

History of Mechanical Circulatory Support Devices
Advanced heart failure (HF) is a major cause of morbidity and mortality in the United States and heart transplantation remains as the gold standard therapy. Due to a scarcity of donor organs, the application of mechanical circulatory support devices (MCSDs) has become a crucial approach in HF therapy as a bridge to transplantation (BTT). Therefore, MCSDs have existed both conceptually and experimentally for more than 40 years, along which an exponential evolution of MCSD technology has occurred. To mimic human physiology, the first generation of left ventricular assist devices (LVADs) were pulsatile volume displacement pumps (HeartMate® XVE LVAS, Thoratec® PVAD and Novacor®, etc.). Insights demonstrating the inessentiality of the pulsatile nature of LVADs for survival from a physiologic standpoint propelled the design of second generation continuous flow devices [HeartMate II® (Thoratec Inc), the Micromed® Debakey VAD, Berlin Heart Incor® (Berlin Heart AG) and the Jarvik 2000® (Jarvik Heart Inc)], which emerged with superior safety and durability. Consequently the Heartmate® II was approved for BTT and destination therapy (DT) in the US 1. Recent reports 2, 3 have demonstrated a two-year survival rate of 87% in DT patients under intense surveillance, comparable to open heart transplantation (OHT) survival statistics. In parallel, these promising outcomes of LVADs in HF therapy have spawned the translational research field of left ventricular (LV) “reverse remodeling”, which has already shown great promise for elucidating underlying molecular and cellular mechanisms.
Clinical Insights into Bridge to Recovery
HF is a highly complex clinical syndrome marked by a multitude of derangements, both in adult and pediatric populations 4. The clinical phenotype of HF begins with an injury and/or supranormal stressor on the heart, and through prolonged dyshomeostasis, eventuates in cellular and organ failure. The compensatory adaptive mechanisms including neurohormonal activation (e.g., the adrenergic system along with aldosterone, renin, and angiotensin) may initially maintain cardiac output, but the inability to sustain this eventually results in HF together with the release of pro-inflammatory cytokines. The response to neurohormonal activation, direct mechanical stretch, and LV volume overload results in progressive maladaptive remodeling (Figure 1). These derangements are potentially reversed during LVAD mechanical unloading 5-7.
Figure 1. Pathophysiology of heart failure.

Compensatory mechanisms to maintain cardiac output eventually result in negative remodeling of the ventricles.
The current gold standard therapy for heart failure is OHT, yet it carries a 50% 10-year survival along with all complications related to long-term immunosuppression and increased medical costs. With the evolution of LVAD technology and reliability, there has been anecdotal evidence of sufficient LV recovery by LVAD support to a point allowing device removal (i.e., BTR) 8. The Harefield group was the first to study a homogenous population of HF patients, in which they applied a defined pharmacological protocol along with LVAD unloading to induce recovery. This involved high intensity neurohormonal blockade followed by induction of “physiologic hypertrophy” with a high dose Clenbuterol, a direct β-agonist, to optimize LV recovery. The procedure resulted in 63.2% of patients successfully recovering following LVAD explantation 9.
Reliable and robust predictive parameters and patient selection criteria for successful bridge to recovery (BTR) have not yet been established. Patients with non-ischemic cardiomyopathy (NICM) have been targeted in studies 10, 11 as the group most likely to undergo successful BTR. However, this NICM population itself has heterogeneous underlying etiologies, such as viral myocarditis, postpartum cardiomyopathy, direct toxin or radiation exposure, and congenital cardiomyopathies. Pre-existing clinical determinants, such as older age, larger LV diameters prior to LVAD implantation, higher degree of myocardial fibrosis, and a prolonged duration of HF greater than six months have been predictive of decreased success of recovery in NICM patients and higher risk of developing HF following LVAD explantation 12. In the HMII bridge-to-transplant and destination therapy trials enrolling 1,108 NICM patients, HF of less than 1 year, and age of 45 or less were predictors of successful BTR and 2-year transplant-free survival 13.
Furthermore, echocardiographic measurements such as ejection fraction (EF), left ventricular end diastolic dimension (LVEDD), right ventricular size and wall thickness, and degree of mitral and aortic insufficiency have been used as critical parameters to predict myocardial recovery following explantation. Dandel et al. 12 found that EF greater than 50% in NICM patients prior to LVAD explantation correlated with a 91.7% chance of cardiac stability lasting ≥5 years after LVAD removal. In contrast, EF below 50% correlated with a 1.5 fold increase in the recurrence of HF at 5 years 12. The positive predictive value of EF could be enhanced if additional parameters like pre-explantation LV size and geometry, stability of unloading-induced cardiac improvement before LVAD removal, and HF duration before LVAD implantation, were also considered. The recovery rate of LVEF after LVAD implantation has been associated with prolonged HF recovery post LVAD explantation as well 14.
A growing area of interest is the determination of cardiac reserves and functional capacity beyond the scope of echocardiography parameters, in order to facilitate further prognostication of patients' recoverability after LVAD implantation 15, 16. MVO2 max and hemodynamic data, such as cardiac output, PA pressures and left ventricular end diastolic pressure at low level flows of HMII have been studied. Exercise capacity was assessed through 6-minute walk tests at minimal flow rates to determine inotropic reserve. In the second Harefield study 10, several patients underwent explantation due to other complications, which necessitated LVAD removal prior to achieving certain echo parameters. These patients performed remarkably well, however detailed molecular assessments of the cardiac recovery in these successful cases of BTR were not performed 11, 12.
Morphological, Molecular and Cellular Insights of Reverse Remodeling after LVAD Support
Morphological and Molecular Insights
Over the past 2 decades, molecular and cellular recovery after LVAD mechanical unloading (Figure 2) has been extensively investigated, in an effort to understand the pathophysiology and critical pathways for potential therapeutic applications (Table 1) 5-7, 17. The current consensus is that morphological, cellular and molecular reversal after LVAD implantation precedes clinical recovery. However, these features alone have not been able to reliably predict clinical recovery 7. With regard to morphologic changes, several early histological studies described a conclusive decrease in cell volume and morphologic restructuring to a pre-diseased state 5, 6, 17. Accordingly, there are reports demonstrating the normalization of cardiac sarcomeric proteins, including Vinculin, Desmin, Tubulin, Tropomyosin, Titin, Actinin, and α-spectrin 5, 6, 17. LVAD support also led to enhancements in cardiomyocyte force in conjunction with accelerated contraction and relaxation times 6. Nevertheless, whether interstitial connective tissue remodeling can be reversed after LVAD support remains controversial 5, 6.
Figure 2. Main levels of reverse remodeling after LVAD.
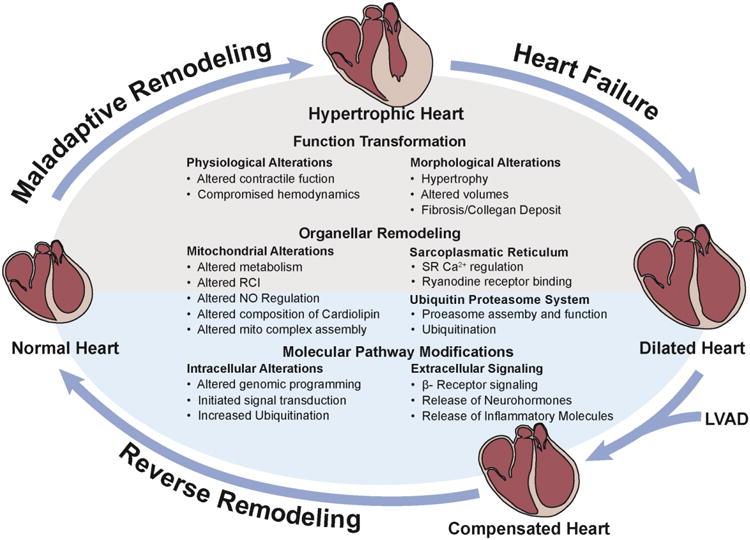
Functional, organellar, and molecular recovery in reversed ventricular remodeling.
Table 1. Summary of Molecular/Cellular Constituents in Reverse Ventricular Remodeling after LVAD.
| Cardiac feature | Cellular/molecular constituent | Effect of LVAD Therapy | Relevance |
|---|---|---|---|
| Extracellular matrix, cytoskeleton and sarcomere biogenesis | Collagen/Fibrosis | Decreased or increased deposit | Extracellular matrix remodeling is key in HF and myocardial recovery, contradicting reports |
| Metalloproteinases/matrix metalloproteinases | Alterations | Enzymes involved in breakdown of collagen in HF and reverse remodeling. | |
| Dystrophin | Disruption is reversed | Role of reversal is still unclear | |
| Myosin heavy chain | Increased | Important for functional contractile recovery | |
| Troponin T, Troponin C, Actin, Smooth muscle α actin | Increased | Important for functional contractile recovery | |
| Vinculin, Desmin, Tubulin | Abundance changes after PTM | Indicates a restored contractile apparatus | |
| Tropomyosin, Titan | Recovered | Only partially recovered despite improvement of the myocardial size, important during cardiac contraction | |
| Actinin, α-spectrin | Increased | Indicates restored shape and structure of cardiomyocytes | |
| Inflammatory response | TNF-α, Il-8, Complement C3a, glycoprotein 130 | Decreased | Both circulating as well as in LV tissue, Indicates a significant role of inflammation in HF progression |
| Il-6 | Increased | IL-6 increased in heart failure and contributes to myocardial injury | |
| Neurohormonal release | Epinephrine and Norepinephrine | Decreased | Significant in myocardial recovery due to deleterious effect on myocardium if chronically elevated as in HF |
| Renin, AT-II, Aldosteron, Arginin Vasopressin | Decreased | Significant in myocardial recovery due to deleterious effect on myocardium if chronically elevated as in HF | |
| ANP, BNP | Decreased in both circulating as well as cardiac tissue | Prominent HF biomarker, both act through guanylcyclase-A | |
| Calcium handling | Sarcolemmal calcium flux through sarcoplasmatic channel | Increased calcium pumping function | Restored after LVAD but requires optimal timing of LV-unloading |
| Sarcoplasmic reticulum calcium content | Increased | Leading to shorter action potential durations | |
| SERCA2a | Increased gene expression in LV (not in RV) | Protein abundance of SERCA is unclear (different studies show discrepancy) | |
| Na+/Ca2+ exchanger | Increased expression and function | Leads to better contractility and inotropy | |
| Phospholamban | Unchanged | Ratio of SERCA/phospholamban may improve which is important in functional recovery and contractility | |
| L-type calcium channel | Flux improved after PTM | Indicates improved contractility | |
| Ryanodine receptor | Function in LV and RV improved after PTM | Indicates improved contractility | |
| Beta adrenergic signaling | β-receptor density and location | Reversal of receptor down regulation and improved density | Correlated to reverse PI3K-ɣ activation and an increase of adenyl cyclase |
| β-receptors location | Normalized in plasma membranes | Depletion of receptors in endosomes | |
| Adenyl cyclase | Increased after LVAD | Significant component in β-adrenergic signal transduction | |
| Metabolism | Creatinine kinase | Increased back to normal levels | Indicates reversed cell damage |
| Cardiolipin | Normalized composition in ischemic cardiomyopahty | Indicates a significant role in ischemic heart disease | |
| Signal transduction | PI3K/Akt/GSK-3β pathway, IGF, FGF, RTKs, Her2/neu | Unregulated or no changes, Her2/neu upregulated | Potential targets for pharmacotherapy, IGF is correlated to stem cell recruitment |
| MAP/Erk/JNK/P38 pathway | Erk down regulated, Erk no change | Potential targets for pharmacotherapy | |
| Apoptosis | Bcl-2 | Normalized | Discrepancy whether apoptosis is increased or decreased |
| Myocyte nucleus | Cell cycle | Possible reactivation | May allow cardiac regeneration |
| Cardiomyocyte DNA content and number of polyploid cells | Decline after LVAD | May allow cardiac regeneration | |
| Number of diploid and binucleated myocytes | Increased after LVAD | May allow cardiac regeneration | |
| Gene expression | Many altered genes during HF | Only small percentage of transcripts show reversal | Reverse remodeling may occur without normalization of abnormal gene expression |
| MicroRNA expression | Many microRNAs in HF are up- or down regulated | Many microRNAs show normalization | May be more sensitive to study reverse remodeling than genes |
LVAD, left ventricular assist device; HF, heart failure; TNF, tumor necrosis factor; Il, interleukin; AT, angiotensin; ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; SERCA2a, sarco/endoplasmic reticulum Ca2+-ATPase; PI3K, phosphoinositide 3-kinase; Akt, also protein kinase B; GSK-3β, glycogen synthase kinase 3 beta, IGF, insulin like growth factor; FGF, fibroblast growth factor; RTK, receptor tyrosine kinase; Her2/neu, human epidermal growth factor receptor 2; MAP, mitogen activated protein; Erk, extracellular signal-regulated kinases; JNK, c-Jun N-terminal kinases
As a governing determinant of cardiac function, calcium (Ca2+) handling of the cell is disturbed during HF, which may contribute to the decreased contractility. In this regard, there appears to be increased gene expression of the sarcolemmal Na+-Ca2+ exchanger and sarco-endoplasmic reticular Ca2+ ATPase subtype 2a (SERCA 2a) following LVAD implantation, but corresponding protein expression remains unclear. Furthermore, levels of the Ca2+ regulatory protein phospholamban appears to be unchanged after LVAD, while L-type Ca2+ channel function and ryanodine receptor function are improved through post-translational modifications following LVAD 5, 6.
Cardiac function is also orchestrated by the neurohormonal system, and there is clear evidence that increased levels of neurohormones during HF (such as Epinephrine, Norepinephrine, Renin, AT-II, Aldosterone and Arginine Vasopressin) are decreased with successful LVAD support 5, 6, 17. Other key hormonal players in maintaining arterial blood pressure and volume homeostasis are cardiac atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP) 5, 6, 17. Both ANP and BNP are important HF biomarkers that are released by the atrium and LV, respectively, upon pressure overload. Since LVAD therapy immediately relieves the burden on the overloaded heart, both ANP and BNP levels show a downward trend. Additionally, β–adrenergic receptor density and response to stimulation can be restored after LVAD support, most likely through alterations in intracellular rather than hemodynamic factors 5-7, and also reversed PI3K-γ activation and alterations in adenylyl cyclase.
Certain aspects of mitochondrial structure and function have been investigated. Metabolism is disrupted during HF, yet studies on the effects of LVAD support on metabolic pathways are limited. Decreased creatine kinase activity during HF is restored after LVAD, indicating improved energy production. Moreover, the composition of cardiolipin, an integral component of the mitochondrial inner membrane which facilitates the function of numerous energetic enzymes, is normalized, following LVAD support in ischemic cardiomyopathy, but not in dilated cardiomyopathy 5, 6, 17.
The inflammatory response characteristic of HF progression, including the upregulation of pro-inflammatory cytokines such as TNF-α, Interleukin-6, Interleukin-8 and several heat shock proteins, was reversed after LVAD implantation 6. Additionally, apoptotic cell death during HF can be attenuated by LVAD support, though controversial results from different studies have been reported 5, 6, 17.
Transcriptional modifications
As gene expression and protein expression are not always synchronized, maladaptive hypertrophy and HF are also characterized on a transcriptional and epigenetic level targeting the activation of gene expression 5. Hypertrophy and HF are both correlated to the activation of genes encoding transcriptional factors (c-jun, c-fos, c-myc), hormonal passengers (ANP, BNP), β-myosin heavy chain, and skeletal α-actin. One of the first studies measuring the expression of cardiac genes in HF samples found that SERCA 2a, the ryanodine receptor, and the sarcolemmal Na+-Ca2+ exchanger were all upregulated after LVAD support 18. A similar study by Hall et al. using microarray analysis of mRNA transcripts in HF patients following LVAD support in combination with pharmacological therapy 19 revealed a significant association of both the integrin and cAMP pathways with the functional recovery in cardiac contractility and metabolism. Nevertheless, some studies have failed to demonstrate a normalized gene expression during mechanical unloading 20. Margulies et al. analyzed 199 human myocardial specimens and found that the abundant changes in transcripts during HF did not respond to LVAD support.
Intracellular Transduction Pathways
Several signal transduction pathways implicated in the progression of HF and LVAD-induced recovery are summarized in Table 1 5-7, 17. One of the first signaling pathways that provided insight as an underlying mechanism of HF is the Ca2+/calmodulin-activated protein kinase and phosphatase (calcineurin) 5. Another prominent signaling pathway is the phosphoinositide 3-kinase (PI3K)/Akt pathway, which stimulates several tyrosine kinase receptors such as insulin-like growth factor (IGF), fibroblast growth factor, transforming growth factor, as well as G-protein coupled receptors. Lastly, the Mitogen-activated protein (MAP) kinase cascade, which includes extracellular signal-related kinases (Erk), c-Jun N-terminal protein kinases (JNK), and p38 MAPK subfamilies is a highly conserved signal transduction pathway 5. Moreover, receptor tyrosine kinases are involved in the transmission of hypertrophic and survival signals in the cardiomyocyte.
LVAD induces significant perturbations of these key signal transduction pathways 21. For example, the activation of Akt, GSK-3β and phosphorylation of P70S6K (as part of a pro-hypertrophy signaling pathway) in patients with HF are downregulated after LVAD, while both JNK and p38 mediated signaling remained unchanged by LVAD. Erk is downregulated by mechanical unloading, while JNK signal transduction exhibited no change 5-7, 17. Lastly, unloading of the heart resulted in an upregulation of Her2/neu and Her4, particularly in patients with ischemic cardiomyopathy. At the same time, glycoprotein 130, the common signal transducer of interleukin-6 cytokines 22, 23, which plays an important role in receptor tyrosine kinase signal transduction, was decreased following LVAD implantation.
In conclusion, a comprehensive profile of the pivotal elements in signal transduction pathways (and their posttranslational modifications) in maladaptive hypertrophy and HF progression are only beginning to be discovered, and future investigations in this area will likely unveil many potential therapeutic targets.
Organelle Specific Insights in Heart Failure and Reverse Remodeling
Mitochondria in Heart Failure and Reverse Remodeling
Many reports from both animal and human clinical studies strongly indicate that mitochondrial dysfunction is crucial in the pathophysiology of HF 24. Next to supplying energy in the form of ATP, mitochondria are also involved in the regulation of intracellular Ca2+ fluxes, redox potential, biosynthesis, reactive oxygen species (ROS) generation/signaling, cell death pathways, and protection against stressors such as ischemia reperfusion injury 25. Mitochondrial energy supply involves the coupling of electron transfer and oxygen consumption through the mitochondrial electron transport chain (ETC) complexes I, II, III, and IV with the phosphorylation of ADP to ATP by F0F1-ATP synthase, also known as complex V. In the process of oxidative phosphorylation, the coupling efficiency between respiration and phosphorylation is often measured as the respiratory control index (RCI). In isolated mitochondria, the active respiring state is often referred to as “state 3” respiration, while the slowest rate when all the ADP has been phosphorylated to ATP is referred to as “state 4”. The RCI can be determined as a ratio of state 3/state 4. A lower RCI indicates a disturbed coupling of oxidation and phosphorylation, causing an inefficient energy production during which oxygen is prematurely reduced as ROS. Under basal conditions, the catabolism of fatty acids through β-oxidation provides approximately 90% of the total ATP used in the heart. In addition to fatty acids, other substrates used for oxidative phosphorylation include glucose, pyruvate, lactate and ketone bodies 24. Thus, several mitochondrial energy pathways including oxidative phoshorylation, the TCA cycle, and fatty acid oxidation are essential for maintaining the contractile function of the heart 24. Both basic research and clinical studies have reported a shift towards glucose oxidation at the expense of fatty acid oxidation in hypertrophied and failing hearts 24. Consequently, ATP levels in failing hearts rapidly decline, leading to cardiac energy deficits. Concomitantly, studies involving transgenic alterations in mitochondrial proteins (e.g., ANT and respiratory complex enzymes) during the progression of HF indicate a crucial role for mitochondria 24.
Supranormal mitochondrial ROS production is implicated in many pathological conditions such as contractile dysfunction, calcium dysregulation, cell death and ventricular hypertrophy and dilation. ROS, including superoxide, hydroxyl radicals, and hydrogen peroxide, are normal byproducts during mitochondrial metabolism. The majority of reports describing pathological ROS release in HF have come from studies conducted in animal models. In agreement, it has been demonstrated that ROS levels are elevated in the failing human heart 26. Contractile dysfunction can result from disturbed mitochondrial oxidative phosphorylation with inefficient energy production, where oxygen is prematurely and incompletely reduced, causing increased ROS release from ETC complex I and III. During HF, elevated ROS release by itself can further induce more ROS release 27. The release of ROS from the mitochondria can lead to extensive oxidative damage to a variety of intracellular molecules such as proteins (e.g., mitochondrial respiratory enzymes, matrix enzymes), DNA, and specific lipids (e.g., membrane phospholipids such as cardiolipin) 24. Furthermore, ROS also modulate multiple overlapping signaling pathways in the progression of ventricular hypertrophy. It is evident that an excessive generation of ROS serves as an important mechanism in the pathophysiology and progression of HF.
More recently, it has become apparent that cell death by apoptosis and necrosis are both important contributors in HF 5. Multiple studies have demonstrated the significant importance and magnitude of cardiac apoptosis during HF. The intrinsic apoptotic pathway features profound interactions between mitochondria, nucleus, and other subcellular organelles. The release of specific mitochondrial proteins from the intermembrane space (e.g., cytochrome-c, endonuclease G, apoptosis inducing factor, and Smac) triggers these events that subsequently cause DNA fragmentation and the activation of Apaf-1 and caspases. This chain of events ultimately culminates in cell death. In contrast to apoptosis, the exact contribution of necrosis in HF has not been extensively studied. However, evidence has shown that HF could be rescued by deletion of the pro-necrotic factor cyclophilin D, but not by the anti-apoptotic factor Bcl-2, implicating that necrosis is also a major component in the progression of HF. Consequently, the mitochondrial release of these pro-cell-death factors also involves the mitochondrial permeability transition (MPT) pore 25. MPT is related to several detrimental mitochondrial events such as mitochondrial membrane potential (ΔΨ) deterioration, ROS overproduction, Ca2+ overload, and impaired nitric oxide signaling 25. Accordingly, MPT pore opening has previously been reported in HF resulting from Ca2+-induced cardiomyopathy 28 and diabetic cardiomyopathy 29.
Surprisingly, there are relatively few studies available that investigate the role of mitochondrial function in reverse ventricular remodeling by LVAD support. An early study by Lee et al. investigated mitochondrial respiratory function in advanced HF patients before and after LVAD support 30. Progression of HF was associated with extremely low respiratory control indexes (RCI). However, the RCIs were significantly improved after LVAD support 30, suggesting an important role of the device in benefiting oxidative phosphorylation and electron transport. Another study by Mital et al. examined the effects of NO on mitochondrial respiratory control during reverse ventricular remodeling, a coupling that is usually disrupted during HF progression 31. The study reported that chronic LVAD support potentiates endogenous NO-mediated regulation of mitochondrial respiration as measured by improved MVO2 consumption. This effect could in turn be abrogated by NO synthase inhibition 31. As aforementioned, Heerdt et al. observed that LVAD supported hearts exhibited a normalization of cardiolipin content within the inner mitochondrial membrane 32. Cardiolipin is essential for normal functionality of the mitochondrial respiratory chain, as well as substrate transport 33. In conclusion, it is evident that the pathological proteomic mitochondrial phenotype is reversible, which may be an important underlying mechanism in reverse ventricular remodeling by LVAD.
The Proteasome in Heart Failure and Reverse Remodeling
The proteasome is usually referred to as the 26S complex, which consists of a 20S catalytic core particle, and one or two 19S regulatory particles 34. The main function of the 26S proteasome is to degrade proteins that are damaged or have reached the end of their functional lifetime 35. As a part of the ubiquitin-proteasome system (UPS), the proteasome maintains cardiac protein homeostasis and thus plays a significant role in left ventricular remodeling and HF 35, 36, 37.
Over the years, clinical studies have observed detrimental side effects in the heart when proteasome inhibition was employed for the treatment of cancer. For example, a large clinical trial of 315 patients using the proteasome inhibitor Bortezomib to treat multiple myeloma revealed significant cardiotoxicity and occurrence of HF 38. During ventricular remodeling and HF, ubiquitinated proteins accumulated, suggesting impaired protein degradation capacity of the UPS system 39. Further investigation on human pressure-overloaded hearts also revealed a correlation between ventricular hypertrophy and a depressed proteasomal activity 36. In a rodent model, our group recently found that ventricular hypertrophy by prolonged β-adrenergic stimulation is concomitant with decreased 20S caspase-like and trypsin-like activities, with an unchanged chymotrypsin-like activity. This functional alteration may be attributed to an increased incorporation of inducible subunits in 20S proteasomes 40. Besides removal of damaged proteins, proteasomes also play critical regulatory roles by degrading pivotal components of biological pathways, such as pro-hypertrophic signals (e.g., Akt and Erk1/2) 41. Inhibition of the proteasome pathway in sympathetically-stimulated mice resulted in the prevention 42 or regression of maladaptive ventricular hypertrophy 43, 44.
Although there is increasing evidence that the UPS plays a significant role in ventricular remodeling and HF, there are still relatively few studies describing if and how the UPS is affected by ventricular unloading after LVAD implantation. Kassiotis et al. 45 reported that the progression of autophagy in the failing heart is reversed following LVAD support. In this study, the investigators further observed that 20S proteasome activity was increased by mechanical unloading 45. Subsequently, Wohlschlaeger et al. 46 reported a depression of the UPS during HF, which was reversed after LVAD therapy 46. Both studies strongly suggest a significant role for the UPS in ventricular hypertrophy and HF, as well as in reverse cardiac remodeling. Although the UPS plays a crucial role in the pathophysiology of maladaptive ventricular remodeling and HF, further studies are necessary to define its causative 42 or compensatory 38 role. LVAD therapy offers a platform to increase our understanding of the UPS in maladaptive remodeling and HF.
Future Perspectives and Directions
Standard HF therapy has shifted from traditional heart transplantation to LVAD implantation with a focus on DT and BTR. Nevertheless, clinical parameters to predict LV recovery after LVAD support are still ill-defined. Hence, we believe that the development of novel patient selection criteria requires that HF is targeted by a systems biology approach incorporating transcriptomics, proteomics, metabolomics, cell biology and bioinformatics (Figure 3). A plausible initial approach would be the exploration of molecular or organellar assays in combination with pre-existing clinical parameters as predictive measures. Collection of blood and ventricular tissue samples during LVAD implantation are suitable means for accomplishing this (Figure 3). In addition to molecular and cellular alterations (Table 1), there is growing evidence that mitochondria and the UPS have important roles in LV remodeling, HF progression, and reverse cardiac remodeling. A plausible initial method for integrating organellar-based profiles would be to implement mitochondrial functional assays (e.g., mitochondrial ΔΨ, susceptibility to MPT, RCI and ETC activities) 47 and proteasome activity assays 47 into the current panel of clinical parameters. In line with mitochondrial function, metabolism is also an important determinant of HF. Accordingly, we propose a comprehensive comparison of essential metabolites using blood samples 48 before and after LVAD implantation. These “Omics” data can be further interrogated using state-of-the-art bioinformatics tools to provide insights into disease 49. This bi-directional translational medicine approach will enable us to study HF and identify patients that would benefit from BTR, DT, or BTT. This molecular and organelle based predictive paradigm may advance personalized medicine through individual patient profiling.
Figure 3. Translational heart failure research.

A bi-directional bench-to-bedside approach combining a systems biology strategy with clinical patient management offers new possibilities for the development of novel therapeutics and clinical parameters.
Acknowledgments
Sources of Funding: This work and the authors are supported by the NHLBI Proteomics Center Award (HHSN268201000035C) to Dr. Ping as well as by the NIH MPI R01 Award (HL01228) to Dr. Ping and Dr. Weiss.
Non-standard Abbreviations and Acronyms
- ANP
Atrial natriuretic peptide
- BNP
Brain natriuretic peptide
- BTR
Bridge to recovery
- BTT
Bridge to transplantation
- DT
Destination therapy
- Erk
Extracellular signal-related kinases
- HF
Heart failure
- IGF
Insulin-like growth factor
- JNK
c-jun N-terminal protein kinases
- LV
Left ventricular
- LVAD
Left ventricular assist device
- LVEDD
Left ventricular end diastolic dimension
- MAP
Mitogen-activated protein
- MCSD
Mechanical circulatory support devices
- MPT
Mitochondrial permeability transition
- NICM
Non-ischemic cardiomyopathy
- OHT
Open heart transplantation
- RCI
Respiratory control index
- ROS
Reactive oxygen species
- UPS
Ubiquitin-proteasome system
Footnotes
Disclosures: None.
References
- 1.Frazier OH, Myers TJ. Left ventricular assist system as a bridge to myocardial recovery. Ann Thorac Surg. 1999;68:734–741. doi: 10.1016/s0003-4975(99)00801-2. [DOI] [PubMed] [Google Scholar]
- 2.Pamboukian SV, Tallaj JA, Brown RN, Holman WL, Blood M, George JF, Costanzo MR, Cadeiras M, Smallfield MC, McGiffin DC, Kirklin JK. Improvement in 2-year survival for ventricular assist device patients after implementation of an intensive surveillance protocol. J Heart Lung Transplant. 2011;30:879–887. doi: 10.1016/j.healun.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Kirklin JK, Naftel DC, Kormos RL, Stevenson LW, Pagani FD, Miller MA, Timothy Baldwin J, Young JB. Fifth intermacs annual report: Risk factor analysis from more than 6,000 mechanical circulatory support patients. J Heart Lung Transplant. 2013;32:141–156. doi: 10.1016/j.healun.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Rosenthal D, Bernstein D. Pediatric mechanical circulatory support: Challenges and opportunities. Circulation. 2006;113:2266–2268. doi: 10.1161/CIRCULATIONAHA.106.623488. [DOI] [PubMed] [Google Scholar]
- 5.Drakos SG, Kfoury AG, Selzman CH, Verma DR, Nanas JN, Li DY, Stehlik J. Left ventricular assist device unloading effects on myocardial structure and function: Current status of the field and call for action. Curr Opin Cardiol. 2011;26:245–255. doi: 10.1097/HCO.0b013e328345af13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klotz S, Jan Danser AH, Burkhoff D. Impact of left ventricular assist device (lvad) support on the cardiac reverse remodeling process. Prog Biophys Mol Biol. 2008;97:479–496. doi: 10.1016/j.pbiomolbio.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Mann DL, Barger PM, Burkhoff D. Myocardial recovery and the failing heart: Myth, magic, or molecular target? J Am Coll Cardiol. 2012;60:2465–2472. doi: 10.1016/j.jacc.2012.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frazier OH. Left ventricular assist device as a bridge to partial left ventriculectomy. Eur J Cardiothorac Surg. 1999;15(Suppl 1):S20–25. discussion S39-43. [PubMed] [Google Scholar]
- 9.Bruckner BA, Stetson SJ, Perez-Verdia A, Youker KA, Radovancevic B, Connelly JH, Koerner MM, Entman ME, Frazier OH, Noon GP, Torre-Amione G. Regression of fibrosis and hypertrophy in failing myocardium following mechanical circulatory support. J Heart Lung Transplant. 2001;20:457–464. doi: 10.1016/s1053-2498(00)00321-1. [DOI] [PubMed] [Google Scholar]
- 10.Birks EJ, George RS, Hedger M, Bahrami T, Wilton P, Bowles CT, Webb C, Bougard R, Amrani M, Yacoub MH, Dreyfus G, Khaghani A. Reversal of severe heart failure with a continuous-flow left ventricular assist device and pharmacological therapy: A prospective study. Circulation. 2011;123:381–390. doi: 10.1161/CIRCULATIONAHA.109.933960. [DOI] [PubMed] [Google Scholar]
- 11.Birks EJ, Tansley PD, Hardy J, George RS, Bowles CT, Burke M, Banner NR, Khaghani A, Yacoub MH. Left ventricular assist device and drug therapy for the reversal of heart failure. N Engl J Med. 2006;355:1873–1884. doi: 10.1056/NEJMoa053063. [DOI] [PubMed] [Google Scholar]
- 12.Dandel M, Weng Y, Siniawski H, Stepanenko A, Krabatsch T, Potapov E, Lehmkuhl HB, Knosalla C, Hetzer R. Heart failure reversal by ventricular unloading in patients with chronic cardiomyopathy: Criteria for weaning from ventricular assist devices. Eur Heart J. 2011;32:1148–1160. doi: 10.1093/eurheartj/ehq353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldstein DJ, Maybaum S, MacGillivray TE, Moore SA, Bogaev R, Farrar DJ, Frazier OH HeartMate IICI. Young patients with nonischemic cardiomyopathy have higher likelihood of left ventricular recovery during left ventricular assist device support. J Card Fail. 2012;18:392–395. doi: 10.1016/j.cardfail.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 14.Muller J, Wallukat G, Weng YG, Dandel M, Spiegelsberger S, Semrau S, Brandes K, Theodoridis V, Loebe M, Meyer R, Hetzer R. Weaning from mechanical cardiac support in patients with idiopathic dilated cardiomyopathy. Circulation. 1997;96:542–549. doi: 10.1161/01.cir.96.2.542. [DOI] [PubMed] [Google Scholar]
- 15.Deng MC, Young JB, Stevenson LW, Oz MC, Rose EA, Hunt SA, Kirklin JK, Kobashigawa J, Miller L, Saltzberg M, Konstam M, Portner PM, Kormos R Board of Directors of the International Society for H, Lung T. Destination mechanical circulatory support: Proposal for clinical standards. J Heart Lung Transplant. 2003;22:365–369. doi: 10.1016/s1053-2498(03)00073-1. [DOI] [PubMed] [Google Scholar]
- 16.von Bayern MP, Cadeiras M, Deng MC. Destination therapy: Does progress depend on left ventricular assist device development? Heart Fail Clin. 2007;3:349–367. doi: 10.1016/j.hfc.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 17.Ambardekar AV, Buttrick PM. Reverse remodeling with left ventricular assist devices: A review of clinical, cellular, and molecular effects. Circ Heart Fail. 2011;4:224–233. doi: 10.1161/CIRCHEARTFAILURE.110.959684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heerdt PM, Holmes JW, Cai B, Barbone A, Madigan JD, Reiken S, Lee DL, Oz MC, Marks AR, Burkhoff D. Chronic unloading by left ventricular assist device reverses contractile dysfunction and alters gene expression in end-stage heart failure. Circulation. 2000;102:2713–2719. doi: 10.1161/01.cir.102.22.2713. [DOI] [PubMed] [Google Scholar]
- 19.Hall JL, Birks EJ, Grindle S, Cullen ME, Barton PJ, Rider JE, Lee S, Harwalker S, Mariash A, Adhikari N, Charles NJ, Felkin LE, Polster S, George RS, Miller LW, Yacoub MH. Molecular signature of recovery following combination left ventricular assist device (lvad) support and pharmacologic therapy. Eur Heart J. 2007;28:613–627. doi: 10.1093/eurheartj/ehl365. [DOI] [PubMed] [Google Scholar]
- 20.Margulies KB, Matiwala S, Cornejo C, Olsen H, Craven WA, Bednarik D. Mixed messages: Transcription patterns in failing and recovering human myocardium. Circulation research. 2005;96:592–599. doi: 10.1161/01.RES.0000159390.03503.c3. [DOI] [PubMed] [Google Scholar]
- 21.Drakos SG, Terrovitis JV, Anastasiou-Nana MI, Nanas JN. Reverse remodeling during long-term mechanical unloading of the left ventricle. Journal of molecular and cellular cardiology. 2007;43:231–242. doi: 10.1016/j.yjmcc.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 22.Deng MC, Erren M, Tjan TD, Tamminga N, Werntze B, Zimmermann P, Weyand M, Hammel D, Schmidt C, Scheld HH. Left ventricular assist system support is associated with persistent inflammation and temporary immunosuppression. Thorac Cardiovasc Surg. 1999;47(Suppl 2):326–331. doi: 10.1055/s-2007-1013192. [DOI] [PubMed] [Google Scholar]
- 23.Plenz G, Baba HA, Erren M, Scheld HH, Deng MC. Reversal of myocardial interleukin-6-mrna expression following long-term left ventricular assist device support for myocarditis-associated low output syndrome. J Heart Lung Transplant. 1999;18:923–924. doi: 10.1016/s1053-2498(99)00054-6. [DOI] [PubMed] [Google Scholar]
- 24.Marin-Garcia J, Goldenthal MJ. Mitochondrial centrality in heart failure. Heart Fail Rev. 2008;13:137–150. doi: 10.1007/s10741-007-9079-1. [DOI] [PubMed] [Google Scholar]
- 25.Korge P, Ping P, Weiss JN. Reactive oxygen species production in energized cardiac mitochondria during hypoxia/reoxygenation: Modulation by nitric oxide. Circulation research. 2008;103:873–880. doi: 10.1161/CIRCRESAHA.108.180869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, Shah AM. Increased myocardial nadph oxidase activity in human heart failure. J Am Coll Cardiol. 2003;41:2164–2171. doi: 10.1016/s0735-1097(03)00471-6. [DOI] [PubMed] [Google Scholar]
- 27.Dai DF, Chen T, Szeto H, Nieves-Cintron M, Kutyavin V, Santana LF, Rabinovitch PS. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol. 2011;58:73–82. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BH, Hewett TE, Robbins J, Houser SR, Molkentin JD. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–2444. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oliveira PJ, Seica R, Coxito PM, Rolo AP, Palmeira CM, Santos MS, Moreno AJ. Enhanced permeability transition explains the reduced calcium uptake in cardiac mitochondria from streptozotocin-induced diabetic rats. FEBS Lett. 2003;554:511–514. doi: 10.1016/s0014-5793(03)01233-x. [DOI] [PubMed] [Google Scholar]
- 30.Lee SH, Doliba N, Osbakken M, Oz M, Mancini D. Improvement of myocardial mitochondrial function after hemodynamic support with left ventricular assist devices in patients with heart failure. The Journal of thoracic and cardiovascular surgery. 1998;116:344–349. doi: 10.1016/s0022-5223(98)70136-9. [DOI] [PubMed] [Google Scholar]
- 31.Mital S, Loke KE, Addonizio LJ, Oz MC, Hintze TH. Left ventricular assist device implantation augments nitric oxide dependent control of mitochondrial respiration in failing human hearts. J Am Coll Cardiol. 2000;36:1897–1902. doi: 10.1016/s0735-1097(00)00948-7. [DOI] [PubMed] [Google Scholar]
- 32.Heerdt PM, Schlame M, Jehle R, Barbone A, Burkhoff D, Blanck TJ. Disease-specific remodeling of cardiac mitochondria after a left ventricular assist device. Ann Thorac Surg. 2002;73:1216–1221. doi: 10.1016/s0003-4975(01)03621-9. [DOI] [PubMed] [Google Scholar]
- 33.Ohtsuka T, Nishijima M, Suzuki K, Akamatsu Y. Mitochondrial dysfunction of a cultured chinese hamster ovary cell mutant deficient in cardiolipin. J Biol Chem. 1993;268:22914–22919. [PubMed] [Google Scholar]
- 34.Gomes AV, Zong C, Edmondson RD, Berhane BT, Wang GW, Le S, Young G, Zhang J, Vondriska TM, Whitelegge JP, Jones RC, Joshua IG, Thyparambil S, Pantaleon D, Qiao J, Loo J, Ping P. The murine cardiac 26s proteasome: An organelle awaiting exploration. Ann N Y Acad Sci. 2005;1047:197–207. doi: 10.1196/annals.1341.018. [DOI] [PubMed] [Google Scholar]
- 35.Powell SR. The ubiquitin-proteasome system in cardiac physiology and pathology. American journal of physiology Heart and circulatory physiology. 2006;291:H1–H19. doi: 10.1152/ajpheart.00062.2006. [DOI] [PubMed] [Google Scholar]
- 36.Hein S, Arnon E, Kostin S, Schonburg M, Elsasser A, Polyakova V, Bauer EP, Klovekorn WP, Schaper J. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: Structural deterioration and compensatory mechanisms. Circulation. 2003;107:984–991. doi: 10.1161/01.cir.0000051865.66123.b7. [DOI] [PubMed] [Google Scholar]
- 37.Patterson C, Ike C, Willis PWt, Stouffer GA, Willis MS. The bitter end: The ubiquitin-proteasome system and cardiac dysfunction. Circulation. 2007;115:1456–1463. doi: 10.1161/CIRCULATIONAHA.106.649863. [DOI] [PubMed] [Google Scholar]
- 38.Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, San-Miguel JF, Blade J, Boccadoro M, Cavenagh J, Dalton WS, Boral AL, Esseltine DL, Porter JB, Schenkein D, Anderson KC Assessment of Proteasome Inhibition for Extending Remissions I. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352:2487–2498. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- 39.Weekes J, Morrison K, Mullen A, Wait R, Barton P, Dunn MJ. Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics. 2003;3:208–216. doi: 10.1002/pmic.200390029. [DOI] [PubMed] [Google Scholar]
- 40.Drews O, Tsukamoto O, Liem D, Streicher J, Wang Y, Ping P. Differential regulation of proteasome function in isoproterenol-induced cardiac hypertrophy. Circulation research. 2010;107:1094–1101. doi: 10.1161/CIRCRESAHA.110.222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Predmore JM, Wang P, Davis F, Bartolone S, Westfall MV, Dyke DB, Pagani F, Powell SR, Day SM. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation. 2010;121:997–1004. doi: 10.1161/CIRCULATIONAHA.109.904557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Depre C, Wang Q, Yan L, Hedhli N, Peter P, Chen L, Hong C, Hittinger L, Ghaleh B, Sadoshima J, Vatner DE, Vatner SF, Madura K. Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation. 2006;114:1821–1828. doi: 10.1161/CIRCULATIONAHA.106.637827. [DOI] [PubMed] [Google Scholar]
- 43.Stansfield WE, Tang RH, Moss NC, Baldwin AS, Willis MS, Selzman CH. Proteasome inhibition promotes regression of left ventricular hypertrophy. American journal of physiology Heart and circulatory physiology. 2008;294:H645–650. doi: 10.1152/ajpheart.00196.2007. [DOI] [PubMed] [Google Scholar]
- 44.Hedhli N, Lizano P, Hong C, Fritzky LF, Dhar SK, Liu H, Tian Y, Gao S, Madura K, Vatner SF, Depre C. Proteasome inhibition decreases cardiac remodeling after initiation of pressure overload. American journal of physiology Heart and circulatory physiology. 2008;295:H1385–1393. doi: 10.1152/ajpheart.00532.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kassiotis C, Ballal K, Wellnitz K, Vela D, Gong M, Salazar R, Frazier OH, Taegtmeyer H. Markers of autophagy are downregulated in failing human heart after mechanical unloading. Circulation. 2009;120:S191–197. doi: 10.1161/CIRCULATIONAHA.108.842252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wohlschlaeger J, Sixt SU, Stoeppler T, Schmitz KJ, Levkau B, Tsagakis K, Vahlhaus C, Schmid C, Peters J, Schmid KW, Milting H, Baba HA. Ventricular unloading is associated with increased 20s proteasome protein expression in the myocardium. J Heart Lung Transplant. 2010;29:125–132. doi: 10.1016/j.healun.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 47.Lau E, Wang D, Zhang J, Yu H, Lam MP, Liang X, Zong N, Kim TY, Ping P. Substrate- and isoform-specific proteome stability in normal and stressed cardiac mitochondria. Circulation research. 2012;110:1174–1178. doi: 10.1161/CIRCRESAHA.112.268359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan CX, Khan AA, Choi JH, Ng CD, Cadeiras M, Deng M, Ping P. Technology platform development for targeted plasma metabolites in human heart failure. Clin Proteomics. 2013;10:7. doi: 10.1186/1559-0275-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zong N, Li H, Li H, Lam MP, Jimenez RC, Kim CS, Deng N, Kim AK, Choi JH, Zelaya I, Liem DA, Meyer D, Odeberg J, Fang C, Lu HJ, Xu T, Weiss JN, Duan H, Uhlen M, Yates J, 3rd, Apweiler R, Ge J, Hermjakob H, Ping P. Integration of cardiac proteome biology and medicine by a specialized knowledgebase. Circ Res. 2013;113:1043–53. doi: 10.1161/CIRCRESAHA.113.301151. [DOI] [PMC free article] [PubMed] [Google Scholar]