

Abstract
Neuronal hyperexcitability occurs early in the pathogenesis of Alzheimer's disease (AD) and contributes to network dysfunction in AD patients. In other disorders with neuronal hyperexcitability, dysfunction in the dendrites often contributes, but dendritic excitability has not been directly examined in AD models. We used dendritic patch-clamp recordings to measure dendritic excitability in the CA1 region of the hippocampus. We found that dendrites, more so than somata, of hippocampal neurons were hyperexcitable in mice overexpressing Aβ. This dendritic hyperexcitability was associated with depletion of Kv4.2, a dendritic potassium channel important for regulating dendritic excitability and synaptic plasticity. The antiepileptic drug, levetiracetam, blocked Kv4.2 depletion. Tau was required, as crossing with tau knock-out mice also prevented both Kv4.2 depletion and dendritic hyperexcitability. Dendritic hyperexcitability induced by Kv4.2 deficiency exacerbated behavioral deficits and increased epileptiform activity in hAPP mice. We conclude that increased dendritic excitability, associated with changes in dendritic ion channels including Kv4.2, may contribute to neuronal dysfunction in early stages AD.
Keywords: Alzheimer, amyloid-beta, dendrites, excitability, Kv4.2, tau
Introduction
The incidence of Alzheimer's disease (AD) is increasing with the aging population, and an astonishing 5.2 million Americans are affected by AD, the most common cause of dementia. Dysregulation of neuronal excitability plays an important role in the pathogenesis of AD. Although neuronal activity declines in the later stages of AD, a variety of evidence indicates that it is increased in the early stages of the disease. The hippocampus is hyperactive in functional imaging studies of patients in the mild cognitive impairment stage of AD (Dickerson et al., 2005), the earliest symptomatic stage, and even in the asymptomatic preclinical stage of AD (Reiman et al., 2012). Cortical neurons in early AD patients are hyperexcitable by transcranial magnetic stimulation (Olazarán et al., 2010). Seizures and subclinical epileptiform activity are also more frequent in early AD stages, including mild cognitive impairment (Palop and Mucke, 2009; Vossel et al., 2013).
The cellular basis of AD-related neuronal hyperexcitability is unclear, but many features are replicated in mouse models, enabling mechanistic studies (Hall and Roberson, 2012). For example, electrophysiological recordings of spontaneous activity onto hippocampal neurons show excitation/inhibition imbalance, with a shift toward more excitatory activity (Roberson et al., 2011). In vivo imaging of neuronal activity patterns confirms early increases in hippocampal neuron firing attributable to soluble Aβ (Busche et al., 2012). And many mouse models of AD also show subclinical epileptiform activity and corresponding hallmarks of compensatory remodeling, such as interneuron sprouting and depletion of calbindin (Palop et al., 2003; Palop et al., 2007).
Dendritic excitability has important effects on synaptic plasticity, and abnormal dendritic excitability contributes to many neurological disorders (Nestor and Hoffman, 2012). For example, aberrant dendritic morphology and/or alterations in dendritic ion channels contribute to dendritic hyperexcitability and thus improper integration of synaptic signals in mouse models of Fragile X syndrome, Rett syndrome, autism, and epilepsy (Nestor and Hoffman, 2012). Therefore, we asked whether AD-related neuronal hyperexcitability could involve changes in dendritic excitability.
Materials and Methods
Mice.
The hAPPJ20 and hAPPJ9 lines overexpress an hAPP minigene with Swedish (K670M/N671L) and Indiana (V717F) mutations under control of the PDGF promoter, with higher levels in the hAPPJ20 line than in hAPPJ9 (Mucke et al., 2000). Both of these lines were on a congenic C57BL/6J background (>15 backcrosses). hAPPJ20 mice were crossed with Tau−/− mice (Dawson et al., 2001), also on a congenic C57BL/6J background (>15 backcrosses). Kv4.2−/− mice (Guo et al., 2005) were on a congenic 129SvEv background. The hAPPJ9/Kv4.2−/− cross was thus on C57BL/6J/129SvEv mixed background. APPswe/PS1dE9 double-transgenic mice (Jankowsky et al., 2001, 2004) were originally obtained from the The Jackson Laboratory (stock #004462) and express a chimeric mouse/human APP transgene that contains the Swedish mutations (K595N/M596L) and a mutant human PS1 transgene carrying the deleted exon 9 variant under control of mouse prion promoter elements. These mice were backcrossed >12 generations to the C57BL/6 background.
Male and female mice were used for all experiments, except for the first cohort of hAPPJ20 mice for dendritic patch-clamp (see Fig. 1) and for the experiments with hAPPswe/PS1dE9 mice (see Fig. 2E), which were all male. Mice were kept on a 12 h light/12 h dark cycle and had ad libitum access to food and water. The studies were approved by the Institutional Animal Care and Use Committees of the University of Alabama at Birmingham and the National Institute of Child Health and Human Development, and were conducted in compliance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals.
Figure 1.
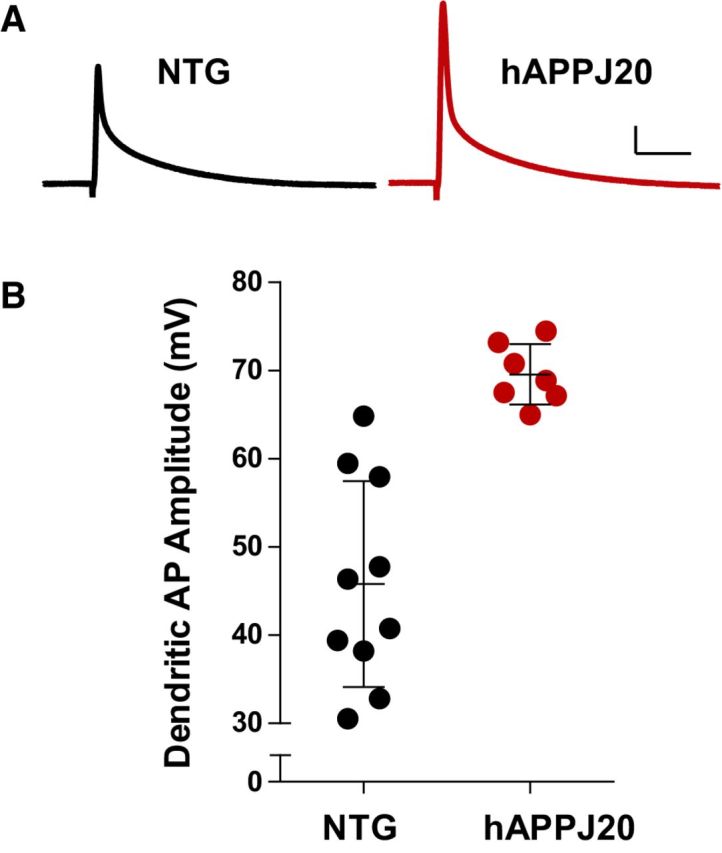
Increased dendritic excitability in hAPPJ20 mice. A, Representative back-propagating dendritic APs recorded in apical dendrites of CA1 pyramidal neurons in NTG and hAPPJ20 mice. Calibration: 10 mV, 12.5 ms. B, Dendritic AP amplitude was increased by 52 ± 3% in hAPPJ20 mice, indicating increased dendritic excitability (age 3 months; p < 0.0001, t test).
Figure 2.
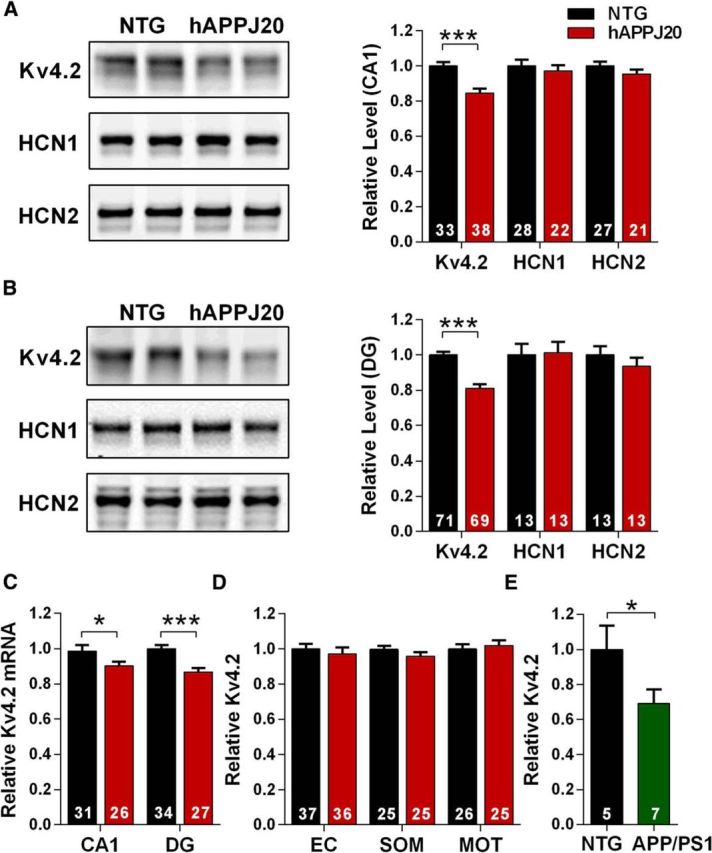
Kv4.2 is depleted in hippocampal regions in hAPPJ20 mice. A, Immunoblots for dendritic ion channels in area CA1. Kv4.2 was decreased in hAPPJ20 mice (ANOVA: hAPP × channel interaction, p < 0.05). ***p < 0.0001 (post hoc test; age 4.5–7 months). B, Immunoblots for dendritic ion channels in dentate gyrus. Kv4.2 was decreased in hAPPJ20 mice (ANOVA: hAPP × channel interaction, p < 0.05). ***p < 0.0001 (post hoc test; age 4.5–7 months). C, Kv4.2 mRNA levels were decreased in area CA1 and DG of hAPPJ20 mice. *p < 0.05 (one-tailed t test). ***p < 0.0001 (one-tailed t test; age 4.5–7 months). D, Kv4.2 levels were unchanged in cortex in hAPPJ20 mice. EC, Entorhinal cortex; SOM, somatosensory cortex; MOT, motor cortex (age 4.5–7 months). E, Kv4.2 was depleted in the hippocampus of hAPPswe/PS1dE9 mice by immunoblot. *p < 0.05 (t test; age 6 months).
Electrophysiology.
Experimenters were blind to genotype for all electrophysiological data acquisition and analysis.
For recordings from CA1 pyramidal neurons, mice were anesthetized with isoflurane, transcardially perfused with oxygenated cutting solution, and decapitated. Perfusion and cutting stock solution contained the following (in mm): 2.5 KCl, 1.25 NaH2PO4, 28 NaHCO3, 7 dextrose, 0.5 CaCl2, and 7 MgCl2, to which 235 sucrose, 1 ascorbic acid, and 3 sodium pyruvate were added immediately before use, pH 7.3. Transverse hippocampal slices (250 μm thick) were prepared using a vibratome (Leica VT1200S) and incubated in ACSF for 30 min at 37°C before being transferred to room temperature. ACSF contained the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 dextrose, 2 CaCl2, and 1 MgCl2, pH 7.3. Ascorbic acid (1 mm) and sodium pyruvate (3 mm) were added to the incubating solution immediately before use.
Slices were transferred to the recording chamber with warmed (31°C-33°C), oxygenated ACSF. DNQX (10 μm), d-AP5 (50 μm), and bicuculline (20 μm) were added to the ACSF to block ligand-gated channels for all recordings. CA1 pyramidal neuron cell bodies and primary apical dendrites were visualized using a Zeiss Axioskop 2 FS plus microscope fit with differential interference contrast optics (Carl Zeiss). Patch pipettes were pulled from borosilicate filaments (3–5 mΩ for soma, 10–12 mΩ for dendrite) and filled with internal solution containing the following (in mm): 20 KCl, 125 K-gluconate, 10 HEPES, 4 NaCl, 0.5 EGTA, 4 Mg-ATP, 0.3 Tris-GTP, and 10 phosphocreatine, pH 7.2. All neurons had resting membrane potentials between −60 and −75 mV. Series resistances ranged from 6 to 15 mΩ and from 15 to 35 mΩ for somatic and dendritic recordings, respectively, and were monitored throughout the experiments. All recordings were made using a Multiclamp 700B amplifier and Clampex 10.2 software. Signals were digitized at 10 kHz with a Digidata 1440A and filtered at 10 kHz for recordings of back-propagating action potentials (APs) and 50 kHz for recordings of firing properties. All recordings were analyzed using IGOR Pro (WaveMetrics).
Single back-propagating APs measured in the soma and primary apical dendrite were stimulated every 5 s for 10 sweeps by a 0.2 ms constant current pulse through a stimulus isolator (WPI, A385) using bipolar glass-stimulating electrodes placed in the alveus. Minimal stimulation to reliably elicit back-propagating APs was used. Series resistance was monitored using a 100 ms, 20 mV hyperpolarizing step 1 s before stimulation. The maximum back-propagating AP amplitude was calculated from the resting potential for each sweep and averaged together for each cell.
Somatic properties were recorded using a 1 s current injection starting with a −200 pA sweep increasing to 200 pA in 20 pA steps with 8 s between sweeps. Neurons were held at −65 mV, and series resistance was monitored between each trial. Three trials were recorded per cell, and the results were averaged by cell. Input resistance was calculated from the slope of the IV curve between −20 and 20 pA. Firing properties (number of spikes, threshold, after-hyperpolarization potential [AHP], etc.) were calculated from the first AP during the 200 pA current injection. Threshold was defined as the point when the slope reached 10 mV/ms. Time to onset was the time between initiating the current injection and threshold. Maximum AHP and amplitude of APs were measured relative to threshold, and spike half-width was measured at 50% maximum amplitude.
For recordings from dentate granule cells, mice were anesthetized and perfused transcardially with ice-cold modified ACSF containing the following (in mm): 110 choline chloride, 26 d-glucose, 2.5 MgCl2, 2.5 KCl, 1.25 Na2PO4, 0.5 CaCl2, 1.3 sodium ascorbate, 3 sodium pyruvate, and 25 NaHCO3, bubbled with 95% O2/5% CO2. The brain was removed, and 350-μm-thick hippocampal slices were prepared using a vibratome (Vibratome 3000EP). Slices were incubated at 37°C for ∼30 min in recording solution containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 Na2PO4, 2 CaCl2, 1 MgCl2, 25 NaHCO3, and 25 d-glucose bubbled with 95% O2/5% CO2, and then transferred to room temperature in the same solution. Slices were bathed in 100 μm picrotoxin, 2 μm NBQX, 1 μm TTX, 10 μm d-APV, and 100 μm CdCl2 to isolate potassium currents. Patch pipettes were filled with an intracellular solution containing the following (in mm): 129 K-gluconate, 20 KCl, 10 HEPES, 10 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid, 2 MgCl2, 2 Na2-ATP, 2 glutathione (reduced or oxidized form), pH 7.3. All recordings were done at 22°C. Series resistance was uncompensated (10–25 mΩ), and experiments were discarded if substantial changes (>15%) were observed.
Immunoblots.
Saline-perfused mouse brains were stored at 80°C and then thawed, and hemibrains were chopped into 450 μm slices (McIlwain tissue chopper); brain regions were microdissected. Tissue was homogenized in a lysis buffer (50 mm Tris base, 150 mm NaCl, 0.5% sodium deoxycholate, 5 mm EDTA, 1 mm DTT, 0.1% Triton X-100), protease inhibitors (Halt), and phosphatase inhibitors (Sigma). Samples were placed in sample buffer and DTT and were heated for 10 min at 70°C, then separated in reducing NuPAGE gels (Life Technologies) and transferred to PVDF Immobilon-FL membrane (Millipore). Membranes were incubated with primary antibody (Kv4.2, 1:2000 from Alomone Labs; α-tubulin, 1:2000 from Sigma; HCN1, 1:3000 DMC51.1; HCN2, 1:2000 DMC52.2 gifts from Dr. Dane Chetkovich, Northwestern University Medical School, Chicago, IL). Membranes were incubated with fluorescent secondary antibody (LI-COR) at 1:20,000 for 1 h at room temperature, scanned by Odyssey Imager (LI-COR), and analyzed using Odyssey Imager software. For hAPPswe/PS1dE9 experiments, mice were perfused with PBS and the hippocampus extracted. The hippocampus was homogenized in carbonate buffer (100 mm Na2CO3, 50 mm NaCl, pH 11.5. 2× lysis buffer, 75 mm NaCl, 75 mm Tris-HCl, pH 6.8, 15% glycerol, 3% SDS) and samples were triturated 10 times with a 25 G needle. Lysates were then centrifuged at 14,000 rpm for 30 min at 4°C. Supernatants were collected and separated by SDS-PAGE. After transfer, membranes were probed with primary antibody for Kv4.2 (1:2000 from Sigma P0233) as outlined above.
Immunohistochemistry.
Saline-perfused brains were fixed for 24 h in 4% PFA and then sectioned at 30 μm on a sliding microtome (Leica SM200R). One set of serial sections was first treated with 3% H2O2 for 10 min and then blocked with 1% milk and 10% normal goat serum for 1 h at room temperature. Then the sections were incubated with anti-calbindin antibody (1:30,000, Swant) overnight at 4°C. This was followed by incubation for 1 h at room temperature with biotinylated goat anti-rabbit antibody (1:500, Vector Laboratories) and then for another hour in avidin-biotin complex (Vectastain Elite ABC Kit, Vector Laboratories). Peroxidase activity was detected with 3,3′-diaminobenzidine and 0.036% H2O2 in 100 mm Tris-HCl for 6 min at room temperature. Sections were mounted on slides and coverslipped using Vectashield HardSet mounting medium with DAPI (Vector Laboratories). Sections were imaged using a Nikon H600L microscope. Calbindin was quantified using ImageJ to measure staining intensity of the dentate gyrus and CA1. The ratio of dentate/CA1 immunoreactivity was graphed (Palop et al., 2011).
qRT-PCR.
RNA was isolated using TRIzol (Life Technologies). cDNA was made from RNA with the SuperScriptIII First Strand Synthesis System (Life Technologies). cDNAs were quantified using Bio-Rad iQ SYBER Green and primers sets, Kv4.2, 5′-TCTGTGCACTTACAATGAGCTGATT-3′ and 5′-TGCATCCCCATGAGAAACACT-3′, GAPDH, 5′-GGGAAGCCCATCACCATCTT-3′, 5′-GCCTTCTCCATGGTGGTGAA-3′. Amplification was on the Roche LightCycler 480 and Kv4.2 mRNA was normalized to GAPDH mRNA.
Drug treatment.
Levetiracetam (LEV; Sequoia Research Products) was dissolved in sterile saline solution. For chronic treatment, Model 2004 osmotic minipumps (ALZET) were filled with saline or LEV solution and were implanted subcutaneously in the interscapular region of the mice. The Model 2004 minipump delivers fluid at a rate of 0.25 μl/h for 28 d. Calculations were done to achieve a dose of 75 mg/kg/d. Minipumps were primed for 24 h at 37°C. Then mice were anesthetized with 3% isoflurane, and the pumps were surgically implanted. After surgery, mice were given fluids and pain medicine and placed in a heated cage for recovery. Mice continued to be group-housed after surgery.
Behavioral assessment.
All behavioral testing was conducted during the light cycle, at least 1 h after the lights came on. Mice were transferred to the testing room with dim lights and white noise for acclimation at least 1 h before experiments. Testing apparatuses were cleaned with 75% ethanol between experiments and disinfected with 2% chlorhexidine after experiments were finished each day. Investigators were blind to genotype. For elevated plus maze assessment, mice were placed in the center hub and allowed free access to explore the arms for 10 min (Med Associates). The apparatus was 1 m high with 2-inch-wide arms. Exploration of each arm was determined by the manufacturer's software. For open field assessment, mice were placed into a 40 cm × 40 cm open field apparatus and allowed to explore for 10 min (Med Associates). Ambulatory distance and time were determined by the manufacturer's software.
EEG.
For electroencephalography (EEG) experiments, mice were anesthetized with 2.5% isoflurane. Six small holes were drilled bilaterally through the cranium, ∼2–2.5 mm posterior and lateral to bregma, ∼4 mm posterior to bregma and 5 mm lateral to the midline, and ∼6 mm posterior to bregma and 3–4 mm lateral to midline, using a dental drill with a 1 mm bit. Three 1.6 mm stainless steel screws (Small Parts) were screwed halfway into alternate holes. An EEG electrode (Plastics One) with 2 lead wires and a ground, cut to rest on the dura, were fitted into the remaining holes. The lead wires were placed bilaterally on the cortical surface of the parietal hemispheres in the cortical region over the underlying hippocampi. Once the wires were positioned, the electrode unit was stabilized with dental acrylic, and the scalp closed with skin glue (3M, Vetbond). This two-electrode system did not allow us to identify the anatomical origin of epileptic activity. One week after electrodes were placed, animals were housed individually in specially constructed EEG monitoring cages. EEG data were acquired at a sampling rate of 500 Hz using Biopac EEG100C amplifiers (Biopac Systems) and AcqKnowlege 4.2 EEG Acquisition and Reader Software (Biopac Systems). All data were manually analyzed in digital format by an experienced observer blinded to genotype. In addition, cages were equipped with IR Digital Color CCD cameras (Digimerge Technologies) to visually record animals concurrent with EEG acquisition. Video was acquired using security system hardware and software (L20WD800 Series, Lorex Technology) to compare EEG abnormalities with behavior. Spikes were defined as <200 ms in duration with amplitudes 5× greater than baseline. The corresponding video of the animal was analyzed for associated behavior during abnormal events.
Statistical analysis.
Data were analyzed using the statistical methods indicated in the figure legends (GraphPad, Prism 6). Error bars and ± ranges indicate SEM.
Results
We first determined whether the dendrites in a mouse model of AD are hyperexcitable. Because of their electrotonic properties, accurately measuring dendritic excitability in neurons with large dendritic trees requires directly recording from the dendrites. We measured dendritic excitability in 3-month-old hAPPJ20 mice using patch-clamp recordings from apical dendrites of CA1 pyramidal neurons, ∼150 μm from the soma. APs induced in the axon travel retrogradely into the dendrites depending on the excitability of dendritic membranes. The amplitude of these back-propagating dendritic APs provides a standard measure of dendritic excitability (Magee et al., 1998).
Dendritic AP amplitude was increased by >50% in hAPPJ20 mice compared with wild-type controls (Fig. 1), indicating dendritic hyperexcitability. To determine whether this hyperexcitability was a general property of the cell, we measured AP firing properties (AP onset, threshold, amplitude, half-width, AHP amplitude, and AP number) at the soma and found no differences (Table 1). Thus, somatic excitability of hippocampal neurons in hAPPJ20 mice was unremarkable, whereas the dendrites were hyperexcitable, suggesting that AD-related neuronal hyperexcitability originates in dendrites.
Table 1.
Somatic firing properties of CA1 pyramidal neurons in hAPPJ20 micea
| NTG (n = 31 cells) | hAPPJ20 (n = 23 cells) | |
|---|---|---|
| AP onset (ms) | 18.57 ± 1.45 | 18.52 ± 1.46 |
| AP threshold (mV) | −46.10 ± 0.65 | −46.13 ± 0.46 |
| AP amplitude (mV) | 96.78 ± 1.00 | 96.37 ± 1.04 |
| AP half-width (ms) | 0.76 ± 0.12 | 0.73 ± 0.13 |
| AHP amplitude (mV) | 6.85 ± 0.47 | 7.48 ± 0.45 |
| AP number | 34.84 ± 1.38 | 32.99 ± 1.15 |
aSomatic firing properties were calculated from the first AP elicited upon a 1 S, 200 pA current injection in whole-cell current-clamp recordings from the soma of NTG and hAPPJ20 mice. No significant differences were found between genotypes. Neurons were held at −65 mV, and series resistance was monitored between each trial. Three trials were recorded per cell, and the results were averaged by cell. Threshold was defined as the point when the slope reached 10 mV/ms. AP onset was the time between initiating the current injection and threshold. Maximum AHP and peak AP amplitude were measured relative to threshold, and AP half-width was measured at 50% maximum amplitude. AP number is the total number of APs elicited by the current injection.
We next probed the molecular mechanisms underlying dendritic hyperexcitability in hAPPJ20 mice. Structural changes were unlikely to underlie the effect, as hAPPJ20 have normal dendrites at this age (Moolman et al., 2004), and we observed no changes in dendritic input resistance (211.2 ± 18.9 MΩ in nontransgenic [NTG] vs 219.6 ± 47.2 MΩ in hAPPJ20) or resting membrane potential (−66.0 ± 1.0 in NTG vs −65.8 ± 0.6 in hAPPJ20). Dendritic APs are regulated by several voltage-gated ion channels that are selectively expressed in dendrites (Jinno et al., 2005). In hippocampal neurons, the primary dendritic ion channels regulating dendritic APs are Kv4.2 potassium channels and voltage-gated Na+ channels (Frick and Johnston, 2005). Alterations in voltage-gated Na+ channel levels were unlikely to mediate the effect, as the maximal rate of dendritic AP rise, an indication of maximal Na+ conductance, was normal and a prior study had found no changes in voltage-gated Na+ channels in the hippocampus of hAPPJ20 mice (Verret et al., 2012).
On the other hand, Kv4.2 levels were reduced in area CA1 of hAPPJ20 mice, an effect consistent with increased dendritic excitability (Fig. 2A). A similar pattern of Kv4.2 reduction was observed in the dentate gyrus (Fig. 2B). This was not a generalized effect on dendritic ion channels, as total levels of HCN1 and HCN2 channels, which are also selectively expressed in dendrites, were unchanged (Fig. 2A,B). Levels of Kv4.2 mRNA were decreased in both CA1 and dentate (Fig. 2C), indicating that Kv4.2 depletion occurs due to downregulation of gene expression. We next asked whether these changes were widespread in the brain. Kv4.2 levels were normal in several cortical regions (Fig. 2D), suggesting hippocampal specificity. Finally, we determined whether similar changes were observed in other mouse models of AD, and found a similar decrease in Kv4.2 in the hippocampus of hAPPswe/PS1dE9 mice (Fig. 2E).
Kv4.2 contributes to the rapidly inactivating A-type potassium current, IA. To determine whether the decrease in Kv4.2 levels was associated with a functional change in IA, we measured this current in dentate granule cells in NTG and hAPPJ20 mice, adapting previously published protocols to isolate the Kv4-mediated component of IA (Chen et al., 2006; Rüschenschmidt et al., 2006). Consistent with the biochemical data, the component of IA carried by Kv4.2 was decreased in hAPPJ20 mice (Fig. 3).
Figure 3.

Decreased IA in hAPPJ20 mice. A, Protocol used to isolate Kv4-mediated component of IA, based on Chen et al. (2006) and Rüschenschmidt et al. (2006). The first pass (P1) activates voltage-gated potassium channels (recordings are performed under conditions that block sodium, calcium, and calcium-activated potassium channels, plus glutamate and GABA receptors). The second pass (P2) includes a depolarizing step to inactivate fast-inactivating (A-type) channels. The subtraction (P1–P2) represents IA, the fast-inactivating potassium current. Each pass includes two pulses to distinguish Kv1- and Kv4-mediated currents, which both contribute to IA. Kv4 currents recover from inactivation an order of magnitude more quickly than Kv1 currents, so the peak current after the second pulse represents Kv4-mediated current, when Kv1 has not yet recovered from inactivation. Thus, the peak recovery between the first and second pulses (arrow) in the P1–P2 subtraction trace represents Kv4-mediated IA (Chen et al., 2006). B, Subtraction (P1–P2) traces for NTG and hAPPJ20 mice, showing decreased Kv4-mediated IA (arrows). C, Quantification of data in B showing decreased Kv4-mediated IA in hAPPJ20 mice (N = 11 cells per genotype; age 4–5 months). **p < 0.01 (t test).
To determine why Kv4.2 is depleted in hAPPJ20 mice, we considered factors known to influence Kv4.2 expression, including epileptiform activity and seizures (Tsaur et al., 1992; Pei et al., 1997; Bernard et al., 2004; Lugo et al., 2008; Aronica et al., 2009), which occur in hAPPJ20 mice (Palop et al., 2007). To determine whether epileptiform activity contributes to Kv4.2 depletion, we used LEV, an antiepileptic drug that has been shown to reduce epileptiform activity and behavioral abnormalities in hAPPJ20 mice (Sanchez et al., 2012). First, we pretested mice before LEV treatment to ensure that both saline and LEV-treated groups were comparable at baseline (Fig. 4A). Mice were implanted with osmotic pumps and treated with saline or LEV for 28 d. We confirmed the prior findings of Sanchez et al. (2012) that LEV corrects behavioral abnormalities in hAPPJ20 mice (Fig. 4B) and blocks calbindin depletion (Fig. 4C,D), which serves as an indicator of epileptiform activity in hAPPJ20 mice (Palop et al., 2011). When we measured Kv4.2 levels, we found that LEV treatment blocked the Kv4.2 depletion in hAPPJ20 mice (Fig. 4E). We conclude that epileptiform activity contributes to Kv4.2 depletion in hAPPJ20 mice.
Figure 4.
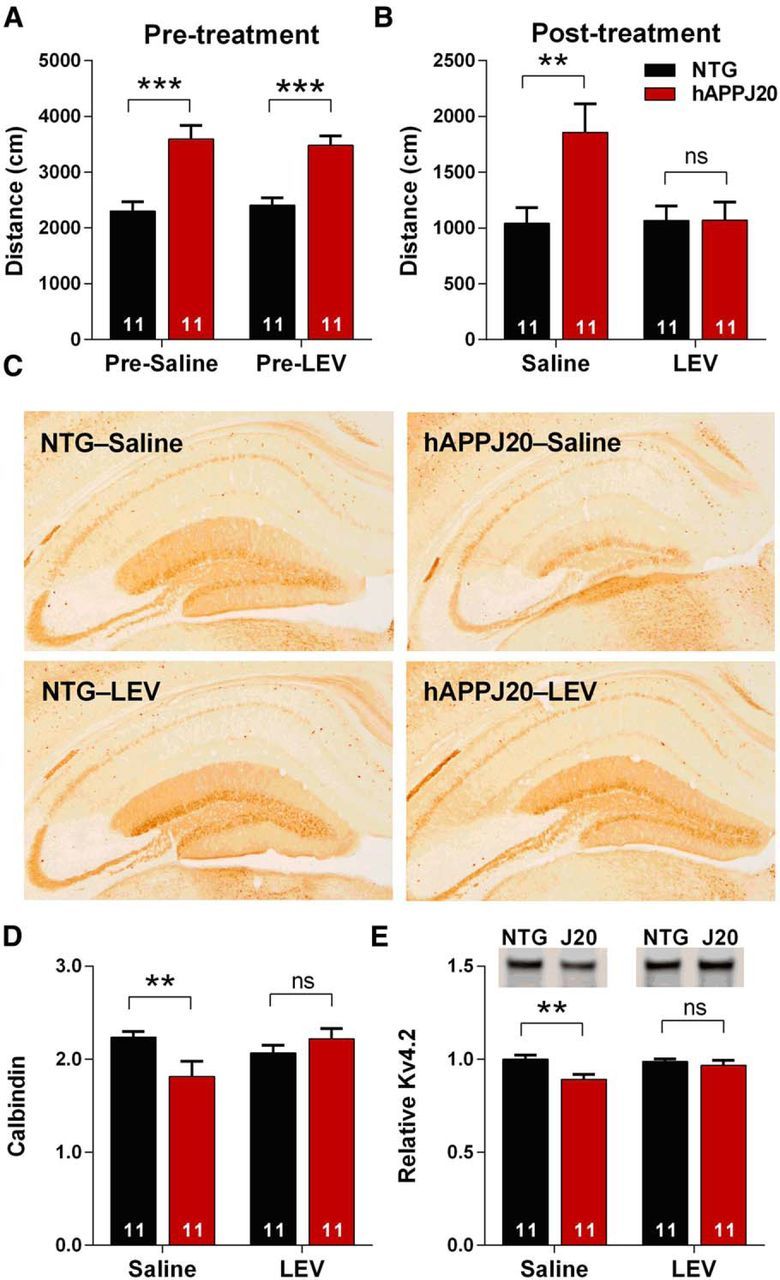
Levetiracetam blocks Kv4.2 depletion in hAPPJ20 mice. A, hAPPJ20 mice showed increased ambulatory distance in open field when pretested before LEV treatment (age 4–5 months, p < 0.0001, t test). B, After 18 d of LEV treatment, hAPPJ20 mice showed normal ambulatory distance (ANOVA: hAPP × treatment interaction, p < 0.05, **p < 0.01 on post hoc test). C, Representative images of calbindin immunohistochemistry after 28 d of LEV treatment. D, Quantification of calbindin immunoreactivity after 28 d of LEV treatment, expressed as the ratio of DG to CA1 immunoreactivity as in Palop et al. (2011). hAPPJ20 mice treated with LEV showed normal levels of calbindin (ANOVA: hAPP × treatment interaction, p < 0.05, **p < 0.01 on post hoc test; age 5–6 months). E, Quantification of Kv4.2 after LEV treatment for 28 d. LEV blocked the Kv4.2 depletion in the hAPPJ20 mice. Representative blots and quantification of Kv4.2 in the DG of hAPPJ20 mice (ANOVA: hAPP effect, p < 0.05, **p < 0.01 on post hoc test; age 5–6 months).
Tau reduction protects against hyperexcitability and seizures in hAPP mice and other models (Roberson et al., 2007, 2011; Ittner et al., 2010; DeVos et al., 2013), so we examined whether the changes in Kv4.2 and dendritic excitability were tau-dependent by crossing hAPPJ20 mice with Tau−/− mice. Using dendritic patch-clamp recordings on CA1 pyramidal neurons, we confirmed the increased dendritic excitability in hAPPJ20/Tau+/+ mice (Fig. 5B), which have normal tau levels and are genetically identical to the hAPPJ20 mice in Figure 1. However, in hAPPJ20/Tau−/− mice, dendritic APs were normal (Fig. 5A,B), indicating that tau is required for the increase in dendritic excitability. Similarly, loss of Kv4.2 was observed in hAPPJ20/Tau+/+ but not in hAPPJ20/Tau−/− mice, both in area CA1 (Fig. 5C) and in dentate (Fig. 5D). Thus, tau is required for Aβ-induced dendritic hyperexcitability and loss of Kv4.2.
Figure 5.

Tau is required for Aβ-induced dendritic hyperexcitability and loss of Kv4.2 in hAPPJ20 mice. A, Representative back-propagating dendritic APs recorded in apical dendrites of CA1 pyramidal neurons from Tau−/− mice. Calibration: 10 mV, 12.5 ms. B, Dendritic AP amplitude was increased in hAPPJ20/Tau+/+ mice, but not in hAPPJ20/Tau−/− mice (ANOVA: hAPP × tau interaction, p < 0.001; on post hoc tests, hAPPJ20/Tau+/+ differed from all other groups, p < 0.0001; age 6.5–8.5 months). C, Tau reduction blocked the loss of Kv4.2 in area CA1 (ANOVA: p < 0.02; *p < 0.05 on post hoc tests, hAPP/Tau+/+ differed from other groups; age 4.5–7 months). D, Tau reduction blocked the loss of Kv4.2 in the dentate gyrus (ANOVA: p < 0.01; **p < 0.01 on post hoc tests, hAPP/Tau+/+ differed from other groups, p < 0. 005; age 4.5–7 months).
If epileptiform activity drives the changes in Kv4.2 and dendritic excitability, these changes could be either part of a protective compensatory response or part of detrimental positive feedback loop. To determine whether loss of Kv4.2 is protective or detrimental in hAPP mice, we crossed Kv4.2-deficient mice with hAPPJ9 mice. hAPPJ9 mice express lower levels of the same hAPP minigene as the hAPPJ20 mice. hAPPJ9 mice show minimal deficits but are sensitized to Aβ-induced dysfunction and are useful for testing hypotheses about exacerbating Aβ toxicity, as they avoid ceiling effects that could occur in crosses with hAPPJ20 mice (Mucke et al., 2000; Chin et al., 2005; Farris et al., 2007). We predicted that if Kv4.2 depletion were detrimental, hAPPJ9/Kv4.2−/− mice would have greater neuronal dysfunction than hAPPJ9/Kv4.2+/+ mice with normal Kv4.2 levels. Kv4.2−/− mice have learning and memory deficits, even without crossing to hAPP mice, that would confound interpretation of the cross, so we focused on abnormalities in the open field and elevated plus maze. Consistent with the hypothesis that loss of Kv4.2 is detrimental, hAPPJ9/Kv4.2−/− mice had increased exploratory activity and spent more time in the center of the open field, changes also seen in hAPP20 mice (Fig. 6A,B). We found that, in the elevated plus maze, hAPPJ9/Kv4.2−/− mice had more total entrances and spent a higher percentage of their time in the open arms compared with hAPPJ9/Kv4.2+/+ mice (Fig. 6C,D).
Figure 6.

Kv4.2 deficiency exacerbates behavioral abnormalities in hAPPJ9 mice. A, In the open field, hAPPJ9/Kv4.2−/− mice showed increased ambulatory distance, an abnormality seen in hAPPJ20 mice (ANOVA: hAPP × Kv4.2 interaction, p < 0.05; *p < 0.05 and ***p < 0.0001 on post hoc test; age 5–7 months). B, In the open field, hAPPJ9/Kv4.2−/− mice showed increased time in the center, an abnormality seen in hAPPJ20 mice (ANOVA: Kv4.2 effect, p < 0.01; *p < 0.05 and **p < 0.01 on post hoc test; age 5–7 months). C, In the elevated plus maze, hAPPJ9/Kv4.2−/− mice showed increased total entrances, an abnormality seen in hAPPJ20 mice (ANOVA: Kv4.2 effect, p < 0.0001; **p < 0.01 and ***p < 0.001 on post hoc test; age 5–7 months). D, In elevated plus maze, hAPPJ9/Kv4.2−/− mice showed increased time in the open arms, an abnormality seen in hAPPJ20 mice (ANOVA: hAPP effect p < 0.0001; Kv4.2 effect p < 0.001; **p < 0.01 and ***p < 0.001 on post hoc test; age 5–7 months).
To determine whether Kv4.2 deficiency also exacerbated epileptiform activity in hAPPJ9 mice, we recorded EEGs and counted epileptiform spikes (Fig. 7). We never observed any epileptiform spikes in NTG mice. Kv4.2−/− mice had infrequent spikes, as did hAPPJ9 mice, which in 8 of 9 mice occurred at a frequency <1/h. The only group with a significant increase in epileptiform spiking was the hAPPJ9/Kv4.2−/− mice. We conclude that Kv4.2 deficiency exacerbates both electroencephalographic and behavioral abnormalities in hAPPJ9 mice.
Figure 7.
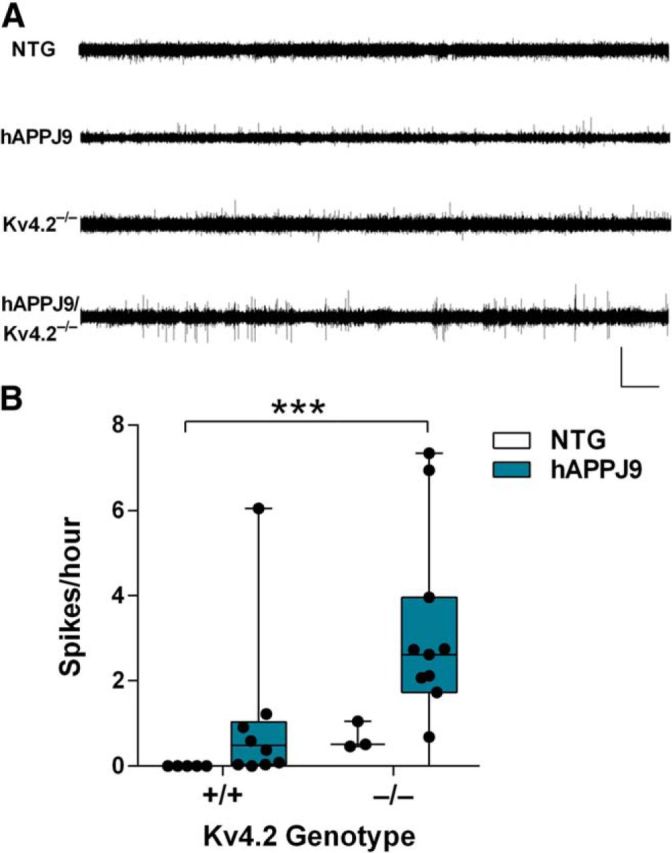
Kv4.2 deficiency exacerbates epileptiform activity in hAPPJ9 mice. A, Representative EEG recordings showing more frequent spikes in hAPPJ9/Kv4.2−/− mice compared with other genotypes. Calibration: 7 mV, 15 min. B, Quantification of epileptiform spikes on EEG showing a significant increase only in hAPPJ9/Kv4.2−/− mice (Kruskal–Wallis test, p < 0.005). ***p < 0.001 (post hoc test; age 10–12 months).
Discussion
In this study, we asked whether AD-related neuronal hyperexcitability involves changes in dendritic excitability. We showed that Aβ-induced hyperexcitability originates in the dendrites in hippocampal neurons and that dendritic hyperexcitability is associated with reduction in the level of Kv4.2 potassium channels. Tau is required for both the decrease in Kv4.2 and the increase in dendritic hyperexcitability. Our data suggest a feedback loop in which Kv4.2 depletion increases excitability and epileptiform activity, which in turn contributes to further reduction in Kv4.2. Loss of Kv4.2 is detrimental and exacerbates deficits in mice expressing hAPP/Aβ.
This is, to our knowledge, the first direct evidence of dendritic hyperexcitability in a model of AD. The ability to patch and directly record from dendrites was critical in this study as dendritic excitability cannot be evaluated using conventional whole-cell patch-clamp or extracellular field recordings because of the electrotonic filtering of dendritic events recorded at the soma.
Kv4.2 is predominantly localized to distal dendrites (Jinno et al., 2005) and has a hyperpolarizing current, so its activation dampens back propagation of APs and dendritic excitability (Yuan et al., 2005; Andrásfalvy et al., 2008). Regulation of the timing of synaptic signals and back-propagating dendritic APs is critical for a form of synaptic plasticity called spike timing-dependent plasticity (Ramakers and Storm, 2002; Kim et al., 2005). Regulation of spike timing-dependent plasticity by Kv4.2 affects induction of LTP at active synapses (Watanabe et al., 2002; Chen et al., 2006). Kv4.2 knock-out mice, which have increased dendritic APs, have impaired learning and memory (Lugo et al., 2012). Thus, lower levels of Kv4.2 in the hippocampus of hAPPJ20 mice might contribute to learning and memory deficits.
To assess the consequences of reduced Kv4.2 levels and increased dendritic excitability on behavior in AD models, we crossed hAPP mice with Kv4.2−/− mice and found that loss of Kv4.2 exacerbated hAPP/Aβ-induced behavioral deficits. The complete deficiency of Kv4.2 in Kv4.2−/− mice obviously represents a larger change in Kv4.2 than the loss we observed in hAPP mice (Fig. 2), and the absence of Kv4.2 is constitutive, not acquired, which are caveats to the interpretation of these data. However, the increase in dendritic excitability we observed in hAPPJ20 mice, measured by amplitude of dendritic AP, is of similar magnitude to that observed in Kv4.2−/− mice (compare Fig. 1 with Chen et al., 2006; their Fig. 4). This suggests either a ceiling effect, whereby even relatively small changes in Kv4.2 produce large effects on dendritic AP amplitude, or a more pronounced effect of acquired (vs. constitutive) loss of Kv4.2 on dendritic excitability due to differences in compensatory mechanisms. Interestingly, modeling studies predict that even relatively small changes in IA, the current carried by Kv4.2, can result in large changes in back-propagating AP amplitude (Migliore et al., 1999).
It is increasingly apparent that dendrites are a key site of AD pathophysiology. Aβ reduces dendritic complexity, induces spine loss, and causes aberrant dendritic signaling through its direct binding to dendritic receptors and downstream effectors (for review, see Cochran et al., 2014). Our data show that Kv4.2 is a dendritic effector downstream of Aβ and that changes in excitability are likely an important aspect of dendritic changes in AD. Previous imaging studies showed increased levels of calcium in dendrites of hAPP mice, consistent with this increased dendritic excitability (Kuchibhotla et al., 2008).
Tau is required for Aβ-induced dysfunction, and tau reduction blocks the increased seizure susceptibility seen in hAPP mice (Roberson et al., 2007, 2011; Ittner et al., 2010). Recent data suggest that tau is present in dendrites and that its dendritic localization is regulated by both Aβ and neuronal activity (Ittner et al., 2010; Zempel et al., 2013; Frandemiche et al., 2014). Our finding that hAPPJ20 mice show tau-dependent dendritic hyperexcitability is consistent with the hypothesis that tau acts in the dendrites to increase excitability, contributing to AD pathophysiology. However, the data are also consistent with a more indirect role for tau in mediating dendritic hyperexcitability, as tau is required for the epileptiform activity in hAPPJ20 mice (Roberson et al., 2011) and thus tau reduction could prevent dendritic hyperexcitability through an antiepileptic effect that may or may not involve dendritically localized tau.
A recent study examined older APP/PS1 mice and found increased neuronal excitability along with dendritic simplification, and used computational modeling to predict that changes in dendritic structure could produce neuronal hyperexcitability (Šišková et al., 2014). This mechanism does not explain the dendritic hyperexcitability we observed. Dendritic arbors of hippocampal neurons in hAPPJ20 mice are normal at 3 months (Moolman et al., 2004), the age at which we first detected increased excitability. Furthermore, there were no differences in input resistance in our dendritic patch-clamp recordings in hAPPJ20 mice. Thus, the dendritic hyperexcitability we observed is likely due to functional changes in channels, including Kv4.2, rather than structural changes, although the dendritic simplification that occurs with aging (Moolman et al., 2004; Šišková et al., 2014) likely adds to these effects. The dendritic hyperexcitability we observed in hAPPJ20 mice is not likely to be related directly to amyloid plaques, as it was observed at 3 months; initial amyloid plaques form at ∼4 months of age in hAPPJ20 mice. This is consistent with other observations indicating that Aβ-induced hyperexcitability and behavioral deficits in hAPP mice do not correlate with plaques (Mucke et al., 2000; Busche et al., 2012). Finally, the dendritic hyperexcitability we observed in hAPPJ20 mice cannot be explained by changes in synaptic inputs, such as inhibitory tone, as the recordings were performed in the presence of glutamate and GABA receptor blockers.
Dendritic abnormalities and hyperexcitability are increasingly being appreciated in CNS diseases, particularly those with cognitive impairment (Nestor and Hoffman, 2012). In Fragile X syndrome, both misregulation of Kv4.2 (Gross et al., 2011; Lee et al., 2011) and improper dendritic arborization (Vetter et al., 2001) have been proposed to contribute to dendritic hyperexcitability and thus improper integration of synaptic signals (Brager and Johnston, 2014). Mouse models of autism and Rett syndrome also show aberrant dendritic morphology (Nestor and Hoffman, 2012). Finally, there is a strong association between traumatic brain injury and risk for Alzheimer's disease and, in the controlled cortical impact traumatic brain injury model, shows lower Kv4.2 levels and current in the hippocampus (Lei et al., 2012), highlighting a point of possible mechanistic overlap.
Our data suggest that increased dendritic excitability may be an early event in AD pathophysiology. Additional studies should further explore the mechanisms underlying this increase, which could potentially be targeted therapeutically early in the course of AD.
Footnotes
This work was supported by the National Institutes of Health Grants R01NS075487, R21NS052595, and T32GM008111 and the National Institute of Child Health and Human Development Intramural Research Program. We thank Dr. Dane Chetkovich for the gift of the HCN antibodies; Dr. Linda Overstreet-Wadiche for help with the dentate granule cell electrophysiology; Dr. Pascal Sanchez for advice on the levetiracetam treatment protocol; and James Black and Miriam Roberson for assistance with the mouse colony.
E.D.R. is an owner of intellectual property related to tau. The remaining authors declare no competing financial interests.
References
- Andrásfalvy BK, Makara JK, Johnston D, Magee JC. Altered synaptic and non-synaptic properties of CA1 pyramidal neurons in Kv4.2 knockout mice. J Physiol. 2008;586:3881–3892. doi: 10.1113/jphysiol.2008.154336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica E, Boer K, Doorn KJ, Zurolo E, Spliet WG, van Rijen PC, Baayen JC, Gorter JA, Jeromin A. Expression and localization of voltage dependent potassium channel Kv4.2 in epilepsy associated focal lesions. Neurobiol Dis. 2009;36:81–95. doi: 10.1016/j.nbd.2009.06.016. [DOI] [PubMed] [Google Scholar]
- Bernard C, Anderson A, Becker A, Poolos NP, Beck H, Johnston D. Acquired dendritic channelopathy in temporal lobe epilepsy. Science. 2004;305:532–535. doi: 10.1126/science.1097065. [DOI] [PubMed] [Google Scholar]
- Brager DH, Johnston D. Channelopathies and dendritic dysfunction in fragile X syndrome. Brain Res Bull. 2014;103:11–17. doi: 10.1016/j.brainresbull.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, Konnerth A. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2012;109:8740–8745. doi: 10.1073/pnas.1206171109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, Schwarz TL, Sweatt JD, Johnston D. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:12143–12151. doi: 10.1523/JNEUROSCI.2667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Puoliväli J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, Mucke L. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2005;25:9694–9703. doi: 10.1523/JNEUROSCI.2980-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran JN, Hall AM, Roberson ED. The dendritic hypothesis for Alzheimer's disease pathophysiology. Brain Res Bull. 2014;103:18–28. doi: 10.1016/j.brainresbull.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci. 2001;114:1179–1187. doi: 10.1242/jcs.114.6.1179. [DOI] [PubMed] [Google Scholar]
- DeVos SL, Goncharoff DK, Chen G, Kebodeaux CS, Yamada K, Stewart FR, Schuler DR, Maloney SE, Wozniak DF, Rigo F, Bennett CF, Cirrito JR, Holtzman DM, Miller TM. Antisense reduction of tau in adult mice protects against seizures. J Neurosci. 2013;33:12887–12897. doi: 10.1523/JNEUROSCI.2107-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson BC, Salat DH, Greve DN, Chua EF, Rand-Giovannetti E, Rentz DM, Bertram L, Mullin K, Tanzi RE, Blacker D, Albert MS, Sperling RA. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology. 2005;65:404–411. doi: 10.1212/01.wnl.0000171450.97464.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris W, Schütz SG, Cirrito JR, Shankar GM, Sun X, George A, Leissring MA, Walsh DM, Qiu WQ, Holtzman DM, Selkoe DJ. Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am J Pathol. 2007;171:241–251. doi: 10.2353/ajpath.2007.070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frandemiche ML, De Seranno S, Rush T, Borel E, Elie A, Arnal I, Lanté F, Buisson A. Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J Neurosci. 2014;34:6084–6097. doi: 10.1523/JNEUROSCI.4261-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick A, Johnston D. Plasticity of dendritic excitability. J Neurobiol. 2005;64:100–115. doi: 10.1002/neu.20148. [DOI] [PubMed] [Google Scholar]
- Gross C, Yao X, Pong DL, Jeromin A, Bassell GJ. Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J Neurosci. 2011;31:5693–5698. doi: 10.1523/JNEUROSCI.6661-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Jung WE, Marionneau C, Aimond F, Xu H, Yamada KA, Schwarz TL, Demolombe S, Nerbonne JM. Targeted deletion of Kv4.2 eliminates Ito,f and results in electrical and molecular remodeling, with no evidence of ventricular hypertrophy or myocardial dysfunction. Circ Res. 2005;97:1342–1350. doi: 10.1161/01.RES.0000196559.63223.aa. [DOI] [PubMed] [Google Scholar]
- Hall AM, Roberson ED. Mouse models of Alzheimer's disease. Brain Res Bull. 2012;88:3–12. doi: 10.1016/j.brainresbull.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/S1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: evidence for augmentation of a 42-specific γ secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Jinno S, Jeromin A, Kosaka T. Postsynaptic and extrasynaptic localization of Kv4.2 channels in the mouse hippocampal region, with special reference to targeted clustering at GABAergic synapses. Neuroscience. 2005;134:483–494. doi: 10.1016/j.neuroscience.2005.04.065. [DOI] [PubMed] [Google Scholar]
- Kim J, Wei DS, Hoffman DA. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J Physiol. 2005;569:41–57. doi: 10.1113/jphysiol.2005.095042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Aβ plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Ge WP, Huang W, He Y, Wang GX, Rowson-Baldwin A, Smith SJ, Jan YN, Jan LY. Bidirectional regulation of dendritic voltage-gated potassium channels by the fragile X mental retardation protein. Neuron. 2011;72:630–642. doi: 10.1016/j.neuron.2011.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Z, Deng P, Li J, Xu ZC. Alterations of A-type potassium channels in hippocampal neurons after traumatic brain injury. J Neurotrauma. 2012;29:235–245. doi: 10.1089/neu.2010.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugo JN, Barnwell LF, Ren Y, Lee WL, Johnston LD, Kim R, Hrachovy RA, Sweatt JD, Anderson AE. Altered phosphorylation and localization of the A-type channel, Kv4.2 in status epilepticus. J Neurochem. 2008;106:1929–1940. doi: 10.1111/j.1471-4159.2008.05508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugo JN, Brewster AL, Spencer CM, Anderson AE. Kv4.2 knockout mice have hippocampal-dependent learning and memory deficits. Learn Mem. 2012;19:182–189. doi: 10.1101/lm.023614.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee J, Hoffman D, Colbert C, Johnston D. Electrical and calcium signaling in dendrites of hippocampal pyramidal neurons. Annu Rev Physiol. 1998;60:327–346. doi: 10.1146/annurev.physiol.60.1.327. [DOI] [PubMed] [Google Scholar]
- Migliore M, Hoffman DA, Magee JC, Johnston D. Role of an A-type K+ conductance in the back-propagation of action potentials in the dendrites of hippocampal pyramidal neurons. J Comput Neurosci. 1999;7:5–15. doi: 10.1023/A:1008906225285. [DOI] [PubMed] [Google Scholar]
- Moolman DL, Vitolo OV, Vonsattel JP, Shelanski ML. Dendrite and dendritic spine alterations in Alzheimer models. J Neurocytol. 2004;33:377–387. doi: 10.1023/B:NEUR.0000044197.83514.64. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestor MW, Hoffman DA. Aberrant dendritic excitability: a common pathophysiology in CNS disorders affecting memory? Mol Neurobiol. 2012;45:478–487. doi: 10.1007/s12035-012-8265-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olazarán J, Prieto J, Cruz I, Esteban A. Cortical excitability in very mild Alzheimer's disease: a long-term follow-up study. J Neurol. 2010;257:2078–2085. doi: 10.1007/s00415-010-5663-8. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 2009;66:435–440. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc Natl Acad Sci U S A. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L, Roberson ED. Quantifying biomarkers of cognitive dysfunction and neuronal network hyperexcitability in mouse models of Alzheimer's disease: depletion of calcium-dependent proteins and inhibitory hippocampal remodeling. Methods Mol Biol. 2011;670:245–262. doi: 10.1007/978-1-60761-744-0_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Q, Burnet PW, Grahame-Smith DG, Zetterström TS. Differential effects of acute and chronic electroconvulsive shock on the abundance of messenger RNAs for voltage-dependent potassium channel subunits in the rat brain. Neuroscience. 1997;78:343–350. doi: 10.1016/S0306-4522(96)00574-X. [DOI] [PubMed] [Google Scholar]
- Ramakers GM, Storm JF. A postsynaptic transient K+ current modulated by arachidonic acid regulates synaptic integration and threshold for LTP induction in hippocampal pyramidal cells. Proc Natl Acad Sci U S A. 2002;99:10144–10149. doi: 10.1073/pnas.152620399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Quiroz YT, Fleisher AS, Chen K, Velez-Pardo C, Jimenez-Del-Rio M, Fagan AM, Shah AR, Alvarez S, Arbelaez A, Giraldo M, Acosta-Baena N, Sperling RA, Dickerson B, Stern CE, Tirado V, Munoz C, Reiman RA, Huentelman MJ, Alexander GE, et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer's disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol. 2012;11:1048–1056. doi: 10.1016/S1474-4422(12)70228-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüschenschmidt C, Chen J, Becker A, Riazanski V, Beck H. Functional properties and oxidative modulation of A-type K+ currents in hippocampal granule cells of control and chronically epileptic rats. Eur J Neurosci. 2006;23:675–685. doi: 10.1111/j.1460-9568.2006.04608.x. [DOI] [PubMed] [Google Scholar]
- Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu GQ, Palop JJ, Mucke L. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc Natl Acad Sci U S A. 2012;109:E2895–E2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šišková Z, Justus D, Kaneko H, Friedrichs D, Henneberg N, Beutel T, Pitsch J, Schoch S, Becker A, von der Kammer H, Remy S. Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer's disease. Neuron. 2014;84:1023–1033. doi: 10.1016/j.neuron.2014.10.024. [DOI] [PubMed] [Google Scholar]
- Tsaur ML, Sheng M, Lowenstein DH, Jan YN, Jan LY. Differential expression of K+ channel mRNAs in the rat brain and down-regulation in the hippocampus following seizures. Neuron. 1992;8:1055–1067. doi: 10.1016/0896-6273(92)90127-Y. [DOI] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012;149:708–721. doi: 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter P, Roth A, Häusser M. Propagation of action potentials in dendrites depends on dendritic morphology. J Neurophysiol. 2001;85:926–937. doi: 10.1152/jn.2001.85.2.926. [DOI] [PubMed] [Google Scholar]
- Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, Hegde M, Cornes SB, Henry ML, Nelson AB, Seeley WW, Geschwind MD, Gorno-Tempini ML, Shih T, Kirsch HE, Garcia PA, Miller BL, Mucke L. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013;70:1158–1166. doi: 10.1001/jamaneurol.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Hoffman DA, Migliore M, Johnston D. Dendritic K+ channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2002;99:8366–8371. doi: 10.1073/pnas.122210599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Burkhalter A, Nerbonne JM. Functional role of the fast transient outward K+ current IA in pyramidal neurons in (rat) primary visual cortex. J Neurosci. 2005;25:9185–9194. doi: 10.1523/JNEUROSCI.2858-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Luedtke J, Kumar Y, Biernat J, Dawson H, Mandelkow E, Mandelkow EM. Amyloid-β oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 2013;32:2920–2937. doi: 10.1038/emboj.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]