

Abstract
Hyperalgesic priming, a form of neuroplasticity in nociceptors, is a model of the transition from acute to chronic pain in the rat, which involves signaling from the site of an acute tissue insult in the vicinity of the peripheral terminal of a nociceptor to its cell body that, in turn, induces a signal that travels back to the terminal to mediate a marked prolongation of prostaglandin E2-induced hyperalgesia. In the present experiments, we studied the underlying mechanisms in the cell body and compared them to the mechanisms in the nerve terminal. Injection of a cell-permeant cAMP analog, 8-bromo cAMP, into the dorsal root ganglion induced mechanical hyperalgesia and priming with an onset more rapid than when induced at the peripheral terminal. Priming induced by intraganglion 8-bromo cAMP was prevented by an oligodeoxynucleotide antisense to mRNA for a transcription factor, cAMP response element-binding protein (CREB), and by an inhibitor of importin, which is required for activated CREB to get into the nucleus. While peripheral administration of 8-bromo cAMP also produced hyperalgesia, it did not produce priming. Conversely, interventions administered in the vicinity of the peripheral terminal of the nociceptor that induces priming—PKCε activator, NGF, and TNF-α—when injected into the ganglion produce hyperalgesia but not priming. The protein translation inhibitor cordycepin, injected at the peripheral terminal but not into the ganglion, reverses priming induced at either the ganglion or peripheral terminal of the nociceptor. These data implicate different mechanisms in the soma and terminal in the transition to chronic pain.
Keywords: dorsal root ganglion, hyperalgesic priming, mechanical hyperalgesia, nociceptor, rat
Introduction
We have developed a preclinical model of chronic pain, hyperalgesic priming (Aley et al., 2000; Reichling and Levine, 2009), in which a prior acute inflammatory or neuropathic insult can produce a form of neuroplasticity in isolectin B4 (IB4)-positive (IB4+) nociceptors (Joseph and Levine, 2010), which contributes to the transition from acute to chronic pain. A key feature of the primed nociceptor is that it responds to pronociceptive mediators with a markedly prolonged mechanical hyperalgesia, is mediated by a novel second messenger signaling pathway involving an inhibitory G-protein-coupled receptor, coupled to phospholipase C β3 and protein kinase Cε (PKCε; Aley et al., 2000; Parada et al., 2005; Joseph et al., 2007; Khasar et al., 2008; Dina et al., 2009; Reichling and Levine, 2009; Ferrari et al., 2013a). After the initiating acute insult, in a hindpaw, there is a 3 d delay to the onset of the primed state (Aley et al., 2000; Bogen et al., 2012). This delay is due to the time required for a signal generated in the terminal of the nociceptor to travel from the site of the acute insult to the cell body, activation of a transcription factor, followed by the generation of a signal in the cell body that travels to the peripheral terminal, where cytoplasmic polyadenylation element-binding protein (CPEB)-dependent protein translation occurs (Bogen et al., 2012; Ferrari et al., 2015).
The induction of the primed state is triggered by pronociceptive mediators that have in common the ability to activate PKCε (Aley et al., 2000; Parada et al., 2005) and its downstream signaling molecules, including α calmodulin-dependent protein kinase II (αCaMKII) and the ryanodine receptor (Ferrari et al., 2013b). As the administration of a translation inhibitor in the periphery can fully reverse, as well as prevent, priming (Bogen et al., 2012; Ferrari et al., 2013c), the translation of mRNAs in the peripheral terminal of the nociceptor (Bogen et al., 2012) participates in the maintenance as well as the induction. Priming, induced by an acute insult at the peripheral terminal of the nociceptor can also be prevented, but not reversed, by intrathecal administration of an oligodeoxynucleotide (ODN) antisense (AS) to mRNA for a nuclear transcription factor, cAMP response element-binding protein (CREB; Ferrari et al., 2015), supporting the suggestion that nuclear mechanisms also play a role in hyperalgesic priming. In the present experiments, we determined whether similar or different mechanisms in the cell body and peripheral terminal of the nociceptor contribute to hyperalgesic priming.
Materials and Methods
Animals.
Experiments were performed on adult male Sprague Dawley rats (220–400 g; Charles River Laboratories). Animals were housed three per cage under a 12 h light/dark cycle in a temperature- and humidity-controlled vivarium at the University of California, San Francisco animal care facility. Food and water were available to animals ad libitum. All nociceptive testing was performed between 10:00 A.M. and 5:00 P.M. Experimental protocols were approved by the Institutional Animal Care and Use Committee at The University of California, San Francisco, and adhered to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Every effort was made to minimize the number of animals used and their suffering.
Testing mechanical nociception.
The mechanical nociceptive threshold was quantified using an Ugo Basile Analgesymeter (Randall–Selitto paw-withdrawal test; Stoelting), which uses a dome-shaped plinth to apply a linearly increasing mechanical force to the dorsum of the rat hindpaw, as previously described (Randall and Selitto, 1957; Taiwo and Levine, 1989; Taiwo et al., 1989). The nociceptive threshold was defined as the force (in grams) at which the rat withdrew its paw from the plinth; the baseline threshold was defined as the mean of the last three readings taken before test agents were injected. Each paw was treated as an independent measure of each experiment performed on a separate group of rats. Data are presented as the mean change from baseline mechanical nociceptive threshold.
Drugs.
The following drugs were used in these experiments: the hyperalgesia-inducing pronociceptive inflammatory mediator prostaglandin E2 (PGE2), rat recombinant monocyte chemotactic protein 1 (MCP-1), cordycepin from Cordyceps militaris (a protein translation inhibitor), the protein transcription inhibitor actinomycin D, the importin inhibitor ivermectin, and nerve growth factor (NGF), all from Sigma-Aldrich; the highly potent membrane-permeable cAMP analog 8-bromo cAMP sodium salt (Tocris Bioscience); the αCaMKII inhibitor peptide CaM2INtide (GenScript); the PKCε-specific translocation inhibitor peptide PKCεV1–2 (PKCε-I; Johnson et al., 1996; Khasar et al., 1999; Calbiochem); the selective activator of PKCε, psi ε receptor for activated C kinase (ψεRACK; Biomatik); and rat recombinant tumor necrosis factor-α (TNF-α; R&D Systems). The selection of the drug doses used in these experiments was based on our published studies (Taiwo et al., 1990; Ouseph et al., 1995; Khasar et al., 1999; Aley et al., 2000; Parada et al., 2005; Ferrari et al., 2013c, 2015).
Stock solutions of PGE2 in absolute ethanol (1 μg/μl) were further diluted in 0.9% NaCl (1:50, Cfinal = 0.02 μg/μl) immediately before injection. The ethanol concentration of the final PGE2 solution was ∼2%, and the injection volume was 5 μl. Actinomycin D and ivermectin were first dissolved in dimethylsulfoxide (DMSO) 100% and then diluted in 0.9% NaCl with 10% DMSO. NGF was dissolved in sterile PBS containing 0.5% bovine serum albumin and diluted in 0.9% NaCl at the time of injection. All other drugs were dissolved in 0.9% NaCl.
The intradermal administration of drugs on the dorsum of the rat hindpaw was performed via a beveled 30 gauge hypodermic needle that was attached to a Hamilton 50 μl syringe, by a short length of polyethylene (PE-10) tubing. The administration of cordycepin, CaM2INtide, PKCεV1–2, and ψεRACK was preceded by a hypotonic shock to facilitate cell permeability to these agents (2 μl of distilled water, separated by a bubble of air to avoid mixing in the same syringe) and to facilitate the movement of compounds across the plasma membrane of the nociceptor into the cytoplasm of the peripheral terminal (Borle and Snowdowne, 1982; Burch and Axelrod, 1987).
Hyperalgesic priming.
Hyperalgesic priming, a model of the transition from acute to chronic pain, was produced by intrathecal injection of MCP-1 (Ferrari et al., 2015) or intraganglion injection of 8-bromo cAMP. To prime the sensory neuron, the agents were injected either intrathecally into the spinal cord or into the fifth lumbar (L5) dorsal root ganglion (DRG). The acute mechanical hyperalgesia produced by the injection of agents was no longer present at the point in time when the intradermal injection of PGE2 on the dorsum of the hindpaw, to verify the presence of priming, was performed. In the primed condition, PGE2 produces mechanical hyperalgesia that lasts >4 h and is still significant at 24 h, as opposed to the effect of intradermal injection of PGE2 into a nonprimed paw, which lasts ∼1 h (Aley et al., 2000; Reichling and Levine, 2009; Ferrari et al., 2013a).
Oligodeoxynucleotide antisense to CREB and aCaMKII.
The ODN AS sequence for CREB mRNA, 5′-TGG TCA TCT AGT CAC CGG TG-3′ (Invitrogen), was directed against a unique region of the rat mRNA sequence. That ODN AS to CREB mRNA, with this sequence, can be used to downregulate the expression of CREB has been shown previously (Widnell et al., 1996; Lane-Ladd et al., 1997; Ma et al., 2003). The ODN mismatch (MM) sequence, 5′-GAC CTC AGG TAG TCG TCG TT-3′, is a scrambled sequence of the AS sequence.
The ODN AS sequence for the α-subunit of CaMKII, 5′-GGTAGCCATCCTGGCACT-3′ (Invitrogen), was directed against a unique region of the rat mRNA sequence. The corresponding National Center for Biotechnology Information GenBank accession number and ODN position within the mRNA sequence are NM_012920 and 33–50, respectively. That this ODN AS can be used to downregulate the expression of αCaMKII has been shown previously (Churn et al., 2000; Ferrari et al., 2013b). The ODN MM sequence 5′-GGTAGCCATAAGGGCACT-3′ corresponds to the AS sequence with three bases mismatched (denoted in bold).
Before use, the ODN, AS or MM, was lyophilized and reconstituted in 0.9% NaCl to a concentration of 2 μg/μl, and was administered by spinal intrathecal injection. During each injection of ODN, rats were briefly anesthetized with 2.5% isoflurane in 95% O2. A 29 gauge hypodermic needle was then inserted into the subarachnoid space on the midline, between the L4 and L5 vertebrae, and a 40 μg dose of ODN, in a volume of 20 μl, was slowly injected. The intrathecal site of injection was confirmed by checking for a flick of the tail, a reflex that is evoked by accessing the subarachnoid space and injection of the ODN (Parada et al., 2003b; Ferrari et al., 2013b, 2015). Animals regained consciousness ∼1 min after the injection of the ODNs. The use of AS to manipulate the expression of proteins in nociceptors, which is important for their role in nociceptor sensitization, is well supported by a large number of previous studies by others (Song et al., 2009; Su et al., 2011; Quanhong et al., 2012; Sun et al., 2013) as well as by our group (Parada et al., 2003b; Ferrari et al., 2010, 2012; Bogen et al., 2012).
Intraganglion drug administration.
Injection of compounds into the L5-DRG was performed with a gingival needle (30 gauge), prepared as previously described (Ferrari et al., 2007; Araldi et al., 2013), attached to a 50 μl Hamilton syringe by a short length of PE-10 tubing. This modification of the injecting needle decreases the risk of tissue damage when its tip penetrates the DRG. Animals were lightly anesthetized by inhalation of isoflurane, and the fur over the lower back was then shaved. The site of skin penetration was 1.5 cm lateral to the vertebral column and ∼0.5 cm caudal from a virtual line passing between the rostral edges of the iliac crests. To facilitate the insertion of the injecting needle through the skin, an initial cutaneous puncture was made with a larger (18 gauge) needle. In sequence, the injecting needle was inserted through the puncture wound in the skin, and was oriented toward the region of the intervertebral space between the fifth and sixth lumbar vertebrae, until the tip touched the lateral region of the vertebrae. To reach the space between the transverse processes of the fifth and sixth lumbar vertebrae, small incremental movements of the needle tip were performed, until the resistance provided by the bone was diminished. When it reached the right position, the tip of the needle felt “locked in place” (through the intervertebral space) and a flinch of the ipsilateral hindpaw was observed (Araldi et al., 2013), indicating that it had penetrated the DRG of the fifth lumbar spinal nerve (Ferrari et al., 2007). The injection procedure from the beginning of anesthetic inhalation until withdrawal of the needle took ∼3 min. Animals regained consciousness ∼1 min after anesthesia was discontinued. Importantly, no change in the nociceptive mechanical paw-withdrawal threshold of the ipsilateral hindpaw, after a single injection or daily repeated injections in the L5-DRG, was observed, and immunofluorescence analysis of the L5-DRG after injections has shown no signs of cell damage (Araldi et al., 2013). Likewise, no significant change in the mechanical threshold in the hindpaw after intraganglion injection of vehicles in our experiments (single or repeated injections) was observed, indicating a lack of damage produced by the injection.
Similar to our studies using spinal intrathecal drug administration (Tambeli et al., 2002; Parada et al., 2003a,b; Miao et al., 2004), we chose to use larger doses of the drugs for the intraganglion injections to be sure that all the cell bodies would be reached by the compound even if there was some diffusion away from the DRG.
Statistics.
In all experiments, the dependent variable was the mechanical paw-withdrawal threshold, expressed as the percentage change from preintervention baseline. The average paw-withdrawal thresholds before the injection of a priming stimulus (MCP-1, 8-br cAMP, ψεRACK, NGF, or TNF-α, depending on the experiment) and before the tests with PGE2 were 119.9 ± 0.5 and 118.7 ± 0.2 g, respectively; no significant difference between these values was observed (t(179) = 2.265, p = 0.2047, paired Student's t test). A total of 180 paws were used in this study. In the experiments in which ODN AS or MM was used (see Fig. 4 for CREB experiments, and see Fig. 7 for αCaMKII experiments), the ODN treatments did not induce a significant change in the mechanical nociceptive threshold (data not shown). To compare the hyperalgesia induced by PGE2 injection in different groups, unpaired Student's t test or two-way repeated-measures ANOVA, followed by Bonferroni post-test, was performed, depending on the experiment. Prism version 5.0 (GraphPad Software) was used for graphics and to perform the statistical analyses; p < 0.05 was considered to be statistically significant. Data are presented as the mean ± SEM.
Figure 4.

CREB antisense prevents (A), but does not reverse (B), hyperalgesic priming induced by intraganglion (i.gl.) injection of 8-bromo cAMP. A, Rats were treated with daily spinal intrathecal injections of ODN AS (black bars) for CREB mRNA, for 3 consecutive days, which decreases its levels in the sensory neuron, and prevents its activation by the priming inducer 8-bromo cAMP (10 μg, injected into the DRG, i.gl.), injected on the fourth day. Control animals were treated, following the same protocol, with ODN MM (white bars). To prevent the possibility that prolonged activation of a signaling pathway might produce priming following the administration of 8-bromo cAMP, the ODN treatment continued through day 5, when the mechanical nociceptive paw-withdrawal thresholds were not significantly (NS) different from pre 8-bromo cAMP baseline values (t(5) = 2.445; p = 0.0583, NS, for the MM group; t(5) = 0.1117; p = 0.9154, NS, for the AS group; paired Student's t test). The presence of hyperalgesic priming was assessed by intradermal injection of PGE2 (100 ng) into the dorsum of the hindpaw. Mechanical hyperalgesia was evaluated 30 min and 4 h later, by the Randall–Sellitto paw-withdrawal test. Average paw-withdrawal thresholds before the injection of 8-bromo cAMP and before the injection of PGE2 (1 d later) were as follows: 119.0 ± 2.7 and 114.3 ± 2.0 g, respectively, for the CREB MM-treated group; and 118.0 ± 2.0 and 118.3 ± 2.0 g, respectively, for the AS-treated group. Two-way repeated-measures ANOVA followed by Bonferroni post-test showed significant mechanical hyperalgesia induced by PGE2, measured 30 min after injection, in both groups. However, while in the MM-treated group the magnitude of PGE2 hyperalgesia was still significant at the fourth hour, in the AS-treated group it was strongly attenuated (***p < 0.001 when the hyperalgesia in those groups was compared at that time point). When tested again for priming with PGE2 1 week after the last treatment with ODN AS or MM, the prolongation of PGE2-induced hyperalgesia was still attenuated (at the 4 h time point) in the ODN AS-treated group, but not in the ODN MM-treated group, indicating a role of CREB in the induction of hyperalgesic priming by i.gl. injection of 8-bromo cAMP (***p < 0.001 when the MS- and the AS-treated groups are compared; N = 6 paws per group). Of note, no difference in the mechanical thresholds was observed at this time point, when compared with prepriming stimuli baseline thresholds: 119.0 ± 2.7 and 116.3 ± 3.1 g, respectively, for the CREB MM-treated group (t(5) = 2.169; p = 0.0822, NS), and 118.0 ± 2.0 and 115.3 ± 2.2 g, respectively, for the AS-treated group (t(5) = 1.019; p = 0.3548, NS; paired Student's t test). B, Rats that were treated with i.gl. injection of 8-bromo cAMP (10 μg) 5 d before were treated with ODN AS (black bars) for CREB mRNA for 3 consecutive days, to decrease the levels of CREB in the nociceptor. Control animals were treated, following the same protocol, with MM (white bars). On the fourth day, PGE2 (100 ng) was injected intradermally into the dorsum of the hindpaw, and the mechanical threshold was evaluated 30 min and 4 h later. The average paw-withdrawal thresholds before the injection of 8-bromo cAMP and before the injection of PGE2 were 118.3 ± 3.0 and 120.3 ± 3.6 g, respectively, for the CREB MM-treated group (t(5) = 0. 0000; p = 1.0000, NS), and 123.3 ± 3.6 and 122.6 ± 2.1 g, respectively, for the AS-treated group (t(5) = 0.6742; p = 0.5301, NS). Paired Student's t test showed no significant (NS) difference between these two values. Two-way repeated-measures ANOVA followed by Bonferroni post-test showed no difference in the magnitude of the PGE2-induced hyperalgesia at 30 min and 4 h between the ODN AS- and MM-treated groups (p > 0.05, NS), indicating that CREB does not play a role in the maintenance of hyperalgesic priming induced by 8-bromo cAMP (N = 6 paws per group).
Figure 7.

Expression of hyperalgesic priming induced by intraganglion injection of 8-bromo cAMP is dependent on PKCε and αCaMKII. Rats primed with intraganglion (i.gl.) injection of 8-bromo cAMP (10 μg) 1 week prior received intradermal injection of PGE2 (100 ng) into the dorsum of the hindpaw in the presence or absence of inhibitors of the second messengers, PKCε (PKCε-I; 1 μg/5 μl, gray bars) and αCaMKII (CaMINtide; 1 μg/5 μl, black bars). Of note, in order to achieve a stronger inhibition of αCaMKII, rats were treated, for 3 consecutive days, prior to injection of the inhibitor plus PGE2, with ODN AS for αCaMKII. Mechanical nociceptive thresholds were evaluated 30 min and 4 h after PGE2, injected 5 min after the inhibitors, by the Randall–Selitto paw-withdrawal test. We observed, in both cases, significant attenuation of the hyperalgesia induced by PGE2 at the fourth hour (***p < 0.001) in the groups treated with the PKCε inhibitor or αCaMKII ODN AS plus CaM2INtide when compared with the vehicle groups (PKCε-I: F(1,10) = 29.55, p = 0.0003; αCaMKII ODN AS plus CaM2INtide: F(1,10) = 54.61, p < 0.0001, two-way repeated-measures ANOVA followed by Bonferroni post-test, N = 6 paws per group), indicating a role of PKCε and αCaMKII in the prolongation of PGE2 hyperalgesia in the i.gl. 8-bromo cAMP-induced hyperalgesic priming (A). B shows that, when PGE2 was injected again at the same site 1 week later, it produced prolonged hyperalgesia, evaluated at the fourth hour, indicating that the reversal of hyperalgesic priming induced by i.gl. 8-bromo cAMP by the inhibition of PKCε or αCaMKII is not permanent (PKCε-I: t(5) = 3.072, p = 0.2770; αCaMKII ODN AS + CaM2INtide: t(5) = 0.6532, p = 0.5424, paired Student's t test, no difference when the 30 min and 4 h time points are compared in both cases; N = 6 paws per group).
Results
Induction of hyperalgesic priming is dependent on gene transcription
We have recently shown that spinal intrathecal administration of an ODN AS to mRNA for CREB prevented, but did not reverse, hyperalgesic priming (Ferrari et al., 2015), which is compatible with the suggestion that gene transcription in the cell body plays a role in the induction of priming. To provide confirmation for the role of gene transcription, we evaluated whether the intraganglion injection of an inhibitor of gene transcription, in the vicinity of the nociceptor cell body, would prevent the development of priming by a stimulus applied at the peripheral or central terminal of the nociceptor. Rats pretreated by intraganglion injection of the transcription inhibitor actinomycin D then received intrathecal injection of the priming inducer MCP-1 (Alvarez et al., 2014; Ferrari et al., 2015). Daily treatment with intraganglion actinomycin D continued until the fourth day after MCP-1 injection, when its acute hyperalgesic effect had disappeared. We then evaluated whether priming had developed by injecting PGE2 intradermally into the dorsum of the hindpaw. While in the rats that received intraganglion injection of vehicle, the mechanical hyperalgesia induced by PGE2 was still significant at the fourth hour after injection, in the paws ipsilateral to the DRGs treated with actinomycin D, the PGE2-induced hyperalgesia was significantly attenuated by the fourth hour, indicating that an inhibitor of transcription injected adjacent to the cell body of the nociceptor prevents the development of priming (Fig. 1).
Figure 1.
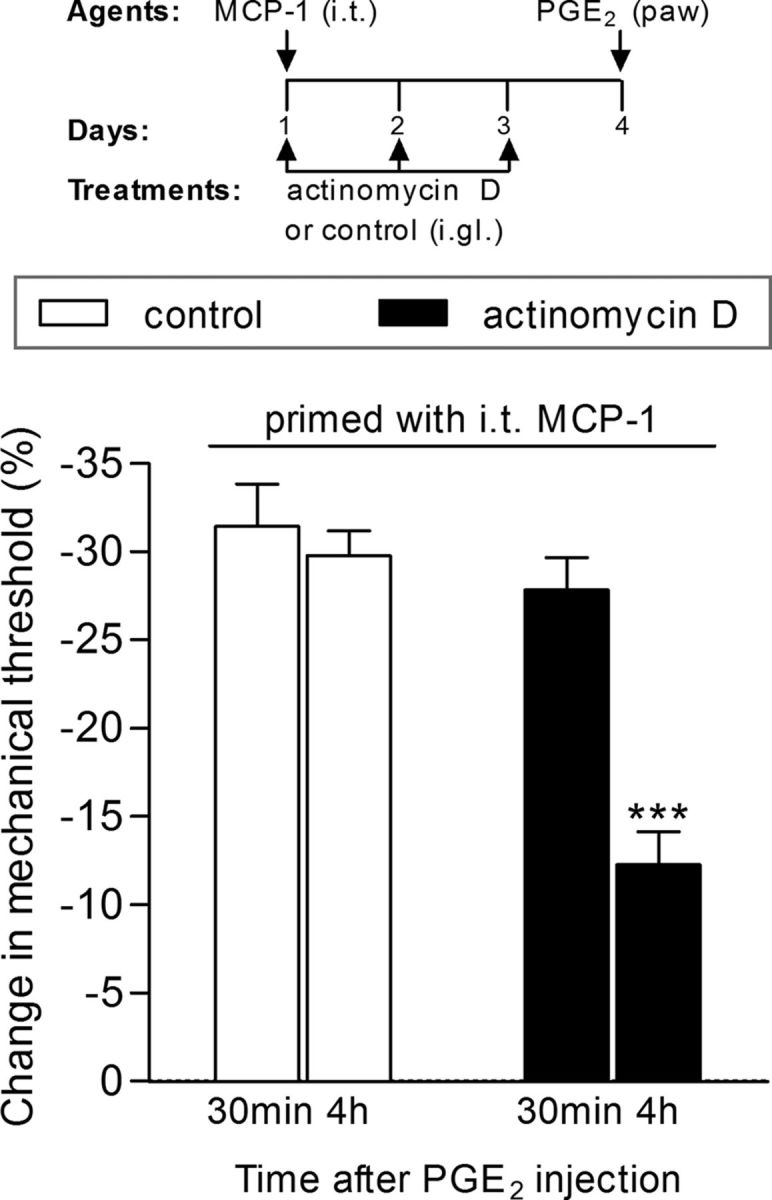
Induction of hyperalgesic priming by intrathecal (i.t.) injection of MCP-1 is dependent on gene transcription. Rats received three daily intraganglion (i.gl.) injections of vehicle (white bars) or the protein transcription inhibitor actinomycin D (10 μg, black bars). Thirty minutes after the first injection of actinomycin D, the central terminal of these nociceptors was stimulated with MCP-1 (20 ng/μl, 20 μl), injected intrathecally. No effect of actinomycin D on MCP-1-induced hyperalgesia was observed (data not shown). The injections of vehicle or actinomycin D continued, once a day, for 3 d. On the fourth day, PGE2 (100 ng) was injected intradermally into the dorsum of the hindpaw at the site of nociceptive testing to evaluate for the presence of priming. The average paw-withdrawal thresholds before injections of vehicle/MCP-1 and immediately before the injection of PGE2 (4 d later) were 110.0 ± 3.6 and 110.0 ± 2.7 g, respectively; for the groups that received actinomycin D/MCP-1, 113.3 ± 3.3 and 113.5 ± 2.2 g, respectively (t(5) = 0.0000; p = 1.0000, NS in both cases; paired Student's t test). Mechanical hyperalgesia was then evaluated by the Randall–Sellitto paw-withdrawal test 30 min and 4 h after PGE2 injection. We observed that, in the paws ipsilateral to the DRGs that received vehicle, the magnitude of hyperalgesia was still significant at the fourth hour after PGE2 injection, indicating the presence of hyperalgesic priming, while in the paws ipsilateral to the DRGs that received actinomycin D, the PGE2-induced hyperalgesia was significantly attenuated at this time point, which is compatible with the prevention of priming (***p < 0.001, when both groups are compared at the fourth hour; two-way repeated-measures ANOVA followed by Bonferroni post-test, N = 6 paws per group). The results of this experiment support the suggestion that the inhibition of transcription in the cell body prevented the induction of priming by intrathecal MCP-1.
Priming induced by agents administered to the cell body
Since the induction of priming was prevented by intraganglion injection of actinomycin D, which blocks gene transcription (Gniazdowski et al., 2003; Wiegert et al., 2009; Kozyrev and Nikitin, 2013; Hayashi et al., 2014), and, as previously shown, the nuclear transcription factor CREB plays a role in this process (Ferrari et al., 2015), we next determined whether an activator of CREB in the cell body induces priming. We injected the potent highly membrane-permeable cAMP analog 8-bromo cAMP into the DRG or, as a control, into the peripheral receptive field of primary afferent nociceptors at the site of nociceptive testing. While both intraganglion (10 μg/5 μl) and intradermal (10 μg/5 μl) injection of 8-bromo cAMP produced robust mechanical hyperalgesia (Fig. 2, first two bars on the left), only the intraganglion route of administration produced priming (Fig. 2, last two bars on the right). The latency to the onset of priming induced by the intraganglion injection of 8-bromo cAMP was between 6 and 12 h (Fig. 3A,B), which was markedly shorter than the ∼72 h required when priming was induced by the injection of an inflammatory mediator at the nociceptor's peripheral receptive field (Bogen et al., 2012; Ferrari et al., 2015). Intrathecal administration of ODN AS to CREB mRNA prevented (Fig. 4A), but could not reverse (Fig. 4B), the hyperalgesic priming induced by intraganglion 8-bromo cAMP. In contrast, while intraganglion administration of substances that produce mechanical hyperalgesia and priming when injected into the peripheral receptive field of nociceptors—ψεRACK, NGF, and TNF-α (Parada et al., 2003a, 2005; Reichling and Levine, 2009; Ferrari et al., 2010; Bogen et al., 2012)—produce mechanical hyperalgesia when injected into the ganglion, indicating that they reached the cell body of the primary afferent nociceptors, when injected at this site, they did not produce priming (Fig. 5).
Figure 2.

Intraganglion, but not intradermal, injection of 8-bromo cAMP induces hyperalgesic priming. Rats received injection of 8-bromo cAMP in the DRG (10 μg, black bars) or intradermally in the dorsum of the hindpaw (10 μg, white bars). Robust mechanical hyperalgesia, evaluated 30 min later, by the Randall–Sellitto paw-withdrawal test, was observed in both groups (p > 0.05 when compared). One week later, a time point when the mechanical thresholds were not significantly different from pre-8-bromo cAMP baseline thresholds (data not shown), PGE2 (100 ng) was injected intradermally into the dorsum of the hindpaw. The average paw-withdrawal thresholds before the injection of 8-bromo cAMP and immediately before the injection of PGE2 (1 week later) were as follows: 120.6 ± 1.6 and 116.3 ± 2.0 g, respectively, for the DRG group (t(5) = 1.904; p = 0.1152, NS); 122.3 ± 3.0 and 116.3 ± 2.3 g, respectively, for the paw group (t(5) = 1.861; p = 0.1219, NS); no significant difference (NS) between these two pairs of values was observed (paired Student's t test). Although the mechanical hyperalgesia induced by PGE2 at the 30 min time point was not different between the groups, 4 h after injection, the PGE2 hyperalgesia was still significant in the group previously treated with intraganglion 8-bromo cAMP, but not in the group that received an injection of 8-bromo cAMP in the hindpaw (***p < 0.001, when both groups are compared at the fourth hour; two-way repeated-measures ANOVA followed by Bonferroni post-test, N = 6 paws per group), indicating that 8-bromo cAMP induces priming by acting at the level of the cell body of the nociceptor in the DRG.
Figure 3.

Time course for the development of hyperalgesic priming induced by intraganglion injection of 8-bromo cAMP. Groups of rats received intraganglion (i.gl.) injection of 8-bromo cAMP (10 μg, black bars) or vehicle. A–C, PGE2 (100 ng) was then injected intradermally into the dorsum of the hindpaw 6 (A), 12 (B), or 24 (C) hours later. Mechanical nociceptive paw-withdrawal thresholds were evaluated before, and 30 min and 4 h after the intradermal injection of PGE2 by the Randall–Selitto paw-withdrawal test. Average paw-withdrawal thresholds measured before, and 6, 12, and 24 h after i.gl. injection of 8-bromo cAMP were as follows: 112.6 ± 0.8 and 116.6 ± 1.7 g, respectively (6 h group); 129.0 ± 3.0 and 128.3 ± 4.0 g, respectively (12 h group); and 126.6 ± 3.9 and 122.6 ± 3.7 g, respectively (24 h group); no significant difference (NS) among these values was observed: after 6 h: t(5) = 2.582; p = 0.4930; NS; after 12 h: t(5) = 0.1147; p = 0.9131, NS; after 24 h: t(5) = 1.054; p = 0.3401, NS; paired Student's t test. For the groups that received vehicle, the average paw-withdrawal thresholds measured before, and 6, 12, and 24 h after i.gl. injection were as follows: 115.6 ± 3.2 and 117.0 ± 2.1 g, respectively (6 h group); 128.3 ± 1.8 and 126.6 ± 3.3 g, respectively (12 h group); and 128.0 ± 4.1 and 118.0 ± 1.2 g, respectively (24 h group). No significant differences among these values were observed: after 6 h: t(5) = 0.7559; p = 0.4838; NS; after 12 h: t(5) = 0.5207; p = 0.6248, NS; after 24 h: t(5) = 2.348; p = 0.0657, NS; paired Student's t test. A, In the group treated with i.gl. injection of 8-bromo cAMP, followed by PGE2 injection into the hindpaw 6 h later, the PGE2-induced hyperalgesia was no longer present when evaluated 4 h after injection (no significant difference was observed when compared with the i.gl.-vehicle group; p > 0.05, two-way repeated-measures ANOVA followed by Bonferroni post-test, N = 6 paws per group), which is compatible with the absence of priming. B, C, However, when PGE2 was injected 12 h (B) or 24 h (C) after i.gl. injection of 8-bromo cAMP, the hyperalgesia observed was still present at the 4 h time point, indicating that hyperalgesic priming was established (***p < 0.001 when compared the vehicle-treated group at the fourth hour; two-way repeated-measures ANOVA followed by Bonferroni post-test, N = 6 paws per group).
Figure 5.

Hyperalgesic agents that also induce priming when injected into the paw produce hyperalgesia, but not priming, when injected in the DRG. A–C, Groups of rats received intraganglion (i.gl.) injection of the hyperalgesic mediators ψεRACK (10 μg; A), NGF (1 μg; B), or TNF-α (10 μg; C). The mechanical threshold was evaluated 60 min and 24 h later. A–C, The paws ipsilateral to the DRGs that received injection of these mediators showed significant mechanical hyperalgesia when compared with the control paws (ipsilateral to the DRGs treated with vehicles; A, **p < 0.01; B, ***p < 0.001, **p < 0.01; C, p < 0.001, when the change in mechanical threshold induced by the i.gl. priming inducers or control is compared over 24 h; two-way repeated-measures ANOVA followed by Bonferroni post-test). One week later, when the mechanical thresholds were not significantly (NS) different from the baseline nociceptive thresholds evaluated before the injection of the mediators (A: ψεRACK group, t(5) = 0.8402, p = 0.4391; control group, t(5) = 1.164, p = 0.2968; B: NGF group, t(5) = 0.9543, p = 0.3837; control group, t(5) = 0.4740, p = 0.6554; C: TNF-α group, t(5) = 0.7559, p = 0.4838; control group, t(5) = 0.9764, p = 0.3737; paired Student's t test), PGE2 (100 ng) was injected intradermally into the dorsum of the hindpaw, and mechanical hyperalgesia was evaluated 30 min and 4 h later. Average paw-withdrawal thresholds before the injection of the mediators and before the injection of PGE2 (1 week later) were as follows: 123.0 ± 2.3 and 119.0 ± 2.7 g, respectively, for the ψεRACK-treated group, and 117.6 ± 1.8 and 115.0 ± 1.0 g, for the control-treated group; 118.6 ± 1.8 and 116.3 ± 1.7 g, respectively, for the NGF-treated group, and 117.6 ± 2.9 and 119.0 ± 1.9 g, for the control-treated group; and 118.3 ± 1.8 and 121.0 ± 2.5 g, respectively, for the TNF-α-treated group; and 117.3 ± 2.1 and 119.6 ± 2.0 g, for the control-treated group. Evaluation of the mechanical hyperalgesia induced by PGE2 showed significant hyperalgesia at 30 min after injection, which was no longer present at the fourth hour time point, in all groups. Two-way repeated-measures ANOVA followed by Bonferroni post-test showed no difference between the controls and the groups treated with the mediators (NS), indicating that i.gl. injection of ψεRACK, NGF, or TNF-α did not produce hyperalgesic priming (N = 6 paws per group).
Role of importin in the induction of priming
Once the transcription factor CREB is activated by cAMP, it is transported into the nucleus by a transport protein, importin, where it can stimulate gene transcription (Forwood et al., 2001; Ahluwalia et al., 2014). Therefore, we next determined whether intraganglion injection of ivermectin, an inhibitor of importin (Wagstaff et al., 2012; Slońska et al., 2013; Fraser et al., 2014; Lokich et al., 2014), prevents the induction of priming. Treatment with ivermectin (10 μg/5 μl), injected 30 min before MCP-1, administered intrathecally to the spinal cord (Fig. 6A), or 8-bromo cAMP, injected into the L5-DRG (Fig. 6B), and for 3 additional days to avoid further activation of the mechanisms involved in priming induction, prevented the development of priming.
Figure 6.

Transport into the nucleus plays a role in the induction of hyperalgesic priming. Rats received intraganglion (i.gl.) injection of vehicle (white bars) or ivermectin (10 μg, black bars), an inhibitor of the nuclear transporter importin. Thirty minutes later, MCP-1 [20 ng/μl, 20 μl, injected intrathecally (i.t.) into the spinal cord, A] or 8-bromo cAMP [10 μg, injected into the DRG, B] was administered. No effect of ivermectin on MCP-1- or 8-bromo cAMP-induced hyperalgesia was observed (data not shown). The injection of vehicle or ivermectin continued daily for 3 d. On the fourth day, PGE2 (100 ng) was injected intradermally into the dorsum of the hindpaw. A, Average paw-withdrawal thresholds before injection of vehicle/MCP-1 and immediately before the injection of PGE2 (4 d later) were 121.3 ± 2.1 and 118.6 ± 1.6 g, respectively (t(5) = 0.9325; p = 0.3939, NS); for the groups that received ivermectin/MCP-1, they were 123.0 ± 2.5 and 123.5 ± 1.6 g, respectively (t(5) = 0.0000; p = 1.0000, NS); no significant (NS) difference between these two values was observed (paired Student's t test). B, Average paw-withdrawal thresholds before injection of vehicle/8-bromo cAMP and immediately before the injection of PGE2 (4 d later) were 117.3 ± 2.1 and 114.6 ± 1.8 g, respectively (t(5) = 0.9035; p = 0.4077, NS); for the groups that received ivermectin/8-bromo cAMP, they were 122.3 ± 1.6 and 122. 5 ± 2.6 g, respectively (t(5) = 0.0000; p = 1.0000, NS); no significant (NS) difference between these two values was observed (paired Student's t test). Mechanical hyperalgesia was then evaluated 30 min and 4 h after PGE2 injection using the Randall–Sellitto paw-withdrawal test. In both A and B, in the paws ipsilateral to the DRGs that received vehicle, the magnitude of hyperalgesia was still significant at the fourth hour after PGE2 injection. However, in the paws ipsilateral to the DRGs that received ivermectin, the prolongation of PGE2-induced hyperalgesia was significantly attenuated (***p < 0.001, A and B, when vehicle and ivermectin groups are compared at the fourth hour; two-way repeated-measures ANOVA followed by Bonferroni post-test), indicating that the induction of priming by i.t. MCP-1 or i.gl. 8-bromo cAMP requires transport into the nucleus. When tested again with PGE2 1 week later; attenuation of the hyperalgesia induced by PGE2 at the fourth hour was still observed (***p < 0.001, A and B, when vehicle and ivermectin groups are compared at the fourth hour), indicating that ivermectin prevented the development of hyperalgesic priming (N = 6 paws per group).
Distinct peripheral terminal and nuclear mechanisms for induction of priming
Finally, we determined whether hyperalgesic priming—the presence of which is evaluated by the prolongation of the hyperalgesia induced by PGE2 (Reichling and Levine, 2009; Ferrari et al., 2013a, 2014)—induced by the intraganglion injection of 8-bromo cAMP, was mediated by the same mechanisms in the peripheral terminal of the nociceptor as when priming was induced by a stimulus applied at the peripheral terminal. One week after the injection of 8-bromo cAMP into the DRG, we injected substances into the peripheral receptive field of primed nociceptors that we have previously shown inhibited, but did not permanently reverse, hyperalgesic priming when priming was induced by an inflammatory stimulus administered into the peripheral receptive field of the nociceptors (Parada et al., 2003b; Ferrari et al., 2013b). The intradermal injection of the selective PKCε inhibitor PKCε-I (1 μg/5 μl) or treatment with ODN AS against αCaMKII for 3 d combined with the αCaMKII inhibitor CaMINtide (1 μg/5 μl) reversibly inhibited the prolongation of PGE2 hyperalgesia induced by the prior intraganglion injection of 8-bromo cAMP (Fig. 7). We next determined whether an intervention that has been shown to reverse the priming induced in the periphery (i.e., treatment with protein translation inhibitors such as cordycepin; Ferrari et al., 2013c) could reverse priming when it was induced by intraganglion administration of 8-bromo cAMP. Cordycepin (1 μg/5 μl), injected into the peripheral receptive field of the nociceptors, at the site of nociceptive testing, was able to reverse the priming induced by intraganglion injection of 8-bromo cAMP (Fig. 8). However, when cordycepin (10 μg/5 μl) was administered into the DRG, it failed to reverse the priming induced by intraganglion injection of 8-bromo cAMP (Fig. 8).
Figure 8.

Protein translation inhibitor reverses priming when it is administered at the terminal of the nociceptor, but not at its cell body. The effect of the protein translation inhibitor cordycepin on priming was evaluated in rats that had received intraganglion (i.gl.) injection of 8-bromo cAMP (10 μg) 2 weeks before. The average paw-withdrawal thresholds before and 2 weeks after 8-bromo cAMP (when the experiments were performed) were 121.6 ± 1.4 and 120.0 ± 1.4 g, respectively; no significant (NS) difference between these two values was observed (t(17) = 1.062, p = 0.3032, NS; paired Student's t test, data not shown). Rats were then divided into a control group (white bars) and two experimental groups, one that received cordycepin intradermally on the dorsum of the hindpaw (1 μg, gray bars) and the other that received i.gl. injection to the DRG (10 μg, black bars). Fifteen minutes later, PGE2 (100 ng) was injected into the dorsum of the hindpaw, and the paw-withdrawal threshold was evaluated 30 min and 4 h later. We observed that, while PGE2-induced hyperalgesia was still significant at the fourth hour in the control group and the experimental group that received cordycepin in the DRG (p > 0.05, when both groups are compared), in the group that received cordycepin in the paw it was significantly attenuated at the fourth hour (***p < 0.001, when compared with the control group), indicating a role of protein translation in the terminal of the nociceptor, but not in the DRG, in hyperalgesic priming (two-way repeated-measures ANOVA followed by Bonferroni post-test; N = 6 paws per group).
Discussion
We have recently proposed two classes of mechanisms for chronic pain in the nociceptor (Reichling and Levine, 2009; Ferrari et al., 2012). In the first class, mediating what we have referred to as type I chronic pain, an ongoing inflammatory process in a “painful tissue” produces pronociceptive mediators that continuously sensitize the peripheral terminals of nociceptors (Feldmann and Maini, 2001; El Bahri et al., 2007). This sensitization underlies the mechanical hyperalgesia reported by many patients with inflammatory diseases. With a more intense sensitization, spontaneous activity develops, which underlies their ongoing pain. Treatment of type I chronic pain is the elimination of the underlying disease process and/or attenuation of the production of the pronociceptive mediators responsible for nociceptor sensitization. A commonly encountered clinical example of treatment for type 1 chronic pain is the use of nonsteroidal anti-inflammatory drugs (NSAIDs), which inhibit the production of prostaglandins (Ferreira, 1972; Kapoor et al., 2005; Woolf, 2010; Ricciotti and FitzGerald, 2011; Chen et al., 2013). Of note in this regard, NSAIDs are the most commonly used agents for the treatment of chronic arthritis pain (American College of Rheumatology, 2000; Bingham, 2002; Martel-Pelletier et al., 2003; Ochi et al., 2003; Hochberg et al., 2012).
In the second class of chronic pain mechanisms, producing what we have referred to as type II chronic pain, there is a neuroplastic change in the nociceptors, such that the patient's pain has become independent of the ongoing disease. In addition, type II chronic pain may be resistant to analgesic agents that were effective during an acute phase of the disease, due to a change in the underlying mechanism. Recently, we have developed a model of type II chronic pain, hyperalgesic priming, in which a prior insult produces a neuroplastic change in IB4+ nociceptors (Joseph and Levine, 2010), such that prolonged hyperalgesia is observed extending long past the half-life of the eliciting mediator in the peripheral terminal of the nociceptor (Reichling and Levine, 2009; Ferrari et al., 2010; Joseph and Levine, 2010). In primed nociceptors, a subsequent exposure to a pronociceptive mediator, prototypically PGE2, whose synthesis is inhibited by NSAIDs, produces markedly prolonged hyperalgesia (Aley et al., 2000; Reichling and Levine, 2009). The prolongation phase of PGE2 hyperalgesia in the primed nociceptor is PKCε mediated; in the acute phase, PGE2 hyperalgesia is mediated by protein kinase A (PKA; Aley and Levine, 1999; Aley et al., 2000; Ferrari et al., 2013a). Thus, in the setting of priming, pain would be resistant to a class of drugs that would work in the acute phase (i.e., inhibitors of PKA). Hyperalgesic priming can be induced by an acute inflammatory or neuropathic insult, as well as by nonpainful insults, such as exposure to unpredictable sound stress or to stress hormones (Khasar et al., 2005, 2008, 2009). The primed state persists without attenuation for at least 2 months after its induction (Aley et al., 2000; Reichling and Levine, 2009).
There are three distinct facets to hyperalgesic priming: (1) induction; (2) expression; and (3) maintenance (i.e., long-term persistence). Priming is induced by the activation of receptors on the peripheral terminals of nociceptors, which have in common the ability to activate PKCε-dependent sensitization of nociceptors (Aley et al., 2000; Parada et al., 2003a; Dina et al., 2008; Ferrari et al., 2010; Alvarez et al., 2014), and inhibition of PKCε prevents the induction of priming by activation of these receptors (Parada et al., 2003a,b, 2005; Reichling and Levine, 2009). Direct activation of PKCε with the selective activator ψεRACK also produces hyperalgesic priming (Aley et al., 2000; Parada et al., 2005). Activated αCaMKII and agonists at the ryanodine receptor, downstream of PKCε, also induce priming (Ferrari et al., 2013b, 2014). Activation of this pathway in the peripheral terminal of the nociceptor generates a signal that travels to the cell body to activate CREB-mediated transcription, which in turn generates a signal that is transported back to the peripheral terminal of the nociceptor (Ferrari et al., 2015). PKCε and αCaMKII mediate the expression of hyperalgesic priming, the dramatic prolongation of mechanical hyperalgesia induced by pronociceptive mediators such as PGE2. Inhibition of these signaling molecules reversibly inhibits the prolongation of PGE2-induced hyperalgesia in the primed nociceptor, but does not eliminate priming (Joseph et al., 2007; Ferrari et al., 2013b, 2014). Therefore, these mediators are not involved in its maintenance. Additionally, activation of protein translation in the nociceptor terminals is also involved in priming induction (Ferrari et al., 2013c, 2015).
The maintenance of priming is dependent on ongoing protein translation in the peripheral terminal of the nociceptor (Ferrari et al., 2013c). To date, only a single intervention has been shown to eliminate the primed state; protein translation inhibitors, such as cordycepin or rapamycin, at the peripheral terminal of the primed nociceptor, permanently reverse hyperalgesic priming (Ferrari et al., 2013c). Conversely, none of the inhibitors (PKCε-I or CaMINtide) that reduced the expression of hyperalgesic priming (Fig. 7; i.e., the prolongation of PGE2-induced hyperalgesia) were able to permanently reverse this neuroplastic change in the nociceptor.
While at the level of the nociceptor cell body, intraganglion injection of 8-bromo cAMP induces mechanical hyperalgesia and hyperalgesic priming, which is reflected by alterations in the peripheral terminal of the nociceptor [i.e., the development of markedly prolonged hyperalgesia induced by subsequent intradermal (or intrathecal) injection of PGE2], intradermal injection of 8-bromo cAMP produces mechanical hyperalgesia, but not priming (Fig. 2). The activation of CREB in the cell body, which is induced by the local increase in cAMP using intraganglion injection of 8-bromo cAMP, can alone induce priming (Fig. 4), with the same underlying mechanism in the peripheral terminal as priming induced by stimuli that activate PKCε in the peripheral terminal of the nociceptors (Fig. 7). Thus, as suggested by our previous study (Ferrari et al., 2015), there appear to be distinct mechanisms in the cell body and the peripheral terminal of the nociceptor that interact to induce hyperalgesic priming.
While hyperalgesic priming induced by a peripheral insult requires ∼72 h to develop (Bogen et al., 2012; Ferrari et al., 2015), that induced by intraganglion injection of 8-bromo cAMP only takes 6–12 h to develop (Fig. 3). Priming induced by intraganglion injection of 8-bromo cAMP is, however, dependent on the same mechanisms in the peripheral terminal of the nociceptor involved in priming induced by interventions at the peripheral terminal; thus, hyperalgesic priming induced by intraganglion 8-bromo cAMP is also reversed by peripheral administration of a protein translation inhibitor (Fig. 8). Based on the length of the peripheral axon in the hindleg of the rat, the only mechanism that could produce such a short latency to onset of the primed state would be rapid axonal transport (Wu et al., 1994; Shubayev and Myers, 2001; Lund et al., 2005; Chidlow et al., 2011). We currently do not know the nature of the signal that is transmitted from the cell body to the peripheral terminal of the nociceptor that is involved in the induction of hyperalgesic priming. It could be a CREB-dependent translated protein that is able to establish the ongoing protein translation. Such a protein would have to normally be in low abundance in the peripheral terminal of the nociceptor. Thus, it would either function to trigger the peripheral translation dependent mechanism or have to be continuously supplied to maintain ongoing peripheral translation. Alternatively, an mRNA transcribed in response to CREB, which would be translated at the peripheral terminal of the nociceptor, could initiate peripheral translation to induce the primed state.
Our hypothesis that distinct mechanisms in the terminals and the cell body of the nociceptor contribute to hyperalgesic priming (Ferrari et al., 2015) is confirmed by our finding that intraganglion administration of mediators that induce priming when administered into peripheral tissue does not induce priming (Fig. 5), while, conversely, administration of a mediator (8-bromo cAMP) that does not induce priming in peripheral tissue, does induce priming when injected into the DRG (Fig. 2). However, the actual molecules that signal from the peripheral terminal of the nociceptor to its cell body, to activate the transcription factor CREB, and from the cell body to the peripheral terminal to activate CPEB-dependent peripheral translation of protein (Bogen et al., 2012; Ferrari et al., 2013b, 2015), in the induction of hyperalgesic priming, remain to be elucidated.
The current study demonstrates the involvement of two distinct and complementary processes that take place in different regions of the nociceptor, in the induction of the neuroplastic changes responsible for the transition to chronic pain. The schematic in Figure 9 shows the mechanisms, similar in both terminals of the peripheral sensory neuron, that have been suggested to play a role in the induction (e.g, activation of PKCε downstream inflammatory mediator receptors) and maintenance (local protein translation, inhibited by cordycepin) of hyperalgesic priming (Ferrari et al., 2015), and the signal produced by an inflammatory insult at the nociceptor terminals that triggers gene transcription at the cell body, producing, in turn, another signal that travels back to the terminals, where the neuroplasticity will be produced and maintained by ongoing protein translation, as demonstrated by our current experiments.
Figure 9.

Hyperalgesic priming mechanisms in the nociceptor terminals and cell body. A schematic of the nociceptor with the central terminal in the spinal cord, and the peripheral terminal in the rat hindpaw, is shown. The blue arrows represent the message triggered by stimulation in either of the terminals and directed toward the cell body, and the red arrows represent the message coming from the cell body to the terminals, where the maintenance of the primed state will take place. On the top is described the induction phase of hyperalgesic priming, separated by the events occurring in the nociceptor terminals (1) and in the cell body (2): inflammatory stimuli such as TNF-α, IL-6, or MCP-1, or the administration of neurotrophins (NGF or GDNF) in the terminals of the nociceptor (1) induce the events that lead to the development of hyperalgesic priming. Subsequent activation of PKCε stimulates CPEB, αCaMKII, or the release of calcium (Ca2+; Aley et al., 2000; Reichling and Levine, 2009; Bogen et al., 2012; Ferrari et al., 2013b), which will produce the message directed toward the cell body (Ferrari et al., 2015). a–c, There (2), activation of cAMP (a) activates the transcription factor CREB (b; pCREB), which will be transported into the nucleus by importin, where gene transcription will take place (c). d, The resulting protein or mRNA will then be transported to the terminals of the nociceptor. On the bottom, the maintenance phase of hyperalgesic priming is shown: at the terminals, ongoing protein translation, triggered by the message originated in the cell body (Bogen et al., 2012; Ferrari et al., 2013c, 2015), is responsible for the maintenance of the neuroplasticity observed in the primed state.
Footnotes
This research was supported by National Institutes of Health Grant NS084545.
The authors declare no competing financial interests.
References
- Ahluwalia A, Jones MK, Szabo S, Tarnawski AS. Aging impairs transcriptional regulation of vascular endothelial growth factor in human microvascular endothelial cells: implications for angiogenesis and cell survival. J Physiol Pharmacol. 2014;65:209–215. [PubMed] [Google Scholar]
- Aley KO, Levine JD. Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci. 1999;19:2181–2186. doi: 10.1523/JNEUROSCI.19-06-02181.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Messing RO, Mochly-Rosen D, Levine JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci. 2000;20:4680–4685. doi: 10.1523/JNEUROSCI.20-12-04680.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez P, Green PG, Levine JD. Role for monocyte chemoattractant protein-1 in the induction of chronic muscle pain in the rat. Pain. 2014;155:1161–1167. doi: 10.1016/j.pain.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American College of Rheumatology. Recommendations for the medical management of osteoarthritis of the hip and knee: 2000 update. American College of Rheumatology Subcommittee on Osteoarthritis Guidelines. Arthritis Rheum. 2000;43:1905–1915. doi: 10.1002/1529-0131(200009)43:9<1905::AID-ANR1>3.0.CO%3B2-P. [DOI] [PubMed] [Google Scholar]
- Araldi D, Ferrari LF, Lotufo CM, Vieira AS, Athié MC, Figueiredo JG, Duarte DB, Tambeli CH, Ferreira SH, Parada CA. Peripheral inflammatory hyperalgesia depends on the COX increase in the dorsal root ganglion. Proc Natl Acad Sci U S A. 2013;110:3603–3608. doi: 10.1073/pnas.1220668110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham CO., 3rd Development and clinical application of COX-2-selective inhibitors for the treatment of osteoarthritis and rheumatoid arthritis. Cleve Clin J Med. 2002;69(Suppl 1):SI5–SI12. doi: 10.3949/ccjm.69.suppl_1.si5. [DOI] [PubMed] [Google Scholar]
- Bogen O, Alessandri-Haber N, Chu C, Gear RW, Levine JD. Generation of a pain memory in the primary afferent nociceptor triggered by PKCε activation of CPEB. J Neurosci. 2012;32:2018–2026. doi: 10.1523/JNEUROSCI.5138-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borle AB, Snowdowne KW. Measurement of intracellular free calcium in monkey kidney cells with aequorin. Science. 1982;217:252–254. doi: 10.1126/science.6806904. [DOI] [PubMed] [Google Scholar]
- Burch RM, Axelrod J. Dissociation of bradykinin-induced prostaglandin formation from phosphatidylinositol turnover in Swiss 3T3 fibroblasts: evidence for G protein regulation of phospholipase A2. Proc Natl Acad Sci U S A. 1987;84:6374–6378. doi: 10.1073/pnas.84.18.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Yang G, Grosser T. Prostanoids and inflammatory pain. Prostaglandins Other Lipid Mediat. 2013;104–105:58–66. doi: 10.1016/j.prostaglandins.2012.08.006. [DOI] [PubMed] [Google Scholar]
- Chidlow G, Ebneter A, Wood JP, Casson RJ. The optic nerve head is the site of axonal transport disruption, axonal cytoskeleton damage and putative axonal regeneration failure in a rat model of glaucoma. Acta Neuropathol. 2011;121:737–751. doi: 10.1007/s00401-011-0807-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churn SB, Sombati S, Jakoi ER, Severt L, DeLorenzo RJ, Sievert L. Inhibition of calcium/calmodulin kinase II alpha subunit expression results in epileptiform activity in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 2000;97:5604–5609. doi: 10.1073/pnas.080071697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dina OA, Green PG, Levine JD. Role of interleukin-6 in chronic muscle hyperalgesic priming. Neuroscience. 2008;152:521–525. doi: 10.1016/j.neuroscience.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dina OA, Khasar SG, Gear RW, Levine JD. Activation of Gi induces mechanical hyperalgesia poststress or inflammation. Neuroscience. 2009;160:501–507. doi: 10.1016/j.neuroscience.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bahri DM, Meddeb N, Sellami S. Rheumatoid arthritis: current status of therapy. Tunis Med. 2007;85:1–8. [PubMed] [Google Scholar]
- Feldmann M, Maini RN. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol. 2001;19:163–196. doi: 10.1146/annurev.immunol.19.1.163. [DOI] [PubMed] [Google Scholar]
- Ferrari LF, Cunha FQ, Parada CA, Ferreira SH. A novel technique to perform direct intraganglionar injections in rats. J Neurosci Methods. 2007;159:236–243. doi: 10.1016/j.jneumeth.2006.07.025. [DOI] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Levine JD. Nociceptor subpopulations involved in hyperalgesic priming. Neuroscience. 2010;165:896–901. doi: 10.1016/j.neuroscience.2009.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Alessandri-Haber N, Levine E, Gear RW, Levine JD. Transient decrease in nociceptor GRK2 expression produces long-term enhancement in inflammatory pain. Neuroscience. 2012;222:392–403. doi: 10.1016/j.neuroscience.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Levine E, Levine JD. Role of a novel nociceptor autocrine mechanism in chronic pain. Eur J Neurosci. 2013a;37:1705–1713. doi: 10.1111/ejn.12145. [DOI] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Levine JD. Role of nociceptor αCaMKII in transition from acute to chronic pain (hyperalgesic priming) in male and female rats. J Neurosci. 2013b;33:11002–11011. doi: 10.1523/JNEUROSCI.1785-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Chu C, Levine JD. Peripheral administration of translation inhibitors reverses increased hyperalgesia in a model of chronic pain in the rat. J Pain. 2013c;14:731–738. doi: 10.1016/j.jpain.2013.01.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Levine JD. Second messengers mediating the expression of neuroplasticity in a model of chronic pain in the rat. J Pain. 2014;15:312–320. doi: 10.1016/j.jpain.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari LF, Bogen O, Reichling DB, Levine JD. Accounting for the delay in the transition from acute to chronic pain: axonal and nuclear mechanisms. J Neurosci. 2015;35:495–507. doi: 10.1523/JNEUROSCI.5147-13.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira SH. Prostaglandins, aspirin-like drugs and analgesia. Nat New Biol. 1972;240:200–203. doi: 10.1038/newbio240200a0. [DOI] [PubMed] [Google Scholar]
- Forwood JK, Lam MH, Jans DA. Nuclear import of Creb and AP-1 transcription factors requires importin-beta 1 and Ran but is independent of importin-alpha. Biochemistry. 2001;40:5208–5217. doi: 10.1021/bi002732+. [DOI] [PubMed] [Google Scholar]
- Fraser JE, Rawlinson SM, Wang C, Jans DA, Wagstaff KM. Investigating dengue virus nonstructural protein 5 (NS5) nuclear import. Methods Mol Biol. 2014;1138:301–328. doi: 10.1007/978-1-4939-0348-1_19. [DOI] [PubMed] [Google Scholar]
- Gniazdowski M, Denny WA, Nelson SM, Czyz M. Transcription factors as targets for DNA-interacting drugs. Curr Med Chem. 2003;10:909–924. doi: 10.2174/0929867033457683. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Kuroda T, Kishimoto H, Wang C, Iwama A, Kimura K. Downregulation of rRNA transcription triggers cell differentiation. PLoS One. 2014;9:e98586. doi: 10.1371/journal.pone.0098586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochberg MC, Altman RD, April KT, Benkhalti M, Guyatt G, McGowan J, Towheed T, Welch V, Wells G, Tugwell P. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken) 2012;64:465–474. doi: 10.1002/acr.21596. [DOI] [PubMed] [Google Scholar]
- Johnson JA, Gray MO, Chen CH, Mochly-Rosen D. A protein kinase C translocation inhibitor as an isozyme-selective antagonist of cardiac function. J Biol Chem. 1996;271:24962–24966. doi: 10.1074/jbc.271.40.24962. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Levine JD. Hyperalgesic priming is restricted to isolectin B4-positive nociceptors. Neuroscience. 2010;169:431–435. doi: 10.1016/j.neuroscience.2010.04.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph EK, Bogen O, Alessandri-Haber N, Levine JD. PLC-beta 3 signals upstream of PKC epsilon in acute and chronic inflammatory hyperalgesia. Pain. 2007;132:67–73. doi: 10.1016/j.pain.2007.01.027. [DOI] [PubMed] [Google Scholar]
- Kapoor M, Shaw O, Appleton I. Possible anti-inflammatory role of COX-2-derived prostaglandins: implications for inflammation research. Curr Opin Investig Drugs. 2005;6:461–466. [PubMed] [Google Scholar]
- Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron. 1999;24:253–260. doi: 10.1016/S0896-6273(00)80837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Green PG, Levine JD. Repeated sound stress enhances inflammatory pain in the rat. Pain. 2005;116:79–86. doi: 10.1016/j.pain.2005.03.040. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Burkham J, Dina OA, Brown AS, Bogen O, Alessandri-Haber N, Green PG, Reichling DB, Levine JD. Stress induces a switch of intracellular signaling in sensory neurons in a model of generalized pain. J Neurosci. 2008;28:5721–5730. doi: 10.1523/JNEUROSCI.0256-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Dina OA, Green PG, Levine JD. Sound stress-induced long-term enhancement of mechanical hyperalgesia in rats is maintained by sympathoadrenal catecholamines. J Pain. 2009;10:1073–1077. doi: 10.1016/j.jpain.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozyrev SA, Nikitin VP. Involvement of translation and transcription processes into neurophysiological mechanisms of long-term memory reconsolidation. Bull Exp Biol Med. 2013;154:584–587. doi: 10.1007/s10517-013-2004-9. [DOI] [PubMed] [Google Scholar]
- Lane-Ladd SB, Pineda J, Boundy VA, Pfeuffer T, Krupinski J, Aghajanian GK, Nestler EJ. CREB (cAMP response element-binding protein) in the locus ceruleus: biochemical, physiological, and behavioral evidence for a role in opiate dependence. J Neurosci. 1997;17:7890–7901. doi: 10.1523/JNEUROSCI.17-20-07890.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokich E, Singh RK, Han A, Romano N, Yano N, Kim K, Moore RG. HE4 expression is associated with hormonal elements and mediated by importin-dependent nuclear translocation. Sci Rep. 2014;4:5500. doi: 10.1038/srep05500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund LM, Machado VM, McQuarrie IG. Axonal isoforms of myosin-I. Biochem Biophys Res Commun. 2005;330:857–864. doi: 10.1016/j.bbrc.2005.02.187. [DOI] [PubMed] [Google Scholar]
- Ma W, Hatzis C, Eisenach JC. Intrathecal injection of cAMP response element binding protein (CREB) antisense oligonucleotide attenuates tactile allodynia caused by partial sciatic nerve ligation. Brain Res. 2003;988:97–104. doi: 10.1016/S0006-8993(03)03348-1. [DOI] [PubMed] [Google Scholar]
- Martel-Pelletier J, Pelletier JP, Fahmi H. Cyclooxygenase-2 and prostaglandins in articular tissues. Semin Arthritis Rheum. 2003;33:155–167. doi: 10.1016/S0049-0172(03)00134-3. [DOI] [PubMed] [Google Scholar]
- Miao FJ, Green PG, Benowitz N, Levine JD. Central terminals of nociceptors are targets for nicotine suppression of inflammation. Neuroscience. 2004;123:777–784. doi: 10.1016/j.neuroscience.2003.10.027. [DOI] [PubMed] [Google Scholar]
- Ochi T, Ohkubo Y, Mutoh S. Role of cyclooxygenase-2, but not cyclooxygenase-1, on type II collagen-induced arthritis in DBA/1J mice. Biochem Pharmacol. 2003;66:1055–1060. doi: 10.1016/S0006-2952(03)00420-9. [DOI] [PubMed] [Google Scholar]
- Ouseph AK, Khasar SG, Levine JD. Multiple second messenger systems act sequentially to mediate rolipram-induced prolongation of prostaglandin E2-induced mechanical hyperalgesia in the rat. Neuroscience. 1995;64:769–776. doi: 10.1016/0306-4522(94)00397-N. [DOI] [PubMed] [Google Scholar]
- Parada CA, Yeh JJ, Joseph EK, Levine JD. Tumor necrosis factor receptor type-1 in sensory neurons contributes to induction of chronic enhancement of inflammatory hyperalgesia in rat. Eur J Neurosci. 2003a;17:1847–1852. doi: 10.1046/j.1460-9568.2003.02626.x. [DOI] [PubMed] [Google Scholar]
- Parada CA, Yeh JJ, Reichling DB, Levine JD. Transient attenuation of protein kinase Cepsilon can terminate a chronic hyperalgesic state in the rat. Neuroscience. 2003b;120:219–226. doi: 10.1016/S0306-4522(03)00267-7. [DOI] [PubMed] [Google Scholar]
- Parada CA, Reichling DB, Levine JD. Chronic hyperalgesic priming in the rat involves a novel interaction between cAMP and PKCepsilon second messenger pathways. Pain. 2005;113:185–190. doi: 10.1016/j.pain.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Quanhong Z, Ying X, Moxi C, Tao X, Jing W, Xin Z, Li W, Derong C, Xiaoli Z, Wei J. Intrathecal PLC(beta3) oligodeoxynucleotides antisense potentiates acute morphine efficacy and attenuates chronic morphine tolerance. Brain Res. 2012;1472:38–44. doi: 10.1016/j.brainres.2012.06.030. [DOI] [PubMed] [Google Scholar]
- Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn Ther. 1957;111:409–419. [PubMed] [Google Scholar]
- Reichling DB, Levine JD. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009;32:611–618. doi: 10.1016/j.tins.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shubayev VI, Myers RR. Axonal transport of TNF-alpha in painful neuropathy: distribution of ligand tracer and TNF receptors. J Neuroimmunol. 2001;114:48–56. doi: 10.1016/S0165-5728(00)00453-7. [DOI] [PubMed] [Google Scholar]
- Slońska A, Cymerys J, Skwarska J, Golke A, Bańbura MW. Influence of importin alpha/beta and exportin 1 on equine herpesvirus type 1 (EHV-1) replication in primary murine neurons. Pol J Vet Sci. 2013;16:749–751. doi: 10.2478/pjvs-2013-0106. [DOI] [PubMed] [Google Scholar]
- Song MJ, Wang YQ, Wu GC. Additive anti-hyperalgesia of electroacupuncture and intrathecal antisense oligodeoxynucleotide to interleukin-1 receptor type I on carrageenan-induced inflammatory pain in rats. Brain Res Bull. 2009;78:335–341. doi: 10.1016/j.brainresbull.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Su L, Wang C, Yu YH, Ren YY, Xie KL, Wang GL. Role of TRPM8 in dorsal root ganglion in nerve injury-induced chronic pain. BMC Neurosci. 2011;12:120. doi: 10.1186/1471-2202-12-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JL, Xiao C, Lu B, Zhang J, Yuan XZ, Chen W, Yu LN, Zhang FJ, Chen G, Yan M. CX3CL1/CX3CR1 regulates nerve injury-induced pain hypersensitivity through the ERK5 signaling pathway. J Neurosci Res. 2013;91:545–553. doi: 10.1002/jnr.23168. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD. Prostaglandin effects after elimination of indirect hyperalgesic mechanisms in the skin of the rat. Brain Res. 1989;492:397–399. doi: 10.1016/0006-8993(89)90928-1. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Coderre TJ, Levine JD. The contribution of training to sensitivity in the nociceptive paw-withdrawal test. Brain Res. 1989;487:148–151. doi: 10.1016/0006-8993(89)90950-5. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Heller PH, Levine JD. Characterization of distinct phospholipases mediating bradykinin and noradrenaline hyperalgesia. Neuroscience. 1990;39:523–531. doi: 10.1016/0306-4522(90)90288-F. [DOI] [PubMed] [Google Scholar]
- Tambeli CH, Parada CA, Levine JD, Gear RW. Inhibition of tonic spinal glutamatergic activity induces antinociception in the rat. Eur J Neurosci. 2002;16:1547–1553. doi: 10.1046/j.1460-9568.2002.02204.x. [DOI] [PubMed] [Google Scholar]
- Wagstaff KM, Sivakumaran H, Heaton SM, Harrich D, Jans DA. Ivermectin is a specific inhibitor of importin alpha/beta-mediated nuclear import able to inhibit replication of HIV-1 and dengue virus. Biochem J. 2012;443:851–856. doi: 10.1042/BJ20120150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widnell KL, Self DW, Lane SB, Russell DS, Vaidya VA, Miserendino MJ, Rubin CS, Duman RS, Nestler EJ. Regulation of CREB expression: in vivo evidence for a functional role in morphine action in the nucleus accumbens. J Pharmacol Exp Ther. 1996;276:306–315. [PubMed] [Google Scholar]
- Wiegert JS, Hofmann F, Bading H, Bengtson CP. A transcription-dependent increase in miniature EPSC frequency accompanies late-phase plasticity in cultured hippocampal neurons. BMC Neurosci. 2009;10:124. doi: 10.1186/1471-2202-10-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ. What is this thing called pain? J Clin Invest. 2010;120:3742–3744. doi: 10.1172/JCI45178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Toma JG, Chan H, Smith R, Miller FD. Disruption of fast axonal transport in vivo leads to alterations in Schwann cell gene expression. Dev Biol. 1994;163:423–439. doi: 10.1006/dbio.1994.1159. [DOI] [PubMed] [Google Scholar]