

Abstract
A major hallmark feature of Alzheimer’s disease (AD) is the accumulation of amyloid β (Aβ), whose formation is regulated by the γ-secretase complex and its activating protein (also known as γ-secretase activating protein, or GSAP). Because GSAP interacts with the γ-secretase without affecting the cleavage of Notch, it is an ideal target for a viable anti-Aβ therapy. GSAP derives from a C-terminal fragment of a larger precursor protein of 98kDa via a caspase-3-mediated cleavage. However, the mechanism(s) involved in its degradation are unknown. In the current paper, we show that GSAP has a short half-life of approximately 5 hours. Neuronal cells treated with proteasome inhibitors markedly prevented GSAP protein degradation, which was associated with a significant increment in Aβ levels and γ-secretase cleavage products. By contrast, treatment with calpain blocker and lysosome inhibitors had no effect. In addition, we provide experimental evidence that GSAP is ubiquitinated. Taken together, our findings reveal that GSAP is degraded through the ubiquitin-proteasome system. Modulation of the GSAP degradation pathway may be implemented as a viable target for a safer anti-Aβ therapeutic approach in AD.
Keywords: Alzheimer’s disease, Amyloid β, APP, γ-secretase activating protein, protein degradation
Introduction
Alzheimer’s disease (AD) is the principal cause of dementia among the elderly. With increasing longevity and the absence of a cure, AD has become not only a major health problem but also a heavy economic burden worldwide. Accumulation of neurotoxic Aβ peptides is a major characteristic of the AD brain and in part responsible for its clinical manifestations. Since the formation of Aβ is under the strict control of the γ-secretase complex, its inhibitors are attractive therapeutic tools for lowering Aβ (1, 2). However, this approach has been seriously hampered by the toxic side-effects secondary to the fact that the γ-secretase complex has numerous substrates, particularly Notch, which reduce its therapeutic potential (3). The γ-secretase activating protein (GSAP) facilitates Aβ production by interacting directly with this secretase without affecting the cleavage of Notch (4). Therefore, GSAP is potentially a relevant target for a viable therapeutic strategy aimed at interfering with pro-amyloidogenic effectors. GSAP selectively increases Aβ production through a mechanism involving its interactions with both γ-secretase and its substrate, the amyloid precursor protein C-terminal fragment (APP-CTF) (4). Recently, we identified a caspase-3 processing domain in the GSAP precursor protein sequence and provided experimental evidence that this caspase is essential for the formation of its active form, GSAP 16kDa and subsequent biogenesis of Aβ peptides (5). In addition, we as well as others have shown that GSAP is increased in post-mortem AD brains, and in brains from mouse models of the disease (5, 6). Thus, while we are learning more and more about the neurobiology of GSAP, no information is available yet on its degradation mechanism. With this goal in mind, we embarked on a series of experiments providing evidence that GSAP is ubiquitinated and degraded by the proteasome system.
Methods
Cell culture
The N2A (neuro-2 A neuroblastoma) neuronal cells stably expressing human APP carrying the K670 N, M671 L Swedish mutation (APP swe) were grown in DMEM-Dulbecco’s modified Eagle medium (11965-092, Gibco) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 400 μg/ml G418 (Gibco), at 37°C in the presence of 5% CO2, as previously described (5, 17). The N2A cells were a gift from Dr. N. Robakis, Mount Sinai School of Medicine, New York.
Cell treatment
N2A-APPswe cells were grown to 70% confluence and then treated with the proteasome inhibitors (Z-IE) (1μM, 5μM, 10μM) and (lactacystin) (1μM, 5μM, 10μM), calpain inhibitor (ALLN) (1μM, 5μM, 10μM), and lysosome inhibitor (chloroquine)(1μM, 5μM, 10μM) separately, for 12 hours, after which supernatants were collected, and cells pellets harvested in lytic buffer for biochemical analyses. For protein half-life study, 100μg/ml cycloheximide was added to the medium and cells harvested at 2, 4, 8, 12, 16 and 24 hours. For transfection, cells were grown to 70% confluence and separately transfected with 1 μg of empty vector pcDNA3.1 (Invitrogen, Carlsbad, CA), or GSAP cDNA (Origene, Rockville, MD), or empty vector pRK5 (Addgene, Cambridge MA), or pRK5-HA-Ubiquitin-WT (Addgene, Cambridge MA), or pRK5-HA-Ubiquitin-K48 (Addgene, Cambridge MA), or pRK5-HA-Ubiquitin-KO (Addgene, Cambridge MA), by using Lipofectamine® 2000 Transfection Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions and as previously described (5,7). After 6 h transfection and/or 12h drug treatments, supernatants were collected, and cells pellets harvested for biochemical analyses. All experiments were approved by Temple University.
Western blot analysis
Protein extracts were sonicated, centrifuged at 13,000 rpm for 45 min at 4oC, and supernatants used for immunoblot analysis, as previously described (5, 7, 8). Actin was always used as an internal loading control. Primary antibodies used were as follows: anti GSAP full length (1:200) and GSAP-16kDa (1:150) (Thermo Scientific, Waltham, MA); anti-TMP21 (1:200) (Santa Cruz Biotech, Dallas, Texas); anti-APP (1:200), anti-APH-1 (1: 200) anti- CTF-α and anti-CTF-β(1:200) (Millipore, Billerica, MA, USA); anti-PS1 (1: 200), anti-Nicastrin (1: 200) (Cell signaling, Danvers, MA); anti-Pen-2 (1: 200) (Invitrogen, Carlsbad, CA); anti-β-actin (1:200) (Santa Cruz Biotech, Dallas, Texas). IRDye infrared secondary antibodies were from LI-COR Bioscience (Lincoln, Nebraska).
Quantitative analysis of Aβ levels
Aβ1-40 and Aβ1-42 levels in cells conditioned media were assayed by a sensitive sandwich ELISA kits (WAKO, Richmond, VA, USA). Analyses were always performed in duplicate and in a coded fashion as previously described (7, 8).
Immunofluorescence microscopy
Immunofluorescence studies were performed as previously described (5). Briefly, cells were placed on glass coverslips and the following day fixed in 4% paraformaldehyde in PBS for 15 min at 22°C, after being washed by PBS for 3 times and 5 min for each time, the coverslips were subsequently treated with 3% H2O2 in methanol, After rinsing several times with PBS, cells were incubated in a blocking solution (5% normal serum/0.4% TX-100) for 1 h at 22°C and then with the primary antibody against GSAP or ubiquitin overnight at 4°C. After washing with PBS, samples were incubated for 1 h with a secondary Texas Red, or Alexa Fluor 488-conjugated antibody (Invitrogen, Carlsbad, CA). Coverslips were mounted using VECTASHIELD mounting medium (Vector Laboratories, Burlingame, CA, USA). Control coverslips were processed as described above except that no primary antibodies were added to the solution. Images were acquired using a Zeiss 710 confocal microscope with a 63x oil objective and sequential scanning at 488 nm and 561 nm. Quantification was performed using Zeiss 2010 software colocalization analysis where % colocalization was calculated as abundance of cytosolic Ubiquitin colocalized with GSAP divided by the total abundance of cytosolic Ubiquitin.
Co-immunoprecipitation studies
Cells were grown to 85–90% confluence and then lysed in 50 mM HEPES, 150 mM NaCl, 5 mM MgCl2, 5 mM CaCl2, 1% CHAPSO (3-[(3-Cholamidopropyl) dimethylammonio]-2-hydroxy-1-propanesulfonate]) containing a protease inhibitor mixture. Prior to immunoprecipitation, cell lysates were diluted in lysis buffer lacking CHAPSO to give 0.25% final CHAPSO concentration. Cell lysates were incubated for 3 hr at room temperature with 5 μg of anti-GSAP antibody. DynabeadsM-280 sheep anti-rabbit IgG (50 μl; Invitrogen, Carlsbad, CA) were added and samples were incubated overnight at 4°C. A control incubation of cell lysates with Dynabeads alone was also conducted. Dynabeads were collected and washed 5 times with lysis buffer containing 0.25% CHAPSO. Bound proteins were eluted with SDS sample buffer containing reducing agent and subject to Western blot analysis by using anti-ubiquitin antibody or anti-HA tag antibody as the detecting antibodies. Input cell lysates were probed with anti-HA and anti-GSAP antibodies by regular Western blot.
Data analysis
One-way ANOVA followed by the Bonferroni’s Multiple Comparison tests were performed using GraphPad Prism 5.0. All data are presented as mean±s.e.m. Significance was set at p < 0.05.
Results
GSAP has a half-life of approximately 5 hours
To examine the half-life of the full length GSAP (GSAP FL) we used cycloheximide, a classical protein synthesis inhibitor, and measured the amount of protein remaining at various time-points. Neuronal cells were treated with 100μg/ml cycloheximide and western blot analysis was performed to measure levels of GSAP FL and to calculate the protein turn-over rate. The steady-state level of GSAP was 59.34±2.23 % and 7.19±1.60% of control after 5 and 24 hrs of cycloheximide treatment, respectively (figure 1).
Figure 1. GSAP has a half-life of approximately 5.4 hurs.

N2A-APPswe cells were incubated withy 100 μg/ml of cylcoheximide for 0, 2, 4, 8, 12, 16 and 24 hours and cells lysates collected for immunoblotting. A. Western blot analysis for full length GSAP (GSAP-FL) levels in lysate form cells incubated with cycloheximide. B. Quantification of the densitometry of the immunoreactivities to the antibody shown in panel A (n=3 per each time point; results are mean ± s.e.m.).
The degradation of GSAP is mediated by the proteasome pathway
To investigate whether the degradation of GSAP protein is regulated by the ubiquitin-proteasome pathway, cells were treated with two distinct proteasome inhibitors, lactacystin and Z-IE (9). At the end of the incubation, steady-state levels of GSAP full length (FL), GSAP 16kDa fragment, and another γ-secretase modulator TMP21 (10) were analyzed by immunoblotting. Z-IE significantly increased the protein levels of both endogenous GSAP full length and its 16kDa fragment in a dose-dependent manner (Fig. 2A, B). Z-IE treatment also increased the level of TMP21 protein levels (Fig. 2A, B). In a similar manner, the other proteasome inhibitor, lactacystin, significantly increased endogenous GSAP full length, 16kDa fragment, and TMP21 proteins levels (Fig. 2C, D).
Figure 2. Proteasome inhibition increases GSAP protein levels.

N2A-APPswe cells were separately treated with the proteasome inhibitors, calpain inhibitor, or the lysosomal inhibitor for 12h, and cell lysates collected for immunoblot analyses. A. Representative western blot analysis of full length GSAP (GSAP-FL), GSAP-16kDa, and TMP21 protein levels in lysates from cells treated with vehicle (Control) or the proteasome inhibitor, Z-IE (1μM, 5μM, and 10μM). B. Densitometric analyses of the immunoreactivities to the antibodies shown in panel A (*p 0.0001) (n=3; results are mean ± s.e.m.). C. Representative western blot analysis of GSAP-FL, GSAP-16kDa, and TMP21 in lysates from cells treated with the vehicle (Control) or proteasome inhibitor, lactacystin (1μM, 5μM, and 10μM). D. Densitometric analyses of the immunoreactivities to the antibodies shown in panel C (*p 0.0002) (n=3; results are mean ± s.e.m.). E. Representative western blot analysis of GSAP-FL, GSAP-16kDa, and TMP21 in lysates from cells treated with vehicle (Control) or the cell-permeable inhibitor of calpain, ALLN (1μM, 5μM, and 10μM). F. Densitometric analyses of the immunoreactivities to the antibodies shown in panel E. G. Representative western blot analysis of GSAP-FL, GSAP-16kDa, and TMP21 in lysates from cells treated with vehicle (Control) or the lysosomal inhibitor chloroquine (1μM, 5μM, and 10μM). H. Densitometric analyses of the immunoreactivities to the antibodies shown in panel G (n=3; results are mean ± s.e.m.).
To investigate whether the effect on GSAP protein levels secondary to the pharmacological inhibition of the ubiquitin-proteasome pathway was specific, cells were separately treated with the membrane permeable competitive calpain inhibitor, ALLN, and the lysosomal inhibitor, chloroquine (11, 12). As shown in Figure 2E–H no significant changes for GSAP full length, GSAP 16kDa fragment, and TMP21 proteins levels were observed in cells treated with these inhibitors (Fig. 2E–H).
Regulation of γ-secretase cleavage of APP and Aβ generation by proteasome inhibition
To examine whether modulation of GSAP levels secondary to the inhibition of the uniquitin-proteasome system resulted in subsequent alteration in the APP proteolytic processing at the γ-secretase site and Aβ production, next we assayed steady-state levels of APP, sAPPα, sAPPβ, the four components of the γ-secretase complex, and APP γ-secretase cleavage products, CTF-β and CTF-α in cell lysates, and Aβ levels in conditioned media. Both of Aβ1-40 and Aβ1-42 increased significantly in the conditioned media of cells treated with Z-IE (5μM) (p<0.0001) and lactacystin (5μM) (p<0.0001) (Figure 3A, B). While no differences were observed for total APP, sAPPα, sAPPβ, or the four components of the γ-secretase complex, a significant decrease in the levels of both CTF-β and CTF-α, was observed in cells treated with Z-IE and lactacystin (Figure 3C, D).
Figure 3. Proteasome inhibition modulates APP cleavage and Aβ generation.

N2A-APPswe cells were treated for overnight with increasing concentration of proteasome inhibitors and supernatant and cell lysates collected for analysis. A. Levels of Aβ 1-40 in conditioned media from cells treated with vehicle (Control), the proteasome inhibitors, Z-IE(*p<0.0001) and lactacystin (#p<0.0001) (n=3 ; results are mean ± s.e.m.). B. Levels of Aβ 1-42 in conditioned media from N2A-APP cells treated with vehicle (Control), the proteasome inhibitors, Z-IE(*p<0.0002) and lactacystin (#p<0.0003) (n=3 ; results are mean ± s.e.m.). C. Representative Western blot analysis of APP, sAPPα, sAPPβ, PS-1, Nicastrin, APH-1, Pen-2, CTFα and CTFβ in lysates from cells treated with vehicle (Control), the proteasome inhibitors, Z-IE and lactacystin. D. Densitometric analyses of the immunoreactivities to the antibodies shown in panel C (*p<0.0001; #p<0.0002) (n=3 ; results are mean ± s.e.m.).
GSAP is ubiquitinated
To investigate whether GSAP protein is ubiquitinated, we first examined the co-localization of GSAP and ubiquitin by performing immunoflurorescence double staining assays. GSAP protein was detected by red fluorescent signal and the ubiquitin was detected as the green fluorescent signal under a fluorescent microscope. The merged image (Figure 4A) showed that the red fluorescent GSAP proteins co-localized with the green fluorescent ubiquitin. Quantitative analysis showed a significant cytosolic co-localization for the two proteins (81.5± 2 %) (Figure 4C). As controls, the slides incubated with only secondary antibodies showed no detectable immunofluorescence (Figure 4B).
Figure 4. Co-localization of Ubiquitin and GSAP protein in neuronal cells.

Images were acquired using a Zeiss 710 confocal microscope with a 63x oil objective and sequential scanning at 488 nm and 561 nm. Quantification was performed using Zeiss 2010 software co-localization analysis where % co-localization was calculated as abundance of cytosolic Ubiquitin co-localized with GSAP divided by the total abundance of cytosolic Ubiquitin. A. Representative images of immunofluorescence analysis of N2A-APPswe cells incubated with primary antibody for ubiquitin (green) and GSAP (red). B. Representative images of immunofluorescence analysis of N2A-APPswe cells incubated only with secondary antibody. C. Quantitative analysis of the immune co-localization signal for GSAP and ubiquitin as obtained in panel A.
To further support the ubiquitination of GSAP, a co-immunoprecipitation assay studies were performed. Cells were transfected with GSAP plasmid, and then treated with the selective E3 ubiquitin ligase inhibitor, SMER3, the reversible and broad-spectrum inhibitor of the deubiquitinilating enzymes (DUBs), PR-619, and the proteasome inhibitor, Z-IE, for 12 hours. Cells only transfected with GSAP plasmid or empty vector were used as control groups in this assay. As shown in the upper panel of Figure 5A, cell lysates were immunoprecipitated with anti-GSAP antibody to pull down GSAP-associated proteins, and the immunoprecipitates were immunoblotted with anti-ubiquitin antibody. Under this experimental condition we observed that ubiquitinated-GSAP bands were increased in GSAP over expressing cells when compared to cells transected with empty vector. However, in the other groups, all of which over-expressed GSAP, the ubiquitinated-GSAP bands decreased in the presence of E3 ubiquitin ligase inhibitor, whereas they were increased in cells treated with DUBs inhibitor and the proteasome inhibitor (Figure 5A). As control, the lower panel of Figure 5A, which represents a western blot analysis of the same cell lysates without the immunoprecipitation, shows as expected the multiple bands of ubiquitin proteins. To further confirm the observation that GSAP is ubiquitinated, cells were co-transfected with GSAP and HA-tagged ubiquitin wild type (WT) or two different ubiquitin mutant plasmids. For our study, we choose the ubiquitin mutant contains arginine substitutions on all of its lysine residues except the one at position 48 (K48), and the one where all of its lysine residues were substituted with arginines (KO). Next, co-immunoprecipitation studies were performed by using anti-GSAP capture antibody for immunoprecipitation and anti-HA tag antibody for immune-blot detection. As shown in the upper panel of Figure 5B, anti-HA immunoblotting of the GSAP immunoprecipitates revealed that co-expression with WT or the K48 ubiquitin mutant yielded an ubiquitinated protein band, which was absent in both the control group where no HA-tagged protein was transfected, and in the cells transfected with HA-tagged KO ubiquitin mutant. The lower panel of Figure 5B shows control western blot analyses using cell lysates without immunoprecipitation and probed with anti-HA and anti-GSAP antibodies respectively.
Figure 5. GSAP is ubiquitinated.
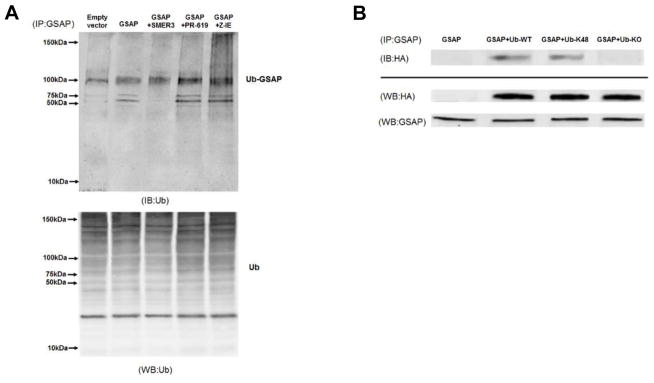
A. N2A-APPswe were cells transfected with empty vector or GSAP plasmid, then treated with the selective E3 UB ligase inhibitor SMER3 (15μM), the pan reversible inhibitor of deubiquitinases (DUBs) PR-619(10μM), and the proteasome inhibitors Z-IE (5μM) overnight. Upper panel. Cell lysates were immunoprecipitated (IP) with anti-GSAP antibody to pull down GSAP-associated proteins, the precipitates were then assayed by immunoblot analysis (IB) with anti-ubiquitin antibody to detect all the ubiquitinated GSAP. Lower panel. Total cell lysates from the condition described in A are directly assayed by Western blot (WB) with an antibody against ubiquitin. B. N2A-APPswe cells were transiently co-transfected with GSAP plus empty vector pRK5, or HA-tagged ubiquitin wilde type (WT), or HA-tagged ubiquitin mutant with only lysine 48 and mutation of all other lysines to arginines (K48), or HA-tagged ubiquitin mutant in which all lysines were mutated to arginines (KO). Upper panel. Cell lysates were first immunoprecipitated (IP) with anti-GSAP antibody to pull down GSAP-associated proteins, then the resultant immunoprecipated product was immunoblotted (IB) with an anti-HA antibody to detect all the ubiquitin in GSAP-containing immunoprecipitates. Lower panel. Total cell lysates from the condition described in B that are directly analyzed by Western Blot (WB) with anti-HA antibody and anti-GSAP antibody.
Discussion
In the current paper, we show for the first time that GSAP has a half-life of approximately 5 hours, is ubiquitinated and then degraded via the proteasome pathway. GSAP is a key molecule responsible for the rate-limiting step in Aβ production by directly interacting with the γ-secretase complex. Because it does not alter Notch processing, GSAP is potentially an ideal target molecule for therapeutic strategy aimed at interfering with pro-amyloidogenic effectors without the side-effects of the classical γ-secretase inhibition. However, despite its implication in AD pathogenesis, many aspects of its neurobiology, which includes its degradation pathway, are still unknown.
Protein degradation systems play a vital role and are essential for maintaining cellular protein homeostasis through their ability to destroy unwanted intracellular peptides. To this end, eukaryotic cells have developed three major proteolytic pathways: the ubiquitin–proteasome complex, the lysosome, and the calpains (13–15). Herein we embarked on a series of experiments testing which one of these main pathways was involved in GSAP degradation. Initially, we showed that when neuronal cells were treated with two distinct and specific proteasome inhibitors, they manifested an accumulation of GSAP. By contrast, when the lysosome system or the calpains were blocked, no effect was observed on GSAP levels. As a positive control, we assayed the effects of these diverse inhibitors on TMP21, another γ-secretase modulator also known to be degraded by the proteasome pathway (16). Thus, we found that while treatment with proteasome inhibitors increased TMP21 protein levels, both lysosome and calpains inhibition failed to produce any significant changes.
To further examine whether changes in proteasomal degradation of GSAP results in modulation of its biological targets, we investigated the effect on APP proteolytic processing and Aβ formation. In these experiments we observed that proteasomal inhibition of GSAP degradation resulted in more γ-cleavage products CTF-β and CTF-α, and higher Aβ1-40 and Aβ1-42 levels, but no changes were detected for the γ-secretase complex and the two APP secreted forms, sAPPα and sAPPβ. This observation is in contrast with previous studies showing that at least 2 components of the γ-secretase complex, namely Nicastrin and APH-1, can also be degraded by the proteasome (17,18). However, the discrepancy with our findings could be ascribed to the fact that, for instance, one study used non neuronal cells (i.e., HEK cells), and both used concentrations of the proteasome inhibitor much higher than the ones we used.
Over recent years, consistent evidence has accumulated showing some indispensable steps that any protein degraded via the proteasome pathway needs to undertake for an efficient process. In particular, it is known that before a protein can be committed to this type of degradation it needs to be covalently bound to several ubiquitin molecules in a process also known as ubiquitination (19). To prove that this was also the case for our protein, we first conducted double staining immunofluorescence assay and observed that GSAP co-localizes with ubiquitin. The physical interaction between GSAP and ubiquitin was then supported by co-immunoprecipitation studies in conjunction with a pharmacological approach by using a selective E3 ubiquitin ligase inhibitor, a broad-spectrum inhibitor of de-ubiquitinase, and a proteasome inhibitor, separately. Under these experimental conditions, as expected, we showed that inhibition of the E3 ubiquitin ligase decreased, whereas inhibition of de-ubiquitinase and pharmacological blockade of the proteasome activity significantly increased ubiquitinated GSAP in the system.
To further confirm that GSAP is ubiquitinated before being degraded through the proteasome pathway, we co-transfected cells with HA-tagged ubiquitin wild type or two ubiquitin mutants: one with conserved ability to promote protein degradation and the other without it. To reach this goal, we took advantage of the notion that within the 76 amino acids forming the ubiquitin protein, the lysine (K) in position 48, but none of the other lysines, is required for its covalent binding to the target protein and the following degradation by the 26S proteasome system (20). Thus, in our studies we used HA-tagged ubiquitin in which all lysines were mutated to arginines, and a tagged ubiquitin with a K48 conserved but the mutation of all other lysines to arginines, and then examined the interaction of GSAP and ubiquitin by co-immunoprecipitation. The results showed that the complete absence of lysine prevented GSAP ubiquitination, while the simple presence of the necessary K48 resulted in GSAP ubiquitination similar to the wild type ubiquitinin. Taken together, these data strongly support that GSAP is ubiquitinated and then specifically degraded through the proteasome pathway.
The finding that GSAP is degraded via the proteasome brings additional relevance to the fact that a proper control of protein catabolism and turnover is essential for the health of any given cells. This is particularly true in neural cells where a malfunction of this pathway could compromise their highly specialized functions. Thus, it is not surprising that besides AD, many other chronic neurodegenerative diseases (i.e., Parkinson’s Disease) are associated with the accumulation of intracellular proteins with very low solubility, a phenomenon that has been at least in part considered the result of an impaired protein digestion and recycling process within a particular subset of neurons (21).
Despite the fact that in most cases we still do not know how the proteasome system becomes impaired in AD, what we are starting to realize is that induction of this pathway may offer a rational therapeutic strategy for the disease.
In summary, our study demonstrates for the first time that GSAP is specifically degraded through the ubiquitin-proteasome system. Because the system can also affect other major components of the Aβ metabolic pathway besides GSAP, its dysregulation most likely constitutes an important event in the pathogenesis of sporadic AD. Taken together, our findings further support the concept that modulation of this pathway may be implemented as a viable target for a safer anti-Aβ therapeutic approach in AD.
Acknowledgments
This work was supported by grants from the National Institute of Health, HL086699 and 1S10RR027327 (to MM), and the Alzheimer Art Quilt Initiative (to DP).
Abbreviations
- AD
Alzheimer’s disease
- Aβ
Amyloid beta
- GSAP
γ-secretase activating protein
- GSAP FL
GSAP full length
- Z-IE
Z-Ile-Glu(OtBu)-Ala-Leu-CHO
- ALLN
Ac-LLnL-CHO
- APP
Amyloid precursor protein
- APP-CTF
Amyloid precursor protein C-terminal fragment
- sAPPα
Soluble amyloid precursor protein alpha
- sAPPβ
Soluble amyloid precursor protein beta
- CTF-α
alpha-C-terminal fragment
- CTF-β
beta-C-terminal fragment
- SMER3
9H-Indeno[1,2-e][1,2,5]oxadiazolo[3,4-b]pyrazin-9-one
- DUBs
Deubiquitinylating enzymes
- PR-619
3,5-dithiocyanatopyridine-2,6-diamine
- WT
Wild-type
- KO
Knock out
- DMEM
Dulbecco’s modified Eagle medium
- G418
Geneticin selective antibiotic
- CO2
Carbon dioxide
- N2A
Neuro-2 A neuroblastoma
- APP swe
Human APP carrying the K670 N, M671 L Swedish mutation
- APH-1
Anterior pharynx defective 1 homolog A
- PS1
Presenilin-1
- Pen-2
Presenilin enhancer protein 2
- ELISA
Enzyme-linked immunosorbent assay
- PBS
Phosphate buffered saline
- H2O2
Hydrogen peroxide
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- NaCl
Sodium chloride
- MgCl2
Magnesium chloride
- CaCl2
Calcium chloride
- CHAPSO
3-[(3-Cholamidopropyl) dimethylammonio]-2-hydroxy-1-propanesulfonate]
- SDS
Sodium dodecyl sulfate
Footnotes
Authors’ contributions
JC and DP conceived and designed the experiments. JC, JGL and NEH performed the experiments. JC and DP analyzed the data. JC, JGL, NEH, MM contributed reagents/materials/analysis tools. JC and DP wrote the paper.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Gandy S. Lifelong management of amyloid beta metabolism to prevent Alzheimer’s disease. N Engl J Med. 2012;367(9):864–866. doi: 10.1056/NEJMe1207995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolfe MS. Inhibition and modulation of γ-secretase for Alzheimer’s disease. Neurotherapeutics. 2008;5:391–398. doi: 10.1016/j.nurt.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deatherage CL, Hadziselimovic A, Sanders CR. Purification and characterization of human γ-secretase activating protein. Biochemistry. 2012;51:5153–5159. doi: 10.1021/bi300605u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He G, Luo W, Remmers C, Netzer WJ, Hendrick J, Bettayeb K, Flajolet M, Gorelick F, Wennogle LP, Greengard P. Gamma-secretase activating protein, a therapeutic target for Alzheimer’s disease. Nature. 2010;467:95–98. doi: 10.1038/nature09325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chu J, Li JG, Joshi YB, Giannopoulos PF, Hoffman NE, Madesh M, Praticò D. Gamma Secretase-Activating Protein Is a Substrate for Caspase-3: Implications for Alzheimer’s disease. Biol Psychiatry. 2014 doi: 10.1016/j.biopsych.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Satoh J, Tabunoki H, Ishida T, Saito Y, Arima K. Immunohistochemical characterization of γ-secreatse activating protein expression in Alzheimer’s disease brain. Neuropathol App Neurobiol. 2012;38:132–141. doi: 10.1111/j.1365-2990.2011.01206.x. [DOI] [PubMed] [Google Scholar]
- 7.Chu J, Praticò D. 5-Lipoxygenase as an endogenous modulator of amyloid-β formation in vivo. Ann Neurol. 2011;69:34–46. doi: 10.1002/ana.22234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu J, Giannopoulos PF, Ceballos-Diaz C, Golde TE, Praticò D. 5-Lipoxygenase gene transfer worsens memory, amyloid and tau brain pathologies in a mouse model of Alzheimer disease. Ann Neurol. 2012;72:442–454. doi: 10.1002/ana.23642. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Azakir BA, Di Fulvio S, Kinter J, Sinnreich M. Proteasomal inhibition restores biological function of Mis-sense mutated dysferlin in patient derived muscle cells. J Biol Chem. 2012;287:10344–10354. doi: 10.1074/jbc.M111.329078. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Chen F, Hasegawa H, Schmitt-Ulms G, Kawarai T, Bohm C, Katayama T, Gu Y, Sanjo N, Glista M, Rogaeva E, Wakutani Y, Pardossi-Piquard R, Ruan X, Tandon A, Checler F, Marambaud P, Hansen K, Westaway D, St George-Hyslop P, Fraser P. TMP21 is a presenilin complex component that modulates gamma-secretase but not epsilon-secretase activity. Nature. 2006;440:1208–1212. doi: 10.1038/nature04667. [DOI] [PubMed] [Google Scholar]
- 11.Shang L, Huang JF, Ding W, Chen S, Xue LX, Ma RF, Xiong K. Calpain: a molecule to induce AIF-mediated necroptosis in RGC-5 following elevated hydrostatic pressure. BMC Neuroscience. 2014;15:63. doi: 10.1186/1471-2202-15-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Long L, Yang X, Southwood M, Lu J, Marciniak SJ, Dunmore BJ, Morrell NW. Chloroquine Prevents Progression of Experimental Pulmonary Hypertension via Inhibition of Autophagy and Lysosomal Bone Morphogenetic Protein Type II Receptor Degradation. Circ Res. 2013;112:1159–1170. doi: 10.1161/CIRCRESAHA.111.300483. [DOI] [PubMed] [Google Scholar]
- 13.Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitinin-proteasome pathway in normal and disease states. J Am Soc Nephrol. 2006;17:1807–1819. doi: 10.1681/ASN.2006010083. [DOI] [PubMed] [Google Scholar]
- 14.Tardif N, Claude M, Lundell L, Thorell A, Rooyackers O. Autophagic-lysosomal pathway is the main proteolytic system modified in the skeletal muscle of esophageal cancer patients. Am J Clin Nutr. 2013;98:1485–1492. doi: 10.3945/ajcn.113.063859. [DOI] [PubMed] [Google Scholar]
- 15.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 16.Liu S, Bromley-Britis K, Xia K, Mittelhotz J, Wang R, Song W. TMP21 degradation is mediated by the ubiquitinin-proteasome pathway. Eur J Neurosci. 2008;28:1980–1988. doi: 10.1111/j.1460-9568.2008.06497.x. [DOI] [PubMed] [Google Scholar]
- 17.He G, Qing H, Tong Y, Cai F, Ishiura S, Song W. Degradation of nicastrin involves proteasome and lysosome. J Neurochem. 2008;101:982–992. doi: 10.1111/j.1471-4159.2007.04449.x. [DOI] [PubMed] [Google Scholar]
- 18.He G, Qing H, Kwok C, Xu X, Yu G, Bernstein A, Song W. Ubiquitin-proteasome pathway mediates degradation of APH-1. J Neurochem. 2007;99:1403–1412. doi: 10.1111/j.1471-4159.2006.04184.x. [DOI] [PubMed] [Google Scholar]
- 19.Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 20.Ciechanover A, Orian A, Schwartz AL. Ubiquitinin-mediated proteolysis: biological regulation via destruction. Bioessays. 2000;22:442–451. doi: 10.1002/(SICI)1521-1878(200005)22:5<442::AID-BIES6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 21.Atkin G, Paulson H. Ubiquitin pathways in neurodegenerative disease. Front Mol Neurosci. 2014;7:63. doi: 10.3389/fnmol.2014.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]