

Abstract
Vascular diseases are usually caused by multifactorial pathogeneses involving genetic and environmental factors. Our current understanding of vascular disease is, however, based on the focused genotype/phenotype studies driven by the “one-gene/one-phenotype” hypothesis. Drugs with “pure target” at individual molecules involved in the pathophysiological pathways are the mainstream of current clinical treatments and the basis of combination therapy of vascular diseases. Recently, the combination of genomics, proteomics, and metabolomics has unraveled the etiology and pathophysiology of vascular disease in a big-data fashion and also revealed unmatched relationships between the omic variability and the much narrower definition of various clinical phenotypes of vascular disease in individual patients. Here, we introduce the phenomics strategy that will change the conventional focused phenotype/genotype/genome study to a new systematic phenome/genome/proteome approach to the understanding of pathophysiology and combination therapy of vascular disease. A phenome is the sum total of an organism’s phenotypic traits that signify the expression of genome and specific environmental influence. Phenomics is the study of phenome to quantitatively correlate complex traits to variability not only in genome, but also in transcriptome, proteome, metabolome, interactome, and environmental factors by exploring the systems biology that links the genomic and phenomic spaces. The application of phenomics and the phenome-wide associated study (PheWAS) will not only identify a systemically-integrated set of biomarkers for diagnosis and prognosis of vascular disease but also provide novel treatment targets for combination therapy and thus make a revolutionary paradigm shift in the clinical treatment of these devastating diseases.
Keywords: Vascular disease, phenomics, hypertension, stroke, combination therapy
Introduction
It is well known that vascular disease, including atherosclerosis, hypertension, stroke, and pulmonary arterial hypertension, are usually caused by multifactorial pathogeneses involving both genetic and environmental factors (Fig. 1) [1]. The conventional hypothesis-driven approaches to vascular research, however, mainly focus on individual factors that are presumably involved in the pathophysiology of vascular disease. While some of these factors have been extensively studied and become the mainstream targets of current clinical treatment of vascular disease, many other factors have been largely neglected or not studied at all [2-5]. A deeper understanding of the genetic basis of vascular disease may provide new insights into the mechanisms underlying the pathophysiology and facilitate development of novel diagnostic and therapeutic modalities. Traditionally, the phenotype-genotype relationship of a disease is studied using strategies of either forward genetics (top-down), which starts with one phenotype and considers its link with one or many genotypes (such as genetic linkage studies), or reverse genetics (bottom-up), which starts with one genotype and see whether its relationship with the phenotype can be confirmed (such as functional studies of gene overexpression or knockout, Fig. 2). In the era of post-genome, the emergence of many omics technologies and platforms, including genomics, proteomics, and metabolomics, has provided many unbiased approaches to unravel the etiology and pathophysiology of many vascular diseases in the fashion of global and integrated biological system rather than single gene-based individual molecular components (Fig. 3). As a consequence of these ‘omics’ studies the current definition of clinical phenotypes of complex disease and combination drug therapy can no longer “match” to the increasingly detailed genomic/ proteomic/metabolomic variants. This has created major challenges for a complete understanding of the relationship between the genomic/proteomic/metabonomic variability and the phenotypic vascular disease in individual patients
Fig. (1).
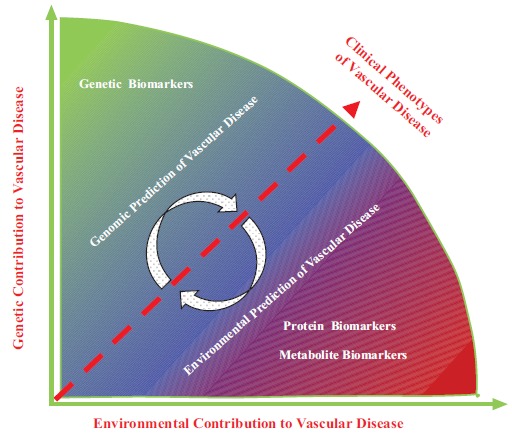
Schematic description of contributions from the interplay between genetic and environmental factors to the development of vascular disease and the role of relative biomarkers in clinical vascular disease phenotypes
Fig. (2).
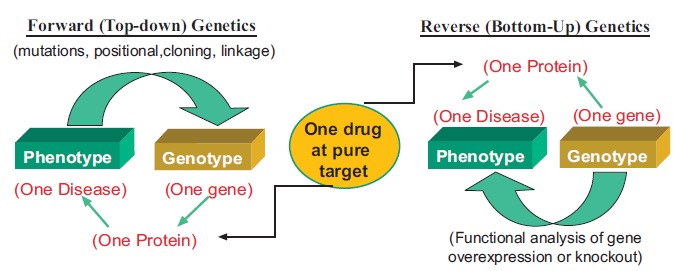
Genetic study of phenotype-genotype association.
Fig. (3).
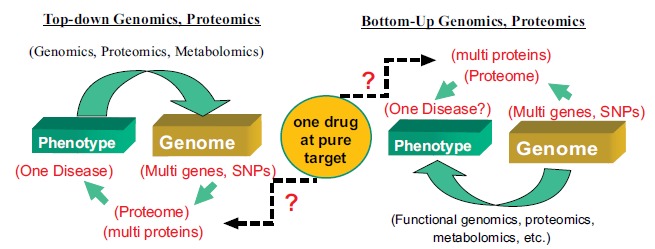
Omic study of phenotype-genotype association.
The color spectrum of development of clinical phenotype of vascular disease schematically shows that in most cases the vascular disease is caused neither solely by genomic factors (green) nor totally by environmental (red) factors. The clinical phenotypes of vascular disease in an individual patient are usually a sum result of the complex of interplay between genetic and environmental contributions. Although in some conditions patients with a positive genetic susceptibility to vascular disease may have a major genetic component, whether the patient will manifest the full clinical phenotypes of the vascular disease is dependent on the contributing environmental factors. Conversely, patients may have different thresholds of an environmentally induced vascular disease depending on the individual’s genetic background. A variety of biomarkers (including genes, microRNAs, proteins, small molecules of metabolites, and imaging) can be deployed to delineate disease progression in vascular disease. Genetic biomarkers (gene mutations, single-nucleotide polymorphisms, microRNAs etc.) can identify an individual’s genetic predisposition. However, it is the environmental imprints on the genetic background that are responsible for the dynamic production of abnormal unique proteins and/or small metabolites that foster disease progression in the susceptible individuals. The environmental prediction of vascular disease can be detected using protein or metabolite biomarkers. As the disease progresses further in the susceptible individual, vascular structural and functional alterations can be detected through imaging technologies.
The traditional phenotype-genotype association study follows the principle of “one gene, one protein, one phenotype (disease)” and uses either forward (top-down) genetics or reverse (bottom-up) genetics strategy to identify the genetic variability responsible for the phenotype or disease. This “one gene, one enzyme, one drug” principle has guided the traditional drug research with identification of “one pure drug target” as the ideal drug with pure therapeutic efficacy and the least side-effect or toxicity.
The post-genome approaches to phenotype-genotype association study use similar strategies to those of genetic approaches but at an “omics” level. The top-down strategy uses a set of technologies and approaches including genomics, transcriptomics, proteomics, interactomes, and metabolomics to study the association of phenotype (a disease) and genome, transcriptome, proteome, interactome, and metabolome. Disease phenotype may be associated with multiple single-nucleotide polymorphisms (SNPs) or mutations of multiple genes, or changes in expression and/or posttranslational modification profiles of multiple proteins. The bottom-up strategy uses functional genomics, functional proteomics and functional metabolomics to test whether the findings from the top-down studies are indeed correlated to the phenotypes. These studies revealed a nonparallel relation between phenotype and genome/proteome. This also leads to current challenges in the definition of clinical phenotypes and “pure” drug targets.
and the translation of the knowledge of genomic/proteomic/ metabonomic research into clinical practice of combination therapy of vascular disease [2, 6, 7]. Clearly, newer approaches are needed for the redefinition of disease using not only the traditional signs and symptoms but also the underlying genomic/proteomic causes and other factors [8]. Here, we introduce the phenomics strategy that will make the transition from the conventional focused genotype/phenotype studies to the global genome/phenome approaches to a systematical understanding of pathophysilogical mechanisms and combination therapy of vascular disease. We also discuss about the advance from the genome-wide association studies (GWAS) to the phenome-wide association studies (PheWAS) in vascular medicine. It is believed that the application of phenomics in the study of vascular disease will identify systemically integrated biomarkers for diagnosis and prognosis and novel treatment targets for combination therapy and thus make a revolutionary paradigm shift in the clinical treatment of vascular disease.
Genetics, Genome, and GWAS in Vascular Disease
A wide variety of approaches have been used to study the association of genetic polymorphisms to phenotypes of vascular disease, evolving from genetics, to genomics and GWAS with the improvement of genotyping analyses. The candidate gene approach is the earliest and simplest method using the strategy of forward genetics based on a priori hypothesis to determine whether a gene is responsible for an intermediate phenotype, such as a molecular and cellular function, or a clinical manifestation. The focus of candidate gene study is on single genes mainly located in exons with polymorphisms that have a major role in determining the gene expression and the transduced protein or changes in cellular function. Cambien et al. published the first study of candidate gene in cardiovascular disease in 1992 [9]. They showed that homozygotes with a deletion in the angiotensin converting enzyme (ACE) gene were at higher risk of coronary artery disease and myocardial infarction. This association was more evident in subjects lacking of some well-established non-genetic risk factors, suggesting the importance of studying both genetic and environmental factors [9]. Later, it was noted that in a gene of 30 Kb there are 10–100 polymorphisms that are potentially responsible for variations of the genetic effect. Therefore, later studies started to genotype more than one polymorphism in the same gene. For example, Iacoviello et al. found that two polymorphisms in distant regions of the gene encoding for coagulation factor VII were associated with myocardial infarction (MI) and suggested a higher gene effect for the presence of both risk alleles [10]. This has increased the analysis complexity and furnished an enormous amount of information in a big-data fashion.
Since the completion of the Human Genome Project in 2000, millions of single nucleotide polymorphisms (SNPs) have been discovered in the human genome. While the SNP is deliberately chosen to mark a gene hypothesized a priori to play a mechanistic role in determining the trait in a candidate gene study, SNPs from across the entire genome are studied agnostically, without premeditation regarding their possible function in a genome-wide association study (GWAS). The patterns of linkage disequilibrium (LD) in the human genome have also been characterized through the International HapMap Project. With the availability of high-throughput genotyping platforms and significantly decreased costs of genotyping and genome sequencing, GWAS has become more affordable and has gradually overwhelmed the analyses on a single candidate gene. GWAS has provided a systematic approach to investigation of the impact of common genomic variations on human vascular disease, such as acute coronary artery syndrome and hypertension [10-14]. While some studies started to use the clustering of common clinical domain phenotypes, most of current GWAS are still using the hypothesis-driven strategy for identifying genetic mechanisms and remain largely focused on clinical categories that do not provide adequate etiological information [15]. In GWAS, a single pre-defined disease or clinical phenotype (trait) is studied and the accrual of a large number of single gene variant–phenotype associations has serendipitously identified one gene affecting or responsible for more than one phenotypic characteristic or single loci associated with multiple diseases (pleiotropy). In addition to the levels of encoded protein in basal conditions, the link between genetic variants and phenotypic diversities could be resulted from different responses to an environmental stimulus (Fig. 1). A final target environmental risk factor can interact with a variety of molecular pathways, which are in turn regulated by multiple genetic factors. Consequently, the effects of environmental risk factors could be modified by different genetic background. As a classic example of how genetic background could impact in disease pathogenesis, it was found that patients with different genotypes showed different levels of monocyte-production of cytokines in response to stimulation with lipopolysaccharide, corresponding to a different response of monocytes to inflammatory stimulation and risk of ischemic vascular disease (MI and stroke) at young age [16]. Actually, a risk factor together with other risk factors such as high salt intake, hypercholesterolaemia, smoking, obesity, sedentary life-style, kidney dysfunction, and vascular inflammation, is very common in the development of hypertension [13, 17-20]. These risk factors lead to early functional and structural changes of the vasculature (e.g. endothelial dysfunction and increased vascular stiffness) that then gradually develop into early and advanced organ damage and failure and hypertension (Fig. 4). During the development of hypertension the genome remains largely unchanged, whereas it is clear that the expression and function of proteins, metabolisms, structure of tissues and organs undergo substantial changes. Therefore, while genetic factors define an individual’s potential or risk for hypertension in early life, their applications in the diagnosis and prognosis of hypertension and prediction of an individual’s risk of developing overt organ damage are very limited. Due to the dynamic nature of disease development, the transcriptome, proteome, metabolome, and interactome will not only change due to disease processes but also in response to therapeutic measures and can therefore be used to unravel the pathophysiological pathways responsible for vascular disease and to monitor treatment effects. In order to gain a more comprehensive and quantitative understanding of the complex interplay of multiple mechanisms in vascular disease the ‘omics’-based studies have been developed in vascular medicine in the past few years [21-24]. These ‘omics’ strategies, including genomics, transcriptomics, proteomics, and metabolomics, however, are still based on the traditional molecular biology principle that DNA is transcribed to RNA and translated to proteins that control an organism’s metabolism. Accordingly, these approaches start from a global study of a large number of molecules and then dissect the changes in the expression of individual genes and proteins under the influence of vascular disease or drug treatment. As a consequence of these ‘omics’ studies the current definition of clinical phenotypes of complex disease and combination drug therapy are not adequately explained by the increasingly detailed genomic/proteomic/metabolomic variants. This has created major challenges for a complete understanding of the relationship between the genomic/proteomic/ metabonomic variability and the phenotypic vascular disease in individual patients and the translation of the knowledge of genomic/proteomic/metabonomic research into the clinical practice of combination therapy of vascular disease [2, 6, 7]. Therefore, current approaches including GWAS for identifying genetic mechanisms or. pleiotropy of vascular diseases remain largely focused on clinical categories that do not adequately reflect the etiology of the diseases [12]. The reason for the limitation is that the conventional concept of vascular disease, for example hypertension, is defined as a clinical phenotype, not a clinical phenome (Fig. 4). Clearly, the current definition of vascular disease is out of date and newer approaches are needed for the redefinition of vascular disease using not only the traditional signs and symptoms but also the underlying molecular causes at the scale of genome/proteome and the interaction with environmental factors and their modifying role over a longer period of time. The newly developed phenomic approach that assembles coherent sets of phenotypic features across individual measurements and diagnostic boundaries provides a new opportunity for systematical investigations of established biological pathways and complements the traditional candidate gene strategy or GWAS focused on single genotype-phenotype association [25, 26]. This new approach provides a novel paradigm for the redefinition of vascular disease as new category of clinical phenome (Fig. 4).
Fig. (4).
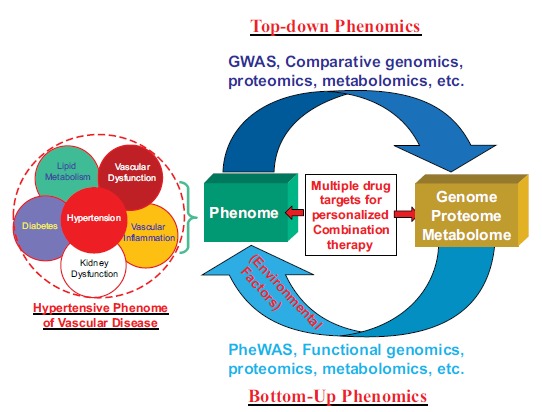
Phenomics approach to the definition of clinical phenome of vascular disease and the identification of multiple drug targets suitable for personalized therapy.
Phenome, Phenomics, and PHEWAS in Vascular Disease
A phenome is the sum of complete phenotypic characteristics (phenomic traits) that signify the expression of the whole genome, proteome, and metabolome under specific environmental influence [26-29]. Phenomics is a recently developed new transdiscipline that studies phenome in order to correlate complex traits to variability not only in genome, but also in proteome, metabolome, interactome, and environmental factors [28-32]. It provides a suite of new technologies and platforms for the transition from focused phenotype-genotype study to a systematic phenome-genome approach, which can be used to redefine the clinical phenotypes of diseases. Phenomics is a multi-phenotype approach that requires collection of a wide breadth of phenotypes with fine resolution, phenomic analysis with constructing heat maps, cluster analysis, pathway analysis, text mining and big data analysis (Fig. 5). Clinical phenomics is the systematic measurement and analysis of qualitative and quantitative clinical phenome (traits), including clinical signs and symptoms, and laboratory results obtained by biochemical, genomic, proteomic, metabolomic, and imaging methods, for the refinement and characterization of a clinical phenome.
Fig. (5).
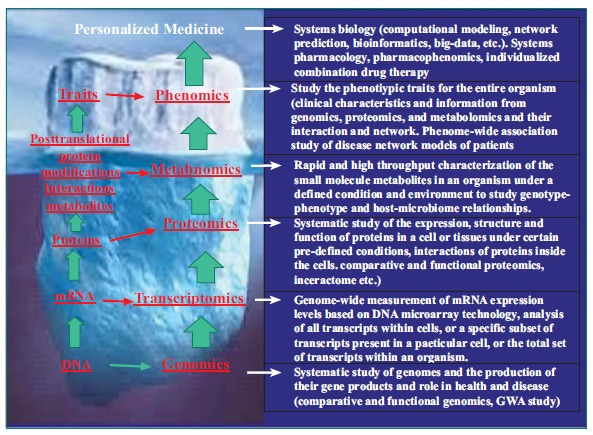
An iceberg model of the relationship of phenomics with other omics approaches and platforms in translational research on personalized medicine.
What will emerge from a phenomics (including clinical phenomics) approach is a more valid and etiologically-based systematic definition of disease phenome (Fig. 4) that may be quite different from those of current disease phenotypes defined by using the clinical symptoms at an organ (e.g., coronary artery, heart, kidney etc.) or system (e.g., cardiovascular or nervous system) level alone, as has been the tradition in clinical practice of Western medicine for the past centuries (Fig. 2). Accordingly, disease should now be defined as a clinical phenome that is the sum total of a patient’s clinical characteristics or phenomic traits that signify the expression of the whole genome, proteome, and metabolome under specific environmental influence (Fig. 4). For example, the identification of multiple disease genes or proteins at the genomic and proteomic levels provide a solid platform for novel definition of the systematically integrated (or clustered) hypertensive phenotypes (hypertension phenome) under the scope of the whole genome/proteome/metabolome (Fig. 4). Therefore, the hypertension phenome will now include not only clinical symptoms of elevated blood pressure (hypertension) but also a series of other systematically-defined phenotypic characteristics at different levels, including vascular morphology and function, heart-kidney-neural circuits/networks, metabolites, proteins, and genes (Fig. 4). It is interesting that in the traditional Chinese medicine (TMC) no hypertension was defined as a single disease (or clinical phenotype). The symptoms of “hypertension” were included in 4 to 5 different “Zheng-Hou” (Fig. 4) defined as “liver-yang hyperactivity (or Gang-Yang-Shang-Kang)”, “liver-qi stagnancy (or Gang-Qi-Yu-Jie)”, “Ying-deficiency with Yang-Hyperactivity (or Yin-Xu-Yang-Sheng)”, or “Abundant Phlegm-dampness (or Tan-Shi-Yong-Sheng)”[33]. In current terms, Zheng-Hou can be explained as a clinical phenome while hypertension is a phenotype [34]. Therefore, phenomics study of TMC may help to redefine these hypertension-associated Zheng-hou at the omics level, i.e., the hypertensive phenome of vascular disease (Fig. 4). With well-defined disease phenomes and a suite of new phenomics technologies and platforms (Fig. 5) the phenomics approach may be used to characterize the phenomes of vascular disease and also to identify the corresponding therapeutic targets for combination therapy at the level of systems biology. Therefore, phenomics will complement other omics approaches, including genomics, proteomics, and metabolomics, and provide a new paradigm for the study of vascular disease and for rationally selecting drug targets and treatments suitable for personalized medicine (Fig. 5). It can be envisaged that phenomics will refine the definition and diagnosis of vascular disease phenome and with systematically-defined therapeutic targets and improve predictive validity for outcomes of drug treatment.
Hypertensive phenotypes will be systematically-integrated and defined as a new hypertensive phenome of vascular disease according to the association with other related (clustered) clinical phenotypes such as vascular dysfunction, vascular inflammation, lipid metabolism, diabetes, and/or kidney dysfunction, multiple disease genes, SNPs, or proteins identified under the scope of the whole genome/proteome/ metabolome, using either the top-down (comparative genomics, proteomics, metabolomics, GWAS, etc.) or bottom-up (functional genomics, proteomics, metabolomics, PheWAS, etc.) or both strategies. In the traditional Chinese medicine (TMC), the symptoms of “hypertension” were included in different “Zheng-Hou” defined as “liver-yang hyperactivity (or Gang-Yang-Shang-Kang)”, “liver-qi stagnancy (or Gang-Qi-Yu-Jie)”, “Ying-deficiency with Yang-Hyperactivity (or Yin-Xu-Yang-Sheng)”, or “Abundant Phlegm-dampness (or Tan-Shi-Yong-Sheng)” [33]. Therefore, the hypertensive phenome will now include not only clinical symptoms and signs of elevated blood pressure (hypertension) but also a series of other systematically-defined phenotypic characteristics at different levels, including vascular morphology and function, heart-kidney-neural circuits/networks, metabolites, proteins, and genes. The phenomics approach will also provide a novel paradigm for drug research and development with the identification of multiple targets corresponding to the disease genes and proteins in the disease phenome, which will be suitable for personalized combination therapy.
As a mimic to the GWAS, Denny et al. proposed in 2010 a novel approach termed phenome-wide association
Phenomics is a novel transdiscipline as a complimentary approach to several other omics approaches, including genomics, transcriptomics, proteomics, and metabolomics in the study of vascular disease and drug research and development, which will ultimately lead to individualized drug therapy and personalized medicine.
study (PheWAS) to screen phenomic data for disease-gene associations [35]. PheWAS utilizes phenomics and big-data technologies to analyze all genetic/proteomic variants and all available phenotypic information from EMRs, electronic health records (EHRs), or observational cohort containing all types of diagnoses of clinical phenotypes such as data from the Clinical Data Warehouse (CDW) in the estimation of association between genome-phenome and detection of pleiotropy [25, 26, 28, 31, 36-38]. With PheWAS, associations between a specific genetic variant and a wide range of physiological and/or clinical outcomes and phenotypes can be explored either by using algorithms to parse the data collected from EHRs, EMRs, or CDW. Denny et al. used the International Classification of Disease (ICD9) billing codes available in most electronic medical records (EMRs) to develop a code translation table for automatic definition of 776 different disease populations and used the prevalent ICD9 codes derived from the EMR database to define their controls. Using this PheWAS algorithm, they genotyped 6005 European-Americans in the BioVU, Vanderbilt's DNA biobank, accrued at five SNPs with previously reported disease associations: coronary artery disease, atrial fibrillation, Crohn's disease, carotid artery stenosis, multiple sclerosis, systemic lupus erythematosus and rheumatoid arthritis. The PheWAS software was used to generate cases and control populations across all ICD9 code groups for each of these five SNPs and to analyze the disease-SNP associations. They found 4 of 7 previously known SNP-disease associations were replicated. In addition, they also identified 19 previously unknown statistical (P < 0.01) SNPs-diseases associations [35]. This study demonstrates the feasibility of using PheWAS analysis as an alternative method that complements GWAS, to investigate genotype-phenotype (SNP-disease) associations. Since 2010, PheWAS has been used to investigate whether SNPs associated with one phenotype are also associated with other phenotypes [35, 39, 40]. More recently, Denny and colleagues expanded their PheWAS using phenotypic data from the EMRs of 13,835 individuals to look for associations between 1,358 phenotypes and 3,144 SNPs that had previously been found to show association with one or more traits in GWAS [41]. In cases for which the PheWAS was sufficiently powered, they found 66% of associations from GWAS were replicated. In addition, they also uncovered 63 cases of previously unknown associations, potentially as pleiotropic effects. EMR-based PheWASs provide a much simpler approach to pleiotropy analysis than the current GWAS-based approach, which requires complex integration of data from multiple studies. Robust test of the EMR/EHR-based PheWAS allows unbiased interrogation across all domains of disease (heart diseases, hypertension, stroke, brain diseases, diabetes, cancers, etc.), and can not only replicate what is known about individual genotype-phenotype associations with various SNPs but also uncover novel associations with a wide range of phenotypes in the EMR/EHR-based cohorts. Larger EMR/EHR-based PheWASs may reveal more pleiotropy than has been estimated from GWAS and have the potential to significantly improve our understanding of the molecular etiologies of diseases. With the fast advance in big-data technology and phenomics, we believe that the application of the EMR/EHR data-based PheWAS in vascular medicine opens important avenues to enhance systematically-integrated analysis of the genomic basis of human cardiovascular disease and responses to drug therapy and to reform our understanding and clinical treatment of these devastating diseases with a new concept of wholism.
ACKNOWLEDGEMENTS
The Duan Laboratory of Cardiovascular Phenomics in the Center for Molecular Medicine and Department of Pharmacology, School of Medicine at University of Nevada is supported by NIH grants #HL63914, #HL106252, and #HL113598. Dr. Hong Yuan and Dr. Ya-ping Zhang are supported by National Basic Research Program of China (No. 2011CB512001), the National Natural Science Foundation of China (NO. 81273594), the National Key Technology R&D Program (2012BAI37B05), International Science and Technological Cooperation Project of Hunan (No. 2010wk2006), Doctoral Research and Innovation Project of Hunan Province (No. CX2011B075), Fundamental Research Funds for the Central South University (2012zzts036).
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Iacoviello L., Donati M.B. Gene-environment interactions: implications for cardiovascular disease. Cardiologia. 1999;44:227–232. [PubMed] [Google Scholar]
- 2.Moxon J.V., Padula M.P., Herbert B.R., Golledge J. Challenges, current status and future perspectives of proteomics in improving understanding, diagnosis and treatment of vascular disease. Eur. J. Vasc. Endovasc. Surg. 2009;38:346–355. doi: 10.1016/j.ejvs.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nordon I., Brar R., Hinchliffe R., Cockerill G., Loftus I., Thompson M. The role of proteomic research in vascular disease. J. Vasc. Surg. 2009;49:1602–1612. doi: 10.1016/j.jvs.2009.02.242. [DOI] [PubMed] [Google Scholar]
- 4.Rafter N., Connor J., Hall J., et al. Cardiovascular medications in primary care: treatment gaps and targeting by absolute risk. N. Z. Med. J. 2005;118:U1676. [PubMed] [Google Scholar]
- 5.Weir M.R. Targeting mechanisms of hypertensive vascular disease with dual calcium channel and renin-angiotensin system blockade. J. Hum. Hypertens. 2007;21:770–779. doi: 10.1038/sj.jhh.1002254. [DOI] [PubMed] [Google Scholar]
- 6.Agarwal R., Gomberg-Maitland M. Current therapeutics and practical management strategies for pulmonary arterial hypertension. Am. Heart J. 2011;162:201–213. doi: 10.1016/j.ahj.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 7.Gudivada R.C., Qu X.A., Chen J., Jegga A.G., Neumann E.K., Aronow B.J. Identifying disease-causal genes using Semantic Web-based representation of integrated genomic and phenomic knowledge. J. Biomed. Inform. 2008;41:717–729. doi: 10.1016/j.jbi.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 8.Toward Precision Medicine . Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease. Washington, D.C. US: National Academies Press; 2011. [PubMed] [Google Scholar]
- 9.Cambien F., Poirier O., Lecerf L., et al. Deletion polymorphism in the gene for angiotensin-converting enzyme is a potent risk factor for myocardial infarction. Nature. 1992;359:641–644. doi: 10.1038/359641a0. [DOI] [PubMed] [Google Scholar]
- 10.Iacoviello L., Di C.A., De K.P., et al. Polymorphisms in the coagulation factor VII gene and the risk of myocardial infarction. N. Engl. J. Med. 1998;338:79–85. doi: 10.1056/NEJM199801083380202. [DOI] [PubMed] [Google Scholar]
- 11.Chen G., Bentley A., Adeyemo A., et al. Genome-wide association study identifies novel loci association with fasting insulin and insulin resistance in African Americans. Hum. Mol. Genet. 2012;21:4530–4536. doi: 10.1093/hmg/dds282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelly T.N., Takeuchi F., Tabara Y., et al. Genome-wide association study meta-analysis reveals transethnic replication of mean arterial and pulse pressure loci. Hypertension. 2013;62:853–859. doi: 10.1161/HYPERTENSIONAHA.113.01148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy D., Ehret G.B., Rice K., et al. Genome-wide association study of blood pressure and hypertension. Nat. Genet. 2009;41:677–687. doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang H.C., Liang Y.J., Wu Y.L., et al. Genome-wide association study of young-onset hypertension in the Han Chinese population of Taiwan. PLoS One. 2009;4:e5459. doi: 10.1371/journal.pone.0005459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muller M.J., Bosy-Westphal A., Krawczak M. Genetic studies of common types of obesity: a critique of the current use of phenotypes. Obes. Rev. 2010;11:612–618. doi: 10.1111/j.1467-789X.2010.00734.x. [DOI] [PubMed] [Google Scholar]
- 16.Iacoviello L., Di C.A., Gattone M., et al. Polymorphisms of the interleukin-1beta gene affect the risk of myocardial infarction and ischemic stro ke at young age and the response of mononuclear cells to stimulation in vitro. Arterioscler. Thromb. Vasc. Biol. 2005;25:222–227. doi: 10.1161/01.ATV.0000150039.60906.02. [DOI] [PubMed] [Google Scholar]
- 17.Ehret G.B. Genome-wide association studies: contribution of genomics to understanding blood pressure and essential hypertension. Curr. Hypertens. Rep. 2010;12:17–25. doi: 10.1007/s11906-009-0086-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ehret G.B., Munroe P.B., Rice K.M., et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–109. doi: 10.1038/nature10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehret G.B., Caulfield M.J. Genes for blood pressure: an opportunity to understand hypertension. Eur. Heart J. 2013;34:951–961. doi: 10.1093/eurheartj/ehs455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Padmanabhan S., Newton-Cheh C., Dominiczak A.F. Genetic basis of blood pressure and hypertension. Trends Genet. 2012;28:397–408. doi: 10.1016/j.tig.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 21.Ferreira A.J., Raizada M.K. Genomic and proteomic approaches for targeting of angiotensin-converting enzyme2 for cardiovascular diseases. Curr. Opin. Cardiol. 2008;23:364–369. doi: 10.1097/HCO.0b013e328303b79b. [DOI] [PubMed] [Google Scholar]
- 22.Padmanabhan S., Hastie C., Prabhakaran D., Dominczak A.F. Genomic approaches to coronary artery disease. Indian J. Med. Res. 2010;132:567–578. [PMC free article] [PubMed] [Google Scholar]
- 23.Geraci M., Meyrick B. Genomics and proteomics of pulmonary vascular disease. Compr. Physiol. 2011;1:467–483. doi: 10.1002/cphy.c100031. [DOI] [PubMed] [Google Scholar]
- 24.Thongboonkerd V. Genomics, proteomics and integrative “omics” in hypertension research. Curr. Opin. Nephrol. Hypertens. 2005;14:133–139. doi: 10.1097/00041552-200503000-00008. [DOI] [PubMed] [Google Scholar]
- 25.Bilder R.M., Sabb F.W., Cannon T.D., et al. Phenomics: the systematic study of phenotypes on a genome-wide scale. Neuroscience. 2009;164:30–42. doi: 10.1016/j.neuroscience.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Houle D., Govindaraju D.R., Omholt S. Phenomics: the next challenge. Nat. Rev. Genet. 2010;11:855–866. doi: 10.1038/nrg2897. [DOI] [PubMed] [Google Scholar]
- 27.Oti M., Huynen M.A., Brunner H.G. Phenome connections. Trends Genet. 2008;24:103–106. doi: 10.1016/j.tig.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 28.Duan D.D. Phenomics of cardiac chloride channels. Compr. Physiol. 2013;3:667–692. doi: 10.1002/cphy.c110014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerlai R. Phenomics: fiction or the future? Trends Neurosci. 2002;25:506–509. doi: 10.1016/s0166-2236(02)02250-6. [DOI] [PubMed] [Google Scholar]
- 30.Bilder R.M. Phenomics: building scaffolds for biological hypotheses in the post-genomic era. Biol. Psychiatry. 2008;63:439–440. doi: 10.1016/j.biopsych.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furbank R.T., Tester M. Phenomics--technologies to relieve the phenotyping bottleneck. Trends Plant Sci. 2011;16:635–644. doi: 10.1016/j.tplants.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 32.Hegele R.A. Phenomics, lipodystrophy, and the metabolic syndrome. Trends Cardiovasc. Med. 2004;14:133–137. doi: 10.1016/j.tcm.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 33.Yang X-C., Xiong X-J., Wang J. Overview and prospect of syndrome differentiation of hypertension in traditional Chinese medicine. China Journal of Chinese Materia Medica. 2014;39:157–161. [PubMed] [Google Scholar]
- 34.Yu Y-N., Liu J., Zhang L., Duan D.D., Wang Z., Wang Y-Y. Clinical Zheng-hou Pharmacology: the Missing Link between Pharmacogenomics and Personalized Medicine? 205. Curr. Vasc. Pharmacol. 2015;13(4):423–432. doi: 10.2174/1570161112666141014144431. [DOI] [PubMed] [Google Scholar]
- 35.Denny J.C., Ritchie M.D., Basford M.A., et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26:1205–1210. doi: 10.1093/bioinformatics/btq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duan D.D., Han Y-S., Li L., Zhao J-Z., Wang Z. Pharmacophenomics: a new paradigm for pharmacology, toxicology, and personalized medicine. Chin J Pharmacol Toxicol. 2014;28:1–9. [Google Scholar]
- 37.Duan D. Phenomics of cardiac chloride channels: the systematic study of chloride channel function in the heart. J. Physiol. 2009;587:2163–2177. doi: 10.1113/jphysiol.2008.165860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lanktree M.B., Hassell R.G., Lahiry P., Hegele R.A. Phenomics: expanding the role of clinical evaluation in genomic studies. J. Investig. Med. 2010;58:700–706. doi: 10.231/JIM.0b013e3181d844f7. [DOI] [PubMed] [Google Scholar]
- 39.Neuraz A., Chouchana L., Malamut G., et al. Phenome-wide association studies on a quantitative trait: application to TPMT enzyme activity and thiopurine therapy in pharmacogenomics. PLOS Comput. Biol. 2013;9:e1003405. doi: 10.1371/journal.pcbi.1003405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pendergrass S.A., Brown-Gentry K., Dudek S., et al. Phenome-wide association study (PheWAS) for detection of pleiotropy within the Population Architecture using Genomics and Epidemiology (PAGE) Network. PLoS Genet. 2013;9:e1003087. doi: 10.1371/journal.pgen.1003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Denny J.C., Bastarache L., Ritchie M.D., et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol. 2013;31:1102–1110. doi: 10.1038/nbt.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]