etiologies for this complex disease, our data demonstrate that arthritic flare may arise by at least two distinct mechanisms. The first is acute RA arthritic flare, at disease onset, which occurs in the setting of increased lymphatic transport from the inflamed joint to “expanded” draining LN. In contrast, we predict that RA flares that take place sporadically in patients with chronic disease are associated with the loss of lymphatic egress from joints, and can be assessed by “collapsed” efferent LN and the absence of a lymphatic pulse. With the recent development of clinical PD-US [41] and NIR-ICG [58] imaging of draining LN instruments to measure and assess the lymphatic pulse are now available providing a unique opportunity to examine the diagnostic and prognostic potential of these biomarkers. Moreover, elucidation of the mechanism of efferent LN collapse may catalyze new drug development to specifically target lymphatics in a population of RA patients with advanced disease for whom effective therapies are limited.
Acknowledgments
This work was supported by research grants from the National Institutes of Health PHS awards (T32 AR053459; R01s AR048697, AR053586 and AR056702; P01 AI078907; DP2OD006501; P30 AR061307 and 1DP2OD006501-10).
Footnotes
Abbreviations: ACPA, anti-citrillunated protein antibodies; B-in, B cells from inflamed nodes; BCDT, B cell depletion therapy; CE-MRI, contrast enhanced magnetic resonance imaging; eNOS, endothelial nitric oxide synthase; FITC-BSA, fluorescein isothiocyanate labeled bovine serum albumin; Gd-DTPA, gadolinium-diethylenetriamine pentaacetic acid; IMID, immune-inflammatory disorder; iNOS, inducible nitric oxid synthase; LPP, lymphatic pumping pressure; LN, lymph node; LNCE, lymph node contrast enhancement; LNCap, lymph node capacity; LNP, lymph node pressure; MP-FRAP, multiphoton fluorescence after photobleaching; NIR-ICG, near infrared-indocyanine green; NPDV, normalized power Doppler volume; NO, nitric oxide; OCPs, osteoclast precursors; PLN, popliteal lymph node; PD-US, power Doppler ultrasound; RA, rheumatoid arthritis; RF, rheumatoid factor; SV, synovial volume; TNF-Tg, tumor necrosis factor alpha transgenic; US, ultrasound; VEGF-C, vascular endothelial growth factor c; VEGFR3, vascular endothelial growth factor receptor 3; WT, wild-type
References
- 1.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–61. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 2.Nishimura K, Sugiyama D, Kogata Y, Tsuji G, Nakazawa T, Kawano S, et al. Meta-analysis: diagnostic accuracy of anti-cyclic citrullinated peptide antibody and rheumatoid factor for rheumatoid arthritis. Annals of internal medicine. 2007;146:797–808. doi: 10.7326/0003-4819-146-11-200706050-00008. [DOI] [PubMed] [Google Scholar]
- 3.Feldmann M, Maini SR. Role of cytokines in rheumatoid arthritis: an education in pathophysiology and therapeutics. Immunol Rev. 2008;223:7–19. doi: 10.1111/j.1600-065X.2008.00626.x. [DOI] [PubMed] [Google Scholar]
- 4.Kuek A, Hazleman BL, Ostor AJ. Immune-mediated inflammatory diseases (IMIDs) and biologic therapy: a medical revolution. Postgrad Med J. 2007;83:251–60. doi: 10.1136/pgmj.2006.052688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MH, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 2004;50:380–6. doi: 10.1002/art.20018. [DOI] [PubMed] [Google Scholar]
- 6.Silman AJ, Newman J, MacGregor AJ. Cigarette smoking increases the risk of rheumatoid arthritis. Results from a nationwide study of disease-discordant twins Arthritis Rheum. 1996;39:732–5. doi: 10.1002/art.1780390504. [DOI] [PubMed] [Google Scholar]
- 7.Mahdi H, Fisher BA, Kallberg H, Plant D, Malmstrom V, Ronnelid J, et al. Specific interaction between genotype, smoking and autoimmunity to citrullinated alpha-enolase in the etiology of rheumatoid arthritis. Nature genetics. 2009;41:1319–24. doi: 10.1038/ng.480. [DOI] [PubMed] [Google Scholar]
- 8.McGonagle D, Ash ZR, Hodgson RJ, Emery P, Radjenovic A. MRI for the assessment and monitoring of RA–what can it tell us? Nature reviews Rheumatology. 2011;7:185–9. doi: 10.1038/nrrheum.2010.172. [DOI] [PubMed] [Google Scholar]
- 9.Smolen JS, Aletaha D, Bijlsma JW, Breedveld FC, Boumpas D, Burmester G, et al. Treating rheumatoid arthritis to target: recommendations of an international task force. Ann Rheum Dis. 2010;69:631–7. doi: 10.1136/ard.2009.123919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quinn MA, Emery P. Window of opportunity in early rheumatoid arthritis: possibility of altering the disease process with early intervention. Clinical and experimental rheumatology. 2003;21:S154–7. [PubMed] [Google Scholar]
- 11.Paavonen K, Mandelin J, Partanen T, Jussila L, Li TF, Ristimaki A, et al. Vascular endothelial growth factors C and D and their VEGFR-2 and 3 receptors in blood and lymphatic vessels in healthy and arthritic synovium. J Rheumatol. 2002;29:39–45. [PubMed] [Google Scholar]
- 12.Wauke K, Nagashima M, Ishiwata T, Asano G, Yoshino S. Expression and localization of vascular endothelial growth factor-C in rheumatoid arthritis synovial tissue. J Rheumatol. 2002;29:34–8. [PubMed] [Google Scholar]
- 13.Huh YM, Kim S, Suh JS, Song H, Song K, Shin KH. The role of popliteal lymph nodes in differentiating rheumatoid arthritis from osteoarthritis by using CE 3D FSPGR MR imaging: relationship of the inflamed synovial volume. Korean J Radiol. 2005;6:117–24. doi: 10.3348/kjr.2005.6.2.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grillet B, Dequeker J. Rheumatoid lymphedema. J Rheumatol. 1987;14:1095–7. [PubMed] [Google Scholar]
- 15.Jayson MI, Cavill I, Barks JS. Lymphatic clearance rates in rheumatoid arthritis. Ann Rheum Dis. 1971;30:638–9. doi: 10.1136/ard.30.6.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olszewski WL, Pazdur J, Kubasiewicz E, Zaleska M, Cooke CJ, Miller NE. Lymph draining from foot joints in rheumatoid arthritis provides insight into local cytokine and chemokine production and transport to lymph nodes. Arthritis Rheum. 2001;44:541–9. doi: 10.1002/1529-0131(200103)44:3<541::AID-ANR102>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 17.Polzer K, Baeten D, Soleiman A, Distler J, Gerlag DM, Tak PP, et al. Tumour necrosis factor blockade increases lymphangiogenesis in murine and human arthritic joints. Ann Rheum Dis. 2008;67:1610–6. doi: 10.1136/ard.2007.083394. [DOI] [PubMed] [Google Scholar]
- 18.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10:4025–31. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–22. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 20.Ritchlin CT, Haas-Smith SA, Li P, Hicks DG, Schwarz EM. Mechanisms of TNF-alpha- and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. The Journal of clinical investigation. 2003;111:821–31. doi: 10.1172/JCI16069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li P, Schwarz EM, O’Keefe RJ, Ma L, Looney RJ, Ritchlin CT, et al. Systemic tumor necrosis factor alpha mediates an increase in peripheral CD11bhigh osteoclast precursors in tumor necrosis factor alpha-transgenic mice. Arthritis Rheum. 2004;50:265–76. doi: 10.1002/art.11419. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Q, Guo R, Lu Y, Zhao L, Zhou Q, Schwarz EM, et al. VEGF-C, a lymphatic growth factor, is a RANKL target gene in osteoclasts that enhances osteoclastic bone resorption through an autocrine mechanism. The Journal of biological chemistry. 2008;283:13491–9. doi: 10.1074/jbc.M708055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Q, Lu Y, Proulx S, Guo R, Yao Z, Schwarz EM, et al. Increased lymphangiogenesis in joints of mice with inflammatory arthritis. Arthritis Res Ther. 2007;9:R118. doi: 10.1186/ar2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van den Berg WB. Animal models of arthritis. What have we learned? J Rheumatol Suppl. 2005;72:7–9. [PubMed] [Google Scholar]
- 25.Proulx ST, Kwok E, You Z, Papuga MO, Beck CA, Shealy DJ, et al. Longitudinal assessment of synovial, lymph node, and bone volumes in inflammatory arthritis in mice by in vivo magnetic resonance imaging and microfocal computed tomography. Arthritis Rheum. 2007;56:4024–37. doi: 10.1002/art.23128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Proulx ST, Kwok E, You Z, Beck CA, Shealy DJ, Ritchlin CT, et al. MRI and quantification of draining lymph node function in inflammatory arthritis. Ann N Y Acad Sci. 2007;1117:106–23. doi: 10.1196/annals.1402.016. [DOI] [PubMed] [Google Scholar]
- 27.Guo R, Zhou Q, Proulx ST, Wood R, Ji RC, Ritchlin CT, et al. Inhibition of lymphangiogenesis and lymphatic drainage via vascular endothelial growth factor receptor 3 blockade increases the severity of inflammation in a mouse model of chronic inflammatory arthritis. Arthritis Rheum. 2009;60:2666–76. doi: 10.1002/art.24764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Q, Wood R, Schwarz EM, Wang YJ, Xing L. Near-infrared lymphatic imaging demonstrates the dynamics of lymph flow and lymphangiogenesis during the acute versus chronic phases of arthritis in mice. Arthritis Rheum. 2010;62:1881–9. doi: 10.1002/art.27464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou Q, Guo R, Wood R, Boyce BF, Liang Q, Wang YJ, et al. Vascular endothelial growth factor C attenuates joint damage in chronic inflammatory arthritis by accelerating local lymphatic drainage in mice. Arthritis Rheum. 2011;63:2318–28. doi: 10.1002/art.30421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J, Kuzin I, Moshkani S, Proulx ST, Xing L, Skrombolas D, et al. Expanded CD23(+)/CD21(hi) B cells in inflamed lymph nodes are associated with the onset of inflammatory-erosive arthritis in TNF-transgenic mice and are targets of anti-CD20 therapy. J Immunol. 2010;184:6142–50. doi: 10.4049/jimmunol.0903489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Ju Y, Bouta EM, Xing L, Wood RW, Kuzin I, et al. Efficacy of B cell depletion therapy for murine joint arthritis flare is associated with increased lymphatic flow. Arthritis Rheum. 2013;65:130–8. doi: 10.1002/art.37709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Zhou Q, Wood R, Kuzin I, Bottaro A, Ritchlin CT, Xing L, Schwarz EM. CD23+/CD21hi B cell translocation and ipsilateral lymph node collapse is associated with asymmetric arthritic flare in TNF-Tg mice. Arthritis Res Ther. 2011;13:R138. doi: 10.1186/ar3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Binder NB, Puchner A, Niederreiter B, Hayer S, Leiss H, Bluml S, et al. Tumor necrosis factor-inhibiting therapy preferentially targets bone destruction but not synovial inflammation in a tumor necrosis factor-driven model of rheumatoid arthritis. Arthritis Rheum. 2013;65:608–17. doi: 10.1002/art.37797. [DOI] [PubMed] [Google Scholar]
- 34.Moshkani S, Kuzin II, Adewale F, Jansson J, Sanz I, Schwarz EM, et al. CD23+ CD21(high) CD1d(high) B cells in inflamed lymph nodes are a locally differentiated population with increased antigen capture and activation potential. J Immunol. 2012;188:5944–53. doi: 10.4049/jimmunol.1103071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J, Zhou Q, Wood R, Kuzin I, Bottaro A, Ritchlin C, et al. CD23+/CD21hi B cell translocation and ipsilateral lymph node collapse is associated with asymmetric arthritic flare in TNF-Tg mice. Arthritis Res Ther. 2011;13:R138. doi: 10.1186/ar3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weinblatt ME, Keystone EC, Furst DE, Moreland LW, Weisman MH, Birbara CA, et al. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum. 2003;48:35–45. doi: 10.1002/art.10697. [DOI] [PubMed] [Google Scholar]
- 37.Weinblatt ME, Kremer JM, Bankhurst AD, Bulpitt KJ, Fleischmann RM, Fox RI, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. The New England journal of medicine. 1999;340:253–9. doi: 10.1056/NEJM199901283400401. [DOI] [PubMed] [Google Scholar]
- 38.Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793–806. doi: 10.1002/art.22025. [DOI] [PubMed] [Google Scholar]
- 39.Leandro MJ, de la Torre I. Translational Mini-Review Series on B Cell-Directed Therapies: The pathogenic role of B cells in autoantibody-associated autoimmune diseases–lessons from B cell-depletion therapy. Clinical and experimental immunology. 2009;157:191–7. doi: 10.1111/j.1365-2249.2009.03978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galli SJ, Borregaard N, Wynn TA. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nature immunology. 2011;12:1035–44. doi: 10.1038/ni.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manzo A, Caporali R, Vitolo B, Alessi S, Benaglio F, Todoerti M, et al. Subclinical remodelling of draining lymph node structure in early and established rheumatoid arthritis assessed by power Doppler ultrasonography. Rheumatology (Oxford) 2011;50:1395–400. doi: 10.1093/rheumatology/ker076. [DOI] [PubMed] [Google Scholar]
- 42.Ju Y, Rahimi H, Li J, Wood RW, Xing L, Schwarz EM. Validation of 3-dimensional ultrasound versus magnetic resonance imaging quantification of popliteal lymph node volume as a biomarker of erosive inflammatory arthritis in mice. Arthritis Rheum. 2012;64:2048–50. doi: 10.1002/art.34448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bouta EM, Ju Y, Rahimi H, de Mesy-Bentley KL, Wood RW, Xing L, et al. Power Doppler Ultrasound Phenotyping of Expanding versus Collapsed Popliteal Lymph Nodes in Murine Inflammatory Arthritis. PLoS One. 2013;8:e73766. doi: 10.1371/journal.pone.0073766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bouta EM, Wood RW, Perry SW, Brown EB, Ritchlin CT, Xing L, et al. Measuring intranodal pressure and lymph viscosity to elucidate mechanisms of arthritic flare and therapeutic outcomes. Ann N Y Acad Sci. 2011;1240:47–52. doi: 10.1111/j.1749-6632.2011.06237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bouta EM, Wood RW, Brown EB, Rahimi H, Ritchlin CT, Schwarz EM. In vivo Quantification of Lymph Viscosity and Pressure in Lymphatic Vessels and Draining Lymph Nodes of Arthritic Joints in Mice. J Physiol. 2014;592:1213–23. doi: 10.1113/jphysiol.2013.266700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sullivan KD, Sipprell WH, 3rd, Brown EB, Jr, Brown EB., 3rd Improved model of fluorescence recovery expands the application of multiphoton fluorescence recovery after photobleaching in vivo. Biophysical journal. 2009;96:5082–94. doi: 10.1016/j.bpj.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmid-Schonbein GW. Nitric oxide (NO) side of lymphatic flow and immune surveillance. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:3–4. doi: 10.1073/pnas.1117710109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liao S, Cheng G, Conner DA, Huang Y, Kucherlapati RS, Munn LL, et al. Impaired lymphatic contraction associated with immunosuppression. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18784–9. doi: 10.1073/pnas.1116152108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mathias R, von der Weid PY. Involvement of the NO-cGMP-K(ATP) channel pathway in the mesenteric lymphatic pump dysfunction observed in the guinea pig model of TNBS-induced ileitis. American journal of physiology Gastrointestinal and liver physiology. 2013;304:G623–34. doi: 10.1152/ajpgi.00392.2012. [DOI] [PubMed] [Google Scholar]
- 50.Padera TP, Kadambi A, di Tomaso E, Carreira CM, Brown EB, Boucher Y, et al. Lymphatic metastasis in the absence of functional intratumor lymphatics. Science (New York, NY) 2002;296:1883–6. doi: 10.1126/science.1071420. [DOI] [PubMed] [Google Scholar]
- 51.Hoshida T, Isaka N, Hagendoorn J, di Tomaso E, Chen YL, Pytowski B, et al. Imaging steps of lymphatic metastasis reveals that vascular endothelial growth factor-C increases metastasis by increasing delivery of cancer cells to lymph nodes: therapeutic implications. Cancer Res. 2006;66:8065–75. doi: 10.1158/0008-5472.CAN-06-1392. [DOI] [PubMed] [Google Scholar]
- 52.Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, et al. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med. 2001;7:192–8. doi: 10.1038/84643. [DOI] [PubMed] [Google Scholar]
- 53.Wu TF, Carati CJ, Macnaughton WK, von der Weid PY. Contractile activity of lymphatic vessels is altered in the TNBS model of guinea pig ileitis. American journal of physiology Gastrointestinal and liver physiology. 2006;291:G566–74. doi: 10.1152/ajpgi.00058.2006. [DOI] [PubMed] [Google Scholar]
- 54.Hos D, Cursiefen C. Lymphatic vessels in the development of tissue and organ rejection. Advances in anatomy, embryology, and cell biology. 2014;214:119–41. doi: 10.1007/978-3-7091-1646-3_10. [DOI] [PubMed] [Google Scholar]
- 55.Martel C, Li W, Fulp B, Platt AM, Gautier EL, Westerterp M, et al. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. The Journal of clinical investigation. 2013;123:1571–9. doi: 10.1172/JCI63685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lim HY, Rutkowski JM, Helft J, Reddy ST, Swartz MA, Randolph GJ, et al. Hypercholesterolemic mice exhibit lymphatic vessel dysfunction and degeneration. The American journal of pathology. 2009;175:1328–37. doi: 10.2353/ajpath.2009.080963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15:545–52. doi: 10.1038/nm.1960. [DOI] [PubMed] [Google Scholar]
- 58.Rasmussen JC, Tan IC, Marshall MV, Adams KE, Kwon S, Fife CE, et al. Human Lymphatic Architecture and Dynamic Transport Imaged Using Near-infrared Fluorescence. Transl Oncol. 2010;3:362–72. doi: 10.1593/tlo.10190. [DOI] [PMC free article] [PubMed] [Google Scholar]