

Abstract
About one third of the world’s population carries Staphylococcus aureus. The recent emergence of extreme drug resistant strains that are resistant to the “antibiotic of last resort”, vancomycin, has caused a further increase in the pressing need to discover new drugs against this organism. The S. aureus enoyl reductase, saFabI, is a validated target for drug discovery. In order to drive the development of potent and selective saFabI inhibitors, we have studied the mechanism of the enzyme and analyzed the interaction of saFabI with triclosan and two related diphenyl ether inhibitors. Results from kinetic assays reveal that saFabI is NADPH-dependent, and prefers acyl carrier protein substrates carrying fatty acids with a long acyl chains. Based on product inhibition studies, we propose that the reaction proceeds via an ordered sequential ternary complex, with the ACP substrate binding first, followed by NADPH. The interaction of NADPH with the enzyme has been further explored by site-directed mutagenesis, and residues R40 and K41 have been shown to be involved in determining the specificity of enzyme for NADPH compared to NADH. Finally, in preliminary inhibition studies, we have shown that triclosan, 5-ethyl-2-phenoxyphenol (EPP) and 5-chloro-2-phenoxyphenol (CPP) are all nanomolar slow-onset inhibitors of saFabI. These compounds inhibit the growth of S. aureus with MIC values of 0.03-0.06 µg/mL. Upon selection for resistance, three novel safabI mutations, A95V, I193S and F204S, were identified. Strains containing these mutations had MIC values ~100 fold larger than the wild-type strain whereas the purified mutant enzymes had Ki values 5 to 3000 fold larger than the wild-type saFabI. The increase in both MIC and Ki values caused by the mutations supports the proposal that saFabI is the intracellular target for the diphenyl ether-based inhibitors.
Staphylococcus aureus is one of the most common causative agents of hospital-acquired infections (1, 2). It is estimated that 30% of healthy people carry S. aureus, usually in the anterior nares, providing a ready reservoir for future infection (3). S. aureus is able to acquire resistance to antibiotics rapidly, and penicillin-resistant strains developed after only a few months of clinical trials (4). After methicillin was introduced to treat penicillin-resistant S. aureus in 1960, methicillin-resistant S. aureus (MRSA) was isolated within one year (5). The percentage of MRSA has been increasing continuously in the last few decades. In the USA, MRSA increased from 2.4% in 1975 (6) to 35% in 1996 (7). Currently, glycopeptide antibiotics, such as vancomycin and teicoplanin, are considered to be the only antibiotics for treating MRSA infections. However, the recent emergence of clinical isolates of vancomycin-resistant S. aureus (VRSA) (8, 9) reaffirm the pressing need to continuously discover new antibiotics against this bacterium.
Fatty acid biosynthesis is divided into two types, FASI and FASII, based on whether the reactions are performed by a single polypeptide or by individual enzymes. The FASI pathway usually exists in vertebrates and certain bacteria, whereas the FASII pathway, as shown in Scheme 1, is usually found in plants and bacteria. The inhibition of the FASII pathway in bacteria causes the breakdown of the cell wall and the disruption of the cell membrane (10, 11), demonstrating its importance for bacterial survival. A number of studies suggest that the enoyl reductase (FabI), which catalyzes the final and rate-limiting step in each cycle, is a regulator of the FASII pathway, and is essential for the viability of bacteria (12). Due to its necessity and low sequence homology to the mammalian FASI reductase, FabI is an attractive target for novel antibiotic discovery. Isoniazid, which has been used in tuberculosis (TB) chemotherapy for decades, is now known to target InhA, the FabI homologue in Mycobacterium tuberculosis (13). A number of inhibitors against FabI have been reported recently (14-19). In the present study we have examined the mechanism of saFabI, as a prelude to the development of potent inhibitors of this enzyme.
Although kinetic studies on saFabI have been conducted previously, a detailed mechanistic analysis of the enzyme has so far not been reported. In addition, existing studies are based on N-acetylcysteamine (NAC) or CoA based-substrates, rather than the natural acyl carrier protein (ACP) substrates (20, 21). In the present work we have compared the kinetic properties of NAC, CoA and ACP-based substrates with saFabI, explored the molecular basis for cofactor specificity, and also studied the mechanism of the saFabI-catalyzed reaction. In addition, we have also analyzed the inhibition of saFabI by triclosan and two related diphenyl ethers, 5-ethyl-2-phenoxyphenol (EPP) and 5-chloro-2-phenoxyphenol (CPP) (Scheme 2). Triclosan is a broad-spectrum antimicrobial that is present in a wide variety of consumer products, such as toothpaste, mouthwashes and hand soaps (22). It has been reported that triclosan can inhibit the growth of S. aureus (23), and this compound is recommended as a method of controlling MRSA in hospitals (24, 25). In order to extend previous studies concerning the mode of action of triclosan (20, 21, 26, 27), we performed selection experiments and identified several saFabI mutations that correlated with an increase in resistance to the three inhibitors. Analysis of the impact of these mutations on cell growth and enzyme inhibition strongly suggests that the diphenyl ether based inhibitors target saFabI within the bacterium.
MATERIALS AND METHODS
Materials
trans-2-Dodecenoic acid was purchased from TCI. His-bind Ni2+-NTA resin was obtained from Invitrogen, and centriplus units were from Millipore. QuikChange site-directed mutagenesis kit was obtained from Stratagene. Triclosan was a gift from Ciba whereas 5-ethyl-2-phenoxyphenol (EPP) and 5-chloro-2-phenoxyphenol (CPP) were available from a previous study (28, 29). All other chemical reagents were purchased from Sigma-Aldrich.
Synthesis of trans-2-dodecenoyl CoA and trans-2-dodecenoyl N-acetylcysteamine
trans-2-Dodecenoyl CoA (DDCoA) was synthesized by using the mixed anhydride method as described previously (30). This method was also used to synthesize trans-2-dodecenoyl N-acetylcysteamine (DDNAC) with minor modifications. Briefly, 4 mmol N-acetylcysteamine was dissolved in 10 mL anhydrous THF, and then 4 mmol of the mixed anhydride was added. After stirring at room temperature for 2 hours, DDNAC was purified by silica gel chromatography using 1:1 ethyl acetate:hexane as the eluant. The identity of the product was confirmed by NMR spectroscopy and ESI mass spectrometry. 1H NMR: δ 0.880 (3H, t), 1.265 (12H, m), 1.964 (3H, s), 2.165-2.237 (2H, q), 3.090 (2H, t), 3.435-3.496 (2H, q), 6.094-6.156 (1H, m), 6.886-6.984 (1H, m). Mass: m/z 300.2 (M+H)+.
Expression and Purification of saFabI
The fabI gene from the methicillin-sensitive S. aureus strain NCTC 8325 was amplified using the KOD DNA polymerase (Novagen) and the primers listed in Table S1. The 0.77 kb PCR product was digested with NdeI and BamHI, and cloned into the same restriction sites in pET-16b. The sequence of the construct was confirmed by DNA sequencing (Sequetech) and the plasmid was then transformed into Escherichia coli strain BL21(DE3)plysS. A single colony was used to inoculate 10 mL of LB medium containing 50 µg/mL ampicillin (LB/AMP) and the culture was grown at 37 ºC overnight. The cells in the starter culture were collected by centrifugation, resuspended in fresh media, transferred to 500 mL LB/Amp medium, and grown at 37 ºC until an optical density of 0.8 at 600 nm had been reached. Subsequently, 0.5 mM IPTG was added to induce the expression of saFabI and the culture was shaken overnight at 25 ºC. The cells were harvested, and resuspended in 30 mL His binding buffer (20 mM Tris HCl, 500 mM NaCl, and 5 mM imidazole, pH 7.9). After cell disruption by three passages through a French press cell (1000 psi), the cell lysate was centrifuged at 120,000 x g for 1 h, and the supernatant was loaded onto a 5 mL Ni2+-NTA His-bind column, which had been preincubated with 20 mL binding buffer. The His-bind column was washed with 40 mL binding buffer, followed by 30 mL of wash buffer (20 mM Tris HCl, 500 mM NaCl, and 60 mM imidazole, pH 7.9), and saFabI was then eluted using 30 mL of elute buffer (20 mM Tris HCl, 500 mM NaCl, and 500 mM imidazole, pH 7.9). SDS-PAGE was used to identify those fractions containing saFabI, and these fractions were combined and subjected to size exclusion chromatography on a G25 column using 30 mM PIPES, 150 mM NaCl, and 100 mM EDTA, pH 7.8 buffer as the mobile phase. Fractions containing saFabI were pooled and concentrated using a centriplus 30.
Site-directed mutagenesis, expression and purification of FabI mutants
Site-directed mutagenesis was performed using the QuikChange mutagenesis kit from Stratagene using the primers listed in Table S1. The sequence of each mutant plasmid was confirmed by ABI DNA sequencing, and the expression and purification of each saFabI mutant followed the same protocol as that described above for the wild type saFabI protein.
Cloning, expression and purification of saACP
The S. aureus acp gene was PCR amplified from NCTC 8325 genomic DNA using the primers listed in Table S1, digested with NdeI and XhoI, then ligated into the pET23b expression vector so that the coding region of the gene was in-frame with a C-terminal His tag. After DNA sequencing, saACP was expressed and purified using the procedure described above for saFabI. After purification, saACP was dialyzed into 75 mM Tris-HCl, 10 mM MgCl2, pH 7.5 and the protein concentration determined using the Bradford method (31).
Enzymatic preparation of crotonyl saACP and dodecenoyl saACP
Conformationally sensitive SDS PAGE and ESI-MS revealed that saACP expressed and purified from E. coli was obtained primarily in the apo form. This protein was converted to the required acyl-saACP using sfp, a phosphopantetheinyl transferase from Bacillus subtilis (32). Briefly, 25 mg/L apo-saACP was incubated with 80 μM crotonyl CoA or dodecenoyl CoA in the presence of 1 μM sfp for 2 h at 37 ºC. The reaction mixture was then loaded onto a Q-sepharose column, and eluted with a gradient consisting of buffer A (20 mM Bis-Tris, pH 7.0) and buffer B (20 mM Bis-Tris, 800 mM NaCl, pH 7.0). Fractions were analyzed by SDS-PAGE and those containing crotonyl-saACP or DDsaACP were pooled and dialyzed into 20 mM Tris-HCl, pH 7.0 buffer. The final products were characterized by ESI mass spectrometry.
Steady-state kinetic assays
Steady-state kinetics were performed at 25 °C in 100 mM Na2HPO4 buffer, pH 7.8. Initial velocities were measured after addition of 50 nM saFabI to give a final assay volume of 500 μL.
For DDsaACP, the reactions were carried out by varying the concentration of DDsaACP at several fixed concentrations of NADPH (50, 100 and 250 μM) or by varying the NADPH concentration at several fixed concentrations of DDsaACP (4.4, 8.2 and 15.0 μM). Kinetic parameters were obtained by fitting the data to equation 1 for a sequential mechanism with GraFit 4.0.
| (1) |
Kma and Kmb, are the Km values of DDsaACP and NADPH, respectively, whereas Kia is the dissociation constant for DDsaACP.
For crotonyl-saACP, as well as the NAC and CoA-based substrates, the kinetic studies employed a concentration range for one substrate (0.3 Km – 4 Km) at near saturating concentration of the second substrate. Kinetic parameters were calculated by fitting the data to the Michaelis-Menten equation (2) using GraFit 4.0.
| (2) |
kcat values were obtained by using the relationship between kcat and Vmax: Vmax = kcat [E].
Product inhibition assays
Product inhibition studies were conducted by varying the concentration of DDsaACP at a fixed concentration of NADPH (250 μM) and at different fixed concentrations of NADP+ (0, 1.4 and 2.7 mM) or by varying the concentration of NADPH at a fixed concentration of DDsaACP (20 μM) and at different fixed concentrations of NADP+ (0, 0.7 and 1.4 mM).
The type of inhibition was determined using Lineweaver-Burk plot analysis. Inhibition constants for competitive, uncompetitive, and non-competitive inhibitor were determined by globally fitting all the data points to the equations 3-5, respectively, by using GraFit 4.0.
| (3) |
| (4) |
| (5) |
The abbreviations Kis and Kii represent the Ki slope and intercept in the double-reciprocal plot according to the nomenclature of Cleland (33).
Fluorescence titration
Equilibrium fluorescence titrations were performed using a Fluorolog-3-21 fluorimeter (Spex) at 25°C by making microliter additions of ligand to a solution containing 1 µM saFabI. In the fluorescence titration of saFabI with ACP or the diphenyl ethers, the excitation wavelength was 290 nm and the emission wavelength was 335 nm. When titrating saFabI with NADPH, the excitation wavelength was 350 nm, and the emission wavelength was 460 nm. A control experiment was performed using an identical procedure except that enzyme was omitted from the cuvette. Kd values were calculated by fitting the data to equation 6.
| (6) |
Selection for resistance
Cells from the methicillin-resistant S. aureus strain N315 were incubated at 37°C in Mueller-Hinton broth (Becton-Dickinson) containing 0.02 µM triclosan, 0.08 µM EPP, or 0.08 µM CPP, respectively. After 48 h, 100 µl of each culture was plated onto Mueller-Hinton agar medium containing 0.08 µM triclosan, 0.15 µM EPP, and 0.15 µM CPP, respectively. The plates were incubated at 37 °C for 24 h. Resistant colonies were picked and their phenotype confirmed by regrowth on the same media containing selective concentrations of triclosan, EPP, and CPP, respectively. The S. aureus fabI gene from the diphenyl ether-resistant mutants was characterized by double-strand nucleotide sequencing of PCR products amplified with Ex-taq polymerase (Takara Bio, Shiga, Japan) and the following primers: SafabI-F (5'-GTCATCATGGGAATCGCTAA-3') and SafabI-R (5'-GCGTGGAATCCGCTATCTAC-3'). Sequencing reactions were performed using the above SafabI-F and SafabI-R primers with the ABI PRISMTM BigDyeTM Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Foster City, Calif), and sequencing data were obtained using an Applied Biosystems 3100 DNA sequencer (Applied Biosystems).
MIC measurement
The MIC values were determined by the microbroth dilution assay according to the Clinical and Laboratory Standards Institute methods for antimicrobial susceptibility tests for aerobically growing bacteria (34).
Preincubation inhibition assays for slow-binding inhibitors
Kinetic assays were performed essentially as described previously (28). Briefly, a 500 μL reaction mixture containing 50 nM saFabI, 250 μM NADPH, and various concentrations of diphenyl ether inhibitors (0 – 2 μM) at different fixed concentrations of NADP+ (20 – 800 μM) were preincubated at 4 ºC for 3 hours. The reaction mixture was then warmed to room temperature, and the reaction was initiated by the addition of 20 μM DDsaACP. Apparent inhibition constants (Ki,app) were calculated by fitting the data to equation 7 at each concentration of NADP+
| (7) |
Subsequently, the series of Ki,app values were fit to equation 8 to obtain K1 and K2, which represent the inhibition constants for inhibitor binding to NADP+ bound or NADPH bound forms of the enzyme, respectively.
| (8) |
Steady-state inhibition studies of diphenyl ethers
Steady-state inhibition experiments were performed by varying the concentration of NADPH at a fixed concentration of DDsaACP (20 μM) and at different fixed concentrations of triclosan, EPP or CPP. Reactions were initiated by the addition of the enzyme. Unless noted, the enzyme concentration was 50 nM. The data analysis was performed as described for the product inhibition assays.
RESULTS AND DISCUSSION
Expression and purification of wild-type and mutant saFabI proteins
The protein was purified using His-tag affinity chromatography, providing homogeneously pure recombinant protein with the predicted molecular mass (~30 kDa) as determined by SDS-PAGE. As previously reported (20), the protein precipitated if 100 mM EDTA was omitted from the G25 buffer. In addition, we also observed that saFabI would precipitate even in 100 mM EDTA when the enzyme concentration was higher than 40 μM.
Preparation of ACP substrates
saACP was purified by His-tag affinity chromatography, and only one form of ACP was observed on a 15% Tris-glycine gel, unlike the ACP (AcpM) from Mycobacterium tuberculosis which is obtained in three forms (apo, holo and acyl form) (35). The ESI mass of saACP after deconvolution was 9711.6 ± 0.4, which is close to the calculated mass of apo-saACP (9707.7), suggesting that only the apo form was expressed. Subsequently, apo-saACP was converted into crotonyl saACP or dodecenoyl saACP (DDsaACP) by sfp, the phosphopantetheine transferase from Bacillus subtilis. According to analysis by SDS-PAGE, the apparent molecular mass of DDsaACP is smaller than apo-ACP, probably due to favorable hydrophobic interactions between the dodecenoyl acyl chain in DDsaACP and the SDS. The MW of DDsaACP was confirmed as correct by ESI mass analysis.
Substrate specificity
As shown in Table 1, saFabI has a 180-fold higher kcat/Km with NADPH than with NADH, suggesting that saFabI is an NADPH-dependent enzyme, which is consistent with previous results when trans-2-octenoyl N-acetylcysteamine was used as the substrate (20). As expected, saFabI also has a preference for ACP-linked substrates compared to those attached to NAC or CoA, indicating that saFabI is specific for its natural substrate. While we were able to detect binding of apo-saACP to saFabI (Kd = 18.2 µM) by fluorescence titration (Figure S1), no change in fluorescence could be detected upon addition of NAC or CoA, suggesting that these carriers do not bind strongly to the enzyme in agreement with the kinetic data. In addition, the enzyme also had a preference for a long chain (C12) substrate compared to the shorter (C4) acyl group, with DDsaACP having a kcat/Km value ~30 fold larger than for the corresponding crotonyl-ACP substrate. This effect is also observed for the NAC or CoA-based substrates (Table 1). Interestingly, the effect of the acyl chain length on kcat/Km is primarily an effect on catalytic efficiency (kcat) rather than Km, suggesting that remote interactions between the longer acyl chain and the enzyme modulate the precise orientation of the catalytic groups in the active site. Finally, both the NAC and CoA-based substrates suffered from some disadvantages, the NAC substrates possessing limited aqueous solubility even in the presence of 5% DMSO, whereas DDCoA exhibited substrate inhibition at high concentrations (60 μM). Substrate inhibition was not observed with DDNAC or DDsaACP, which leads to the hypothesis that the adenosine portion of CoA can bind non-productively in the NADPH binding-site.
Table 1.
Kinetic Parameters for the Reduction of Different Substrates with saFabI
| Substrates |
kcat
(min−1) |
Km
(μM) |
kcat/Km
(min−1 μM−1) |
|---|---|---|---|
| Crotonyl NACa | 1.5 ± 0.5 | 8.0 ± 0.3 | 0.20 ± 0.06 |
| Crotonyl CoAb,c | - | - | - |
| Crotonyl saACPc | 11.6 ± 1.1 | 11.5 ± 2.0 | 1.1 ± 0.2 |
| Dodecenoyl NACc | 30.4 ± 1.6 | 23.3 ± 2.9 | 1.3 ± 0.2 |
| Dodocenoyl CoA c | 18.0 ± 0.8 | 24.1 ± 2.5 | 0.8 ± 0.1 |
| Dodecenoyl saACP d | 130.2 ± 9.1 | 4.5 ± 0.5 | 29.5 ± 5.0 |
| NADHe | - | - | 0.010 ± 0.001 |
| NADPH d,e | 130.2 ± 9.1 | 70.8 ± 6.0 | 1.840 ± 0.311 |
Parameters were measured by Heath et al. (20);
no enzymatic activity was observed;
reactions were carried out with 300 μM NADPH;
Km and kcat values were obtained by fitting data into the sequential mechanism equation;
reactions were carried out with 20 μM DDsaACP.
It is interesting that saFabI is NADPH-dependent, whereas the FabI homologues from E. coli, B. subtilis, Haemophilus influenzae and M. tuberculosis, are all reported to be NADH-dependent ACP reductases (36-38). According to the ecFabI-NAD+ crystal structure (Figure 1, pdb code 1DFI), Q40 is very close (2.8 Å) to the 2'-hydroxyl group of the NAD+ adenosine moiety. Sequence alignment of the FabI proteins from different organisms (Figure 2), indicates that two positively-charged residues, R40 and K41, appear close to the position of Q40 in saFabI, whereas nonpolar residues (L, F), or a negatively charged residue (E) is present in the cases of the FabIs from B. subtilis, H. influenzae and M. tuberculosis, respectively. Basic residues, such as R or K, in this position are expected to interact strongly with the 2’-phosphate of NADPH. In order to investigate the importance of R40 and K41 in the interaction of NADPH with saFabI, we constructed two single-mutants, R40Q and K41N, and found that both mutants show at least a 50-fold decrease in kcat/Km for NADPH, whereas kcat/Km for NADH increases by 5 to 7 fold (Table 2). The double mutation R40Q/K41N further decreases kcat/Km for NADPH by 10-fold, proving that both R40 and K41 are involved in interactions with 2’-phosphate of NADPH. Since all the residues apart from Q40 which have polar interactions with NAD+ in the ecFabI-NAD+ crystal structure are highly conserved in saFabI, it might be expected that the cofactor specificity would be completely reversed in the saFabI double mutant. However, the NADH-specificity of the double mutant remains almost the same as the single mutants, only 7 fold higher than that of the wild type saFabI, indicating that other factors are critical for optimal binding of NADH.
Figure 1. Structure of the ecFabI:NAD+ Complex.

Structure of ecFabI complexed with NAD+ (pdb code 1DFI) showing interactions between the protein and the NAD+ ribose. ecFabI is colored yellow and polar interactions between the protein residues (green) and NAD+ (cyan) are indicated with red dashed lines. Q40 interacts with the 2’-hydroxyl group in the adenosine moity of NAD+. The Figure was made using pymol (44).
Figure 2. FabI Sequence Alignment.

Sequence alignment of FabIs from E. coli, S. aureus, H. influenzae, M. tuberculosis and B. subtilis performed using ClustalW. The red-colored amino acids are proposed to interact with the cofactor, according to the X-ray structure of ecFabI:NAD+ complex.
Table 2.
Effect of Mutagenesis on the Specificity of saFabI for NADH and NADPH
| Enzyme |
kcat/Km (min−1 μM−1) |
|
|---|---|---|
| NADPH | NADH | |
| WT | 1.840 ± 0.311 | 0.010 ± 0.001 |
| R40Q | 0.020 ± 0.001 | 0.053 ± 0.003 |
| K41N | 0.036 ± 0.002 | 0.072 ± 0.002 |
| R40Q/K41N | 0.004 ± 0.001 | 0.076 ± 0.005 |
Enzymatic mechanism of the saFabI-catalyzed reaction
As expected, double-reciprocal plots of the enzyme kinetic data generated intersecting lines (Figure S2A and S2B), which is a characteristic of a ternary complex mechanism in which NADPH transfers a hydride directly to the substrate. In order to determine the order of substrate binding, the effect of the product NADP+ on the reaction was analyzed. Initial velocities were measured at various concentrations of DDsaACP and a fixed concentration of NADPH and at different fixed concentrations of NADP+. The parallel lines in the double reciprocal plot that were obtained (Figure S3A) suggest that NADP+ is an uncompetitive inhibitor with respect to DDsaACP. However, the assays were also performed with NADPH as the varied substrate. As shown in Figure S3B, the lines intersect to the left of the y-axis, indicating that the NADP+ is a non-competitive inhibitor with respect to NADPH. The inhibition patterns of NADP+ against the two substrates, are only consistent with an ordered bi-bi mechanism, in which DDsaACP binds first, followed by NADPH.
In fluorescence titration experiments, no fluorescence change was observed when titrating saFabI with NADPH, suggesting that NADPH does not bind to saFabI, or that binding does not affect the fluorescence of the NADPH fluorophore. In contrast, a Kd value of 2.3 μM was obtained when saFabI was titrated with DDsaACP (Figure S4). These results provide further support for the proposed ordered bi-bi mechanism.
Antimicrobial activity of the diphenyl ether saFabI inhibitors
The molecular basis for the antibacterial activity of triclosan against S. aureus has attracted much attention. It was proposed that triclosan inhibits S. aureus by targeting the enoyl reductase in this organism (20, 21, 26), since it is known that triclosan inhibits the FabI homologue in E. coli (39-41), and that saFabI shows 43% sequence identity to ecFabI. However, the observation that small-colony variants of S. aureus have increased resistance to triclosan suggests more complex underlying mode of action of this antimicrobial agent (27).
As a first step in developing novel inhibitors against S. aureus, we synthesized two diphenyl ether analogues of triclosan: 5-ethyl-2-phenoxy-phenol (EPP) and 5-chloro-2-phenoxy-phenol (CPP). Both compounds have similar MIC values to triclosan against different strains of S. aureus (Table S2). It is very encouraging that the MIC values are similar for both methicillin-sensitive (ATCC 29213) and methicillin-resistant S. aureus strains (N315, Mu50), suggesting that the diphenyl ethers are orthogonal to current antibiotics. In order to probe the mechanism of action of the diphenyl ether inhibitors, selection experiments resulted in the identification of three novel mutations in the fabI gene, A95V, I193S and F204S (Table 3), suggesting that saFabI is the intracellular target for these compounds. Based on the ecFabI/NAD/triclosan crystal structure (42), the three residues are thought to lie in the cofactor binding pocket of saFabI (Figure 3). Interestingly, the residues corresponding to A95 and F204 in ecFabI (G93 and F203), were also found to be mutated in triclosan-resistant E. coli. In addition, the residue corresponding to I193 in ecFabI (I192) interacts with the cofactor, as shown in Figure 3. As expected, the three saFabI mutations affected the kinetic parameters associated with NADPH more significantly than the parameters for the ACP substrate (Table 4). In particular, the kcat/Km value for NADPH decreased by 240 fold for the A95V mutant, while the kcat/Km value for DDsaACP decreased only 18 fold, confirming that the mutation has a major impact on the interaction of the cofactor with the enzyme.
Table 3.
Selection for Resistance to the Diphenyl Ether saFabI Inhibitors
| Mutants derived from S. aureus N315 |
Selections for mutants resistant to compound |
fabI gene |
|
|---|---|---|---|
| Nucleotide changes a | Amino acid changes | ||
| N315Tr.4 | Triclosan | GCA → GTA | Ala95 → Val |
| N315Tr.5 | Triclosan | GCA → GTA | Ala95 → Val |
| N315PT01.1 | EPP | GCA → GTA | Ala95 → Val |
| N315PT01.2 | EPP | GCA → GTA | Ala95 → Val |
| N315PT01.3 | EPP | GCA → GTA | Ala95 → Val |
| N315PT01.4 | EPP | GCA → GTA | Ala95 → Val |
| N315PT52.1 | CPP | ATC → AGC | Ile 193 → Ser |
| N315PT52.2 | CPP | TTC → TCC | Phe204 → Ser |
| N315PT52.3 | CPP | TTC → TCC | Phe204 → Ser |
| N315PT52.4 | CPP | TTC → TCC | Phe204 → Ser |
Mutation sites are shown in underline.
Figure 3. Structure of ecFabI Complexed with NAD+ and Triclosan.

Structure of triclosan bound to ecFabI (pdb code 1D8A) showing the proximity of residues G93, I192 and F203 to the inhibitor binding site. The corresponding residues in saFabI were found to be mutated in the diphenyl ether resistant S. aureus strains. ecFabI is colored yellow, while the polar interactions between the residues (green) in ecFabI and NAD/triclosan (cyan), are indicated with red dashed lines. The Figure was made using pymol (44).
Table 4.
Kinetic Parameters for the Reduction of DDsaACP by Wild Type and Mutant FabIs
| Enzyme | kcat (min−1) | NADPH |
DDsaACP |
||
|---|---|---|---|---|---|
|
Km
(μM)a |
kcat/Km
(min−1 μM−1)a |
Km
(μMb |
kcat/Km
(min−1 μM−1)b |
||
| WT | 130.2 ± 9.1 | 70.8 ± 6.0 | 1.8 ± 0.3 | 4.5 ± 0.5 | 29.5 ± 5.0 |
| A95V | 18.4 ± 0.1 | 269.4 ± 29.2 | 0.007 ± 0.001 | 11.4 ± 1.6 | 1.7 ± 0.2 |
| I193S | - | > 1000 | 0.13 ± 0.02 | - | 5.1 ± 0.4 |
| F204S | 157.3 ± 7.2 | 429.6 ± 45.0 | 0.17 ± 0.02 | 18.4 ± 1.3 | 7.8 ± 0.7 |
Reactions were carried out with 20 μM DDsaACP;
reactions were carried out with 300 μM NADPH.
To analyze further the affect of the mutations on enzyme inhibition, we quantified the interaction of the three diphenyl ether inhibitors with both wild-type and mutant saFabIs. In the case of wild-type saFabI, all three compounds were slow-onset inhibitors, binding preferentially to the enzyme-NADP+ product complex. The slow step in the formation of the final enzyme-inhibitor complex is believed to result from the ordering of a flexible loop that covers the active site, as seen for the inhibition of ecFabI by triclosan. Since the loop in ecFabI (residues 193-202) is highly conserved in saFabI, we propose that the slow step during the inhibition of saFabI by the diphenyl ethers is also caused by the loop ordering. We used the preincubation method to measure K1 and K2, which represent the equilibrium dissociation constants of the compounds from the E-NADP+ and E-NADPH forms of the enzyme, respectively. According to the inhibition results (Figure 4A-D), the compounds are uncompetitive inhibitors of NADP+, and competitive inhibitors of NADPH. K1 is 20 to 40 fold smaller than K2, suggesting that the diphenyl ethers bind preferentially to E-NADP+. In addition, the K1 values correlate well with the MIC data, suggesting that saFabI is the target of this class of inhibitor. The use of equation 6 to analyze the inhibition data is based on the assumption that the inhibitors do not bind tightly to the free enzyme. In agreement with this assumption, fluorescence titrations of saFabI with the three inhibitors (Figure S5A-S5C) gave Kd values between 50 and 200 µM, at least 200-fold higher than either K1 or K2.
Figure 4. Inhibition of Wild Type saFabI by the Diphenyl Ether Inhibitors.

(A) Dependence of Ki,app on [NADP+] for inhibition by triclosan. (B) Dependence of Ki,app on [NADPH] for inhibition by triclosan. (C) Dependence of Ki,app on [NADP+] for inhibition by EPP. (D) Dependence of Ki,app on [NADP+] for inhibition by CPP.
In contrast to the wild-type enzyme, the diphenyl ether compounds are classical reversible inhibitors of the saFabI mutant enzymes (Figures S6-S8). Triclosan, CPP and EPP are uncompetitive or noncompetitive inhibitors of the mutant saFabIs with respect to NADPH, with Ki values 5 to 3000 fold higher than for the wild-type enzyme. This increase in Ki correlates with the increase in the MIC values observed for the S. aureus strains harboring the safabI mutations (Table 5). This further substantiates the hypothesis that saFabI is the target of diphenyl ethers within this bacterium. Interestingly, for the mutant I193S, Ki values of triclosan, EPP and CPP increase only by 5 to 25 fold, but the MICs increase by more than 60 fold. This suggests that additional mechanisms of resistance are operative, such as the overexpression of the mutant saFabI as reported by Fan et al. (26). We also observed that the frequency with which each mutation occurred also correlated with the effect of that mutation on enzyme inhibition. Thus, A95V, the most frequent mutation in the resistant strains, had the largest impact on inhibition of saFabI by the diphenyl ether inhibitors.
Table 5.
Inhibition of Wild Type and Mutant FabIs by the Diphenyl Ether Inhibitors
| Enzyme | Compound | K1 (nM) | K2 (nM) | Inhibition pattern | MIC against S.
aureus N315 μg/mL (μM) |
|---|---|---|---|---|---|
| WT | Triclosan | 4.9 ± 0.6a | 375.1 ± 20.3a | UC with respect to NADP+. | 0.03 (0.10) |
| 5.6 ± 0.2b | 216.4 ± 7.5b | C with respect to NADPH. | |||
|
| |||||
| EPPa | 8.3 ± 0.8 | 273.1 ± 13.2 | UC with respect to NADP+. | 0.03 (0.14) | |
| CPPa | 2.0 ± 0.2 | 48.7 ± 1.6 | UC with respect to NADP+. | 0.06 (0.27) | |
|
| |||||
| Triclosanc | 8100 ± 1190 | - | UC with respect to NADPH. | 2.80 (10) | |
| A95V | EPPc | 1660 ± 50 | - | UC with respect to NADPH. | 4.40 (20) |
| CPPc | 6150 ± 160 | - | UC with respect to NADPH. | 8.80 (40) | |
|
| |||||
| Triclosand | 35 ± 18 | 473 ± 196 | NC with respect to NADPH. | 2.80 (10) | |
| I193S | EPPd | 41 ± 5 | 783 ± 281 | NC with respect to NADPH. | 8.80 (40) |
| CPPd | 52 ± 2 | 690 ± 320 | NC with respect to NADPH. | 17.60 (80) | |
|
| |||||
| Triclosanc | 210 ± 20 | - | UC with respect to NADPH. | 0.70 (2.5) | |
| F204S | EPPd | 160 ± 60 | 300 ± 110 | NC with respect to NADPH. | 2.20 (10) |
| CPPc | 570 ± 30 | - | UC with respect to NADPH. | 8.80 (40) | |
NADP+ was varied;
NADPH was varied;
Uncompetitive. Kii has been listed under K1. Enzyme concentration was 200 nM;
noncompetitive inhibition. Kii has been listed under K1, and Kis has been listed under K2. Kii and Kis were defined by Cleland previously (33).
Finally, we also analyzed the ability of the three compounds to inhibit the growth of Enterococcus faecalis. This organism contains both a FabI enoyl reductase as well as a FabK homologue that is not sensitive to triclosan (43). In Table S3 it can be seen that the three diphenyl ethers have MIC values against E. faecalis that are ~1000-fold higher than for S. aureus again supporting the contention that the antibacterial action of these compounds is directed against FabI.
CONCLUSION
In summary, we have studied the mechanism of the reaction catalyzed by saFabI using substrates based on the natural ACP carrier molecule. From the results of the kinetic assays, we propose that the binding of substrates in the ternary complex mechanism is ordered, with DDsaACP binding first followed by NADPH. Two residues, R40 and K41 are shown to be critical for cofactor specificity. In addition, we investigated the mechanism of action of three diphenyl ether-based saFabI inhibitors. All three compounds are slow-onset inhibitors of saFabI in vitro, binding preferentially to the E-NADP+ product complex with K1 values of 2-8 nM. These compounds have potent antibacterial activity, with MIC values against S. aureus of 0.03-0.06 µg/mL. Selection experiments led to the identification of three mutations in the safabI gene, A95V, I193S and F204S, which correlate with resistance to the diphenyl ethers and cause a significant reduction in the affinity of the inhibitors for the enzyme. These experiments confirm the hypothesis that the diphenyl-ether-based inhibitors target saFabI in live cells, and substantiate the view that saFabI is a novel target for antibacterial drug discovery.
Supplementary Material
Scheme 1.
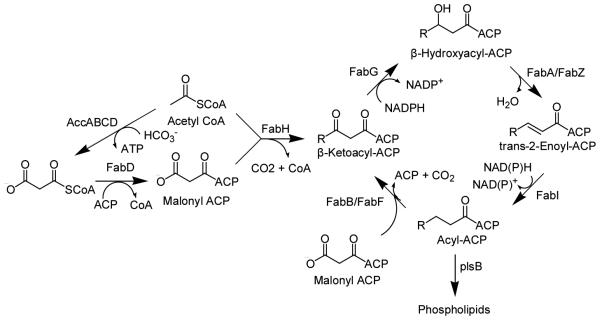
The Type II Fatty Acid Biosynthesis Pathway.
Scheme 2.
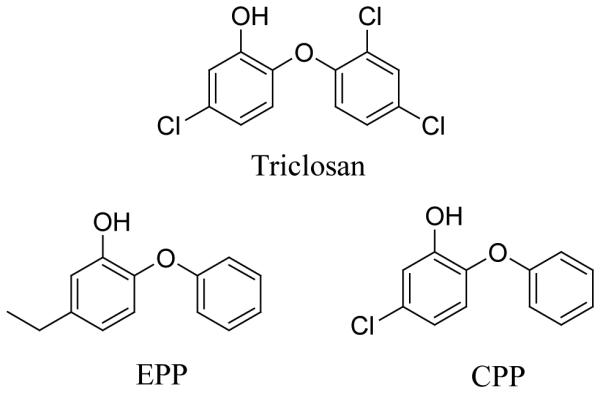
The Diphenyl Ether saFabI Inhibitors
ACKNOWLEDGMENTS
We are grateful to Dr. Michael Burkart (UCSD) for providing the sfp plasmid.
Footnotes
This work was supported in part by NIH grants AI44639 and AI70383.
- FAS
- fatty acid biosynthesis
- ACP
- acyl carrier protein
- NAC
- N-acetylcysteamine
- CoA
- coenzyme A
- DDsaACP
- dodecenoyl saACP
- MIC
- minimum inhibitory concentration
- EPP
- 5-ethyl-2-phenoxyphenol
- CPP
- 5-chloro-2-phenoxyphenol
SUPPORTING INFORMATION AVAILABLE
Double reciprocal plots for the kinetic and inhibition studies and the MIC data for the diphenyl ethers against the S. aureus mutant strains and E. faecalis. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Diekema DJ, Pfaller MA, Schmitz FJ, Smayevsky J, Bell J, Jones RN, Beach M. Survey of infections due to Staphylococcus species: frequency of occurrence and antimicrobial susceptibility of isolates collected in the United States, Canada, Latin America, Europe, and the Western Pacific region for the SENTRY Antimicrobial Surveillance Program, 1997-1999, Clin. Infect. Dis. 2001;32(Suppl 2):S114–132. doi: 10.1086/320184. [DOI] [PubMed] [Google Scholar]
- 2.Pfaller MA, Jones RN, Doern GV, Sader HS, Kugler KC, Beach ML. Survey of blood stream infections attributable to gram-positive cocci: frequency of occurrence and antimicrobial susceptibility of isolates collected in 1997 in the United States, Canada, and Latin America from the SENTRY Antimicrobial Surveillance Program. Diagn. Microbiol. Infect. Dis. 1999;33:283–297. doi: 10.1016/s0732-8893(98)00149-7. SENTRY Participants Group. [DOI] [PubMed] [Google Scholar]
- 3.Peacock SJ, de Silva I, Lowy FD. What determines nasal carriage of Staphylococcus aureus? Trends Microbiol. 2001;9:605–610. doi: 10.1016/s0966-842x(01)02254-5. [DOI] [PubMed] [Google Scholar]
- 4.Bradley SF. Methicillin-resistant Staphylococcus aureus infection. Clin. Geriatr. Med. 1992;8:853–868. [PubMed] [Google Scholar]
- 5.Barber M. Methicillin-resistant staphylococci. J. Clin. Pathol. 1961;14:385–393. doi: 10.1136/jcp.14.4.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Panlilio AL, Culver DH, Gaynes RP, Banerjee S, Henderson TS, Tolson JS, Martone WJ. Methicillin-resistant Staphylococcus aureus in U.S. hospitals, 1975-1991. Infect. Control Hosp. Epidemiol. 1992;13:582–586. doi: 10.1086/646432. [DOI] [PubMed] [Google Scholar]
- 7.Gaynes R, Culver D. Annual Meeting of the Infectious Disease Society of America. IDSA; San Francisco: 1997. Nosocomial methicillin-resistant Staphylococcus aureus (MRSA) in the United States, 1975-1996. National Nosocomial Infection Surveillance (NNIS) System. [Google Scholar]
- 8.From the Centers for Disease Control and Prevention Vancomycin resistant Staphylococcus aureus--Pennsylvania. Jama. 20022002:2116. [PubMed] [Google Scholar]
- 9.Hiramatsu K, Hanaki H, Ino T, Yabuta K, Oguri T, Tenover FC. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J. Antimicrob. Chemother. 1997;40:135–136. doi: 10.1093/jac/40.1.135. [DOI] [PubMed] [Google Scholar]
- 10.Egan AF, Russell RR. Conditional mutations affecting the cell envelope of Escherichia coli K-12. Genet. Res. 1973;21:139–152. doi: 10.1017/s001667230001332x. [DOI] [PubMed] [Google Scholar]
- 11.Turnowsky F, Fuchs K, Jeschek C, Hogenauer G. envM genes of Salmonella typhimurium and Escherichia coli. J. Bacteriol. 1989;171:6555–6565. doi: 10.1128/jb.171.12.6555-6565.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heath RJ, Rock CO. Enoyl-acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J. Biol. Chem. 1995;270:26538–26542. doi: 10.1074/jbc.270.44.26538. [DOI] [PubMed] [Google Scholar]
- 13.Rawat R, Whitty A, Tonge PJ. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: adduct affinity and drug resistance. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13881–13886. doi: 10.1073/pnas.2235848100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baldock C, Rafferty JB, Sedelnikova SE, Baker PJ, Stuitje AR, Slabas AR, Hawkes TR, Rice DW. A mechanism of drug action revealed by structural studies of enoyl reductase. Science. 1996;274:2107–2110. doi: 10.1126/science.274.5295.2107. New York, N.Y. [DOI] [PubMed] [Google Scholar]
- 15.Karlowsky JA, Laing NM, Baudry T, Kaplan N, Vaughan D, Hoban DJ, Zhanel GG. In vitro activity of API-1252, a novel FabI inhibitor, against clinical isolates of Staphylococcus aureus and Staphylococcus epidermidis. Antimicrob. Agents Chemother. 2007;51:1580–1581. doi: 10.1128/AAC.01254-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitagawa H, Kumura K, Takahata S, Iida M, Atsumi K. 4-Pyridone derivatives as new inhibitors of bacterial enoyl-ACP reductase FabI. Bioorg. Med. Chem. 2007;15:1106–1116. doi: 10.1016/j.bmc.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 17.Ling LL, Xian J, Ali S, Geng B, Fan J, Mills DM, Arvanites AC, Orgueira H, Ashwell MA, Carmel G, Xiang Y, Moir DT. Identification and characterization of inhibitors of bacterial enoyl-acyl carrier protein reductase. Antimicrob. Agents Chemother. 2004;48:1541–1547. doi: 10.1128/AAC.48.5.1541-1547.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Payne DJ, Miller WH, Berry V, Brosky J, Burgess WJ, Chen E, DeWolf Jr WE, Jr., Fosberry AP, Greenwood R, Head MS, Heerding DA, Janson CA, Jaworski DD, Keller PM, Manley PJ, Moore TD, Newlander KA, Pearson S, Polizzi BJ, Qiu X, Rittenhouse SF, Slater-Radosti C, Salyers KL, Seefeld MA, Smyth MG, Takata DT, Uzinskas IN, Vaidya K, Wallis NG, Winram SB, Yuan CC, Huffman WF. Discovery of a novel and potent class of FabI-directed antibacterial agents. Antimicrob. Agents Chemother. 2002;46:3118–3124. doi: 10.1128/AAC.46.10.3118-3124.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang YM, Rock CO. Evaluation of epigallocatechin gallate and related plant polyphenols as inhibitors of the FabG and FabI reductases of bacterial type II fatty-acid synthase. J. Biol. Chem. 2004;279:30994–31001. doi: 10.1074/jbc.M403697200. [DOI] [PubMed] [Google Scholar]
- 20.Heath RJ, Li J, Roland GE, Rock CO. Inhibition of the Staphylococcus aureus NADPH-dependent enoyl-acyl carrier protein reductase by triclosan and hexachlorophene. J. Biol. Chem. 2000;275:4654–4659. doi: 10.1074/jbc.275.7.4654. [DOI] [PubMed] [Google Scholar]
- 21.Slater-Radosti C, Van Aller G, Greenwood R, Nicholas R, Keller PM, DeWolf WE, Jr., Fan F, Payne DJ, Jaworski DD. Biochemical and genetic characterization of the action of triclosan on Staphylococcus aureus. J. Antimicrob. Chemother. 2001;48:1–6. doi: 10.1093/jac/48.1.1. [DOI] [PubMed] [Google Scholar]
- 22.Russell AD. Whither triclosan? J. Antimicrob. Chemother. 2004;53:693–695. doi: 10.1093/jac/dkh171. [DOI] [PubMed] [Google Scholar]
- 23.Regos J, Zak O, Solf R, Vischer WA, Weirich EG. Antimicrobial spectrum of triclosan, a broad-spectrum antimicrobial agent for topical application. II. Comparison with some other antimicrobial agents. Dermatologica. 1979;158:72–79. doi: 10.1159/000250746. [DOI] [PubMed] [Google Scholar]
- 24.Ayliffe GA, Buckles A, Casewell MW, Cookson BD, Cox RA, Duckworth GJ, Griffiths-Jones A, Heathcock R, Humphreys H, Keane CT, Marples RR, Shanson DC, Slack R, Tebbs E. Revised guidelines for the control of methicillin-resistant Staphylococcus aureus infection in hospitals. British Society for Antimicrobial Chemotherapy, Hospital Infection Society and the Infection Control Nurses Association. J. Hosp. Infect. 1998;39:253–290. doi: 10.1016/s0195-6701(98)90293-6. [DOI] [PubMed] [Google Scholar]
- 25.Bamber AI, Neal TJ. An assessment of triclosan susceptibility in methicillin-resistant and methicillin-sensitive Staphylococcus aureus. J. Hosp. Infect. 1999;41:107–109. doi: 10.1016/s0195-6701(99)90047-6. [DOI] [PubMed] [Google Scholar]
- 26.Fan F, Yan K, Wallis NG, Reed S, Moore TD, Rittenhouse SF, DeWolf WE, Jr., Huang J, McDevitt D, Miller WH, Seefeld MA, Newlander KA, Jakas DR, Head MS, Payne DJ. Defining and combating the mechanisms of triclosan resistance in clinical isolates of Staphylococcus aureus. Antimicrob. Agents Chemother. 2002;46:3343–3347. doi: 10.1128/AAC.46.11.3343-3347.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seaman PF, Ochs D, Day MJ. Small-colony variants: a novel mechanism for triclosan resistance in methicillin-resistant Staphylococcus aureus. J. Antimicrob. Chemother. 2007;59:43–50. doi: 10.1093/jac/dkl450. [DOI] [PubMed] [Google Scholar]
- 28.Sivaraman S, Sullivan TJ, Johnson F, Novichenok P, Cui G, Simmerling C, Tonge PJ. Inhibition of the bacterial enoyl reductase FabI by triclosan: a structure-reactivity analysis of FabI inhibition by triclosan analogues. J. Med. Chem. 2004;47:509–518. doi: 10.1021/jm030182i. [DOI] [PubMed] [Google Scholar]
- 29.Sullivan TJ, Truglio JJ, Boyne ME, Novichenok P, Zhang X, Stratton CF, Li HJ, Kaur T, Amin A, Johnson F, Slayden RA, Kisker C, Tonge PJ. High affinity InhA inhibitors with activity against drug-resistant strains of Mycobacterium tuberculosis. ACS Chem. Biol. 2006;1:43–53. doi: 10.1021/cb0500042. [DOI] [PubMed] [Google Scholar]
- 30.Parikh S, Moynihan DP, Xiao G, Tonge PJ. Roles of tyrosine 158 and lysine 165 in the catalytic mechanism of InhA, the enoyl-ACP reductase from Mycobacterium tuberculosis. Biochemistry. 1999;38:13623–13634. doi: 10.1021/bi990529c. [DOI] [PubMed] [Google Scholar]
- 31.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 32.Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P, Walsh CT. Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry. 1998;37:1585–1595. doi: 10.1021/bi9719861. [DOI] [PubMed] [Google Scholar]
- 33.Cleland WW. Statistical analysis of enzyme kinetic data. Methods Enzymol. 1979;63:103–138. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- 34.CLSI . Approved Standard M7-A6. 6th Clinical and Laboratory Standards Institute; Wayne, PA: 2006. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically. [Google Scholar]
- 35.Schaeffer ML, Agnihotri G, Kallender H, Brennan PJ, Lonsdale JT. Expression, purification, and characterization of the Mycobacterium tuberculosis acyl carrier protein, AcpM. Biochim. Biophys. Acta. 2001;1532:67–78. doi: 10.1016/s1388-1981(01)00116-0. [DOI] [PubMed] [Google Scholar]
- 36.Bergler H, Wallner P, Ebeling A, Leitinger B, Fuchsbichler S, Aschauer H, Kollenz G, Hogenauer G, Turnowsky F. Protein EnvM is the NADH-dependent enoyl-ACP reductase (FabI) of Escherichia coli. J. Biol. Chem. 1994;269:5493–5496. [PubMed] [Google Scholar]
- 37.Heath RJ, Su N, Murphy CK, Rock CO. The enoyl-[acyl-carrier-protein] reductases FabI and FabL from Bacillus subtilis. J. Biol. Chem. 2000;275:40128–40133. doi: 10.1074/jbc.M005611200. [DOI] [PubMed] [Google Scholar]
- 38.Marcinkeviciene J, Jiang W, Kopcho LM, Locke G, Luo Y, Copeland RA. Enoyl-ACP reductase (FabI) of Haemophilus influenzae: steady-state kinetic mechanism and inhibition by triclosan and hexachlorophene. Arch. Biochem. Biophys. 2001;390:101–108. doi: 10.1006/abbi.2001.2349. [DOI] [PubMed] [Google Scholar]
- 39.Heath RJ, Rubin JR, Holland DR, Zhang E, Snow ME, Rock CO. Mechanism of triclosan inhibition of bacterial fatty acid synthesis. J. Biol. Chem. 1999;274:11110–11114. doi: 10.1074/jbc.274.16.11110. [DOI] [PubMed] [Google Scholar]
- 40.Heath RJ, Yu YT, Shapiro MA, Olson E, Rock CO. Broad spectrum antimicrobial biocides target the FabI component of fatty acid synthesis. J. Biol. Chem. 1998;273:30316–30320. doi: 10.1074/jbc.273.46.30316. [DOI] [PubMed] [Google Scholar]
- 41.McMurry LM, Oethinger M, Levy SB. Triclosan targets lipid synthesis. Nature. 1998;394:531–532. doi: 10.1038/28970. [DOI] [PubMed] [Google Scholar]
- 42.Levy CW, Roujeinikova A, Sedelnikova S, Baker PJ, Stuitje AR, Slabas AR, Rice DW, Rafferty JB. Molecular basis of triclosan activity. Nature. 1999;398:383–384. doi: 10.1038/18803. [DOI] [PubMed] [Google Scholar]
- 43.Heath RJ, Rock CO. A triclosan-resistant bacterial enzyme. Nature. 2000;406:145–146. doi: 10.1038/35018162. [DOI] [PubMed] [Google Scholar]
- 44.Delano WL. The PyMOL Molecular Graphics System. DeLano Scientific LLC; San Carlos, CA, USA: 2002. http://www.pymol.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.