

Abstract
Mechanisms underlying motor neuron degeneration in spinal muscular atrophy (SMA), the leading inherited cause of infant mortality, remain largely unknown. Many studies have established the importance of hyperphosphorylation of the microtubule-associated protein tau in various neurodegenerative disorders, including Alzheimer's and Parkinson's diseases. However, tau phosphorylation in SMA pathogenesis has yet to be investigated. Here we show that tau phosphorylation on serine 202 (S202) and threonine 205 (T205) is increased significantly in SMA motor neurons using two SMA mouse models and human SMA patient spinal cord samples. Interestingly, phosphorylated tau does not form aggregates in motor neurons or neuromuscular junctions (NMJs), even at late stages of SMA disease, distinguishing it from other tauopathies. Hyperphosphorylation of tau on S202 and T205 is mediated by cyclin-dependent kinase 5 (Cdk5) in SMA disease condition, because tau phosphorylation at these sites is significantly reduced in Cdk5 knock-out mice; genetic knock-out of Cdk5 activating subunit p35 in an SMA mouse model also leads to reduced tau phosphorylation on S202 and T205 in the SMA;p35−/− compound mutant mice. In addition, expression of the phosphorylation-deficient tauS202A,T205A mutant alleviates motor neuron defects in a zebrafish SMA model in vivo and mouse motor neuron degeneration in culture, whereas expression of phosphorylation-mimetic tauS202E,T205E promotes motor neuron defects. More importantly, genetic knock-out of tau in SMA mice rescues synapse stripping on motor neurons, NMJ denervation, and motor neuron degeneration in vivo. Altogether, our findings suggest a novel mechanism for SMA pathogenesis in which hyperphosphorylation of non-aggregating tau by Cdk5 contributes to motor neuron degeneration.
Keywords: Cdk5, motor neuron, neurodegeneration, SMA, tau
Introduction
Spinal muscular atrophy (SMA), the leading inherited cause of infant mortality, is an autosomal recessive genetic disorder characterized by the loss of spinal motor neurons (Lunn and Wang, 2008; Burghes and Beattie, 2009). SMA is caused by mutations in the survival motor neuron SMN1 gene, leading to reduced levels of SMN protein (Lefebvre et al., 1995). Although SMN is expressed ubiquitously in all tissues, spinal motor neurons are particularly vulnerable to diminished levels of SMN (Monani, 2005; Burghes and Beattie, 2009). SMN protein has been implicated in several functional processes, including small nuclear ribonucleoprotein (snRNP) biogenesis and pre-mRNA splicing (Pellizzoni et al., 1998; McWhorter et al., 2003; Rossoll et al., 2003). Despite extensive efforts to characterize the disease molecularly and cellularly, mechanisms that underlie motor neuron degeneration in SMA remain essentially unknown. Currently, there is no effective therapy for SMA; therefore, a better understanding of the pathogenic mechanism is needed in developing therapeutics for this devastating disorder.
The microtubule-associated protein tau, identified originally as a microtubule-assembly factor (Weingarten et al., 1975; Witman et al., 1976), is a known critical regulator of intracellular trafficking and signal transduction protein complex formation (Morris et al., 2011). Site-specific phosphorylation of tau regulates its physiological roles in microtubule dynamics, axonal transport, and neurite outgrowth (Biernat et al., 1993; Mandell and Banker, 1996; Tatebayashi et al., 2004). However, hyperphosphorylation of tau results in self-assembly of neurofibrillary tangles of filaments, which are involved in pathogenesis of Alzheimer's disease and other neurodegenerative tauopathies (Grundke-Iqbal et al., 1986; Wood et al., 1986; Lee et al., 1991; Vanderweyde et al., 2012). Phosphorylation of serine 202 (S202) and threonine 205 (T205), located in the proline-rich (PR) domain of tau, has been implicated in the pathogenesis of Parkinson's and Alzheimer's diseases (Paudel et al., 1993; Bailey et al., 2013). In both physiological and pathological conditions, tau can be phosphorylated by multiple kinases, including cyclin-dependent kinase 5 (Cdk5; Baumann et al., 1993; Michel et al., 1998). Cdk5 is a proline-directed serine/threonine kinase that critically regulates many neuronal functions, such as neuronal migration, axonal morphogenesis, and synaptic activity (Fu et al., 2001; Su and Tsai, 2011). Dysregulation of Cdk5 activity can lead to tau hyperphosphorylation and is implicated in a number of neurodegenerative disorders, including Alzheimer's disease and amyotrophic lateral sclerosis (Patrick et al., 1999; Nguyen et al., 2001; Wong et al., 2011). However, the role of tau phosphorylation by Cdk5 in SMA pathogenesis has not been explored.
In this study, we report that hyperphosphorylation of non-aggregating tau on S202 and T205 is increased significantly in motor neurons of SMA animal models and human patients. Expression of tau with enhanced phosphorylation on S202 and T205 by Cdk5 leads to motor neuron defects in zebrafish and mouse motor neuron degeneration, whereas tau deficient for phosphorylation alleviates motor neuron defects. Genetic knock-out of tau in SMA mice rescues synaptic stripping, neuromuscular junction (NMJ) defects, and motor neuron degeneration phenotypes in vivo. Altogether, our findings suggest a potential novel mechanism underlying SMA pathogenesis.
Materials and Methods
Mice and human samples.
The Δ7 SMA mouse model (Smn−/−;SMN2tg/tg;SMNΔ7tg/tg; catalog #005025), the Hung-Li SMA mouse model (Smn−/−;SMN2Hungtg/−; catalog #005058), the tau knock-out mice (catalog #007251), the p35 knock-out mice (catalog #004163), the Cdk5 conditional knock-out mice (Cdk5F/F; catalog #014156), and the Nestin–Cre mice (catalog #003771) were obtained from the Jackson Laboratory. Genomic DNA extracted from tail samples was used for PCR-based genotyping as reported previously (Chae et al., 1997; Tronche et al., 1999; Hsieh-Li et al., 2000; Le et al., 2005; Samuels et al., 2007). Both male and female mice were used. SMA and control human tissue was collected at autopsy after parental informed consent as approved by the Johns Hopkins University School of Medicine Institutional Review Board. Some control human tissues were obtained from the Human Development Brain and Tissue Bank for Developmental Disorders at the University of Maryland (Table 1).
Table 1.
Characteristics of patient spinal cord samples
| Control thoracic spinal cord |
SMA thoracic spinal cord |
||||||
|---|---|---|---|---|---|---|---|
| Case ID | Age (months) | PMI (h) | COD | Case ID | Age (months) | PMI (h) | SMA type |
| CNTL 07_01 | 0.03 | 69 | Meconium aspiration | SMA 11_01 | 1.8 | 7 | I |
| MBB 1296 | 3.3 | 16 | Congenital heart defect | SMA 12_01 | 2.5 | 7 | I |
| MBB 86 | 4.6 | 11 | Congenital heart defect | SMA 08_01 | 4.5 | 14.5 | I |
| MBB 1055 | 8 | 12 | Broncho-pneumonia | SMA 07_01 | 11 | 24 | I |
| MBB 195 | 4.1 | 16 | Complications of prematurity | SMA 10_14 | 7 | 25 | I |
PMI, Postmortem interval; COD, cause of death.
Immunohistochemistry.
SMA and wild-type (WT) mice were perfused with PBS and 4% PFA. Spinal cords were isolated and fixed in freshly made 4% PFA overnight. Postmortem human spinal cord samples were fixed in 4% PFA for 12 h. All samples were washed extensively with PBS and treated sequentially with 15 and 30% sucrose before embedding into OCT. Cryosections of 18 μm thickness were then prepared with a Leica CM1950 cryostat. For immunostaining, tissue sections were first treated with 10 mm citric acid antigen retrieval solution (Dako) at 98°C for 20 min, then permeabilized in 0.25% Triton X-100, and blocked with 5% donkey serum and 5% goat serum in PBST buffer (PBS with 0.05% Tween 20). Samples were then incubated with primary antibodies overnight at 4°C, washed with PBST, incubated with secondary antibodies, washed with PBST, mounted in Aquamount (Thermo Fisher Scientific), and imaged with Zeiss LSM510 confocal microscope. Primary antibodies used in this study are as follows: AT-8 phospho-tau S202&T205 (1:500, mouse monoclonal; Autogen Bioclear), AT-100 phospho-tau T212&S214 (1:100, mouse monoclonal; catalog #MN1060; Pierce), tau (1:100, Tau-5 mouse monoclonal; 1:250, Tau C-17 goat polyclonal; both from Santa Cruz Biotechnology), P35/P25 (1:500, rabbit monoclonal; catalog #2680; Cell Signaling Technology), HB9 (1:10,000, rabbit polyclonal; Dr. Samuel Pfaff, The Salk Institute, La Jolla, CA), Cux1 (1:20, rabbit polyclonal; catalog #SC-13024; Santa Cruz Biotechnology), Isl1 (1:750, mouse monoclonal; clone 40.2D6; Developmental Studies Hybridoma Bank), SV2 (1:500, mouse monoclonal; Developmental Studies Hybridoma Bank), choline acetyltransferase (ChAT; 1:100, goat polyclonal; catalog #AB144P; Millipore Bioscience Research Reagents), vesicular glutamate transporter 1 (VGluT1; 1:500, rabbit polyclonal; catalog #135303; Synaptic Systems), neurofilament (1:3000, chicken polyclonal; catalog #AB5539; Millipore), α-bungarotoxin (BTX; 1:1000, Alexa Fluor 594 conjugated; Invitrogen), Tuj1 (1:1000, mouse monoclonal; catalog #MRB-435P; Covance), synaptophysin (1:200, rabbit polyclonal; catalog #18-0130; Invitrogen), O4 (1:25; catalog #MAB345; Millipore), glial fibrillary acidic protein (GFAP; 1:500, rabbit polyclonal; catalog #Z0334; Dako), cleaved caspase-3 (1:500, rabbit polyclonal; catalog #PC679; Calbiochem), and Iba1 (1:1000, goat polyclonal; catalog #ab107159; Abcam). Secondary antibodies are from Jackson ImmunoResearch and used at 1:500 dilution.
To stain NMJs, flexor digitorum brevis (FDB), longissimus, and extensor digitorum longus (EDL) muscles were isolated and teased into layers of 5–10 fibers thick to facilitate penetration of antibodies. Presynaptic nerve terminals were labeled with anti-neurofilament and anti-synaptophysin or anti-tau antibodies. Acetylcholine receptors (AChRs) were labeled by BTX. Z-stack images of fluorescently labeled NMJs were captured at sequential focal planes 1 μm apart using a Zeiss LSM 510 META confocal microscope. All confocal images were taken using the same imaging parameters, including laser intensities, amplification gains, and offsets. To study excitatory synapse formation on spinal motor neurons, lumbar spinal cord segments (L1–L2) were dissected and processed for 80-μm-thick vibratome (Leica) sections. Motor neurons were labeled with anti-ChAT antibody and excitatory presynaptic terminals with anti-VGluT1. Glutamatergic synapses were identified as boutons apposed to the membrane of motor neuron soma and proximal dendrites. Statistical significance was determined using one-way ANOVA analysis with Tukey's HSD post hoc analysis.
Western blotting and immunoprecipitation.
Spinal cord samples or motor neurons were lysed in RIPA buffer (150 mm NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mm Tris, pH 7.5, 1 mm EDTA, 5 mm DTT, 10 mm NaF, and 1 mm Na3VO4) plus proteinase inhibitors (Roche) and PhosSTOP phosphatase inhibitors (Roche) to be used for Western blotting or immunoprecipitation buffer (200 mm NaCl, 0.4% Triton X-100, 0.7% CHAPS, 50 mm Tris, pH 8.0, 5 mm EDTA, and 5 mm DTT) for immunoprecipitation. Samples were homogenized by a Polytron handheld homogenizer (Kinematica) and quantified using the BCA protein assay kit (Pierce). To detect tau phosphorylation by Western blot analysis, 50 μg of tissue lysate in RIPA buffer were used. Flag-tagged WT and mutant tau were immunoprecipitated with anti-Flag M2 magnetic beads (catalog #M8823; Sigma) following the instructions of the manufacturer. All proteins were separated with 10% SDS-PAGE, transferred to PVDF membrane, and incubated with the indicated antibodies. Primary antibodies were diluted in TBS containing 0.05% Tween 20 and 5% bovine serum albumin (BSA) as follows: AT-8 anti-phospho-tau S202&T205 (1:500, mouse monoclonal; Autogen Bioclear), AT-100 anti-phospho-tau T212&S214 (1:100, mouse monoclonal; catalog #MN1060; Pierce), anti-tau (1:100, Tau-5 mouse monoclonal; 1:1000, Tau C-17 goat polyclonal; Santa Cruz Biotechnology), anti-Cdk5 (1:5, C-17 mouse monoclonal; Dr. Li-Huei Tsai, Massachusetts Institute of Technology, Boston, MA), and anti-GAPDH (1:1000, rabbit polyclonal; catalog #sc-25778; Santa Cruz Biotechnology). Horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10,000; Jackson ImmunoResearch) and Femto LUCENT plus HRP reagent kit (G-Biosciences) were used for exposure and quantification.
Morpholino knockdown and mRNA expression in zebrafish.
An antisense morpholino (MO1) was designed against the 5′ start sequence of zebrafish smn gene: 5′-CGACATCTTCTGCACCATTGGC-3′ (Gene Tools). An additional MO2 was designed against the 5′ UTR sequence of the smn gene: 5′-TTTAAATATTTCCCAAGTCCAACGT-3′ (McWhorter et al., 2003). WT tau, tau S202A,T205A, and tau S202E,T205E in pCS2 vector were linearized with NotI, and capped RNAs were generated using the SP6 mMESSAGE mMACHINE kit (Ambion). One-cell stage Tg(olig2:egfp) fish (Shin et al., 2003) embryos were injected with 4 ng of smn MO1 and 200 pg of WT or mutant tau mRNA. Embryos were fixed with 4% PFA at ∼28 h after fertilization. GFP-positive motor axons between myotomes 7 and 16 were imaged using a Zeiss LSM510 fluorescence confocal microscope. Similar to previous studies (Akten et al., 2011), we classify truncations or truncation plus abnormal branching of fish motor neuron axons as severe defects, abnormal branching without truncations as moderate defects lacking stereotyped morphology, but not branched or truncated as mild defects. The percentage of fish with at least 50% of their motor axons display severe or moderate defects was quantified. Fish with gross morphological defects were not included. Both sides of the fish trunk were scored. Statistical significance was determined using one-way ANOVA analysis with Tukey's HSD post hoc analysis.
Mouse primary spinal cord neuron and NSC-34 cell culture.
Primary neurons from mouse spinal cords were cultured in Neurobasal medium (Life Technologies) supplemented with B27 (Life Technologies). Briefly, spinal cords from E12.5 mouse embryos were dissected out and dissociated with 0.25% trypsin. After enriching motor neurons with Optiprep density gradient centrifugation and BSA cushion, cells were seeded on glass coverslips coated with 20 μg/ml poly-l-lysine (Sigma) and 8 μg/ml laminin (Sigma) and grown in the presence of 50 μg/ml BDNF, 50 μg/ml CNTF, and 25 μg/ml GDNF (PeproTech). The NSC-34 motor neuron cell line was generously provided by Dr. Neil Cashman (University of British Columbia, Vancouver, Canada; Cashman et al., 1992). Cells were cultured in DMEM (CellGro) supplemented with 3% fetal bovine serum at 37°C with 5% CO2. Cells were transfected with Lipofectamine 2000 (Life Technologies) following the instructions of the manufacturer. Cell death was detected by the In Situ Cell Death Detection TUNEL kit, TMR Red (Roche) 48 h after transfection.
Constructs.
The human tau40 (human 2N/4R tau) construct was used as the template to clone into pCS2–Flag vector for transfection and generating RNA. Mutant tau S202A,T205A and tau S202E,T205E were generated by using the QuickChange site-directed mutagenesis kit (Stratagene) and confirmed by sequencing.
In vitro kinase assay.
In vitro reconstitution kinase assay was performed as described previously (Ma et al., 2000). Bacterial recombinant His–tau was obtained from Prospec (catalog #Pro-295). Flag–tau and Flag–tau S202A,T205A were expressed in the NSC-34 motor neuron cell line. Cells were lysed 2 d after transfection. Anti-Flag M2 magnetic beads (catalog #M8823; Sigma) were used to purify Flag-tagged tau proteins. Cdk5/p25 kinase was purchased from SignalChem (catalog #C33-10G-10). Purified His–tau, Flag–tau, and Flag–tau S202A,T205A proteins were washed and suspended in kinase buffer containing 50 mm HEPES, pH 7.4, 5 mm MnCl2, 5 mm MgCl2, and 2 mm DTT added fresh and incubated with or without Cdk5/p25 in the presence of 100 μm ATP or 10 μm [γ-32P]ATP at 30°C for 15 min. Samples were then denatured in SDS sample buffer, separated on SDS-PAGE, and analyzed by autoradiography or Western blotting.
Results
Microtubule-associated protein tau is hyperphosphorylated in mouse SMA models and human SMA patients
The phosphorylation of tau has been implicated in a number of neurodegenerative disorders (Morris et al., 2011). Because SMA is characterized by spinal motor neuron degeneration, we examined the phosphorylation status of tau in SMA disease conditions, using two widely studied SMA mouse models: the Δ7 SMA mice (Smn−/−;SMN2tg/tg;SMNΔ7tg/tg; Le et al., 2005) and the Hung-Li SMA mice (Smn−/−;SMN2Hungtg/tg; Hsieh-Li et al., 2000). These SMA mouse models have been generated by expressing low levels of human SMN transgene in Smn knock-out mice to closely resemble the genetic state of human disease (Hsieh-Li et al., 2000; Le et al., 2005). Both mouse models recapitulate hallmarks of SMA pathogenesis at the molecular, cellular, and behavioral levels, including reduced SMN protein expression, spinal motor neuron degeneration, and motor behavioral defects (Hsieh-Li et al., 2000; Le et al., 2005). Diseased mice in both SMA models have an average lifespan of ∼13 d. We focused on the AT-8 antibody that recognizes tau phosphorylated on S202 and T205 (Goedert et al., 1994, 1995; Porzig et al., 2007) and postnatal day 9 (P9), when SMA disease symptoms begin to appear in these mouse models. S202 and T205 are located in the PR domain of tau (Fig. 1A), and their phosphorylation has been implicated in the pathogenesis of Parkinson's and Alzheimer's diseases and other tauopathies (Paudel et al., 1993; Bailey et al., 2013; Ferrer et al., 2014). Western blot analyses of spinal cords from P9 Δ7 SMA mice reveal that tau phosphorylation on S202 and T205 is significantly increased in mice affected by SMA (n = 4, p = 0.0006, Student's t test; Fig. 1B,C) but not in healthy littermate control. As a control, we used the AT-100 antibody that recognizes phosphorylated tau on T212 and S214, implicated previously in Alzheimer's disease (Zheng-Fischhöfer et al., 1998). We found that the level of phosphorylation on T212 and S214 of tau is similar between SMA and control mice (Fig. 1B,C), suggesting that increased tau phosphorylation is specific to S202 and T205. To test whether the hyperphosphorylation of tau is unique to the Δ7 SMA mice, we performed similar analyses using a different model, the Hung-Li SMA mice. The phosphorylation of tau on S202 and T205, but not on T212 and S214, is also significantly increased in the Hung-Li SMA mice at P9 (n = 4, p = 0.0003, Student's t test; Fig. 1D,E). These data suggest that increased phosphorylation of tau is likely associated with the general SMA pathology but not limited to a specific SMA mouse model. We also examined tau phosphorylation levels at earlier time points in both SMA mouse models and found increased phosphorylation of tau on S202 and T205, but not on T212 and S214, although this increase was not statistically significant (data not shown). More importantly, to test whether the hyperphosphorylation of tau on S202 and T205 is conserved in human SMA patients, we examined lysates from five postmortem spinal cords of type I human SMA patients and five age-matched control samples (Table 1). We have found that phosphorylation of tau on S202 and T205, but not T212 and S214, is specifically increased in human SMA patients (n = 5, p = 0.0009, Student's t test; Fig. 1F,G). Altogether, these data establish a critical correlation between tau hyperphosphorylation and SMA pathogenesis.
Figure 1.
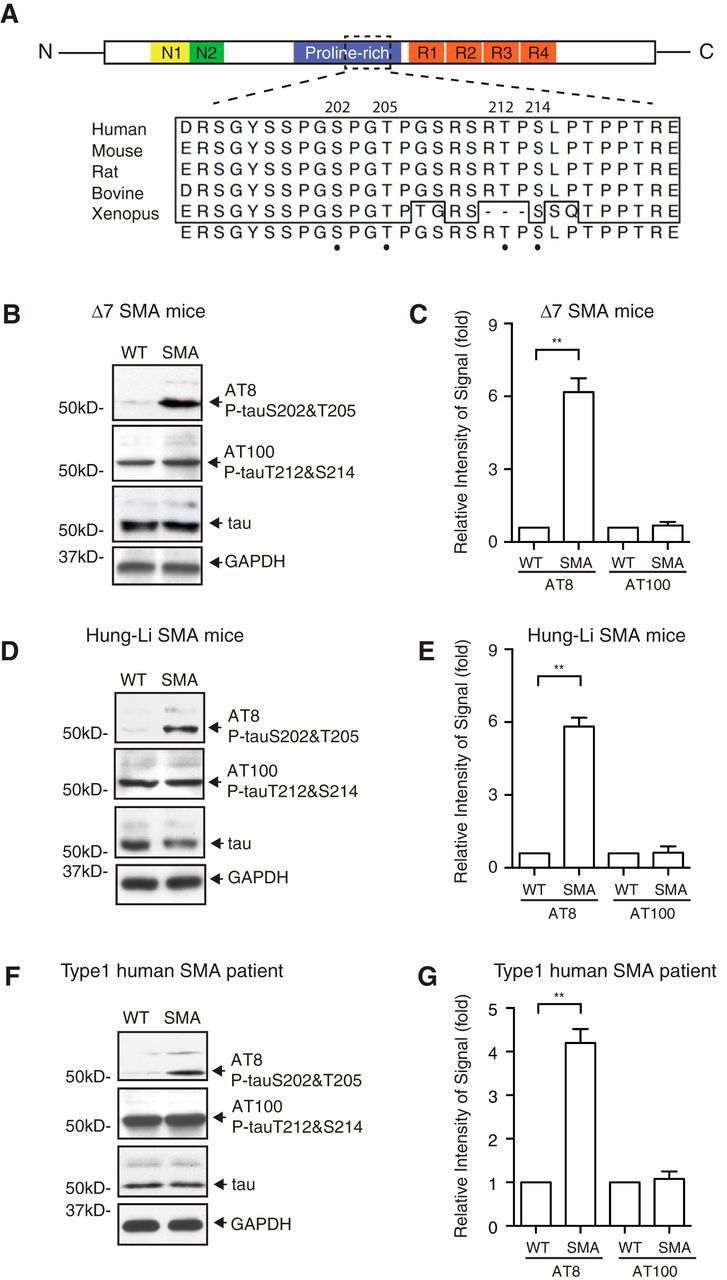
Microtubule-associated protein tau is hyperphosphorylated in SMA mouse models and human SMA patients. A, Functional domains and conserved phosphorylation sites of tau. Phosphorylated S202 and T205 recognized by the AT-8 antibody and phosphorylated T212 and S214 recognized by the AT-100 antibody are located in the PR domain of tau. Amino acid sequence containing S202 and T205 is conserved across species. N1, N2, N-Terminal regions of tau; R1, R2, R3, R4, microtubule-binding repeat sequences; N, N terminus; C, C terminus. Numbering is based on the longest human tau isoform with 441 residues. B, C, Western blot analysis of tau phosphorylation using spinal cord lysates from Δ7 SMA mice (Smn−/−;SMN2tg/tg;SMNΔ7tg/tg) at P9. Phosphorylation of tau on S202 and T205 as recognized by AT-8, but not on T212 and S214 as recognized by AT-100, is increased in mice affected by SMA. Data are quantified by densitometry from four independent experiments and are mean ± SEM. **p < 0.01, Student's t test. D–E, Western blot analysis of tau phosphorylation using spinal cord lysates from Hung-Li SMA mice (Smn−/−;SMN2Hungtg/tg) at P9. The phosphorylation of tau on S202 and T205, but not on T212 and S214, is increased in the Hung-Li SMA mice. Data are quantified by densitometry from four independent experiments and are mean ± SEM. **p < 0.01, Student's t test. F, G, Western blot analysis of tau phosphorylation in type I human SMA patient spinal cord lysates and age-matched control samples. The phosphorylation of tau on S202 and T205, but not on T212 and S214, is upregulated in humans affected by SMA. Data are quantified from three independent experiments and are mean ± SEM. **p < 0.01, Student's t test.
Increased phosphorylation of tau on S202 and T205 occurs specifically in SMA motor neurons without forming aggregates
Our finding that the phosphorylation of tau on S202 and T205 is increased significantly in SMA patients and mouse models prompted us to further investigate the localization of phosphorylated tau, which may shed light on the functional consequence of these phosphorylation events. Hyperphosphorylated tau has been associated with protein filaments that form neurofibrillary tangles in Alzheimer's disease and related neurodegenerative tauopathies (Grundke-Iqbal et al., 1986; Nukina and Ihara, 1986; Wood et al., 1986; Kondo et al., 1988; Lee et al., 1991; Vanderweyde et al., 2012). To examine the localization and potential aggregation of phosphorylated tau in SMA, we performed immunostaining with the AT-8 (phospho-S202&T205) antibody on spinal cord sections from SMA mice and human SMA patients. We have found that, at P9 in both the Δ7 SMA mice (Fig. 2E–H) and the Hung-Li SMA mice (Fig. 2I–L), tau phosphorylation on S202 and T205 is increased significantly in spinal motor neurons, which are marked by the motor neuron-specific transcription factor HB9 (Fig. 2B,F,J,N). However, little tau hyperphosphorylation is detected in layer II/III cortical neurons as marked by Cux1 in P9 Δ7 SMA mice (Fig. 2M′–P′). In addition, using immunostaining and Western blot analysis, we found similar low levels of tau phosphorylation on S202 and T205 recognized by the AT-8 antibody in cultured spinal cord neurons from SMA and control mice (data not shown), suggesting that the in vivo signaling environment is essential for maintaining tau hyperphosphorylation. Previous studies have suggested that tau is located predominantly in neurons and less so in glia (Lee et al., 2001), which we confirmed by performing coimmunostaining using antibodies recognizing tau (Tau-5), phosphorylated tau (AT-8), the astrocyte marker GFAP, the microglia marker IbaI, and the oligodendrocyte marker O4. No hyperphosphorylated tau is observed in astrocytes, microglia, or oligodendrocytes (data not shown). To investigate whether the increased tau phosphorylation is also conserved in humans, we stained human SMA patient spinal cord sections with antibodies for phosphorylated tau and the motor neuron marker ChAT, because the anti-mouse HB9 antibody does not recognize human HB9. Similar to SMA mice, enhanced tau phosphorylation on S202 and T205, but not T212 and S214, is detected specifically in spinal motor neurons from SMA patients (Fig. 2Q–X). Interestingly, none of the hyperphosphorylated tau forms fibrillary tangles or other forms of aggregates, unlike other tauopathies. These findings indicate that tau is hyperphosphorylated without forming aggregates in motor neurons affected by SMA, even at late stages of the disease.
Figure 2.
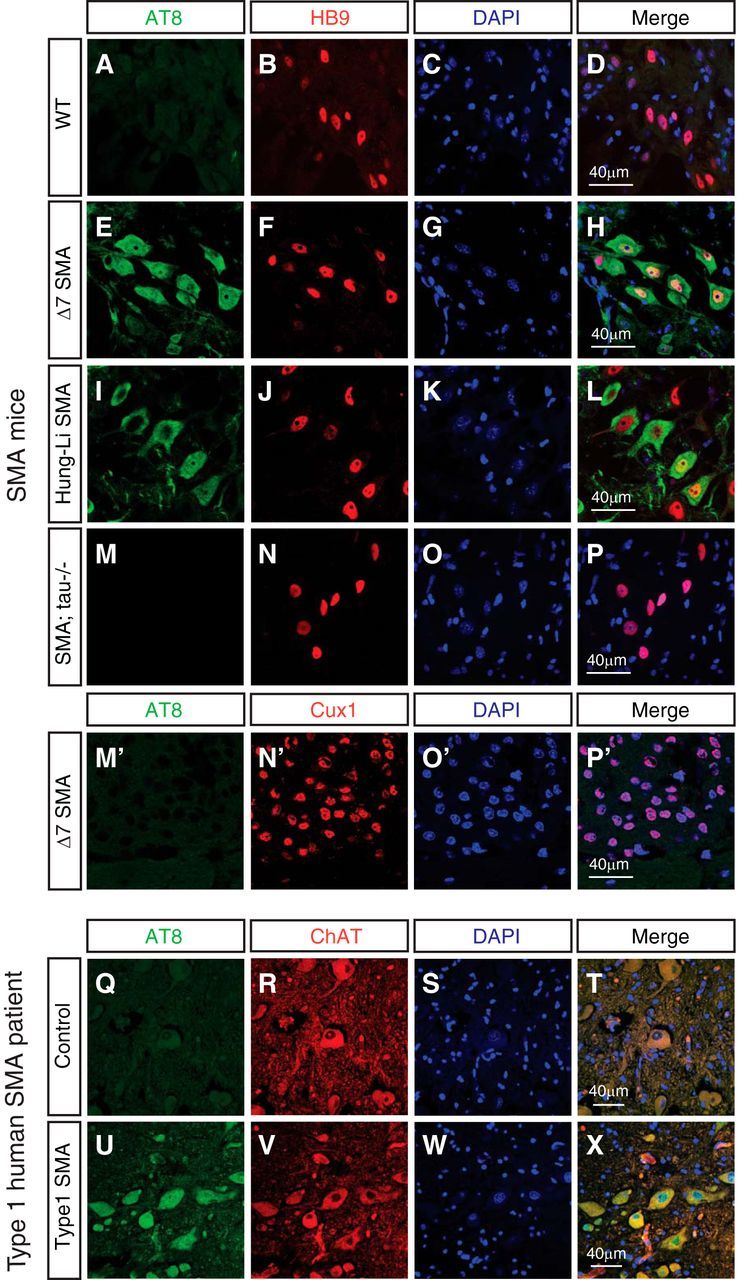
Increased phosphorylation of tau on S202 and T205 occurs specifically in motor neurons affected by SMA without forming aggregates. A–P′, Immunostaining of WT, SMA, and tau−/−;SMA compound mutant mouse spinal cords and cortices with the AT-8 antibody recognizing tau phosphorylated on S202 and T205. Spinal cord sections from P9 WT mice (A–D), Δ7 SMA mice (E–H), Hung-Li SMA mice (I–L), and tau−/−;SMA compound mutant mice (M–P) are stained with AT-8 (green) and the motor neuron-specific HB9 antibody (red). The phosphorylation of tau on S202 and T205 is increased in spinal motor neurons in both SMA mouse models (A–L) but not in the tau−/−;SMA mice (M–P). Immunostaining of cortex sections from P9 Δ7 SMA mice (M′–P′) with AT-8 (green) and the layer II/III cortical neuronal marker Cux1 (red) shows minimal tau hyperphosphorylation detected in layer II/III cortical neurons. Data shown are representative images from eight sets of mice in four independent experiments. Q–X, Immunostaining of spinal cord sections from type I human SMA patients (U–X) and age-matched control samples (Q–T) with the AT-8 (phospho-tauS202&T205) antibody. Tau phosphorylated on S202 and T205 (green) is increased in ChAT-positive (red) motor neurons affected in SMA patients. Data shown are representative images from four sets of samples in four independent experiments.
In addition to expression in neuronal cell bodies, microtubule-associated protein tau is localized in axons. The sciatic nerve is the largest nerve containing the axons of spinal motor neurons that innervate hindlimb muscles. To test whether levels of tau phosphorylated on S202 and T205 are increased in axons in SMA, we performed immunostaining of the sciatic nerve from P9 Δ7 SMA mice and healthy littermate controls using the AT-8 antibody. We found no increased tau phosphorylation in the sciatic nerve (Fig. 3A–H). Similarly, Western blot analysis of the sciatic nerve from P9 Δ7 SMA mice and WT littermates revealed similar low levels of tau phosphorylated on S202 and T205 (data not shown; n = 3, p = 0.46, Student's t test). Spinal motor neurons extend their axons over long distances to innervate muscle fibers at NMJs. In SMA mouse models, neurofilament accumulation and delayed endplate maturation are observed in NMJs (Cifuentes-Diaz et al., 2002; Kariya et al., 2008; Ling et al., 2012). Furthermore, defective colocalization of presynaptic motor axon and postsynaptic AChRs has been identified on muscle fibers of mice with SMA (Cifuentes-Diaz et al., 2002; Kariya et al., 2008; Kong et al., 2009; Ling et al., 2012). To investigate whether phosphorylated tau exists as aggregated protein in SMA mice NMJs, colocalization of phosphorylated tau and AChRs in P12 Δ7 SMA mouse EDL muscle and longissimus muscle was examined. As shown in Figure 3I–P, there is no increase of tau phosphorylation or tau aggregation in SMA mice NMJs compared with non-diseased mice, suggesting that non-aggregating tau phosphorylated on S202 and T205 is increased specifically in spinal motor neurons affected by SMA.
Figure 3.

Phosphorylation of tau on S202 and T205 is not increased in the sciatic nerve or NMJs of SMA mice. A–H, Immunostaining of WT and SMA mouse sciatic nerves with the AT-8 antibody recognizing tau phosphorylated on S202 and T205. Sciatic nerve cross-sections from P9 WT mice (A–D) and Δ7 SMA mice (E–H) are stained with the AT-8 (green) antibody and TuJ1 (red) antibody recognizing neuronal β-III tubulin. The phosphorylation of tau on S202 and T205 is at similarly low levels in the sciatic nerves of SMA and control mice. I–P, Immunostaining of NMJs of Δ7 SMA mice at P12 with antibodies recognizing tau and phosphorylated tau (AT-8). At NMJs of the EDL muscle, little phosphorylation on S202 and T205 (I, M, green) or aggregates of phosphorylated tau is detected in either WT or SMA mice at the end stage of SMA pathology (P12). AChRs labeled by BTX (J, N, red) show poor organization in SMA mice (N) compared with WT mice (J). Arrows highlight the junction point at which motor axons enter NMJs (K–P).
Cdk5 mediates the phosphorylation of tau on S202 and T205 in SMA
In addition to the localization of hyperphosphorylated tau, it is critical to identify the protein kinase that mediates these phosphorylation events, which may shed light on mechanisms underlying motor neuron degeneration in SMA. Previous studies in Alzheimer's disease suggest that the phosphorylation of tau could be mediated by multiple kinases, including glycogen synthase kinase 3 and Cdk5 (Baumann et al., 1993; Paudel et al., 1993; Michel et al., 1998). Recently, we found that Cdk5 kinase activity is upregulated in both SMA mouse models and human SMA-induced pluripotent stem cell-derived motor neurons (N. Miller and Y.-C. Ma, unpublished observations), which motivated us to focus on the role of Cdk5 in mediating the phosphorylation of tau on S202 and T205. To test whether Cdk5 can directly phosphorylate tau, we incubated purified His-tagged human tau protein from bacteria together with Cdk5/p25 kinase in a kinase assay reaction. p25 is a potent Cdk5 activator that is generated during cleavage of activating subunit p35 by Ca2+-dependent protease calpain (Lew et al., 1994; Tsai et al., 1994; Lee et al., 2000). Recombinant Cdk5/p25 robustly phosphorylates WT tau directly on S202 and T205, as recognized by the AT-8 antibody (Fig. 4A). In addition, expression of Cdk5/p25 together with Flag-tagged human tau in mouse primary spinal cord neurons leads to hyperphosphorylation of WT tau on S202 and T205 (Fig. 4B,C). The phosphorylation of WT tau on S202 and T205 in mouse spinal cord neurons can be reduced by the Cdk5 inhibitor roscovitine (n = 4, p < 0.0001, one-way ANOVA with Tukey's HSD post hoc analysis; Fig. 4B,C), suggesting that Cdk5 phosphorylates tau on S202 and T205 in spinal cord neurons. To examine the role of Cdk5 in mediating tau phosphorylation in vivo, we used Cdk5 knock-out mice. Because Cdk5 null mice are perinatal lethal (Ohshima et al., 1996), which precludes the analysis of postnatal motor neurons, we used a conditional Cdk5 knock-out generated by crossing Cdk5F/F mice (Samuels et al., 2007) with Nestin–Cre mice (Tronche et al., 1999), which express Cre in neuronal cells and survive until adulthood. In spinal cord lysates from the conditional knock-out mice (Cdk5F/F;Nestin–Cre), Cdk5 expression and tau phosphorylation on S202 and T205 were reduced significantly (n = 4, p < 0.0001, Student's t test; Fig. 4D,E). Furthermore, to test whether the increased phosphorylation of tau on S202 and T205 in SMA mice can also be rescued by reducing Cdk5 kinase activity, we genetically removed the Cdk5 activating subunit p35 in the Hung-Li SMA mice (Smn−/−;SMN2Hungtg/tg). The kinase activity of Cdk5 is dictated by its regulatory subunit p35 and p25 (Lew et al., 1994; Tsai et al., 1994; Lee et al., 2000; Su and Tsai, 2011). Increased conversion of p35 to p25 has been implicated in pathological conditions, including Alzheimer's disease (Patrick et al., 1999). We found that the level of p25 was increased in the Hung-Li SMA mice (Fig. 4F). In addition, using a compound mutant mouse line in which p35 is removed genetically in the Hung-Li SMA mice (p35−/−;Smn−/−;SMN2Hungtg/tg or p35−/−;SMA), we found that hyperphosphorylation of tau on S202 and T205 is reduced significantly (n = 4, p < 0.0001, one-way ANOVA with Tukey's HSD post hoc analysis; Fig. 4F,G). Together, our genetic, pharmacological, and biochemical evidence demonstrate that Cdk5 mediates the increased phosphorylation of tau on S202 and T205 in SMA disease conditions.
Figure 4.
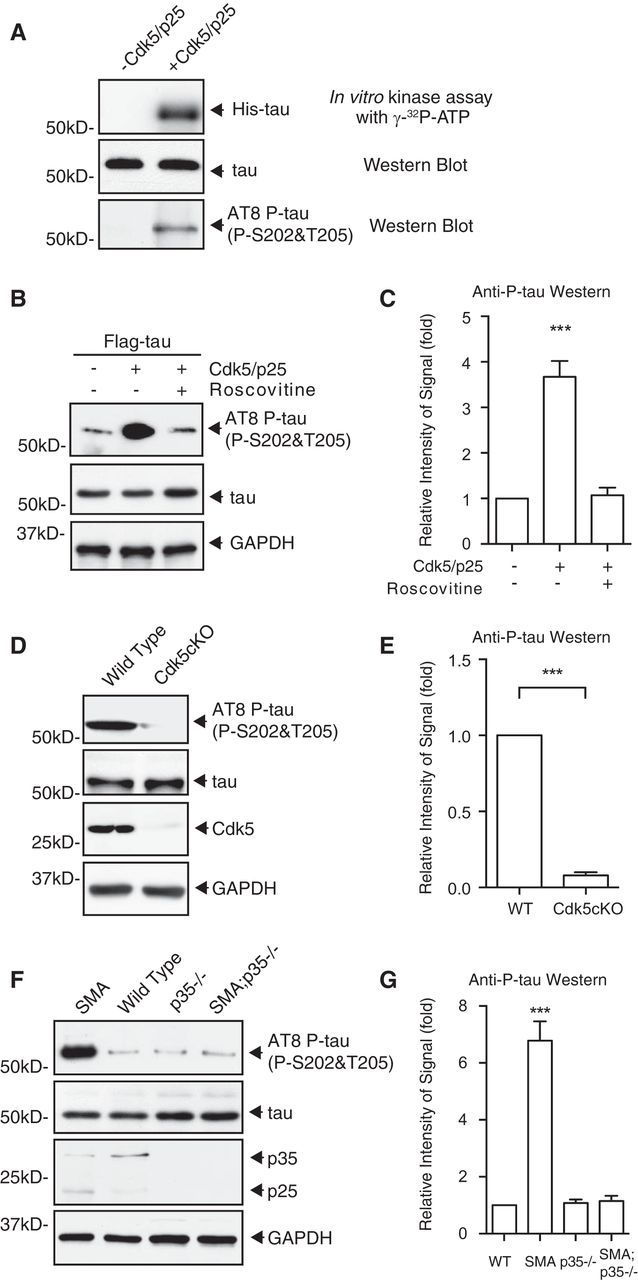
Cdk5 mediates the increased phosphorylation of tau on S202 and T205 in SMA disease conditions. A, Recombinant tau can be phosphorylated directly by Cdk5 on S202 and T205 in vitro. Autoradiograph showing that His-tagged tau purified from bacteria is phosphorylated by recombinant Cdk5/p25 in a reconstitution kinase assay with [γ-32P]ATP. Western blot analysis with the AT-8 antibody reveals that tau is phosphorylated on S202 and T205 by Cdk5/p25 in the in vitro kinase assay. B, C, Western blot analysis of tau phosphorylation on S202 and T205 by Cdk5 in motor neurons. Expression of Cdk5 together with its activating subunit p25 in cultured mouse motor neurons leads to increased phosphorylation of WT tau on S202 and T205 as recognized by the AT-8 antibody but not the tauS202A,T205A mutant. The Cdk5 inhibitor roscovitine reduces the phosphorylation of tau on S202 and T205. Data shown in C are quantified by densitometry from four independent experiments and are mean ± SEM. ***p < 0.0001, one-way ANOVA with Tukey's HSD post hoc analysis. D, E, Western blot analysis of tau phosphorylation on S202 and T205 in neuronal tissue-specific Cdk5 knock-out mice (Cdk5F/F;Nestin–Cre or Cdk5KO). The phosphorylation of tau on S202 and T205 recognized by the AT-8 antibody is reduced significantly in spinal cord lysates of Cdk5 neuronal tissue-specific knock-out mice. Data shown in E are quantified by densitometry from four independent experiments and are mean ± SEM. ***p < 0.0001, Student's t test. F, G, Western blot analysis of tau phosphorylation on S202 and T205 in p35−/−;Smn−/−;SMN2Hungtg/tg or p35−/−;SMA compound mutant mice. The increased phosphorylation of tau on S202 and T205 is significantly rescued by genetic knock-out of Cdk5 activating subunit p35 in Hung-Li SMA mice. Data shown in G are quantified by densitometry from four independent experiments and are mean ± SEM. ***p < 0.0001, one-way ANOVA with Tukey's HSD post hoc analysis.
Cdk5-mediated tau phosphorylation leads to degeneration in cultured mouse spinal cord neurons and motor neuron defects in a zebrafish SMA model in vivo
We next sought to examine whether an increase in Cdk5-mediated tau phosphorylation induces motor neuron degeneration. We expressed Cdk5 and tau together with GFP in cultured mouse primary spinal cord neurons and measured neurodegeneration by TUNEL assay. Neurons transfected with Cdk5, p25, and WT tau show significantly increased cell death (Fig. 5A–E) compared with cells transfected with WT tau alone (p < 0.0001, Student's t test; WT tau, n = 21; WT tau plus Cdk5, n = 31). In contrast, the expression of the phosphorylation-deficient tau S202A,T205A mutant (tau A) together with Cdk5/p25 leads to significantly reduced cell death compared with cells transfected with Cdk5/p25 and WT tau (p < 0.0001, Student's t test; WT tau plus Cdk5, n = 31; tau A plus Cdk5, n = 15; Fig. 5E). To test whether increased tau phosphorylation might be sufficient to promote primary spinal cord neuron death, we overexpressed the phosphorylation mimetic tau S202E,T205E mutant (tau E) and observed significantly increased cell death compared with expressing WT tau alone (p < 0.0001, Student's t test; WT tau, n = 21; tau E, n = 15; Fig. 5E). Altogether, these data suggest that increased phosphorylation of tau on S202 and T205 promotes degeneration in mouse primary spinal cord neurons.
Figure 5.
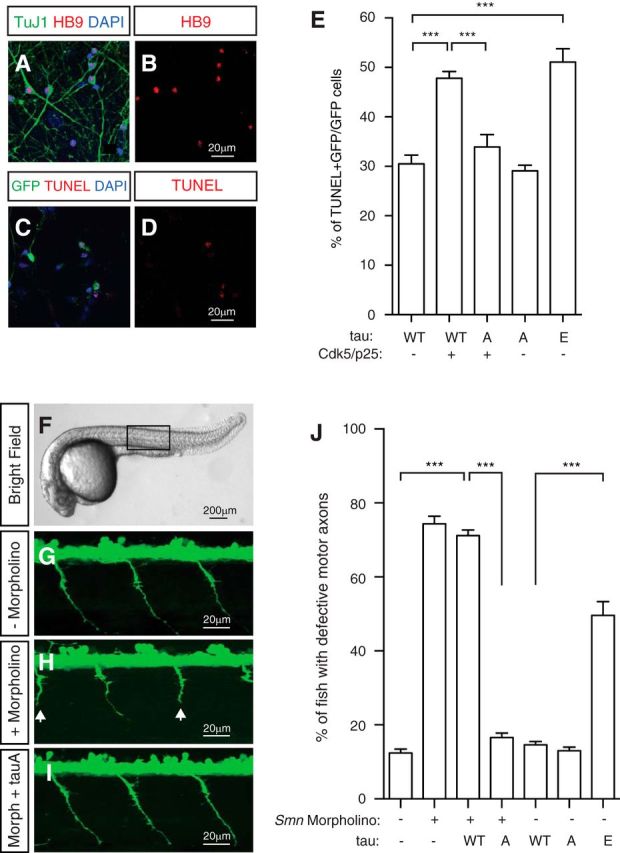
Phosphorylation of tau on S202 and T205 is critical for motor neuron functions in vitro and in vivo. A–E, Increased tau phosphorylation by Cdk5 promotes degeneration of mouse spinal cord neurons. Cultured mouse spinal cord neurons (A, B) can be recognized by antibodies to the motor neuron marker HB9 (A, B, red) and the pan-neuronal marker TuJ1 (A, green). TUNEL staining (C, D, red) shows degeneration induced by expressing tau constructs together with Cdk5/p25 and GFP. Expression of WT tau, but not the phosphorylation-deficient tauS202A,T205A mutant, together with Cdk5/p25 or expressing the phosphorylation mimetic tauS202E,T205E mutant leads to significantly increased neurodegeneration. Quantified results (E) are from five independent experiments and 6116 neurons and are mean ± SEM. ***p < 0.0001, Student's t test. F–J, Phosphorylation-deficient tauS202A,T205A mutant rescues motor neuron defects in a zebrafish SMA model in vivo. Antisense MO knockdown of Smn in Tg(olig2:egfp) transgenic zebrafish leads to truncations (white arrows in H) or abnormal branching of motor axons. G–I correspond to regions similar to the boxed area in F. Motor neuron defects can be rescued by the expression of RNA encoding the phosphorylation-deficient mutant tauS202A,T205A (J). Expression of tauS202E,T205E leads to significantly increased motor axon defects in zebrafish (J). Data are from five independent experiments and 572 fish and are mean ± SEM. ***p < 0.0001, Student's t test.
Next we asked whether Cdk5-mediated tau phosphorylation drives motor neuron defects in an in vivo model of SMA. Antisense MO knockdown of smn in zebrafish recapitulates many aspects of motor neuron defects in SMA disease, including truncation and ectopic branching of motor axons (McWhorter et al., 2003). These motor neuron defects can be rescued by the expression of WT SMN but not the mutated Δ7SMN present in human patients (McWhorter et al., 2003). To examine the effect of tau phosphorylation, we injected RNA encoding the nonphosphorylatable tau S202A,T205A mutant and the phosphorylation mimetic tau S202E,T205E mutant together with smn MO into one-cell stage Tg(olig2:egfp) transgenic zebrafish embryos. Motor neuron axons of Tg(olig2:egfp) fish are labeled specifically by GFP (Shin et al., 2003). Expression of tau S202E,T205E leads to significantly increased motor axon truncation defects (n = 5 experiments; p < 0.0001, Student's t test; Fig. 5F–J), suggesting that increased phosphorylation of S202 and T205 is sufficient for inducing motor neuron deficits in vivo. In addition, the expression of nonphosphorylatable tau S202A,T205A mutant rescues motor axon defects caused by MO knockdown of smn in zebrafish, indicating the importance of tau phosphorylation in mediating motor neuron defects in vivo (n = 5 experiments, p < 0.0001, Student's t test; Fig. 5J).
Genetic knock-out of tau in an SMA mouse model rescues synaptic stripping on motor neurons, NMJ denervation, and motor neuron degeneration in vivo
Phosphorylated tau is a major component of the paired helical filament and neurofibrillary tangles in Alzheimer's disease (Goedert et al., 1988, 2006). Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer's disease mouse model (Roberson et al., 2007). To further explore the role of tau in SMA pathogenesis, we examined the effects of genetically removing tau in SMA mice on disease phenotypes. Because of functional redundancies among different forms of microtubule-associated proteins, tau knock-out mice show a normal lifespan with hyperactivity, impairment of fear conditioning, and defective axonal outgrowth (Ikegami et al., 2000; Dawson et al., 2001). We crossed the tau knock-out mice (Dawson et al., 2001) with the Hung-Li SMA mice to generate the tau−/−;Smn−/−;SMN2Hungtg/tg or tau−/−;SMA compound mutant mice and examined various SMA phenotypes. In SMA pathogenesis, motor neuron degeneration is preceded by other defects, including reduced excitatory synapse formation (synaptic stripping) on motor neurons and NMJ denervation (Kariya et al., 2008; McGovern et al., 2008; Ling et al., 2010, 2012; Mentis et al., 2011). Accordingly, we first tested whether genetic knock-out of tau could rescue the synaptic stripping defects in SMA mice. The number of glutamatergic synapses on L1–L2 level motor neurons was quantified by the colocalization of the excitatory glutamate synapse marker VGluT1 with the motor neuron cell body marker ChAT (Fig. 6A–E). We found that excitatory synaptic boutons on motor neurons were reduced significantly in SMA mice compared with WT littermate controls. Importantly, excitatory synapse formation on the tau−/−;SMA compound mutant mice is restored to the WT level (p < 0.0001, one-way ANOVA with Tukey's HSD post hoc analysis; WT, n = 40; tau−/−, n = 35; SMA, n = 61; SMA;tau−/−, n = 70; Fig. 6A–E), suggesting that tau knock-out rescues the excitatory synapse stripping phenotype in SMA mice. In addition to synaptic defects, another key pathogenic phenotype of SMA disease is innervation deficiencies of the NMJ by motor axons (Kariya et al., 2008; Ling et al., 2010, 2012). In the zebrafish model of SMA, we also observed motor axon defects induced by tau phosphorylation (Fig. 5F–J). These findings prompted us to examine the functional consequence of tau knock-out on NMJ phenotypes in SMA mice. In the Δ7 SMA mouse model, NMJ denervation was found in many muscles, including the FDB controlling the second digit (FDB-2; Ling et al., 2012). Thus, we examined the innervation of FDB-2 NMJ in WT, SMA, tau−/−, and tau−/−;SMA mice. NMJs were stained with anti-synaptophysin and anti-neurofilament antibodies to label presynaptic structures and BTX to mark postsynaptic AChRs (Fig. 6F–I). NMJs on FDB-2 muscles are innervated fully in WT and tau−/−mice but show significantly increased denervation or partial innervation in the Hung-Li SMA mice (Fig. 6J). The levels of partially or fully denervated NMJ in the Hung-Li SMA mice were lower than those reported for the Δ7 SMA mice, likely as a result of genetic background and strain differences between these mouse models (Ling et al., 2012). Importantly, FDB-2 NMJ innervation in tau−/−;SMA mice was rescued back to WT levels, indicating that genetic knock-out of tau rescues NMJ defects in SMA mice (p < 0.0001, one-way ANOVA with Tukey's HSD post hoc analysis; Fig. 6F–J). More than 100 NMJs on the FDB-2 muscle from each mouse were used. Four mice for each genotype were analyzed.
Figure 6.
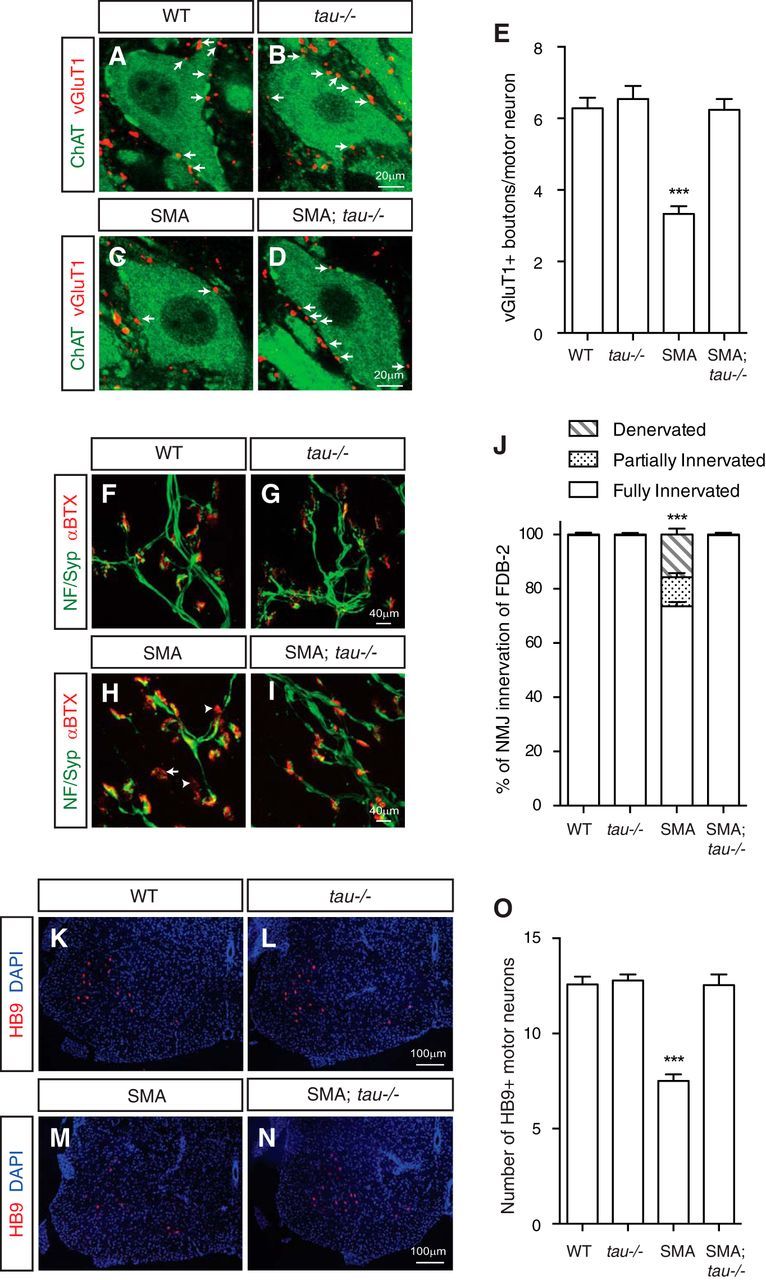
Genetic knock-out of tau rescues synaptic and NMJ defects and motor neuron degeneration in SMA mice. A–E, Immunostaining of excitatory glutamatergic synapse formation on spinal cord motor neurons. Colocalization of the glutamatergic synapse marker VGluT1 (A–D, red) with the motor neuron marker ChAT (A–D, green) shows that excitatory synaptic boutons (arrows) on motor neurons are reduced significantly in SMA mice compared with WT littermates. The synaptic defect is rescued to the WT level by genetic knock-out of tau in SMA mice. Quantification in E is from three to five mice from each genotype and are mean ± SEM. ***p < 0.0001, one-way ANOVA with Tukey's HSD post hoc analysis. F–J, Staining of motor neuron axon innervation of NMJs. Colocalization of presynaptic nerve terminals marked by immunostaining with anti-neurofilament (NF) and anti-synaptophysin (Syp) antibodies (F–I, green) with postsynaptic AChRs labeled by BTX (F–I, red) shows increased denervation (arrowheads) and partial innervation (arrow) of NMJs on the FDB-2 muscle of SMA. The NMJ innervation defect is rescued to the WT level by genetic knock-out of tau in SMA mice. Quantification in J is from >100 NMJs on the FDB-2 muscle from each mouse. Four mice for each genotype were used. Denervated, partially innervated, and fully innervated NMJs were analyzed separately. Results are mean ± SEM. ***p < 0.0001, one-way ANOVA with Tukey's HSD post hoc analysis. K–O, Immunostaining of mouse spinal cord sections with the motor neuron marker HB9 to quantify motor neuron degeneration. Lumbar level spinal cord sections of P9 SMA mice show a near 40% reduction of HB9-positive (K–N, red) motor neurons compared with WT littermates. Genetic knock-out of tau in SMA (tau−/−;SMA compound mutant) mice rescues motor neuron degeneration to the WT level (O). Quantification in O is from 10 to 15 sections of four mice from each genotype in four independent experiments and are mean ± SEM. ***p < 0.0001, one-way ANOVA with Tukey's HSD post hoc analysis.
NMJ denervation and synaptic stripping have been suggested to contribute to motor neuron degeneration in SMA disease (Kariya et al., 2008; McGovern et al., 2008; Ling et al., 2010; Mentis et al., 2011). To examine motor neuron degeneration in SMA mice, lumbar level spinal cord sections were stained with antibodies recognizing motor neuron-specific transcription factor HB9. Quantification of HB9-expressing spinal motor neurons showed a dramatic reduction in SMA mice compared with WT or tau−/− control littermates, reflecting motor neuron degeneration in SMA disease conditions (p < 0.0001, one-way ANOVA with Tukey's HSD post hoc analysis; WT, n = 129; tau−/−, n = 139; SMA, n = 144; SMA;tau−/−, n = 186; Fig. 6K–O). No significant motor neuron degeneration was detected in tau knock-out mice. Remarkably, the number of motor neurons in tau−/−;SMA compound mutant mice is restored to WT levels, suggesting a rescuing effect of tau knock-out on motor neuron degeneration in SMA pathogenesis (Fig. 6O). The survival, body weight, and behavioral defects are not rescued in the tau−/−;SMA double mutant mice, similar to findings of other genetic rescue studies (Paez-Colasante et al., 2013). One potential explanation for the persisting defects is that genetic removal of tau did not rescue tau−/−;SMA mouse defects in other organs, including the heart, caused by ubiquitous reduction of SMN protein, because tau is expressed specifically in neurons (Rudnik-Schöneborn et al., 2008; Bevan et al., 2010; Heier et al., 2010; Shababi et al., 2010). Altogether, results from these experiments suggest that genetic knock-out of tau rescues spinal motor neuron degeneration and defective synapse formation on motor neurons in SMA mice.
Discussion
Although phosphorylated tau has well established roles in tauopathies, including Alzheimer's disease, the phosphorylation of tau in SMA pathogenesis has not been explored. Using two SMA mouse models and human SMA patient spinal cord samples, we show that the phosphorylation of tau on S202 and T205 is significantly increased in motor neurons affected by SMA. The expression of tau with enhanced phosphorylation on S202 and T205 by Cdk5 leads to motor neuron defects in zebrafish and mouse motor neurons, whereas tau deficient for phosphorylation alleviates motor neuron defects. Interestingly, phosphorylated tau does not form aggregates in either human SMA patients or SMA mice even at late stages of the disease, distinguishing it from other tauopathies.
The functions of non-aggregating tau
One of the most unexpected findings from this study is that phosphorylated tau does not form aggregates in SMA disease conditions. Traditionally, tau aggregates are considered a pathologic hallmark of tauopathies. However, increasing evidence suggests that tau inclusions may not be responsible for neurodegeneration. In a regulatable tau transgenic mouse line, inhibiting tau production after filamentous tau inclusion formation reversed learning behavior impairments, although tau inclusion formation progressed (Santacruz et al., 2005). It has also been reported that decreasing the level of soluble tau reduces caspase activation in inclusion-positive neurons without affecting the number or size of tau aggregates, suggesting that non-aggregating tau may activate the proapoptotic pathways (de Calignon et al., 2010). In addition, studies in transgenic flies indicate that soluble tau species, possibly dimers, confer toxicity (Feuillette et al., 2010). Therefore, small soluble tau complexes or tau with aberrant conformation, rather than tau aggregates, may be the real trigger of cell death pathways and neurodegeneration. This is consistent with our finding that tau phosphorylated on S202 and T205 can increase motor neuron degeneration without forming precipitates (Fig. 2).
Motor neuron-specific increase of tau phosphorylation
We have found that increased tau phosphorylation on S202 and T205 is localized primarily in SMA disease motor neurons. This is likely a result of both the neuronal-specific expression of tau and the restricted increase of Cdk5 activity in motor neurons affected by SMA. Previous studies have suggested that tau is expressed primarily in neurons but show limited expression in glial cells (Trojanowski et al., 1989; Tashiro et al., 1997; Klein et al., 2002), which we validated via immunostaining in which we detected little phosphorylated tau detected in glial cells at P9 in the spinal cords of WT or SMA mice. In addition to the neuronal-specific expression of tau, our group recently found that Cdk5 activity was increased in spinal motor neurons affected by SMA (Miller and Y.-C. Ma, unpublished observations), which may also contribute to the motor neuron-specific increase of tau phosphorylation. It is worth noting that some spinal cord sections show high background fluorescent signals in both WT and SMA mice when mouse monoclonal (including the AT-8 anti-phosphorylated tau) antibodies are used. We have occasionally found similar patterns of staining on control samples using just the anti-mouse secondary antibody, suggesting that these background signals are likely from unspecific cross-reactions between secondary antibodies and mouse tissue sections. We found that efficient perfusion of mice before fixation was essential for reducing these unspecific background staining. Regardless of background signals in the neuropil, phosphorylation of tau on S202 and T205 is always significantly higher in motor neurons affected by SMA than those from control mice.
The motor neuron-specific increase of tau phosphorylation in SMA mice does not exclude the possibility that non-neuronal cells contribute to motor neuron degeneration. In SMA conditions, changes in non-neuronal cells, such as microglia, may lead to increased Cdk5 activity and tau phosphorylation in motor neurons in a non-cell-autonomous manner. This possibility is consistent with observations in neuronal-specific Smn knock-out mice, which show NMJ organization defects but not tau phosphorylation changes at P30 (Cifuentes-Diaz et al., 2002). These Nestin–Cre;SmnF/F mice survive until 45 d of age and display milder degeneration of motor neurons: ∼30% decrease at P30 compared with 42% reduction of spinal motor neurons at P9 observed in Hung-Li SMA mice (Fig. 6). The phenotypic differences between these two mouse lines could be attributable to contributions of non-neuronal cells to motor neuron degeneration in Hung-Li SMA mice. Because the best characterized function of SMN is in snRNP biogenesis and pre-mRNA splicing, we are currently using RNA isolated from motor neurons and glia of SMA mice together with high throughput sequencing to study splicing defects that may lead to the disruption of Ca2+ homeostasis and increased Cdk5 phosphorylation of tau.
Motor neuron degeneration caused by tau phosphorylation
How does the hyperphosphorylation of tau contribute to motor neuron degeneration in SMA? Possible explanations include the disruption of tau-interacting pro-survival signaling complexes, increased excitotoxicity induced by tau phosphorylation, and the dysregulation of motor neuron axonal transport. First, increased tau phosphorylation in SMA motor neurons may lead to degeneration by disrupting pro-survival signaling pathways. Tau has been shown to act as a protein scaffold to bind signaling complexes through its PR domain (Lee et al., 1998; Reynolds et al., 2008). Therefore, phosphorylation of tau on S202 and T205 located in its PR domain (Fig. 1A) may alter the localization and activity of tau binding partners, including phosphatidylinositol 3-kinase, the growth factor receptor scaffolding protein Grb2, and Src family kinases that are critical for neuronal survival (Lee et al., 1998; Reynolds et al., 2008). Second, increased phosphorylation of tau could result in excitotoxicity in motor neurons affected by SMA. It has been reported that tau is required for targeting the Src family kinase Fyn to postsynaptic sites to phosphorylate the NR2B subunit of NMDA receptors to prevent NMDA receptor-mediated excitotoxicity (Ittner et al., 2010). In addition, phosphorylation promotes the entrance of tau into dendritic spines to impair miniature EPSCs (Hoover et al., 2010). Thus, increased phosphorylation of tau could disrupt the balance between excitatory and inhibitory synaptic inputs on motor neurons, leading to excitotoxicity in SMA. This possibility is consistent with our observation that defective glutamatergic synapse formation on SMA motor neurons, which may contribute to excitotoxicity in SMA disease condition (Mentis et al., 2011), can be rescued by genetic knock-out of tau in SMA mice (Fig. 6). It is worth noting that the lifespan of SMA mice was not rescued significantly by tau knock-out. This is likely because tau hyperphosphorylation-induced defects are restricted to neurons; other pathological deficiencies in SMA mice, including those in the cardiovascular systems, cannot be alleviated by the genetic removal of tau. Finally, phosphorylation of tau could impair microtubule assembly in axons and microtubule-dependent axonal transport. The phosphorylation of tau on S202 and T205 in sciatic nerve motor axons and NMJs is not upregulated in SMA mice (Fig. 3A–P), suggesting that increased tau phosphorylation in motor neuron somas may contribute to axonal defects. Tau binds microtubules directly through its microtubule-binding repeat sequences. Hyperphosphorylation of somatic tau may neutralize the positive charge within its microtubule-binding repeat sequences (Fischer et al., 2009; Jho et al., 2010), reducing its microtubule-binding affinity, thus affecting microtubule organization and stability in axons (Caceres and Kosik, 1990; Harada et al., 1994; Perez et al., 2009). Additionally, tau phosphorylation could affect activities of microtubule-dependent motor protein dynein and kinesin (Ebneth et al., 1998; Dixit et al., 2008), leading to axonal transport defects. Our finding that expression of tau phosphorylated on S202 and T205 in zebrafish produce a motor axon truncation phenotype may reflect compromised microtubule structure or axonal transport that are critical for supplying building material and energy for axonal outgrowth. The defective motor axon innervation of NMJs in SMA mice may also be related to dysfunction of axonal transport and microtubule assembly caused by tau hyperphosphorylation. In particular, microtubule-dependent defects may be exacerbated in spinal motor neurons that have long axons, contributing to the selective vulnerability of motor neurons in SMA. Altogether, phosphorylated non-aggregating tau could be a nexus for multiple pathways that leads to motor neuron degeneration. Additional exploration of the underlying mechanisms will have implications for understanding the pathogenesis of SMA and other neurodegenerative disorders.
Footnotes
This work was supported by grants from the Schweppe Foundation (Y.C.M.), the Whitehall Foundation (Y.C.M.), Cure SMA (Y.C.M. and C.P.K.), and the Spinal Muscular Atrophy Foundation (C.J.S.), Nationals Institutes of Health Grant R01 NS062869 (C.J.S.) and AG043970 (Y.C.M.), and National Natural Science Foundation of China Grant 31371253 (L.M.). We thank Maria Nguyen, Lauren Geary, and Chaohui Zhao for their technical assistance with immunohistochemistry and Western blot analyses. We thank members of the Ma laboratory and Dr. Han-Xiang Deng for critically reading this manuscript and Dr. Lester Binder for providing the human tau40 construct.
The authors declare no competing financial interests.
References
- Akten B, Kye MJ, Hao le T, Wertz MH, Singh S, Nie D, Huang J, Merianda TT, Twiss JL, Beattie CE, Steen JA, Sahin M. Interaction of survival of motor neuron (SMN) and HuD proteins with mRNA cpg15 rescues motor neuron axonal deficits. Proc Natl Acad Sci U S A. 2011;108:10337–10342. doi: 10.1073/pnas.1104928108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey RM, Covy JP, Melrose HL, Rousseau L, Watkinson R, Knight J, Miles S, Farrer MJ, Dickson DW, Giasson BI, Lewis J. LRRK2 phosphorylates novel tau epitopes and promotes tauopathy. Acta Neuropathologica. 2013;126:809–827. doi: 10.1007/s00401-013-1188-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann K, Mandelkow EM, Biernat J, Piwnica-Worms H, Mandelkow E. Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett. 1993;336:417–424. doi: 10.1016/0014-5793(93)80849-P. [DOI] [PubMed] [Google Scholar]
- Bevan AK, Hutchinson KR, Foust KD, Braun L, McGovern VL, Schmelzer L, Ward JG, Petruska JC, Lucchesi PA, Burghes AH, Kaspar BK. Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum Mol Genet. 2010;19:3895–3905. doi: 10.1093/hmg/ddq300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11:153–163. doi: 10.1016/0896-6273(93)90279-Z. [DOI] [PubMed] [Google Scholar]
- Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. 2009;10:597–609. doi: 10.1038/nrn2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres A, Kosik KS. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature. 1990;343:461–463. doi: 10.1038/343461a0. [DOI] [PubMed] [Google Scholar]
- Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, Dahrouge S, Antel JP. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dyn. 1992;194:209–221. doi: 10.1002/aja.1001940306. [DOI] [PubMed] [Google Scholar]
- Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai LH. Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron. 1997;18:29–42. doi: 10.1016/S0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- Cifuentes-Diaz C, Nicole S, Velasco ME, Borra-Cebrian C, Panozzo C, Frugier T, Millet G, Roblot N, Joshi V, Melki J. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum Mol Genet. 2002;11:1439–1447. doi: 10.1093/hmg/11.12.1439. [DOI] [PubMed] [Google Scholar]
- Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci. 2001;114:1179–1187. doi: 10.1242/jcs.114.6.1179. [DOI] [PubMed] [Google Scholar]
- de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT. Caspase activation precedes and leads to tangles. Nature. 2010;464:1201–1204. doi: 10.1038/nature08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J Cell Biol. 1998;143:777–794. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, López-González I, Carmona M, Arregui L, Dalfó E, Torrejón-Escribano B, Diehl R, Kovacs GG. Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol. 2014;73:81–97. doi: 10.1097/NEN.0000000000000030. [DOI] [PubMed] [Google Scholar]
- Feuillette S, Miguel L, Frébourg T, Campion D, Lecourtois M. Drosophila models of human tauopathies indicate that Tau protein toxicity in vivo is mediated by soluble cytosolic phosphorylated forms of the protein. J Neurochem. 2010;113:895–903. doi: 10.1111/j.1471-4159.2010.06663.x. [DOI] [PubMed] [Google Scholar]
- Fischer D, Mukrasch MD, Biernat J, Bibow S, Blackledge M, Griesinger C, Mandelkow E, Zweckstetter M. Conformational changes specific for pseudophosphorylation at serine 262 selectively impair binding of tau to microtubules. Biochemistry. 2009;48:10047–10055. doi: 10.1021/bi901090m. [DOI] [PubMed] [Google Scholar]
- Fu AK, Fu WY, Cheung J, Tsim KW, Ip FC, Wang JH, Ip NY. Cdk5 is involved in neuregulin-induced AChR expression at the neuromuscular junction. Nat Neurosci. 2001;4:374–381. doi: 10.1038/86019. [DOI] [PubMed] [Google Scholar]
- Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A. 1988;85:4051–4055. doi: 10.1073/pnas.85.11.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Crowther RA, Cohen P, Vanmechelen E, Vandermeeren M, Cras P. Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer's disease: identification of phosphorylation sites in tau protein. Biochem J. 1994;301:871–877. doi: 10.1042/bj3010871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189:167–169. doi: 10.1016/0304-3940(95)11484-E. [DOI] [PubMed] [Google Scholar]
- Goedert M, Klug A, Crowther RA. Tau protein, the paired helical filament and Alzheimer's disease. J Alzheimers Dis. 2006;9:195–207. doi: 10.3233/jad-2006-9s323. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084–6089. [PubMed] [Google Scholar]
- Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, Hirokawa N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369:488–491. doi: 10.1038/369488a0. [DOI] [PubMed] [Google Scholar]
- Heier CR, Satta R, Lutz C, DiDonato CJ. Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum Mol Genet. 2010;19:3906–3918. doi: 10.1093/hmg/ddq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, Li H. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- Ikegami S, Harada A, Hirokawa N. Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neurosci Lett. 2000;279:129–132. doi: 10.1016/S0304-3940(99)00964-7. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Jho YS, Zhulina EB, Kim MW, Pincus PA. Monte carlo simulations of tau proteins: effect of phosphorylation. Biophys J. 2010;99:2387–2397. doi: 10.1016/j.bpj.2010.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariya S, Park GH, Maeno-Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, Landmesser LT, Monani UR. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:2552–2569. doi: 10.1093/hmg/ddn156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Kramer EM, Cardine AM, Schraven B, Brandt R, Trotter J. Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J Neurosci. 2002;22:698–707. doi: 10.1523/JNEUROSCI.22-03-00698.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo J, Honda T, Mori H, Hamada Y, Miura R, Ogawara M, Ihara Y. The carboxyl third of tau is tightly bound to paired helical filaments. Neuron. 1988;1:827–834. doi: 10.1016/0896-6273(88)90130-4. [DOI] [PubMed] [Google Scholar]
- Kong L, Wang X, Choe DW, Polley M, Burnett BG, Bosch-Marcé M, Griffin JW, Rich MM, Sumner CJ. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J Neurosci. 2009;29:842–851. doi: 10.1523/JNEUROSCI.4434-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- Lee G, Newman ST, Gard DL, Band H, Panchamoorthy G. Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci. 1998;111:3167–3177. doi: 10.1242/jcs.111.21.3167. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Lew J, Huang QQ, Qi Z, Winkfein RJ, Aebersold R, Hunt T, Wang JH. A brain-specific activator of cyclin-dependent kinase 5. Nature. 1994;371:423–426. doi: 10.1038/371423a0. [DOI] [PubMed] [Google Scholar]
- Ling KK, Lin MY, Zingg B, Feng Z, Ko CP. Synaptic defects in the spinal and neuromuscular circuitry in a mouse model of spinal muscular atrophy. PLoS One. 2010;5:e15457. doi: 10.1371/journal.pone.0015457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling KK, Gibbs RM, Feng Z, Ko CP. Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum Mol Genet. 2012;21:185–195. doi: 10.1093/hmg/ddr453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120–2133. doi: 10.1016/S0140-6736(08)60921-6. [DOI] [PubMed] [Google Scholar]
- Ma YC, Huang J, Ali S, Lowry W, Huang XY. Src tyrosine kinase is a novel direct effector of G proteins. Cell. 2000;102:635–646. doi: 10.1016/S0092-8674(00)00086-6. [DOI] [PubMed] [Google Scholar]
- Mandell JW, Banker GA. A spatial gradient of tau protein phosphorylation in nascent axons. J Neurosci. 1996;16:5727–5740. doi: 10.1523/JNEUROSCI.16-18-05727.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern VL, Gavrilina TO, Beattie CE, Burghes AH. Embryonic motor axon development in the severe SMA mouse. Hum Mol Genet. 2008;17:2900–2909. doi: 10.1093/hmg/ddn189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhorter ML, Monani UR, Burghes AH, Beattie CE. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J Cell Biol. 2003;162:919–931. doi: 10.1083/jcb.200303168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentis GZ, Blivis D, Liu W, Drobac E, Crowder ME, Kong L, Alvarez FJ, Sumner CJ, O'Donovan MJ. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron. 2011;69:453–467. doi: 10.1016/j.neuron.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel G, Mercken M, Murayama M, Noguchi K, Ishiguro K, Imahori K, Takashima A. Characterization of tau phosphorylation in glycogen synthase kinase-3beta and cyclin dependent kinase-5 activator (p23) transfected cells. Biochim Biophys Acta. 1998;1380:177–182. doi: 10.1016/S0304-4165(97)00139-6. [DOI] [PubMed] [Google Scholar]
- Monani UR. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron. 2005;48:885–896. doi: 10.1016/j.neuron.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MD, Larivière RC, Julien JP. Deregulation of Cdk5 in a mouse model of ALS: toxicity alleviated by perikaryal neurofilament inclusions. Neuron. 2001;30:135–147. doi: 10.1016/S0896-6273(01)00268-9. [DOI] [PubMed] [Google Scholar]
- Nukina N, Ihara Y. One of the antigenic determinants of paired helical filaments is related to tau protein. J Biochem. 1986;99:1541–1544. doi: 10.1093/oxfordjournals.jbchem.a135625. [DOI] [PubMed] [Google Scholar]
- Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, Brady RO, Martin LJ, Kulkarni AB. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci U S A. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Colasante X, Seaberg B, Martinez TL, Kong L, Sumner CJ, Rimer M. Improvement of neuromuscular synaptic phenotypes without enhanced survival and motor function in severe spinal muscular atrophy mice selectively rescued in motor neurons. PLoS One. 2013;8:e75866. doi: 10.1371/journal.pone.0075866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Paudel HK, Lew J, Ali Z, Wang JH. Brain proline-directed protein kinase phosphorylates tau on sites that are abnormally phosphorylated in tau associated with Alzheimer's paired helical filaments. J Biol Chem. 1993;268:23512–23518. [PubMed] [Google Scholar]
- Pellizzoni L, Kataoka N, Charroux B, Dreyfuss G. A novel function for SMN, the spinal muscular atrophy disease gene product, in pre-mRNA splicing. Cell. 1998;95:615–624. doi: 10.1016/S0092-8674(00)81632-3. [DOI] [PubMed] [Google Scholar]
- Perez M, Santa-Maria I, Gomez de Barreda E, Zhu X, Cuadros R, Cabrero JR, Sanchez-Madrid F, Dawson HN, Vitek MP, Perry G, Smith MA, Avila J. Tau—an inhibitor of deacetylase HDAC6 function. J Neurochem. 2009;109:1756–1766. doi: 10.1111/j.1471-4159.2009.06102.x. [DOI] [PubMed] [Google Scholar]
- Porzig R, Singer D, Hoffmann R. Epitope mapping of mAbs AT8 and Tau5 directed against hyperphosphorylated regions of the human tau protein. Biochem Biophys Res Commun. 2007;358:644–649. doi: 10.1016/j.bbrc.2007.04.187. [DOI] [PubMed] [Google Scholar]
- Reynolds CH, Garwood CJ, Wray S, Price C, Kellie S, Perera T, Zvelebil M, Yang A, Sheppard PW, Varndell IM, Hanger DP, Anderton BH. Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cgamma1, Grb2, and Src family kinases. J Biol Chem. 2008;283:18177–18186. doi: 10.1074/jbc.M709715200. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Rossoll W, Jablonka S, Andreassi C, Kröning AK, Karle K, Monani UR, Sendtner M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol. 2003;163:801–812. doi: 10.1083/jcb.200304128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnik-Schöneborn S, Heller R, Berg C, Betzler C, Grimm T, Eggermann T, Eggermann K, Wirth R, Wirth B, Zerres K. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J Med Genet. 2008;45:635–638. doi: 10.1136/jmg.2008.057950. [DOI] [PubMed] [Google Scholar]
- Samuels BA, Hsueh YP, Shu T, Liang H, Tseng HC, Hong CJ, Su SC, Volker J, Neve RL, Yue DT, Tsai LH. Cdk5 promotes synaptogenesis by regulating the subcellular distribution of the MAGUK family member CASK. Neuron. 2007;56:823–837. doi: 10.1016/j.neuron.2007.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shababi M, Habibi J, Yang HT, Vale SM, Sewell WA, Lorson CL. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum Mol Genet. 2010;19:4059–4071. doi: 10.1093/hmg/ddq329. [DOI] [PubMed] [Google Scholar]
- Shin J, Park HC, Topczewska JM, Mawdsley DJ, Appel B. Neural cell fate analysis in zebrafish using olig2 BAC transgenics. Methods Cell Sci. 2003;25:7–14. doi: 10.1023/B:MICS.0000006847.09037.3a. [DOI] [PubMed] [Google Scholar]
- Su SC, Tsai LH. Cyclin-dependent kinases in brain development and disease. Annu Rev Cell Dev Biol. 2011;27:465–491. doi: 10.1146/annurev-cellbio-092910-154023. [DOI] [PubMed] [Google Scholar]
- Tashiro K, Hasegawa M, Ihara Y, Iwatsubo T. Somatodendritic localization of phosphorylated tau in neonatal and adult rat cerebral cortex. Neuroreport. 1997;8:2797–2801. doi: 10.1097/00001756-199708180-00029. [DOI] [PubMed] [Google Scholar]
- Tatebayashi Y, Haque N, Tung YC, Iqbal K, Grundke-Iqbal I. Role of tau phosphorylation by glycogen synthase kinase-3beta in the regulation of organelle transport. J Cell Sci. 2004;117:1653–1663. doi: 10.1242/jcs.01018. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Schuck T, Schmidt ML, Lee VM. Distribution of tau proteins in the normal human central and peripheral nervous system. J Histochem Cytochem. 1989;37:209–215. doi: 10.1177/37.2.2492045. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Tsai LH, Delalle I, Caviness VS, Jr, Chae T, Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- Vanderweyde T, Yu H, Varnum M, Liu-Yesucevitz L, Citro A, Ikezu T, Duff K, Wolozin B. Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J Neurosci. 2012;32:8270–8283. doi: 10.1523/JNEUROSCI.1592-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witman GB, Cleveland DW, Weingarten MD, Kirschner MW. Tubulin requires tau for growth onto microtubule initiating sites. Proc Natl Acad Sci U S A. 1976;73:4070–4074. doi: 10.1073/pnas.73.11.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AS, Lee RH, Cheung AY, Yeung PK, Chung SK, Cheung ZH, Ip NY. Cdk5-mediated phosphorylation of endophilin B1 is required for induced autophagy in models of Parkinson's disease. Nat Cell Biol. 2011;13:568–579. doi: 10.1038/ncb2217. [DOI] [PubMed] [Google Scholar]
- Wood JG, Mirra SS, Pollock NJ, Binder LI. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau) Proc Natl Acad Sci U S A. 1986;83:4040–4043. doi: 10.1073/pnas.83.11.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng-Fischhöfer Q, Biernat J, Mandelkow EM, Illenberger S, Godemann R, Mandelkow E. Sequential phosphorylation of Tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur J Biochem. 1998;252:542–552. doi: 10.1046/j.1432-1327.1998.2520542.x. [DOI] [PubMed] [Google Scholar]