

Abstract
The bacterial type II fatty acid biosynthesis (FASII) pathway is an essential but unexploited target for drug discovery. In this review we summarize SAR studies on inhibitors of InhA, the enoyl-ACP reductase from the FASII pathway in M. tuberculosis. Inhibitor scaffolds that are described include the diaryl ethers, pyrrolidine carboxamides, piperazine indoleformamides, pyrazoles, arylamides, fatty acids, and imidazopiperidines, all of which form ternary complexes with InhA and the NAD cofactor, as well as isoniazid and the diazaborines which covalently modify the cofactor. Analysis of the structural data has enabled the development of a common binding mode for the ternary complex inhibitors, which includes a hydrogen bond network, a large hydrophobic pocket and a third ‘size-limited’ binding area comprised of both polar and non-polar groups. A critical factor in InhA inhibition involves ordering of the substrate binding loop, located close to the active site, and a direct link is proposed between loop ordering and slow onset enzyme inhibition. Slow onset inhibitors have long residence times on the enzyme target, a property that is of critical importance for in vivo activity.
Introduction
The outer layer of the mycobacterial cell wall is comprised predominantly of mycolic acids, very long chain (C60-C90) fatty acids that are important for the ability of Mycobacterium tuberculosis to live and replicate inside macrophages, and also for the inability of many antibiotics to penetrate into the cytosol [1-2]. Consequently, compounds that antagonize the ability of mycobacteria to synthesize mycolic acids are promising leads for developing novel tuberculosis chemotherapeutics. The fatty acid precursors required for mycolic acid synthesis are generated by the mycobacterial type I (FASI) and type II (FASII) biosynthetic pathways (Fig. (1)). The FASI enzyme complex, which is homologous to the synthase found in mammalian cells, catalyzes the de novo synthesis of C20-C24 fatty acyl-CoAs that are subsequently extended by the FASII system producing fatty acids up to C56 in length.
Fig. (1).
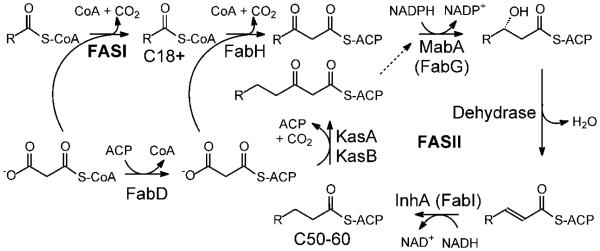
Fatty acid biosynthesis in M. tuberculosis.
The FASI pathway synthesizes C18+ acyl-CoAs that are subsequently extended by the dissociated FASII system. The FASI pathway catalyzes the de novo biosynthesis of fatty acids from acetyl-CoA and malonyl-CoA. In the FASII pathway chain elongation utilizes malonyl-ACP generated from malonyl-CoA by FabD, the malonyl-CoA:ACP transacylase. Entry of fatty acids into the FASII pathway is mediated by the β-ketoacyl-ACP synthase FabH which condenses the acyl-CoA with malonyl-ACP. The resulting β-ketoacyl-ACP is reduced by the NADPH-dependent reductase MabA, dehydrated by an unidentified enzyme, and reduced by the NADH-dependent enoyl-ACP reductase InhA. Thereafter, subsequent rounds of elongation are primed by the β-ketoacyl-ACP synthases KasA and KasB. Note that the acyl carrier protein in M. tuberculosis is sometimes referred to as AcpM. In addition, since InhA is a FabI enoyl-ACP reductase, this enzyme is sometimes referred to as the FabI from M. tuberculosis.
In targeting essential bacterial pathways, there is always a concern that the organism can acquire the required metabolites from the environment and thus evade the impact of pathway inhibition. In this regard, a recent study raised questions about the essentiality of the FASII pathway in Streptococcus agalactiae and, by extension, Gram positive bacteria in general [3]. While doubt has now been cast on the generality of the conclusions reached in this study, at least with regard to the important nosocomial pathogen Staphylococcus aureus [4], it is important to note that mammals do not synthesize mycolic acids and thus the FASII pathway must play an essential role in mycobacteria. This belief is supported by the knowledge that the very effective front-line drug isoniazid targets InhA, the enoyl-ACP reductase in the M. tuberculosis FASII pathway [5-9], while Jacobs and coworkers have demonstrated that inactivation of InhA in M. smegmatis results in cell lysis [10].
The primary mechanism of resistance to isoniazid occurs from mutations in the mycobacterial catalase peroxidase enzyme KatG that is responsible for drug activation and not from mutations in InhA [11-12]. Consequently, inhibitors of InhA that do not require KatG activation should be active against most clinical strains of isoniazid-resistant M. tuberculosis [13]. InhA is a member of the short chain dehydrogenase reductase superfamily and is in the FabI class of enoyl-ACP reductases that are found in bacteria such as Escherichia coli, S. aureus and Francisella tularensis [7, 14]. Given the continuing need to develop antibacterial agents with novel mechanisms of action, there have been a number of efforts to develop FabI inhibitors [14], particularly the enzyme from S. aureus (saFabI) [15], and currently two Phase I trials are in progress for the saFabI inhibitors developed by Affinium Pharmaceuticals Inc. [16-17] and Fab Pharma SA.
In the present review we describe current attempts to develop potent inhibitors of InhA. Three classes of inhibitors are based on isoniazid, the diazaborines and triclosan, antibacterial agents that were subsequently shown to inhibit InhA. In addition we also summarize inhibitor discovery resulting from compounds identified by high-throughput screening (HTS) that includes the pyrrolidine carboxamides, piperazine indoleformamides, pyrazoles and arylamides, and conclude with attempts to use fatty acids to inhibit InhA. Despite the structural diversity in the different InhA inhibitor classes, two generalizations arise from an analysis of the data. Firstly, in almost every case the inhibitors bind to the enzyme in the presence of the oxidized and/or reduced cofactor, albeit in the case of isoniazid and the diazaborines as covalent adducts of the cofactor [14]. And secondly, high affinity inhibition is often coupled to ordering of the ‘substrate binding loop’ that is located close to the active site [14]. Given the importance of loop ordering and its relationship to the residence time of the inhibitor on the enzyme [18-19], we first briefly summarize the structural and mechanistic basis for high affinity inhibition of InhA and the FabI class of enoyl-ACP reductases.
Structural and Mechanistic Basis for High Affinity Inhibition of Enoyl-ACP Reductases
A central theme in FabI inhibitor discovery concerns the role that inhibitor binding plays in ordering of a loop of amino acids close to the active site. This sequence of amino acids is known as the substrate binding loop, and the importance of loop ordering during FabI inhibition was first noted in studies on the inhibition of the E coli FabI by the diazaborine class of compounds [20]. Since then, X-ray crystallography has revealed ordered loops in a number of FabI:inhibitor complexes and those relevant to InhA are described in this review. Based on our own work, we believe that loop ordering is the structural event that occurs when FabI enzymes interact with slow onset inhibitors. In this type of inhibition, the initial rapid association for enzyme and inhibitor is followed by a slow step that results in formation of the final EI* complex (Fig. (2)).
Fig. (2).

Two step mechanism for slow onset enzyme inhibition.
In this form of slow onset inhibition, initial rapid formation of EI is followed by a slow step leading to formation of the final EI* complex. Assuming that k−1>>k2 and k−2, then hence koff ≈ k−2 and residence time = 1/k−2. [21]
A classic example of this effect is given by the inhibition of the E. coli FabI (ecFabI) by the diaryl ether antibacterial agent triclosan [22], which is a slow onset inhibitor of ecFabI [24-25]. The X-ray structures of ecFabI in a binary complex with the oxidized cofactor NAD+ and in a ternary complex with NAD+ and triclosan are shown in Fig. (3), where it can be seen that formation of the ternary enzyme inhibitor complex leads to ordering of the substrate binding loop (red).
Fig. (3).

Structures of ecFabI in the absence and presence of triclosan.
The figure was made using the pdb files 1dfi [20] and 1qsg [22]for the binary and ternary enzyme complexes, respectively. In the binary FabI:NAD+ complex (left panel) the substrate binding loop is disordered and the adjacent chain has been colored red. In the ternary FabI:NAD+:triclosan complex (right panel), the substrate binding loop is ordered (red) and forms a helix that covers the bound inhibitor. The figure was made using PyMol [23].
Importantly, slow onset enzyme inhibitors have long residence times on their enzyme targets which, in a growing number of cases, is thought to be critical for in vivo drug activity [19, 21, 26-27]. In particular, compounds with long residence times will remain bound to their targets even when the free drug concentration is low, thus prolonging their biological activity. Recent studies from our lab on the FabI enzyme from the category A pathogen Francisella tularensis (ftuFabI) have revealed that the antibacterial activity of a series of ftuFabI inhibitors in an animal model of F. tularensis infection correlate with the residence time of the compounds on ftuFabI but not with their Ki values for enzyme inhibition or their MIC (minimum inhibitor concentration) values for inhibiting bacterial growth [18]. Thus in vivo drug activity in this case is mediated by the kinetics but not the thermodynamics of enzyme-inhibitor complex formation. This has important implications for drug discovery programs that are usually driven by thermodynamic estimates of compound potency (Ki or IC50 values), and, in the case of tuberculosis drug discovery, increases the motivation for developing inhibitors that promote loop ordering and are slow onset inhibitors of InhA. In the present review we summarize efforts to develop inhibitors of InhA with particular emphasis on the results of SAR studies within each compound class.
Isoniazid-Based Inhibitors
Isoniazid (INH) has been used as a front-line antitubercular drug for over 50 years [28]. Activation of INH by the endogenous catalase-peroxidase enzyme KatG is essential for INH activity [10-12, 29] and results in the formation of an acyl pyridine adduct with NAD (the INH-NAD adduct) (Fig. (4)). The 4S isomer of the adduct is a nanomolar slow-onset inhibitor of InhA with a residence time of 60 min [9], and the X-ray crystal structure of the enzyme inhibitor complex (1zid.pdb) [8] indicates that the substrate binding loop is ordered in agreement with the above discussion (Fig. (5)). Both enantiomers of the INH-NAD adduct, as well as those formed with NADP+ can undergo tautomerization (Fig. (4), I1-I3) [30], increasing the number of potential targets for the drug in M. tuberculosis. Although InhA was the initial INH target identified [5-7], the 4R INH-NADP adduct is a nanomolar inhibitor of the mycobacterial DHFR enzyme [31], while affinity purification has revealed 16 additional proteins in the M. tuberculosis genome capable of binding to the INH-NAD(P) adduct(s) [32]. There is also evidence that INH can inhibit non-NAD(P) binding proteins such as KasA, the β-ketoacylACP synthase in the FASII pathway [33]. Importantly, since mutations of KatG are the most common mechanisms of resistance to INH [12, 34], inhibitors that bind to the final drug target(s) should have activity against the majority of INH-resistant clinical strains [13].
Fig. (4).

Structure of the INH-NAD adduct
Fig. (5).

X-ray structure of the INH-NAD adduct (cyan) bound to InhA (1zid.pdb) [8]. The substrate binding loop is colored red. This portion of the protein (res 196-211) is disordered in the structure of triclosan bound to InhA (2b35.pdb [13]).
In order to explore SAR involving the INH-NAD inhibitor, the benzoylhydrazine-NAD adduct (BH-NAD) has been synthesized and shown to competitively inhibit InhA with a Ki value < 1 nM [9]. Although this indicates that the acyl-pyridine can be replaced by a benzoyl group, the preliminary studies also showed that benzoic hydrazide is less readily activated than INH. In addition, a series of BH-NAD analogues have been synthesized (Fig. (6), I4, I5) [35], in which the ADPR portion of the molecule has been modified. However none of these analogues significantly affected InhA activity, indicating the importance of the nucleotide for InhA inhibition.
Fig. (6).
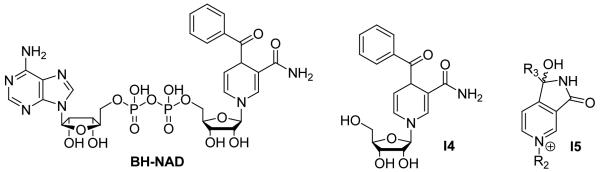
BH-NAD and its derivatives.
A series of 4-phenoxybenzamides have also been prepared in order to mimic key elements of the INH-NAD adduct (Table 1) [36]. However, as seen for the BH-NAD analogues, activity assays indicated that only compounds with a NAD moiety were able to inhibit InhA.
Finally, an iron complex of isoniazid, [FeII(CN)5(INH)]3- (Fig. (7)) was found to inhibit InhA with a Ki value of 70 nM [37]. Interestingly, this complex also inhibited growth of M. tuberculosis with an MIC value of 0.2 μg/ml. The iron complex inhibited InhA without forming an adduct with NADH, consistent with docking results that suggest the inhibitor may bind directly to the active site [38-39]. Additional inorganic complexes also demonstrated antimycobacterial activity [40-42], however the target of these compounds has not been confirmed.
Fig. (7).
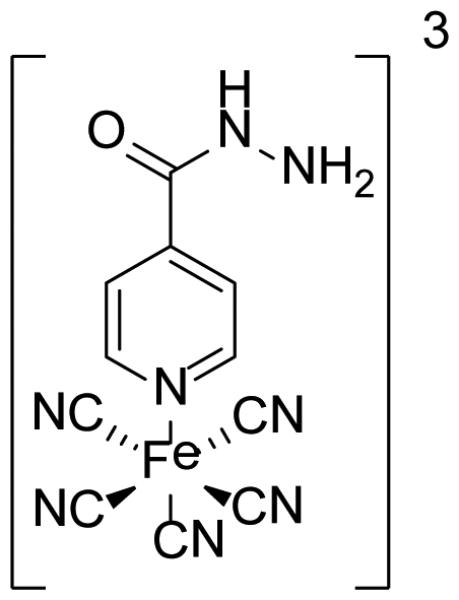
Structure of the [FeII(CN)5(INH)]3- complex
Diaryl-Ether Inhibitors
Triclosan (Fig. (8)) is a biocide which is widely used in consumer products such as toothpaste and deodorant. While at high concentration triclosan may inhibit bacterial growth by acting as a membrane disruptant, the antibacterial activity of this compound at low concentration stems from the inhibition of FabI [22, 43-45]. Although the activity of triclosan against InhA and M. tuberculosis is relatively modest (Ki 0.2 µM, MIC 12.5 µg/ml) [13, 46], the lack of a requirement for KatG activation suggests that triclosan derivatives with increased affinity for InhA should be excellent leads for developing drugs that are active against MDR-TB and XDR-TB.
Fig. (8).

Triclosan
The interaction of triclosan with the FabI enzyme class, and in particular with InhA, has been thoroughly studied (reviewed in ref [14]). The compound is an uncompetitive inhibitor of InhA, binding preferentially to the enzyme:NAD+ product complex with a Ki value of 0.2 µM [46]. Slow onset inhibition is not observed when triclosan inhibits InhA, and, consistent with our hypothesis, the InhA substrate binding loop is thus disordered in the X-ray structure of the InhA:NAD+:triclosan ternary complex (2b35.pdb) (Fig. (9)). [13].
Fig. (9).

X-ray crystal structure of triclosan bound to InhA.
A: Overall structure in which the ends of the disordered substrate binding loop are colored red. B: Hydrogen bonding interactions between triclosan (yellow), NAD+ (cyan) and Tyr158 (gold). The figure was made with PyMol [23].
Key features involved in enzyme inhibitor recognition include hydrogen bonds between the A-ring hydroxyl group, Tyr158 and the NAD+ ribose, a hydrogen bond between the ether oxygen and the NAD+ ribose, and a stacking interaction between the aromatic A ring and the nicotinamide ring of NAD+ [22]. The importance of these interactions for the inhibition of other FabI enzymes by triclosan had been probed by site-directed mutagenesis, synthesis and computational studies [47-49], and the phenolic A-ring and ether oxygen are preserved in the analogues described below. However, ultimately it may be necessary to develop compounds that lack the hydroxyl group due to the likelihood that this functionality is susceptible to Phase II metabolism such as glucuronidation and sulfation [50].
In Table 2 we list the diaryl ethers in which the substituent on the A-ring has been systematically modified while making only minor changes to the B ring (PT01, PT03-PT08 [13, 51]; PT70 [52]; F2-F26, [53]).
Table 2.
Diaryl-Ether InhA inhibitors.
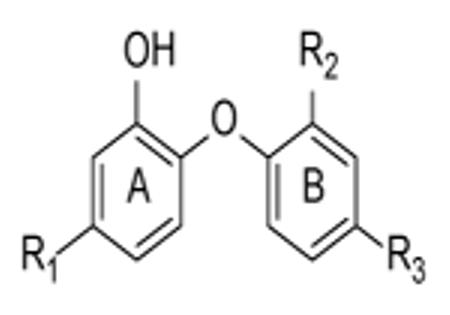
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Compound | R1 | R2 | R3 | IC50 (nM) | Ki (nM) | MIC (μg/ml) |
|
| ||||||
| PT01 (2PP) | -CH2CH3 | -H | -H | 2000 ± 700a | N.D. | 3.8 ± 0 |
| PT06 (4PP) | -(CH2)3CH3 | -H | -H | 80 ± 15a | N.D. | 2.6 ± 0.8d |
| PT03 (5PP) | -(CH2)4CH3 | -H | -H | 17 ± 5a | 11.8 ± 4.5 | 1.0 ± 0 |
| PT04 (6PP) | -(CH2)5CH3 | -H | -H | 11 ± 1a | 9.4 ± 0.5 | 2.1 ± 0.9 |
| PT05 (8PP) | -(CH2)7CH3 | -H | -H | 5.0 ± 0.3a | 1.1 ± 0.2 | 1.9 ± 0.5 |
| PT08 (9PP) | -(CH2)8CH3 | -H | -H | N.D. | N.D. | 14.1 ± 9.2 |
| PT07 (14PP) | -(CH2)13CH3 | -H | -H | 150 ± 24a | 30.3 ± 4.7 | 175 |
| PT70 | -(CH2)5CH3 | -CH3 | -H | <50b | 0.022 ± 0.001 | 3.125 |
| F2 | -CH3 | -CI | -CI | 800 ± 199c | N.D. | N.D. |
| F3 |
|
-CI | -CI | >10,000c | N.D. | N.D. |
| F4 | -COOH | -CI | -CI | >10,000c | N.D. | N.D. |
| F5 | -C(O)NH2 | -CI | -CI | >10,000c | N.D. | N.D. |
| F6 | -Ph | -CI | -CI | >10,000c | N.D. | N.D. |
| F7 | -CH2Cy | -CI | -CI | 110 ± 131c | N.D. | 9.4 |
| F8 | -CH2CH3 | -CI | -CI | 120 ± 19c | N.D. | N.D. |
| F9 | -(CH2)2CH3 | -CI | -CI | 91 ± 15c | N.D. | N.D. |
| F10 | -(CH2)3CH3 | -CI | -CI | 55 ± 20c | N.D. | 9.4 |
| F11 |
|
-CI | -CI | 96 ± 46c | N.D. | 19 |
| F12 |
|
-CI | -CI | 63 ± 9c | N.D. | N.D. |
| F13 |
|
-CI | -CI | 130 ± 56c | N.D. | N.D. |
| F14 | 2-pyridyl | -CI | -CN | >10,000c | N.D. | N.D. |
| F15 | 3-pyridyl | -CI | -CI | >10,000c | N.D. | N.D. |
| F16 | 4-pyridyl | -CI | -CN | >10,000c | N.D. | N.D. |
| F17 | -CH2(2-pyridyl) | -CI | -CI | 29 ± 11c | N.D. | N.D. |
| F18 | -CH2(3-pyridyl) | -CI | -CI | 42 ± 10c | N.D. | N.D. |
| F19 | -CH2(4-pyridyl) | -CI | -CN | 75 ± 16c | N.D. | N.D. |
| F20 | -o-tolyl | -CI | -CI | 1300 ± 77c | N.D. | N.D. |
| F21 | -o-tolyl | -CI | -CN | >10,000c | N.D. | N.D. |
| F22 | -m-tolyl | -CI | -CI | 870 ± 110c | N.D. | N.D. |
| F23 | p-F-Ph- | -CI | -CI | >10,000c | N.D. | N.D. |
| F24 | -Bn | -CI | -CI | 51 ± 6c | N.D. | 9.4 |
| F25 | -(CH2)2Ph | -CI | -CI | 21 ± 8c | N.D. | 19 |
| F26 | -(CH2)3Ph | -CI | -CI | 50 ± 14c | N.D. | 4.7 |
| Triclosan | 1000 ±100a | 220 ± 20 | 12.5 ± 0 | |||
| Isoniazid | N/A | 0.75 ± 0.08e | 0.05 ± 0 | |||
|
| ||||||
IC50 values were determined with inhA at concentration of 1 nM, 100 nM and 5 nM, respectively.
IC50 values were determined with inhA at concentration of 1 nM, 100 nM and 5 nM, respectively.
IC50 values were determined with inhA at concentration of 1 nM, 100 nM and 5 nM, respectively.
unpublished data.
Ki for the inhibition of InhA by the INH-NAD adduct. [9] N.D. = not determined.
Analysis of the SAR data in Table 2 reveals that the size and shape of R1 have the most effect on binding affinity. PT01, PT03-PT08 and F8-F10 indicate that an alkyl substituent of 5-8 carbons gives optimal inhibition of InhA and in vitro antibacterial activity. The X-ray crystal structures of PT03 [13] and C16-N-acetylcysteamine (C16-NAC) [54], a substrate analog, bound to InhA (Fig. (10A)) indicate that the PT03 A-ring can be superimposed on the acyl portion of C16-NAC, and that the last 3 carbon atoms of PT03 mimic the turn in the fatty acid chain of C16-NAC, implying that additional binding space is available near the end of the PT03 alkyl chain. In this regard, it can be seen that F17-F19, and F24-F26, in which an aromatic ring has been attached to the A-ring through a linker have similar IC50 values for InhA inhibition compared to F10. This indicates that additional space is available for inhibitor recognition, as can clearly be seen in the X-ray crystal structure of F7 bound to InhA (Fig. (10B)) [53]. Interestingly, F3, F6, F14-F16, and F20-F23, in which no linker is present between the additional ring and A-ring, all have IC50 values much larger than those with linkers, probably because the additional bulky substituent is too close to Phe149 and results in a steric clash [53].
Fig. (10).

Superposition of the X-ray structures of InhA and NAD+ (cyan) in complex with (A) C16-NAC (slate, 1bvr.pdb) and PT03 (yellow, 2b36.pdb), and (B) C16-NAC (slate) and F7 3fng.pdb) [53]. In each case the surface for 2b36.pdb is shown in which the active site loop is disordered. The figure was made with PyMol [23].
PT03-PT05 were the most potent first generation diaryl ether InhA inhibitors developed in our lab . Compared to triclosan, the Ki value for PT05 has decreased 200-fold while the MIC value has decreased 10-fold. We anticipated that increased contacts between enzyme and ligand might result in loop ordering. However PT03-PT05 are not slow onset inhibitors and X-ray structural studies indicate that they do not promote loop ordering upon binding to the InhA:NAD+ complex. However, importantly these compounds have similar MIC values for drug sensitive and INH-resistant strains of M. tuberculosis, in agreement with the hypothesis that InhA inhibitors that do not require KatG activation should be active against drug-resistant bacteria. We also explored the mode of action of these compounds and demonstrated that overexpression of the putative target (InhA) resulted in a 10-fold increase in MIC. Finally, we compared the gene expression profile for PT04 and PT05 with that for triclosan and showed that two putative drug-resistance mechanisms upregulated by triclosan were not affected by the alkyl diaryl ethers. This suggests that the improved antibacterial activity may result partly from the ability to evade drug detoxification [13, 51].
In addition to SAR studies on the A-ring side chain, modification of the B-ring has also been performed [55]. With PT04 as the lead compound, a variety of substituents have been introduced at the ortho-, meta-, and para- positions on the B-ring. In addition, the B-ring has also been replaced with several heterocyclic aromatic groups (Table 3).
Table 3.
SAR study on B-ring of diaryl ethers.
| Compounc | Structure | IC50 (nM) | MIC90(μg/ml) |
|---|---|---|---|
| PT04 |
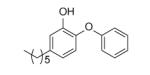
|
11 ± 1 | 2.1 ± 9 |
|
| |||
| PT10 |
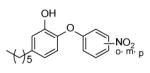
|
100 ± 20 | 12.50 |
| PT11 | 48 ± 6 | 12.50 | |
| PT09 | 90 ± 10 | 25.0 ± 0 | |
|
| |||
| PT13 |
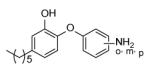
|
62 ± 5 | 3.13 |
| PT14 | 1090 ± 90 | 100 ± 0 | |
| PT15 | 55 ± 6 | 12.50 | |
|
| |||
| PT16 |
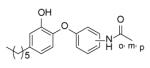
|
1550 ± 460 | > 200 ± 0 |
| PT17 | 5600 ± 770 | 100 | |
| PT18 | 1300 ±200 | 50.0 ± 0 | |
|
| |||
| PT19 |
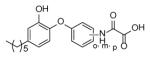
|
2360 ± 200 | 100.00 |
| PT20 | 580 ± 40 | 130 ± 58 | |
| PT21 | 1930 ±90 | > 200 ± 0 | |
|
| |||
| PT28 |
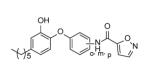
|
3220 ± 550 | > 200 ± 0 |
| PT29 | 1220 ±60 | > 200 ± 0 | |
| PT30 | 130 ±34 | > 200 ± 0 | |
|
| |||
| PT76 |
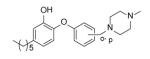
|
1315 ± 256 | |
| PT67 | 306 ± 46 | > 100 | |
|
| |||
| PT40 |
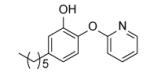
|
11,500 ± 1160 | 50 |
| PT77 |
|
236 ± 31 | 3.13 |
| PT73 |
|
160 ± 16 | 3.13 |
| PT41 |
|
8200 ± 98 | 100.0 ± 0 |
| PT42 |
|
650 ± 60 | 6.25 ± 0 |
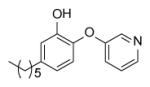
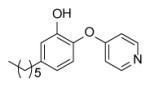
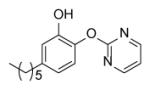
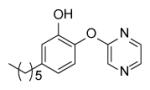
Many of the analogues in Table 3 were synthesized in an attempt to increase solubility of the non-polar lead compound. While decreases in logP were observed for some compounds [55], in general the main conclusion reached from the analoging studies was the inability of the active site to tolerate the introduction of bulky groups into the B-ring. Thus, while replacement of the B-ring with aniline led to only a modest decrease in potency, depending on the position of the amino substituent (e.g. PT13), subsequent derivatization of the amines with acyl groups led to dramatic decreases in both enzyme inhibition and MIC (PT16-PT21, PT28-PT30). A similar result was also observed when the B-ring was functionalized with a piperazine moiety (PT67, PT76). In addition, in most cases replacement of the B ring with a pyridine or pyrimidine ring also led to significant decreases in enzyme inhibition, although interestingly in the case of PT73, PT77 and PT42, without altering the MIC value by more than a factor of 3.
Comparison of the data in Tables 2 and 3 indicates that small substituents on the B-ring are well tolerated. In Table 2 all the substituents are either ortho or para to the ether linkage (F2-F26) and comparison of the biological data for the three aniline analogues (PT13-PT15) indicates that substitution of the meta position is less favorable. Analysis of the structural data reveals that the ortho substituent can occupy a space adjacent to both the nonpolar amino acids in the substrate binding loop and to the polar NAD+ ribose, thereby favoring groups that can participate in both polar and nonpolar interactions. This can clearly be seen in the X-ray structure of F7 bound to InhA where the meta-chloro group is within 4 Å of the backbone amide of Gly96, the methyl group of Ala98 and the 2’ ribose hydroxyl (Fig. (11)).
Fig. (11).

X-ray structure of F7 bound to InhA (3fng.pdb) [53]). Interactions between the B-ring meta-chloro group and the protein/cofactor are shown with dotted black lines. The figure was made with PyMol [23].
The importance of the B-ring meta substituent can also be clearly seen when comparing SAR data for PT04 and PT70. Although the first generation alkyl diaryl ethers developed by us were significantly more potent than triclosan (PT01, PT03-PT08 [13]), in none of the cases were the compounds slow onset enzyme inhibitors. Recognizing the importance of residence time on in vivo activity [18-19, 21, 27], we speculated that introduction of a substituent meta to the ether linkage would limit rotation about the ether bond and thus reduce the entropic penalty for enzyme inhibition. Consequently, we synthesized PT70, which differs from PT04 by the presence of a meta methyl group. Significantly, in contrast to PT04, PT70 is a slow onset inhibitor of InhA with a residence time of 23 min[52]. In addition, PT70 binds to InhA with a K1 value of 22 pM, compared to 11 nM for PT04. Thus, introduction of the methyl group has both increased the thermodynamic affinity of the inhibitor for the enzyme, and resulted in a compound that triggers the slow step that leads to the final EI* complex. In keeping with our expectations, the substrate binding loop in the InhA:NAD+:PT70 is ordered (Fig. (12)) [52], whereas that for the corresponding complex with PT04 is not [13]. Inspection of the X-ray structure reveals similar enzyme-inhibitor interactions to those described for F7, in particular contacts between the B-ring methyl and the cofactor and substrate binding loop (Fig. (12)).
Fig. (12).

X-ray structure of PT70 bound to InhA in the presence of NAD+. The substrate binding loop is shown with a mesh surface, and interactions are shown (red dots) between the B-ring methyl and the cofactor as well as the methyl side chain of Ala198 in the loop. The figure was made with PyMol [23].
PT70 is currently under evaluation in an animal model of tuberculosis infection and additional efforts are currently under way to improve the ADME properties of these compounds.
In summary the diaryl ether skeleton is a promising foundation for the development of novel FabI-directed antibacterial agents. Compounds are already in Phase 1 trials for the treatment of staphylococcal infections, and confidence is high that this success can be repeated with inhibitors that target InhA for treating drug-resistant tuberculosis.
Diazaborines
The diazaborines are bicyclic compounds that contain a boron atom and 1, 2-diazine in one heterocycle (Fig. (13)). Depending on the arene (X), diazaborines can be classified as thieno-, benzo-, furo-, and pyrrolo-diazaborine, when X is thiophene, benzene, furan, and pyrrole, respectively. A diazaborine was first identified in 1981 as a novel inhibitor of lipopolysaccharide biosynthesis synthesis [56] and subsequently the FabI enzyme in E. coli (envM) was shown to be the target for this class of compounds [57].
Fig. (13).
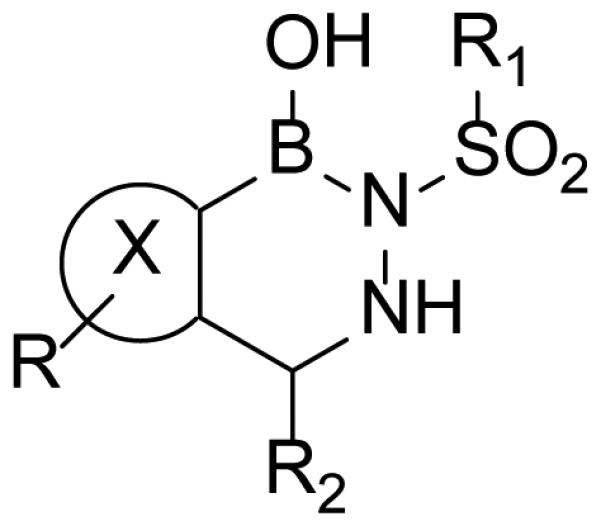
Diazaborine scaffold
Although a large number of diazaborine derivatives have been were synthesized and evaluated for their antibacterial activity [58], SAR studies with M. tuberculosis have not been extensively explored. Davis et al. synthesized 6 compounds and determined their MIC values using two different types of media (Table 4) [59]. In general the antibacterial activity of the compounds was modest, the best MIC being 8 µg/ml. Comparison of B2 with B3 and B4 indicates that large substituents are preferred on N-2, together with a carbonyl (C=O or C=S) at C-3 (B3, B4, B5). Bioisosteres B3 and B4 have the same potency, and while a typical benzodiazaborine B1 has moderate antibacterial activity, surprisingly, the ring-opened amine B6 has similar activity to that of B1.
Table 4.
Antitubercular activity of diazaborine derivatives.
| Compound | Structure | MIC (μg/ml) |
|---|---|---|
| Isoniazid | 0.02 | |
| Pyrazinamide | 197 | |
| B1 |

|
12a/96b |
| B2 |
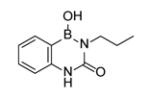
|
>128a/>128b |
| B3 |
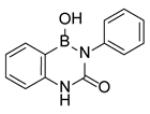
|
8a/16b |
| 84 |
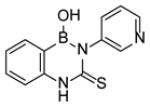
|
8a/16b |
| B5 |
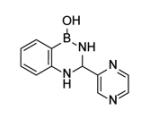
|
> 128a/128b |
| B6 |
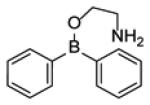
|
16a/64b |
MIC was measured in Bactec 6A media.
MIC was measured in Bactec 12B media.
The mechanism of enzyme inhibition with specific focus on ecFabI has been studied in detail using X-ray crystallography and site-directed mutagenesis [20, 60-63]. The compounds form an adduct with the cofactor through a dative bond between the NAD+ 2’-ribose hydroxyl group and the diazaborine boron atom (Fig. (14)). Thus, in this respect they mirror the mechanism of action of isoniazid, although the diazaborines are not prodrugs. In addition, the boron hydroxyl group is hydrogen bonded to Tyr156 while the NAD+ nicotinamide ring forms a π-π stacking interaction with the bicyclic diazaborine ring which also has van der Waals interactions with Tyr156, Tyr146, Phe203, and Ile200. Extensive SAR studies have been performed on the mechanism of FabI inhibition together with antibacterial activity which have revealed that the 1, 2-diaza-moiety is essential. This raises an interesting question concerning the mode of action of the compounds in mycobacteria since the 1, 2-diaza group is not required for activity against M. tuberculosis (Table 4). One point of interest when comparing the E. coli and M. tuberculosis enzymes is the importance of the residue at position 63 (64 in InhA). Selection for resistance to diazaborine in E. coli results in replacement of Gly93 with a serine in ecFabI [57], while a similar experiment with isoniazid results in a Ser94Ala mutation in InhA [5].
Fig. (14).

X-ray structure of a thienodiazaborine (TDB, yellow) bound to ecFabI [20]. The figure was made with PyMol [23].
Pyrrolidine Carboxamides
As the interest in developing InhA inhibitors has grown, several groups have turned to HTS in order to identify novel leads. He et al. identified 30 hits from a HTS that inhibited InhA but that did not affect the activity of the FabI enzymes from E. coli or Plasmodium falciparum [64]. From these leads pyrrolidine carboxamides (Fig. (15)) were chosen for further optimization.
Fig. (15).
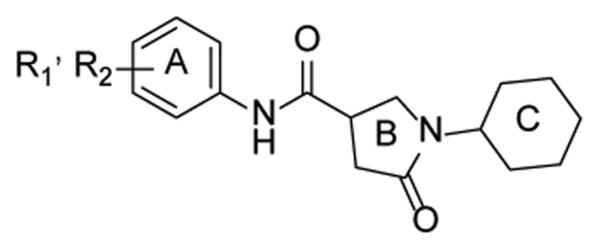
General structure of pyrrolidine carboxamide.
Table 5 contains SAR data from modifications made to the A-ring. Activity data for the various monosubstituted compounds showed that only meta- substitution was tolerated and that small electron withdrawing groups enhanced activity, while the meta-OH, -OCH3, -COCH3, -NHSO2CH3, and -CONH2 analogues having no activity. Consistent with this SAR, compound d11 with dual meta-substitution gave the best activity among all the derivatives. Compared to the corresponding monosubstituted compounds, an additional meta-CH3 or CF3 group did not affect the activity (d7, d12, d14). In addition, derivatives with linkers between the A-ring and the carboxamide (Fig. (16), g47-g49) did not have detectable activity, which suggests extension of the side chain is unfavorable.
Table 5.
Pyrrolidine carboxamide A-ring derivatives.
| Compound | R1 | R2 | IC50 (MM) |
|---|---|---|---|
| s1 | H | H | 10.66 ± 0.51 |
| s2 | H | 2-COOCH3 | 34.88 ± 2.06 |
| s3 | H | 2-Br | >100 |
| s4 | H | 3-Br | 0.89 ± 0.05 |
| s5 | H | 4-Br | 28.02 ± 4.29 |
| s6 | H | 3-CI | 1.35 ± 0.05 |
| s7 | H | 4-CI | >100 |
| s8 | H | 3-1 | 26%a |
| s9 | H | 4-1 | 14.50 ± 0.79 |
| s10 | H | 3-CH3 | 16.79 ± 0.54 |
| s11 | H | 3-CF3 | 3.51 ± 0.09 |
| s12 | H | 3-NO2 | 10.59 ± 0.48 |
| s13 | H | 3-NHAc | >75 |
| s15 | H | 3-CH2(CH3)2 | 5.55 ± 0.21 |
| s16 | H | 4-NHAc | >100 |
| s17 | H | 4-Ac | 73.58 ± 9.97 |
| s18 | H | 4-OEt | >100 |
| s19 | H | 4-OPh | >100 |
| d1 | 2-CI | 4-CI | 56.02 ± 10.23 |
| d2 | 2-CI | 5-CI | 56.50 ± 11.09 |
| d3 | 2-CH3 | 5-CI | 0.97 ± 0.03 |
| d4 | 3-CH3 | 4-Br | 37.41 ± 1.76 |
| d5 | 2-CH3 | 6-CH3 | >100 |
| d6 | 2-CH3 | 5-CH3 | 10.05 ± 0.33 |
| d7 | 3-CH3 | 5-CH3 | 3.14 ± 0.12 |
| d8 | 2-CH3 | 3-CI | 23.12 ± 1.00 |
| d9 | 2-CH3 | 4-NO2 | 31.37 ± 1.45 |
| d10 | 3-F | 5-F | 1.49 ±0.05 |
| d11 | 3-CI | 5-CI | 0.39 ± 0.01 |
| d12 | 3-Br | 5-CF3 | 0.85 ± 0.05 |
| d13 | 3-OMe | 5-CF3 | 1.30 ± 0.04 |
| d14 | 3-CF3 | 5-CF3 | 3.67 ± 0.17 |
| d15 | 2-OCH3 | 5-CI | 1.60 ± 0.06 |
| d16 | 3-CI | 4-F | 14.83 ± 0.98 |
| d17 | 3-Br | 4-CH3 | 51%a |
| d18 | 3, 4-(-OCH2CH2O-) | >100 | |
Percentage of InhA inhibition at 15μM.
Fig. (16).
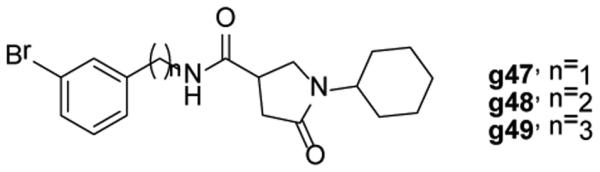
Pyrrolidine carboxamides: Linkers between the A-ring and carboxamide.
Replacement of the A-ring with cyclopentyl, cyclohexyl, isobutyl, piperazine, morpholine, and a variety of other heterocycles resulted in a decrease or total loss of activity, which suggests that aromaticity of the A-ring is important for activity. Only r7 (Fig. (17), IC50 = 5.1 μM) gave better activity than s1, which implies that extension of aromatic ring structure may gain more hydrophobic contacts and thus improve activity.
Fig. (17).
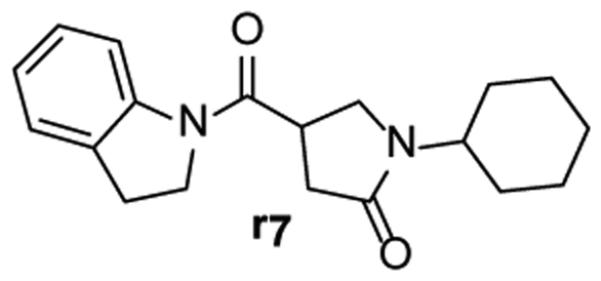
Strucutre of compound r7.
Forty five aromatic A-rings were incorporated into the molecule via microtiter synthesis and screened in situ without isolation. Subsequently, 10 compounds were selected for synthesis and purification in order to permit the determination of IC50 values (Table 6). The results showed that polyaromatic moieties (p20, p21, p24) significantly increased activity. For compound p21 and p24, the substitution position and linkers between the two benzene rings are limited, and changes of position or linker may reduce the activity (data not shown, [64]). Considering the flexibility of the InhA binding pocket, it is plausible that bulky aromatic rings are accommodated by induced conformational changes.
Table 6.
Replacement of A-ring in the pyrrolidine carboxamides.

| ||
|---|---|---|
|
| ||
| Compound | R3 | IC50 (μM) |
| p9 |
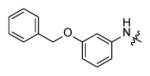
|
3.39 ± 0.10 |
| p20 |
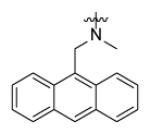
|
0.75 ± 0.04 |
| p21 |
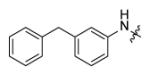
|
0.41 ± 0.01 |
| p24 |
|
0.39 ± 0.01 |
| p27 |
|
5.18 ± 0.34 |
| p28 |

|
6.41 ± 0.12 |
| p31 |
|
1.39 ± 0.02 |
| p33 |
|
2.57 ± 0.17 |
| p36 |

|
5.51 ± 0.22 |
| p37 |

|
4.47 ± 0.28 |
In addition to altering the A-ring, SAR data for the C-ring was also obtained. Replacement of the C-ring with a benzene or substituted benzene ring decreased or totally abolished activity, suggesting that the chair form of the C-ring is preferred over a planar ring. To further explore the C-ring conformation, a combinational library with 15 A-rings and 8 C-rings including saturated rings of 5-8 carbons and multiple rings was prepared through microtiter synthesis and screened in situ for InhA inhibition. Selected compounds were subsequently synthesized in order to facilitate IC50 measurements (Table 7).
Table 7.
IC50 values for selected pyrrolidine carboxamides C-ring analogues.
| Structure |
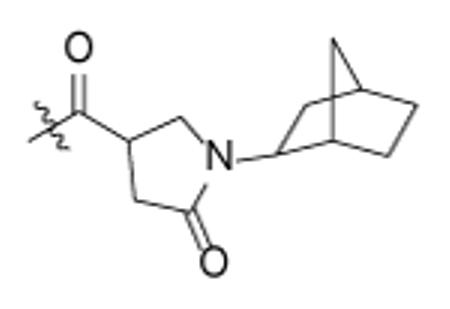
|
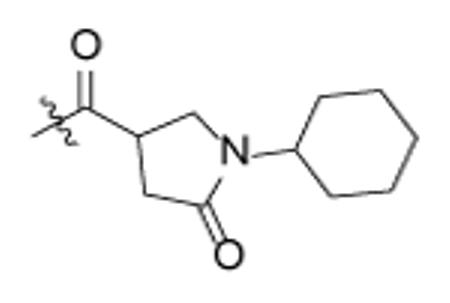
|
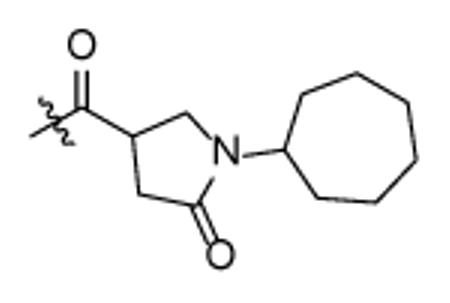
|

|
|---|---|---|---|---|
|
p62
0.46 ± 0.01 |
0.39 ± 0.01 | N.D. | N.D. |
|
p63
0.85 ± 0.05 |
0.75 ± 0.04 |
p65
0.32 ± 0.02 |
p67
0.62 ± 0.05 |
|
N.D. |
p64
0.14 ± 0.01 |
p66
0.27 ± 0.03 |
p68
1.29 ± 0.10 |
N.D. = Not determined.
Analysis of the data in Table 7 indicates that analogues with cyclopentyl, phenyl, adamantyl, or bicyclohexyl C-rings have reduced activity or are inactive, while cycloheptyl (p65, p66) and bicyclo-[2.2.1]-heptyl (p62, p63) analogues have similar activity to the cyclohexyl compounds. p67 and p68 with cyclooctyl C-rings had slightly reduced activity and, taken together, the results suggests that the C-ring binds in a hydrophobic pocket that can only cyclohexyl, cycloheptyl, and bicyclo-[2.2.1]-heptyl groups. For no exception, the meta-biphenyl A-ring analogs are better than corresponding anthracenylmethyl compounds, which suggests more tridimensional space in active site.
The X-ray structure of compound d11 bound to InhA has been determined and reveals how the pyrrolidine carboxamides interact with the active site (Fig. (18)). Consistent with conclusions from molecular modeling, the pyrrolidine ring plays a central role in forming a hydrogen-bonding network with the active site. The pyrrolidine carbonyl is hydrogen-bonded to the 2’-hydroxyl of the nicotinamide ribose and to the hydroxyl of Tyr158; while the cyclohexyl ring makes van der Waals contacts with the side chains of Gly96, Phe97, and the nicotinamide ribose. In addition, the lactam ring interacts with the nicotinamide ring and the side chains of Met161 and Met199, while the meta-halogen is within 4.1 Å of Ala157, Gly104, Gyr158, and Met105. For the dichloro-substituted compound (d11), the second chloro group has hydrophobic contacts with Pro156, Met155, and Leu218 which likely explains the increased potency of this compound compared to the mono-substituted analogues. The structural data also reveal that the substrate binding loop is ordered, although no kinetic data are available to substantiate the possibility that d11 is a slow onset inhibitor.
Fig. (18).

X-ray structure of d11 bound to InhA in the presence of NAD+. The inhibitor is colored yellow. The protein surface is light orange while the surface of the loop is red. The figure was made with PyMol using the pdb file 2h7m [64].
Following chromatographic resolution, only a single enantiomer of p24, d11, and p64 was found to be active, the absolute configuration of which was determined using the X-ray structure of d11 bound to InhA (Fig. (18)). p64b, an enantiomer of p64, was the pyrrolidine carboxamide with the lowest IC50 so far identified (62 nM). However the MIC values of most compounds are larger than 125 μM, with d12 and p67 having the best activity, with MIC value of 62.5μM, while p9, p20, p63, and p65 have MIC values of 125 μM. The low whole cell antibacterial activity may be due to poor membrane permeability and/or efflux.
Piperazine Indoleformamides
Screening has also led to the identification of piperazine indoleformamides has novel leads for developing InhA inhibitors [65-67]. SAR studies indicated that substitution of the indole imine reduced activity and that substitution of the piperazine ring was limited to the 4-N position (data not shown, [65]). Subsequent studies have focused on varying the group attached to the piperazine ring and selected compounds are listed in Table 8. In general, the introduction of bulkier groups resulted in better activity, with a fluorene substituent giving the best activity. Exploration of chemical space around the fluorene revealed that, generally, substituents at the 2- or 3- position are well tolerated, even for some bulky groups (Genz-13100, Genz-13108), while 4- substituent groups reduce binding affinity.
Table 8.
Piperazine indoleformamides.
| Compound |

|
IC50 (μM) |
|---|---|---|
| p56 | 4-CI | 4.2 |
| p58 | 3-CI | 1.21 |
| p59 | 3-CF3 | 1.89 |
|
| ||
| p60 |
|
0.23 |
| p4 |
|
1.04 ± 0.04 |
|
| ||
|
||
|
| ||
| Genz-10850 | H | 0.16 |
| Genz-11918 | 1-CO2CH3 | 0.24 |
| Genz-12637 | 2-CI, 3-NHAc | >10 |
| Genz-12638 | 2, 7-I2 | 0.13 |
| Genz-12639 | 2-NHAc | 0.28 |
| Genz-12640 | 2-NMe2 | 0.91 |
| Genz-12641 | 2-NHCHO | 0.18 |
| Genz-12643 | 2, 4, 7-CI3 | 0.17 |
| Genz-12644 | 2-NO2 | 0.13 |
| Genz-12645 | 3-NO2 | 0.13 |
| Genz-12646 | 2, 7-Br2 | 0.12 |
| Genz-13100 | 2-NHCOBn | 0.59 |
| Genz-13108 | 2-NHCOPr | 0.46 |
| Genz-13347 | 2-NEt2 | >10 |
| Genz-13348 | 2-OCH3 | 0.52 |
| Genz-13349 | 4-OCH3 | 0.82 |
The X-ray structure of Genz-10850 bound to InhA indicates a series of both hydrogen-bonding and hydrophobic interactions between enzyme and inhibitor (Fig. 19). The indole imine is hydrogen bonded to a phosphate oxygen in NAD+ phosphate, which explains why an unsubstituted nitrogen atom is essential for binding. In addition, the amide carbonyl is hydrogen bonded to the 2’-OH of the nicotinamide ribose and also to the phenolic hydroxyl of Tyr158, which supports the importance of the indoleformamide scaffold. Extensive hydrophobic interactions are formed between the fluorenyl ring and Met103, Ala157, Ala198, Met199, Ile202, Ile215, and Leu218, explaining the increase in activity resulting from introduction of the fluorenyl group.
Fig. (19).

X-ray structure of Genz-10850 (yellow) bound to InhA with NAD+ (cyan) (1P44.pdb) [65]. The surface of the substrate binding loop is colored red. The figure was made using PyMol [23].
Although Genz-10850 inhibited InhA with an IC50 value of 160nM, the MIC for inhibiting bacterial growth was larger than 30 μM which may be due to poor membrane permeability [65] or efflux [67].
Pyrazole Derivatives
Pyrazole derivatives were also identified in the same screen that yielded the piperazine indoleformamides [65]. Table 9 lists compounds in which modifications were made primarily on the 1-N and 4-C positions of the pyrazole.
Table 9.
Inhibition percentages of pyrazole derivatives.

| |||
|---|---|---|---|
| Compound | R1 | R2 | % Inhibitiona |
| Genz-5537 |

|
17 | |
| Genz-5538 |

|
14 | |
| Genz-5539 |
|
8 | |
| Genz-5540 |
|
|
10 |
| Genz-7466 |
|
11 | |
| Genz-8575 |
|
91 | |
| Genz-5542 | 82 | ||
| Genz-5984 |
|
10 | |
| Genz-5985 |
|
17 | |
| Genz-5986 |
|
9 | |
| Genz-6341 |

|
|
60 |
| Genz-6356 |
|
54 | |
| Genz-6371 |
|
27 | |
| Genz-6386 |
|
5 | |
Percent inhibition of inhA activity at compound concentration of 40μM.
With R1 fixed as 4-chloro-2-nitrophenyl, trifluoromethylpyrimidine (Genz-5542) is the only R2 substituent that gave InhA inhibition over 80% at compound concentration of 40 μM. R1 was then further explored with substituted benzene rings when trifluoromethylpyrimidine was conserved as R2. Among the derivatives, the dinitrophenyl analog (Genz-8575) provided better activity than the parent compound, giving an IC50 of 2.4 μM and a MIC value of 2.5μM against M. tuberculosis H37Rv. Although the IC50 of Genz-8575 is 15-fold higher than Genz-10850, the MIC value is at least 12-fold smaller, implying that the pyrazole derivatives are a better scaffold than the piperazine indoleformamides.
Arylamides
The HTS performed by He et al.[64] identified 30 hits of which the arylamides (Fig. (20)) were the largest series of compounds. Optimization has primarily focused on the A- and B-rings (Table 10) [67].
Fig. (20).
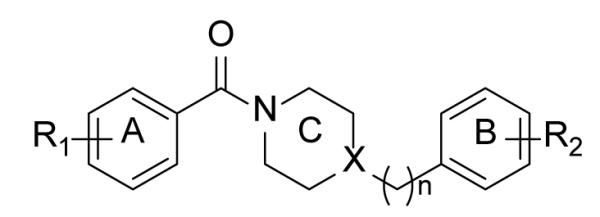
Arylamides
Table 10.
SAR study on arylamides.
| Compound | X | n | R1 | R2 | IC50 (μM) |
|---|---|---|---|---|---|
| a1 | N | 0 | H | H | 38.86 ± 1.35 |
| a2 | N | 0 | 4-CH3 | H | 16.64 ± 0.49 |
| a3 | N | 0 | 4-CH3 | 3-CF3 | 6.26 ± 0.33 |
| a4 | N | 0 | 4-CH3 | 3-CI | 3.07 ± 0.48 |
| a5 | N | 0 | 3-CH3 | 3-CI | 9.43 ± 0.80 |
| a6 | N | 0 | 3-CH3 | 4-NO2 | 15.47 ± 1.52 |
| a7 | N | 0 | 3, 4-Me2 | 3-CI | 0.99 ± 0.03 |
| a8 | N | 0 | 3, 4-Me2 | 3-CF3 | 1.85 ±0.08 |
| a9 | N | 0 | 4-i-Pr | 3-CI | >100 |
| a10 | N | 0 | 4-t-Bu | 3-CI | >100 |
| a11 | N | 0 | 4-t-Bu | 3-CF3 | >100 |
| a12 | N | 0 | 4-t-Bu | 4-CH3, 3-CI |
>100 |
| a13 | N | 0 | 2-F | 3-CI | 13.87 ± 1.03 |
| a14 | N | 0 | 4-F | 3-CI | 9.74 ± 0.62 |
| a15 | N | 0 | 3-CI | 3-CI | 6.73 ± 0.27 |
| a16 | N | 0 | 3, 4-CI2 | 3-CI | 6.05 ± 0.58 |
| a17 | N | 0 | 3, 4-CI2 | H | 17.62 ± 1.24 |
| a18 | N | 1 | H | H | 31.50 ± 1.65 |
| b1 | C | 1 | 3-CI | H | 7.74 ± 0.25 |
| b2 | C | 1 | 2-F | H | 14.11 ± 0.42 |
| b3 | C | 1 | 4-CH3 | H | 5.16 ± 0.45 |
| b4 | C | 1 | 3-CH3 | H | 7.39 ± 0.38 |
Introduction of a para-methyl on the A-ring improved inhibition, and an additional meta-methyl increased activity by 3-fold, while a single meta-methyl reduced activity by 3-fold compared to the mono para-methyl compound. Bulky groups at the para-position of the A-ring abolished activity (a9-a12), which indicates a limit to the size of the group that can be accommodated by the active site at this position. Electron withdrawing meta-substituents are essential for inhibition, while many para- and ortho-substituted compounds showed no activity in the HTS (data not shown, [67]). Replacement of piperazine with piperidine or insertion of a methylene between B-ring and C-ring had no apparent effect on activity.
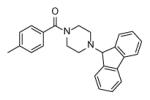
The similarity in structure between the arylamides and piperazine indoleformamides suggested that bulky groups might be tolerated on the piperazine ring of the former compounds. Further SAR work was thus undertaken through the microtiter synthesis of a focused library using a combination of aromatic acids and substituted piperazines. Following in situ screening, selected compounds were resynthesized and evaluated. Examples are shown in Table 11) [67].
Table 11.
Selected compounds and their activity.
| Compound | Structure | IC50 (μM) | MIC (μM) |
|---|---|---|---|
| p1 |
|
0.40 ± 0.02 | >125 |
| p2 |
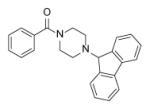
|
0.09 ± 0.00 | >125 |
| p5 |
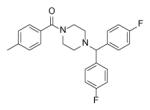
|
1.89 ± 0.11 | >125 |
| p6 |
|
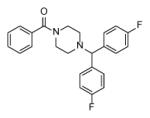
2.04 ± 0.08 | 125 |
The best inhibitors from this series incorporated piperazines with polyaromatic rings, indicating that the B-ring is involved in hydrophobic contacts with the enzyme. The smallest IC50 value (90 nM) was achieved by compound p2 with a benzene A-ring and fluorenyl B-ring.
The X-ray structure of b3 bound to InhA reveals the binding mode of the arylamides (Fig. (21). The amide carbonyl oxygen is hydrogen bonded to the 2’-hydroxyl of the nicotinamide ribose as well as the Tyr158 hydroxyl group. In addition the B-ring interacts with Phe149, Pro193, Leu218, and Val203 through hydrophobic contacts, which are clearly enhanced through the introduction of –Cl or -CF3 groups, or by replacing the B-ring with bulkier groups as shown by the SAR studies.
Fig. (21).

X-ray structure of b3 (yellow) bound to InhA with NAD+ (cyan) (2nsd.pdb) [67]. The surface of the substrate binding loop is colored red. The figure was made using PyMol [23].
MIC values against M. tuberculosis were measured for selected compounds. a6 and p6 have the smallest MIC values (125 μM), while most inhibitors have MIC values above 125 μM. Since the structure of arylamides is very similar to the piperazine indoleformamides, it is likely that the arylamides also suffer from poor antibacterial activity due to problems crossing the membrane and/or efflux.
Fatty Acids
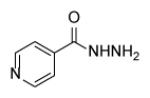
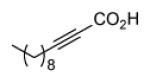
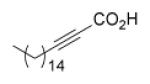
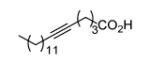
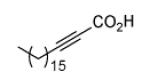
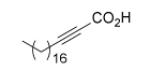
Since InhA catalyzes the reduction of 2-trans-enoyl ACPs, inhibition of the enzyme can in principal be achieved using unreactive analogs of the natural fatty acid substrate. Both 2-trans-hexadecenoyl-CoA and 2-trans-octadecenoyl-CoA are substrates for InhA, and the corresponding alkynoic acids are competitive inhibitors of InhA with complete inhibition achieved at a concentration of 43μM [68]. Interestingly, compared to their corresponding CoA thioesters, 2-hexadecynoic acid (L3) and 2-octadecynoic acid (L7) demonstrated much better inhibitory activity against M. tuberculosis H37Rv, with MIC values of 20 μM and 25 μM, respectively [69]. To generate SAR and explore their mode of action, 13 alkynoic acids were synthesized and MIC values against M. smegmatis mc2155 and M. bovis BCG were determined (Table 12) [69].
Table 12.
Antibacterial activity of alkynoic acids.
| Compound | Structure | M.smegmatAs c2155 MIC(μM) | M. bovis BCG MIC(μM) |
|---|---|---|---|
| Isoniazid |
|
40 | 0.7 |
| L1 |
|
>300 | N.D. |
| L2 |
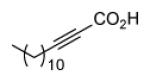
|
268 | N.D. |
| L3 |
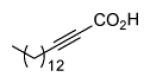
|
10 | 4 |
| L4 |
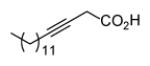
|
20 | N.D. |
| L5 |
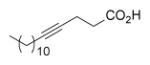
|
30 | N.D. |
| L6 |
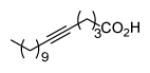
|
80 | N.D. |
| L7 |
|
4 | 9 |
| L8 |
|
36 | N.D. |
| L9 |

|
>360 | N.D. |
| L10 |
|
360 | N.D. |
| L11 |
|
35 | N.D. |
| L12 |
|
>300 | N.D. |
| L13 |
|
>300 | N.D. |
| L14 |
|
>200 | N.D. |
| L15 |
|
>200 | N.D. |
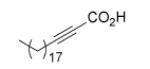
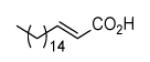
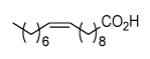
N.D. = not determined.
From Table 12 it can be seen that the antimycobacterial activity of the alkynoic acids is affected by both the position of the triple bond and the chain length of the fatty acid. L7 has the smallest MIC against M. smegmatis, while L14 and L15 have no activity at all, which suggests that the triple bond is essential for inhibitory activity. In addition, comparison of the data for hexadecynoic acids L3-L6 and octadecynoic acids L7-L10 indicates that the MIC values decrease as the triple bond is moved away from the carboxylic acid. This indicates that 2-alkynoic acids are the best scaffold, while fatty acids of 16-19 carbons give optimal antibacterial activity against the mycobacteria studied.
Further studies into the mode of action of the alkynoic acids using immunoblotting revealed that L3 and L7 directly inhibit InhA, while the use of 14C labeled L3 demonstrated the formation of two major metabolites, 3-ketoacyl CoA and 3-alkynoyl CoA, which inhibit fatty acid biosynthesis and β-oxidation respectively [69]. Since the 2-alkynoic acids have alternative effects on eukaryotes [70], there is some promise that they could be developed into specific antimycobacterials.
In addition to the studies with alkynoic acids, other types of fatty acids have been explored as potential leads for developing InhA inhibitors. Tasdemir and coworkers isolated several natural marine fatty acids from the Turkish sponge Agelas oroides with activity against InhA. FAMA, a mixture of 23:2 Δ5, 9 and 24:2 Δ5, 9 has an IC50 of 9.4 μg/ml, and FAMD, a mixture of saturated methyl-branched fatty acids has an IC50 of 8.2 μg/ml [71]. Also, four 2-methoxylated saturated fatty acids (L16-L19, Fig. (22)) were synthesized and tested against M. tuberculosis. Compound L17, with a 10 carbon chain, had the smallest MIC value (239.51 μM). However, the target of these fatty acids derivatives was not clarified [72]. Finally, several saturated and unsaturated fatty acids have also been shown to possess antimycobacterial activity [73-74]. However these compounds were not tested against M. tuberculosis, and their mode of action was proposed to involve insertion of the molecules into the cell membrane with a concomitant change in membrane permeability [70, 73].
Fig. (22).
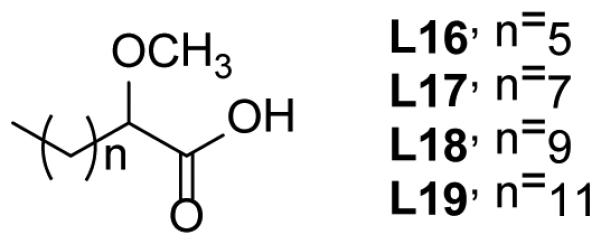
2-Methoxylated saturated fatty acids.
Imidazopiperidines
Wall et al. used screening to identify the imidazopiperidines (Fig. (23a)) as a potential novel class of InhA inhibitors [75]. Sixteen derivatives were obtained through solid-phase synthesis and the biological activity of these compounds is given in Table 13.
Fig. (23).
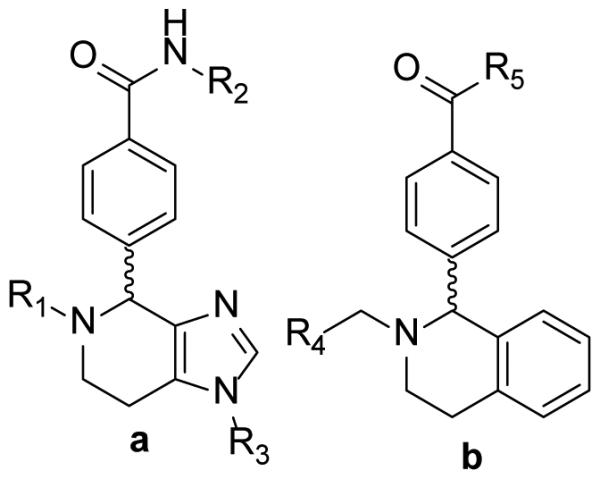
Imidazopiperidines and derivatives (a) Imidazopiperidines (b) analogues in which the imidazole has been replaced.
Table 13.
Enzyme inhibition and whole cell activity for imidazopiperidines.
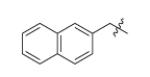
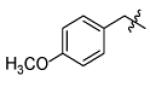
| Compound | R1 | R2 | R3 | IC50 (μM) | MIC50 (μM) |
|---|---|---|---|---|---|
| m1 |
|
|
H | 0.24 | 32 |
| m2 |
|
|
H | 0.41 | 63 |
| m3 |

|
|
H | 1.39 | 32 |
| m4 |
|
|
H | 3.02 | 32 |
| m5 |
|
|
H | 3.5 | 32 |
| m6 |
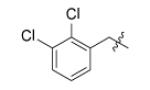
|
|
H | 9.81 | 32 |
| m7 |
|

|
H | 10.18 | 63 |
| m8 |
|
|
H | 13.38 | 16 |
|
m1aa
mlbb |
Enantiomers of m1 | >10 0.08 |
63 63 |
||
|
m2aa
m2bb |
Enantiomers of m2 | >10 0.2 |
32 63 |
||
The enantiomer with retention time < 7min.
The enantiomer with retention time > 7min.
Replacement of the imidazole group with benzene (Fig. (23b)) completely abolished inhibition (data not shown) and Table 13 concentrates on analoging around the imidazopiperidine scaffold. It was found that 4-methoxybenzyl is preferred at R2, while electron withdrawing substituents on the piperidine ring were favored. A combination of 4-methoxybenzyl at R2 and mono- or di-chlorobenzyl at R1 gave the best activity (m1, m2), however the lack of correlation between IC50 and MIC50 suggests that the compounds might have other target(s) in the cell or that uptake and/or detoxification may be an issue.
General Binding Model
Comparison of the structural and mechanistic data enables a common binding mode for InhA inhibitors to be elucidated. With only one exception, the inhibitors described in this review bind in the presence of the cofactor, either in non-covalent ternary complexes or as adducts of the cofactor. For those that form ternary complexes and where Ki values have been determined, there is usually a preference for NAD+, and so these compounds are uncompetitive inhibitors binding to the InhA:NAD+ product complex. In addition, while a clear link exists between slow onset inhibition and ordering of the substrate binding loop for the diaryl ether inhibitors, in most cases the kinetics of enzyme inhibition has not been studied in detail although there are now many InhA:inhibitor X-ray structures in which the loop is ordered.
Of key importance for inhibitor binding is the formation of a hydrogen bonding network between a group on the inhibitor, Tyr158 and the 2’-hydroxyl on the nicotinamide ribose. Inhibitors with hydrogen bonding (HB) functionalities such as hydroxyl or carbonyl groups participate in this network [22, 25, 44, 62, 65]. In addition, since the natural substrates for InhA are significantly longer than for other FabI enzymes (24-56 carbons in length, [76]), the substrate binding loop in InhA is also larger, thereby enabling inhibitors with bulky hydrophobic groups such as fluorene to bind. Extensive hydrophobic contacts in this area include Met103, Gly104, Phe149, Ala157, Ala198, Met199, Ile202, Ile215, and Leu218 [13, 53, 65]. Finally, a third binding area can be identified close to the hydrogen bonding group in the inhibitor. This binding pocket is relatively constrained in size (size limited) and is also exposed to solvent as well as being close to polar groups such as the cofactor phosphodiester bridge and non-polar groups in the substrate binding loop (e.g. Ala198). Not surprisingly then, the SAR data shows that enzyme inhibition is highly sensitive to the chemical nature and size of the inhibitor substituent at this position, and groups that are small and that can also participate in both polar and non-polar contacts are preferred. In Fig. (24) the inhibitors from 7 different structures have been overlayed in order to highlight these three binding regions. Moving forward the challenge will be to use this information to optimize inhibitor affinity whilst at the same time engineering the compounds to reduce metabolic liabilities, improve compound ADME and enhance bioactivity.
Fig. (24).

Common binding mode for InhA inhibitors.
Seven structures have been overlayed in which InhA is in complex with: two triclosan molecules (1p45.pdb), PT03 (2b36.pdb), C16-NAC (1bvr.pdb), F3 (3fng.pdb), Genz-10850 (1p44.pdb), d11 (2h7m.pdb), and b3 (2nsd.pdb). Key areas of interaction are highlighted and include groups involved in the hydrogen bond (HB) network (pink), the hydrophobic pocket (yellow), and the region surrounded by both polar and nonpolar groups (size-limited, slate).
Conclusion
In the past decade, beginning with the discovery that InhA is the target for isoniazid, there have been several efforts to develop potent InhA inhibitors as leads for novel tuberculosis chemotherapeutics. In addition to the approaches described in this review, de novo design [77], computational simulation [78-79], and selective optimization of side activities (SOSA) [80] have also been utilized. These activities are all underpinned by the expectation that InhA inhibitors that do not require activation by KatG will be active against isoniazid-resistant strains of M. tuberculosis. Importantly, at least two studies support this contention [13, 65], and several sub-nanomolar inhibitors of the enzyme have been developed. Current efforts are now focused on optimizing the in vivo properties of these lead compounds with an emphasis both on the chemical properties that affect ADME, as well as factors that affect antibacterial activity such as uptake and detoxification.
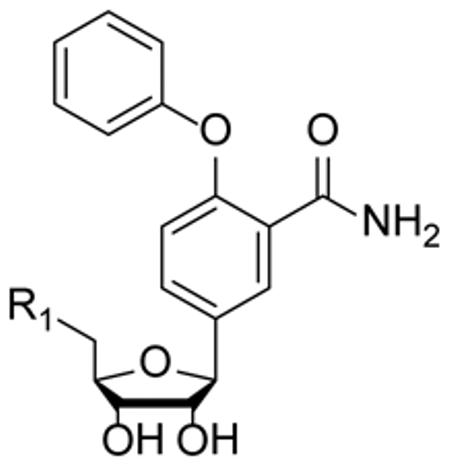
Table 1.
INH-NAD Analogs
| ||
|---|---|---|
|
| ||
| Compound | R1 | IC50 (μM) |
| 16 | -OH | >100 |
| 17 | -OPO(OH)2 | >100 |
| 18 | -ADP | 27 |
|
| ||
References
- [1].Barry CE, Lee RE, Mdluli K, Sampson AE, Schroeder BG, Slayden RA, Yuan Y. Mycolic acids: Structure, biosynthesis and physiological functions. Prog. Lipid. Res. 1998;37(2-3):143–179. doi: 10.1016/s0163-7827(98)00008-3. [DOI] [PubMed] [Google Scholar]
- [2].Brennan PJ, Nikaido H. The Envelope of Mycobacteria. Annu. Rev. Biochem. 1995;64:29–63. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- [3].Brinster S, Lamberet G, Staels B, Trieu-Cuot P, Gruss A, Poyart C. Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature. 2009;458(7234):83–86. doi: 10.1038/nature07772. [DOI] [PubMed] [Google Scholar]
- [4].Balemans W, Lounis N, Gilissen R, Guillemont J, Simmen K, Andries K, Koul A. Essentiality of FASII pathway for Staphylococcus aureus. Nature. 2010;463(7279):E3. doi: 10.1038/nature08667. discussion E4. [DOI] [PubMed] [Google Scholar]
- [5].Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, Wilson T, Collins D, Lisle G.d., Jr inhA, a Gene Encoding a Target for Isoniazid and Ethionamide in Mycobacterium tuberculosis. Science. 1994;263(5144):227–230. doi: 10.1126/science.8284673. W.R.J. [DOI] [PubMed] [Google Scholar]
- [6].Dessen A, Quemard A, Blanchard JS, Jacobs WR, Sacchettini JC. Crystal-Structure and Function of the Isoniazid Target of Mycobacterium-Tuberculosis. Science. 1995;267(5204):1638–1641. doi: 10.1126/science.7886450. [DOI] [PubMed] [Google Scholar]
- [7].Quemard A, Sacchettini JC, Dessen A, Vilcheze C, Bittman R, Jacobs WR, Blanchard JS. Enzymatic Characterization of the Target for Isoniazid in Mycobacterium-Tuberculosis. Biochemistry. 1995;34(26):8235–8241. doi: 10.1021/bi00026a004. [DOI] [PubMed] [Google Scholar]
- [8].Rozwarski DA, Grant GA, Barton DH, Jacobs WR, Jr., Sacchettini JC. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science. 1998;279(5347):98–102. doi: 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]
- [9].Rawat R, Whitty A, Tonge PJ. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: Adduct affinity and drug resistance. Proc. Natl. Acad. Sci. U. S. A. 2003;100(24):13881–13886. doi: 10.1073/pnas.2235848100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Vilcheze C, Morbidoni HR, Weisbrod TR, Iwamoto H, Kuo M, Sacchettini JC, Jacobs WR., Jr. Inactivation of the inhA-encoded fatty acid synthase II (FASII) enoyl-acyl carrier protein reductase induces accumulation of the FASI end products and cell lysis of Mycobacterium smegmatis. J. Bacteriol. 2000;182(14):4059–4067. doi: 10.1128/jb.182.14.4059-4067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang Y, Heym B, Allen B, Young D, Cole S. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature. 1992;358(6387):591–593. doi: 10.1038/358591a0. [DOI] [PubMed] [Google Scholar]
- [12].Musser JM, Kapur V, Williams DL, Kreiswirth BN, van Soolingen D, van Embden JD. Characterization of the catalase-peroxidase gene (katG) and inhA locus in isoniazid-resistant and -susceptible strains of Mycobacterium tuberculosis by automated DNA sequencing: restricted array of mutations associated with drug resistance. J. Infect. Dis. 1996;173(1):196–202. doi: 10.1093/infdis/173.1.196. [DOI] [PubMed] [Google Scholar]
- [13].Sullivan TJ, Truglio JJ, Boyne ME, Novichenok P, Zhang X, Stratton CF, Li HJ, Kaur T, Amin A, Johnson F, Slayden RA, Kisker C, Tonge PJ. High affinity InhA inhibitors with activity against drug-resistant strains of Mycobacterium tuberculosis. ACS Chem. Biol. 2006;1(1):43–53. doi: 10.1021/cb0500042. [DOI] [PubMed] [Google Scholar]
- [14].Lu H, Tonge PJ. Inhibitors of FabI, an enzyme drug target in the bacterial fatty acid biosynthesis pathway. Acc. Chem. Res. 2008;41(1):11–20. doi: 10.1021/ar700156e. [DOI] [PubMed] [Google Scholar]
- [15].Seefeld MA, Miller WH, Newlander KA, Burgess WJ, DeWolf, Elkins PA, Head MS, Jakas DR, Janson CA, Keller PM, Manley PJ, Moore TD, Payne DJ, Pearson S, Polizzi BJ, Qiu X, Rittenhouse SF, Uzinskas IN, Wallis NG, Huffman WF. Indole Naphthyridinones as Inhibitors of Bacterial Enoyl-ACP Reductases FabI and FabK. J. Med. Chem. 2003;46(9):1627–1635. doi: 10.1021/jm0204035. W.E. [DOI] [PubMed] [Google Scholar]
- [16].Abbanat D, Morrow B, Bush K. New agents in development for the treatment of bacterial infections. Curr. Opin. Pharmacol. 2008;8(5):582–592. doi: 10.1016/j.coph.2008.08.001. [DOI] [PubMed] [Google Scholar]
- [17].Karlowsky JA, Kaplan N, Hafkin B, Hoban DJ, Zhanel GG. AFN-1252, a FabI inhibitor, demonstrates a Staphylococcus-specific spectrum of activity. Antimicrob. Agents Chemother. 2009;53(8):3544–3548. doi: 10.1128/AAC.00400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lu H, England H, am Ende CW, Truglio JJ, Luckner S, Reddy BG, Marlenee N, Knudson SE, Knudson DL, Bowen RA, Kisker C, Slayden RA, Tonge PJ. Slow-Onset Inhibition of the FabI Enoyl Reductase from Francisella Tularensis: Residence Time and In Vivo Activity. ACS Chem. Biol. 2009;4:221–231. doi: 10.1021/cb800306y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lu H, Tonge PJ. Residence Time: Critical Information for Lead Optimization. Curr. Opin. Chem. Biol. 2010 doi: 10.1016/j.cbpa.2010.06.176. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Baldock C, Rafferty JB, Sedelnikova SE, Baker PJ, Stuitje AR, Slabas AR, Hawkes TR, Rice DW. A mechanism of drug action revealed by structural studies of enoyl reductase. Science. 1996;274(5295):2107–2110. doi: 10.1126/science.274.5295.2107. [DOI] [PubMed] [Google Scholar]
- [21].Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discov. 2006;5(9):730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- [22].Stewart MJ, Parikh S, Xiao GP, Tonge PJ, Kisker C. Structural basis and mechanism of enoyl reductase inhibition by triclosan. J. Mol. Biol. 1999;290(4):859–865. doi: 10.1006/jmbi.1999.2907. [DOI] [PubMed] [Google Scholar]
- [23].Delano WL. The PyMOL Molecular Graphics System. 2002 http://www.pymol.org.
- [24].Sivaraman S, Zwahlen J, Bell AF, Hedstrom L, Tonge PJ. Structure-Activity Studies of the Inhibition of FabI, the Enoyl Reductase from Escherichia coli, by Triclosan: Kinetic Analysis of Mutant FabIs. Biochemistry. 2003;42(15):4406–4413. doi: 10.1021/bi0300229. [DOI] [PubMed] [Google Scholar]
- [25].Ward WHJ, Holdgate GA, Rowsell S, McLean EG, Pauptit RA, Clayton E, Nichols WW, Colls JG, Minshull CA, Jude DA, Mistry A, Timms D, Camble R, Hales NJ, Britton CJ, Taylor IWF. Kinetic and structural characteristics of the inhibition of enoyl (acyl carrier protein) reductase by triclosan. Biochemistry. 1999;38(38):12514–12525. doi: 10.1021/bi9907779. [DOI] [PubMed] [Google Scholar]
- [26].Tummino PJ, Copeland RA. Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry. 2008;47(32):8465. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- [27].Swinney DC. Biochemical mechanisms of drug action: what does it take for success? Nat. Rev. Drug Discov. 2004;3(9):801–808. doi: 10.1038/nrd1500. [DOI] [PubMed] [Google Scholar]
- [28].Middlebrook G. Sterilization of tubercle bacilli by isonicotinic acid hydrazide and the incidence of variants resistant to the drug in vitro. Am. Rev. Tuberc. 1952;65(6):765–767. [PubMed] [Google Scholar]
- [29].Lei BF, Wei CJ, Tu SC. Action mechanism of antitubercular isoniazid - Activation mycobacterium tuberculosis KatG, isolation, and characterization of InhA inhibitor. J. Biol. Chem. 2000;275(4):2520–2526. doi: 10.1074/jbc.275.4.2520. [DOI] [PubMed] [Google Scholar]
- [30].Ducasse-Cabanot S, Cohen-Gonsaud M, Marrakchi H, Nguyen M, Zerbib D, Bernadou J, Daffe M, Labesse G, Quemard A. In vitro inhibition of the Mycobacterium tuberculosis beta-ketoacyl-acyl carrier protein reductase MabA by isoniazid. Antimicrob. Agents Chemother. 2004;48(1):242–249. doi: 10.1128/AAC.48.1.242-249.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Argyrou A, Vetting MW, Aladegbami B, Blanchard JS. Mycobacterium tuberculosis dihydrofolate reductase is a target for isoniazid. Nat. Struct. Mol. Biol. 2006;13(5):408–413. doi: 10.1038/nsmb1089. [DOI] [PubMed] [Google Scholar]
- [32].Argyrou A, Jin L, Siconilfi-Baez L, Angeletti RH, Blanchard JS. Proteome-wide profiling of isoniazid targets in Mycobacterium tuberculosis. Biochemistry. 2006;45(47):13947–13953. doi: 10.1021/bi061874m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mdluli K, Slayden RA, Zhu Y, Ramaswamy S, Pan X, Mead D, Crane DD, Musser JM, Barry CE., 3rd Inhibition of a Mycobacterium tuberculosis beta-ketoacyl ACP synthase by isoniazid. Science. 1998;280(5369):1607–1610. doi: 10.1126/science.280.5369.1607. [DOI] [PubMed] [Google Scholar]
- [34].Ramaswamy SV, Reich R, Dou SJ, Jasperse L, Pan X, Wanger A, Quitugua T, Graviss EA. Single nucleotide polymorphisms in genes associated with isoniazid resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2003;47(4):1241–1250. doi: 10.1128/AAC.47.4.1241-1250.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Broussy S, Bernardes-Genisson V, Quemard A, Meunier B, Bernadou J. The first chemical synthesis of the core structure of the benzoylhydrazine-NAD adduct, a competitive inhibitor of the Mycobacterium tuberculosis enoyl reductase. J. Org. Chem. 2005;70(25):10502–10510. doi: 10.1021/jo051901z. [DOI] [PubMed] [Google Scholar]
- [36].Bonnac L, Gao G-Y, Chen L, Felczak K, Bennett EM, Xu H, Kim T, Liu N, Oh H, Tonge PJ, Pankiewicz KW. Synthesis of 4-phenoxybenzamide adenine dinucleotide as NAD analogue with inhibitory activity against enoyl-ACP reductase (InhA) of Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2007;17(16):4588–4591. doi: 10.1016/j.bmcl.2007.05.084. [DOI] [PubMed] [Google Scholar]
- [37].Oliveira JS, Sousa EHS, Basso LA, Palaci M, Dietze R, Santos DS, Moreira IS. An inorganic iron complex that inhibits wild-type and an isoniazid-resistant mutant 2-trans-enoyl-ACP (CoA) reductase from Mycobacterium tuberculosis. Chem. Commun. (Camb) 2004;(3):312–313. doi: 10.1039/b313592f. [DOI] [PubMed] [Google Scholar]
- [38].Oliveira JS, de Sousa EHS, de Souza ON, Moreira IS, Santos DS, Basso LA. Slow-onset inhibition of 2-trans-enoyl-ACP (CoA) reductase from Mycobacterium tuberculosis by an inorganic complex. Curr. Pharm. Des. 2006;12(19):2409–2424. doi: 10.2174/138161206777698927. [DOI] [PubMed] [Google Scholar]
- [39].Vasconcelos IB, Meyer E, Sales FAM, Moreira IS, Basso LA, Santos DS. The mode of inhibition of Mycobacterium tuberculosis wild-type and isoniazid-resistant 2-trans-enoyl-ACP(CoA) reductase enzymes by an inorganic complex. Anti-Infect. Agents Med. Chem. 2008;7(1):50–62. [Google Scholar]
- [40].David S, Barros V, Cruz C, Delgado R. In vitro effect of free and complexed indium(III) against Mycobacterium tuberculosis. FEMS Microbiol. Lett. 2005;251(1):119–124. doi: 10.1016/j.femsle.2005.07.044. [DOI] [PubMed] [Google Scholar]
- [41].Razafimahefa D, Ralambomanana DA, Hammouche L, Pélinski L, Lauvagie S, Bebear C, Brocard J, Maugein J. Synthesis and antimycobacterial activity of ferrocenyl ethambutol analogues and ferrocenyl diamines. Bioorg. Med. Chem. Lett. 2005;15(9):2301–2303. doi: 10.1016/j.bmcl.2005.03.004. [DOI] [PubMed] [Google Scholar]
- [42].do Nascimento FB, Von Poelhsitz G, Pavan FR, Sato DN, Leite CQF, Selistre-de-Arújo HS, Ellena J, Castellano EE, Deflon VM, Batista AA. Synthesis, characterization, X-ray structure and in vitro antimycobacterial and antitumoral activities of Ru(II) phosphine/diimine complexes containing the "SpymMe2" ligand, SpymMe2=4,6-dimethyl-2-mercaptopyrimidine. J. Inorg. Biochem. 2008;102(9):1783–1789. doi: 10.1016/j.jinorgbio.2008.05.009. [DOI] [PubMed] [Google Scholar]
- [43].McMurry LM, Oethinger M, Levy SB. Triclosan targets lipid synthesis. Nature. 1998;394(6693):531–532. doi: 10.1038/28970. [DOI] [PubMed] [Google Scholar]
- [44].Levy CW, Roujeinikova A, Sedelnikova S, Baker PJ, Stuitje AR, Slabas AR, Rice DW, Rafferty JB. Molecular basis of triclosan activity. Nature. 1999;398(6726):383–384. doi: 10.1038/18803. [DOI] [PubMed] [Google Scholar]
- [45].McMurry LM, McDermott PF, Levy SB. Genetic evidence that InhA of Mycobacterium smegmatis is a target for triclosan. Antimicrob. Agents Chemother. 1999;43(3):711–713. doi: 10.1128/aac.43.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Parikh SL, Xiao G, Tonge PJ. Inhibition of InhA, the Enoyl Reductase from Mycobacterium tuberculosis, by Triclosan and Isoniazid. Biochemistry. 2000;39(26):7645–7650. doi: 10.1021/bi0008940. [DOI] [PubMed] [Google Scholar]
- [47].Heath RJ, Yu YT, Shapiro MA, Olson E, Rock CO. Broad spectrum antimicrobial biocides target the FabI component of fatty acid synthesis. J. Biol. Chem. 1998;273(46):30316–30320. doi: 10.1074/jbc.273.46.30316. [DOI] [PubMed] [Google Scholar]
- [48].Sivaraman S, Sullivan TJ, Johnson F, Novichenok P, Cui G, Simmerling C, Tonge PJ. Inhibition of the Bacterial Enoyl Reductase FabI by Triclosan: A Structure-Reactivity Analysis of FabI Inhibition by Triclosan Analogs. J. Med. Chem. 2004;47:509–518. doi: 10.1021/jm030182i. [DOI] [PubMed] [Google Scholar]
- [49].Rafi SB, Cui G, Song K, Cheng X, Tonge PJ, Simmerling C. Insight through molecular mechanics Poisson-Boltzmann surface area calculations into the binding affinity of triclosan and three analogues for FabI, the E. coli enoyl reductase. J. Med. Chem. 2006;49(15):4574–4580. doi: 10.1021/jm060222t. [DOI] [PubMed] [Google Scholar]
- [50].Wang LQ, Falany CN, James MO. Triclosan as a substrate and inhibitor of 3 '-phosphoadenosine-5 '-phosphosulfate-sulfotransferase and UDP-glucuronosyl transferase in human liver fractions. Drug Metab. and Dispos. 2004;32(10):1162–1169. doi: 10.1124/dmd.104.000273. [DOI] [PubMed] [Google Scholar]
- [51].Boyne ME, Sullivan TJ, am Ende CW, Lu H, Gruppo V, Heaslip D, Amin AG, Chatterjee D, Lenaerts A, Tonge PJ, Slayden RA. Targeting fatty acid biosynthesis for the development of novel chemotherapeutics against Mycobacterium tuberculosis: evaluation of A-ring-modified diphenyl ethers as high-affinity InhA inhibitors. Antimicrob. Agents Chemother. 2007;51(10):3562–3567. doi: 10.1128/AAC.00383-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Luckner SR, Liu N, am Ende CW, Tonge PJ, Kisker C. A Slow, Tight Binding Inhibitor of InhA, the Enoyl-Acyl Carrier Protein Reductase from Mycobacterium tuberculosis. Journal of Biological Chemistry. 2010;285(19):14330–14337. doi: 10.1074/jbc.M109.090373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Freundlich JS, Wang F, Vilcheze C, Gulten G, Langley R, Schiehser GA, Jacobus DP, Jacobs WR, Jr., Sacchettini JC. Triclosan derivatives: Towards potent inhibitors of drug-sensitive and drug-resistant Mycobacterium tuberculosis. ChemMedChem. 2009;4(2):241–248. doi: 10.1002/cmdc.200800261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Rozwarski DA, Vilcheze C, Sugantino M, Bittman R, Sacchettini JC. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD(+) and a C16 fatty acyl substrate. J. Biol. Chem. 1999;274(22):15582–15589. doi: 10.1074/jbc.274.22.15582. [DOI] [PubMed] [Google Scholar]
- [55].am Ende CW, Knudson SE, Liu N, Childs J, Sullivan TJ, Boyne M, Xu H, Gegina Y, Knudson DL, Johnson F, Peloquin CA, Slayden RA, Tonge PJ. Synthesis and in vitro antimycobacterial activity of B-ring modified diaryl ether InhA inhibitors. Bioorg. Med. Chem. Lett. 2008;18(10):3029–3033. doi: 10.1016/j.bmcl.2008.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hogenauer G, Woisetschlager M. A Diazaborine derivative inhibits lipopolysaccharide biosynthesis. Nature. 1981;293(5834):662–664. doi: 10.1038/293662a0. [DOI] [PubMed] [Google Scholar]
- [57].Bergler H, Hogenauer G, Turnowsky F. Sequences of the Envm Gene and of 2 Mutated Alleles in Escherichia-Coli. J. Gen. Microbiol. 1992;138:2093–2100. doi: 10.1099/00221287-138-10-2093. [DOI] [PubMed] [Google Scholar]
- [58].Grassberger MA, Turnowsky F, Hildebrandt J. Preparation and Antibacterial Activities of New 1,2,3-Diazaborine Derivatives and Analogs. J. Med. Chem. 1984;27(8):947–953. doi: 10.1021/jm00374a003. [DOI] [PubMed] [Google Scholar]
- [59].Davis MC, Franzblau SG, Martin AR. Syntheses and evaluation of benzodiazaborine compounds against M. tuberculosis H37Rv in vitro. Bioorg. Med. Chem. Lett. 1998;8(7):843–846. doi: 10.1016/s0960-894x(98)00126-7. [DOI] [PubMed] [Google Scholar]
- [60].Baldock C, de Boer GJ, Rafferty JB, Stuitje AR, Rice DW. Mechanism of action of diazaborines. Biochem. Pharmacol. 1998;55(10):1541–1550. doi: 10.1016/s0006-2952(97)00684-9. [DOI] [PubMed] [Google Scholar]
- [61].de Boer GJ, Pielage GJA, Nijkamp HJJ, Slabas AR, Rafferty JB, Baldock C, Rice DW, Stuitje AR. Molecular genetic analysis of enoyl-acyl carrier protein reductase inhibition by diazaborine. Mol. Microbiol. 1999;31(2):443–450. doi: 10.1046/j.1365-2958.1999.01182.x. [DOI] [PubMed] [Google Scholar]
- [62].Roujeinikova A, Sedelnikova S, de Boer GJ, Stuitje AR, Slabas AR, Rafferty JB, Rice DW. Inhibitor binding studies on enoyl reductase reveal conformational changes related to substrate recognition. J. Biol. Chem. 1999;274(43):30811–30817. doi: 10.1074/jbc.274.43.30811. [DOI] [PubMed] [Google Scholar]
- [63].Levy CW, Baldock C, Wallace AJ, Sedelnikova S, Viner RC, Clough JM, Stuitje AR, Slabas AR, Rice DW, Rafferty JB. A study of the structure-activity relationship for diazaborine inhibition of Escherichia coli enoyl-ACP reductase. J. Mol. Biol. 2001;309(1):171–180. doi: 10.1006/jmbi.2001.4643. [DOI] [PubMed] [Google Scholar]
- [64].He X, Alian A, Stroud R, Ortiz de Montellano PR. Pyrrolidine Carboxamides as a Novel Class of Inhibitors of Enoyl Acyl Carrier Protein Reductase from Mycobacterium tuberculosis. J. Med. Chem. 2006;49(21):6308–6323. doi: 10.1021/jm060715y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kuo MR, Morbidoni HR, Alland D, Sneddon SF, Gourlie BB, Staveski MM, Leonard M, Gregory JS, Janjigian AD, Yee C, Musser JM, Kreiswirth B, Iwamoto H, Perozzo R, Jacobs WR, Jr., Sacchettini JC, Fidock DA. Targeting Tuberculosis and Malaria through Inhibition of Enoyl Reductase: Compound Activity and Structural Data. J. Biol. Chem. 2003;278(23):20851–20859. doi: 10.1074/jbc.M211968200. [DOI] [PubMed] [Google Scholar]
- [66].Staveski MM, Sneddon SF, Yee C, Janjigian A. InhA Inhibitors and Methods of Use Thereof. 2002 2001-US40045 2001056974, 20010206. [Google Scholar]
- [67].He X, Alian A, Ortiz de Montellano PR. Inhibition of the Mycobacterium tuberculosis enoyl acyl carrier protein reductase InhA by arylamides. Bioorg. Med. Chem. 2007;15(21):6649–6658. doi: 10.1016/j.bmc.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Marrakchi H, Laneelle G, Quemard A. InhA, a target of the antituberculous drug isoniazid, is involved in a mycobacterial fatty acid elongation system, FAS-II. Microbiology. 2000;146(2):289–296. doi: 10.1099/00221287-146-2-289. [DOI] [PubMed] [Google Scholar]
- [69].Morbidoni HR, Vilcheze C, Kremer L, Bittman R, Sacchettini JC, Jacobs WR. Dual inhibition of mycobacterial fatty acid biosynthesis and degradation by 2-alkynoic acids. Chem. Biol. 2006;13(3):297–307. doi: 10.1016/j.chembiol.2006.01.005. [DOI] [PubMed] [Google Scholar]
- [70].Carballeira NM. New advances in fatty acids as antimalarial, antimycobacterial and antifungal agents. Prog. Lipid Res. 2008;47(1):50–61. doi: 10.1016/j.plipres.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Tasdemir D, Topaloglu B, Perozzo R, Brun R, O'Neill R, Carballeira NM, Zhang X, Tonge PJ, Linden A, Ruedi P. Marine natural products from the Turkish sponge Agelas oroides that inhibit the enoyl reductases from Plasmodium falciparum, Mycobacterium tuberculosis and Escherichia coli. Bioorg. Med. Chem. 2007;15(21):6834–6845. doi: 10.1016/j.bmc.2007.07.032. [DOI] [PubMed] [Google Scholar]
- [72].Carballeira NM, Cruz H, Kwong CD, Wan BJ, Franzblau S. 2-Methoxylated fatty acids in marine sponges: Defense mechanism against mycobacteria? Lipids. 2004;39(7):675–680. doi: 10.1007/s11745-004-1281-8. [DOI] [PubMed] [Google Scholar]
- [73].Saito H, Tomioka H, Yoneyama T. Growth of Group-Iv Mycobacteria on Medium Containing Various Saturated and Unsaturated Fatty-Acids. Antimicrob. Agents Chemother. 1984;26(2):164–169. doi: 10.1128/aac.26.2.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kabara JJ, Swieczko Dm, Truant JP, Conley AJ. Fatty-Acids and Derivatives as Antimicrobial Agents. Antimicrob. Agents Chemother. 1972;2(1):23. doi: 10.1128/aac.2.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Wall MD, Oshin M, Chung GAC, Parkhouse T, Gore A, Herreros E, Cox B, Duncan K, Evans B, Everett M, Mendoza A. Evaluation of N-(phenylmethyl)-4-[5-(phenylmethyl)-4, 5, 6, 7-tetrahydro-1H-imidazo[4, 5-c]pyridin-4-yl]benzamide inhibitors of Mycobacterium tuberculosis growth. Bioorg. Med. Chem. Lett. 2007;17(10):2740–2744. doi: 10.1016/j.bmcl.2007.02.078. [DOI] [PubMed] [Google Scholar]
- [76].Slayden RA, Barry CE. The role of KasA and KasB in the biosynthesis of meromycolic acids and isoniazid resistance in Mycobacterium tuberculosis. Tuberculosis. 2002;82(4-5):149–160. doi: 10.1054/tube.2002.0333. [DOI] [PubMed] [Google Scholar]
- [77].Rao GS, Vijayakrishnan R, Kumar M. Structure-based design of a novel class of potent inhibitors of InhA, the enoyl acyl carrier protein reductase from Mycobacterium tuberculosis: a computer modelling approach. Chem. Biol. Drug Des. 2008;72(5):444–449. doi: 10.1111/j.1747-0285.2008.00722.x. [DOI] [PubMed] [Google Scholar]
- [78].Lu X-Y, Chen Y-D, Jiang Y-J, You Q-D. Discovery of potential new InhA direct inhibitors based on pharmacophore and 3D-QSAR analysis followed by in silico screening. Eur. J. Med. Chem. 2009;44(9):3718–3730. doi: 10.1016/j.ejmech.2009.03.043. [DOI] [PubMed] [Google Scholar]
- [79].Kumar A, Siddiqi MI. CoMFA based de novo design of pyrrolidine carboxamides as inhibitors of enoyl acyl carrier protein reductase from Mycobacterium tuberculosis. J. Mol. Model. 2008;14(10):923–935. doi: 10.1007/s00894-008-0326-8. [DOI] [PubMed] [Google Scholar]
- [80].Kinnings SL, Liu N, Buchmeier N, Tonge PJ, Xie L, Bourne PE. Drug discovery using chemical systems biology: repositioning the safe medicine Comtan to treat multi-drug and extensively drug resistant tuberculosis. PLoS Comput. Biol. 2009;5(7):e1000423. doi: 10.1371/journal.pcbi.1000423. [DOI] [PMC free article] [PubMed] [Google Scholar]