

Abstract
Alcohol and nicotine are the two most co-abused drugs in the world. Previous studies have shown that nicotine can increase alcohol drinking in nondependent rats, yet it is unknown whether nicotine facilitates the transition to alcohol dependence. We tested the hypothesis that chronic nicotine will speed up the escalation of alcohol drinking in rats and that this effect will be accompanied by activation of sparsely distributed neurons (neuronal ensembles) throughout the brain that are specifically recruited by the combination of nicotine and alcohol. Rats were trained to respond for alcohol and made dependent using chronic, intermittent exposure to alcohol vapor, while receiving daily nicotine (0.8 mg/kg) injections. Identification of neuronal ensembles was performed after the last operant session, using immunohistochemistry. Nicotine produced an early escalation of alcohol drinking associated with compulsive alcohol drinking in dependent, but not in nondependent rats (air exposed), as measured by increased progressive-ratio responding and increased responding despite adverse consequences. The combination of nicotine and alcohol produced the recruitment of discrete and phenotype-specific neuronal ensembles (∼4–13% of total neuronal population) in the nucleus accumbens core, dorsomedial prefrontal cortex, central nucleus of the amygdala, bed nucleus of stria terminalis, and posterior ventral tegmental area. Blockade of nicotinic receptors using mecamylamine (1 mg/kg) prevented both the behavioral and neuronal effects of nicotine in dependent rats. These results demonstrate that nicotine and activation of nicotinic receptors are critical factors in the development of alcohol dependence through the dysregulation of a set of interconnected neuronal ensembles throughout the brain.
Keywords: addiction, alcohol, compulsivity, Fos, neuronal ensembles, tobacco
Introduction
Alcoholism is a chronically relapsing disorder characterized by compulsive alcohol seeking and taking and loss of control in limiting intake (Koob and Le Moal, 1997; Koob and Le Moal, 2001, 2005, 2008a, b; Spanagel and Hölter, 1999; Heilig et al., 2010). Epidemiological studies indicate that alcoholism is more prevalent in smokers than in nonsmokers (DiFranza and Guerrera, 1990; Britt and Bonci, 2013; Doyon et al., 2013a, b). Evidence has shown that nicotine can increase alcohol drinking in nonalcoholic humans (Kouri et al., 2004; Barrett et al., 2006; Harrison and McKee, 2008) and nondependent rodents (Olausson et al., 2001; Lê et al., 2003, 2010; Alén et al., 2009; Hauser et al., 2012). However, it is unknown whether nicotine facilitates the transition to alcohol dependence and increases compulsive alcohol drinking. Moreover, the neurocircuitry underlying the facilitating effect of nicotine on alcohol drinking remains to be demonstrated.
Characterization of neurons using double-labeling methods with immediate-early genes as neural activity markers and electrophysiology studies have suggested that drug-related behaviors can be encoded by a minority of sparsely distributed neurons, called neuronal ensembles (Mattson et al., 2008; Root et al., 2010; Cruz et al., 2013, 2014). Therefore, investigation of the neuronal ensembles activated by the interaction between alcohol and nicotine can provide a mechanistic framework for understanding how nicotine increases the susceptibility to alcohol dependence.
Given the lack of data on the interaction of nicotine exposure in the development of alcohol addiction, we tested the hypothesis that chronic nicotine increases the escalation of alcohol self-administration and compulsive alcohol drinking in dependent and nondependent rats using progressive-ratio (PR) responding and responding despite adverse consequences (quinine adulteration test; Vendruscolo et al., 2012; Seif et al., 2013). Moreover, we tested whether chronic nicotine recruits specific neuronal ensembles in the nucleus accumbens core (NAc-Core) and shell (NAc-Shell), dorsomedial and ventromedial PFC (dmPFC and vmPFC), orbitofrontal cortex (OFC), bed nucleus of stria terminalis (BNST), BLA and central (CeA) amygdala, and anterior and posterior ventral tegmental area (aVTA and pVTA). Neuronal ensembles activated by nicotine and alcohol were identified by double labeling using fluorescent immunohistochemistry for Fos, a protein induced in strongly activated neurons, and NeuN, a neuronal marker. We then identified the specific cell types that make up these activated neuronal ensembles with antibodies against Fos and calcium/CaMKII (a marker of cortical glutamatergic pyramidal projection neurons), Fos and GAD67 (a marker of GABAergic neurons), and TH (a marker of dopaminergic neurons).
Materials and Methods
Subjects.
Adult male Wistar rats (Charles River), weighing 225–275 g at the beginning of the experiments, were housed in groups of two to three per cage in a temperature-controlled (22°C) vivarium on a 12 h light/dark cycle (lights on at 8:00 P.M.) with ad libitum access to food and water. All of the behavioral tests were conducted during the dark phase of the light/dark cycle. All of the procedures adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute.
Operant self-administration.
Self-administration sessions were conducted in standard operant conditioning chambers (Med Associates). Animals were first trained to self-administer 10% (w/v) alcohol and water solutions until stable responding was maintained. First, to facilitate the acquisition of operant self-administration, rats were initially provided free-choice access to alcohol (10% w/v) and water for 1 d in their home cages to habituate them to the taste of alcohol. Second, the rats were subjected to an overnight session in the operant chambers with access to one lever (right lever) that delivered water (FR1). Food was available ad libitum during this training. Third, after 1 d off, the rats were subjected to a 2 h session (FR1) for 1 d and a 1 h session (FR1) the next day, with one lever delivering alcohol (right lever). All of the subsequent sessions lasted 30 min, and two levers were available (left lever: water; right lever: alcohol) until stable levels of intake were reached. Upon completion of this procedure, the animals were allowed to self-administer a 10% (w/v) alcohol solution and water on an FR1 schedule of reinforcement (i.e., each operant response was reinforced with 0.1 ml of the solution).
Nicotine treatment.
Rats were further subdivided into four groups (vapor-nicotine, vapor-saline, air-nicotine, and air-saline). The rats received a daily nicotine injection (0.8 mg/kg, s.c.) or 0.9% saline (1.0 ml/kg, s.c.) 6–8 h into withdrawal from alcohol vapor exposure (15 min before the alcohol sessions). This procedure was maintained until the end of the experiment. The air groups (air-nicotine and air-saline) received the same treatment (nicotine or saline, under the same time schedule) but were not exposed to alcohol vapor. A different subset of rats received mecamylamine (1.0 mg/kg, s.c.) or 0.9% saline (1.0 ml/kg, s.c.) injections 45 min before each nicotine administration.
Alcohol vapor chambers.
The rats were made dependent by chronic, intermittent exposure to alcohol vapors as previously described (O'Dell et al., 2004; Gilpin et al., 2008). They underwent cycles of 14 h on (blood alcohol levels during vapor exposure ranged between 150 and 250 mg%) and 10 h off, during which behavioral testing for acute withdrawal occurred (i.e., 6–8 h after vapor was turned off when brain and blood alcohol levels are negligible; Gilpin et al., 2009). In this model, rats exhibit somatic withdrawal signs and negative emotional symptoms, reflected by anxiety-like responses and elevated brain reward thresholds (Schulteis et al., 1995; Roberts et al., 2000; Valdez et al., 2002; Rimondini et al., 2003; O'Dell et al., 2004; Sommer et al., 2007; Edwards et al., 2012). Nondependent rats were not exposed to alcohol vapor.
Operant self-administration during alcohol vapor exposure.
Behavioral testing occurred two to four times per week. The rats were tested for alcohol (and water) self-administration on an FR1 schedule of reinforcement in 30 min sessions. Operant self-administration on an FR1 schedule requires minimal effort by the animal to obtain the reinforcement and herein was considered a measure of intake. For five sessions, the rats were tested on a PR schedule, under which the number of lever presses that were necessary to obtain the next reinforcement progressively increased according to the following progression: 1, 1, 2, 2, 3, 3, 4, 4, 5, 5, 7, 7, 9, 9, 11, 11, 13, 13, etc. The PR session stopped after 90 min or when 15 min elapsed without the rat obtaining reinforcement. The rats were then maintained on an FR1 schedule until stable levels of alcohol self-administration were established before testing the effect of quinine on alcohol drinking. The alcohol solution was adulterated with increasing concentrations of quinine (0.005, 0.01, 0.025, and 0.05 g/L) presented between sessions (one concentration per session). This test measures the persistence of animals to consume alcohol despite the aversive bitter taste of quinine that was added to the alcohol solution and has been validated as a measure of compulsive intake (Vendruscolo et al., 2012; Seif et al., 2013).
Finally, to identify the neuronal ensembles recruited by the interaction between nicotine and alcohol, the rats were injected with either nicotine (0.8 mg/kg) or saline, and 15 min later they were tested for alcohol (and water) self-administration on an FR1 schedule of reinforcement for their last 30 min sessions. The naive rats were exposed to alcohol vapor (naive-vapor) or not exposed (naive-air), but were not subjected to operant alcohol self-administration and only received a saline injection 90 min before the perfusion to control for baseline Fos level. Sixty minutes after the end of the test session, the rats were deeply anesthetized with ketamine (56.4 mg/kg) and xylazine (8.2 mg/kg) and then perfused with 100 ml of PBS followed by 400 ml of 4% paraformaldehyde. The brains were postfixed in paraformaldehyde for 90 min and transferred to 30% sucrose in PBS solution at 4°C for 2–3 d. Brains were frozen in powdered dry ice and kept at −80°C until sectioning.
Immunohistochemistry.
Coronal sections were cut 40 μm thick between bregma +4.2 and −6.48 mm (Paxinos and Watson, 2005). Free-floating sections were washed three times in PBS, blocked with 3% NGS in PBS with 0.25% Triton X-100 (PBS-Tx), and incubated for 24 h at 4°C with anti-Fos antibody (sc-52; Santa Cruz Biotechnology) diluted 1:4000 in blocking solution. Sections were washed again with PBS and incubated for 2 h in biotinylated goat anti-rabbit secondary antibody (1:400; Vector Laboratories) in PBS-Tx and 1% NGS. After washing in PBS, sections were incubated for 1 h in avidin-biotin-peroxidase complex (ABC Elite kit, PK-6100; Vector Laboratories) in PBS containing 0.5% Triton X-100. Finally, sections were washed in PBS and developed in DAB for ∼3 min, transferred to PBS, and mounted to chrome-alum gelatin-coated slides. Once dry, the slides were dehydrated through a graded series of alcohol and cleared with CitraSolv (Fisher Scientific) before coverslipping with Permount (Sigma).
Bright-field images of Fos immunoreactivity in the NAc were captured using a CCD camera (CoolSNAP; Photometrics) and QImaging EXi Aqua attached to a Zeiss Axioskop 2 microscope. Images for counting labeled cells were captured at 100× magnification. Labeled cells from two sections per rat were bilaterally and automatically counted using IPLab 3.9.4 r5 software for Macintosh (Scanalytics) and iVision 4.0.15 for Macintosh (BioVision). Counts from all images from each rat were averaged, so that each rat was an n of 1.
Double-labeling immunohistochemistry.
We used double-labeling immunohistochemistry to characterize neurons activated during the alcohol self-administration session. For these experiments, we used three to four animals from each group. Coronal sections were cut 40 μm thick between bregma +4.2 and −6.48 mm (Paxinos and Watson, 2005). We determined the proportion of all neurons expressing Fos during the alcohol self-administration session by double labeling Fos and the neuron-specific protein NeuN. We also assessed the phenotype of Fos-expressing neurons by double labeling against Fos and calcium/CaMKII (a marker of cortical glutamatergic pyramidal projection neurons), Fos and GAD67 (a marker of GABAergic neurons), and TH (a marker of dopaminergic neurons).
For Fos + NeuN labeling, 40 μm sections were washed three times in TBS and permeabilized for 30 min in TBS with 0.2% Triton X-100. Sections were incubated in primary antibodies diluted in TBS with 0.2% Triton X-100 for 24 h on a shaker at 4°C. Primary antibodies were anti-Fos (rabbit, 1:400 dilution, sc-52) and anti-NeuN (mouse, 1:2000 dilution). Sections were washed three times in TBS and incubated with fluorescently labeled secondary antibodies diluted in TBS with 0.2% Triton X-100 for 2 h on a shaker at room temperature. Secondary antibodies were Alexa Fluor 488-labeled donkey anti-rabbit (1:200 dilution, A-10042; Invitrogen) and Alexa Fluor 568-labeled donkey anti-mouse (1:2000 dilution, S-11249; Invitrogen) to label NeuN. After labeling, sections were washed in TBS, mounted on chrome-alum gelatin-coated slides, and coverslipped with VectaShield hard-set mounting media.
For phenotype characterization, 40-μm-thick sections were first washed three times in PBS. Sections were incubated for 1 h in a blocking solution (5% NGS and 2.5% bovine serum albumin in PBS with 0.2% Triton X-100), and then they were incubated for 48 h with the following primary antibodies: anti-Fos antibody (rabbit, 1:400 dilution, sc-52; Santa Cruz Biotechnology) and anti-CaMKII antibody (mouse, 1:100 dilution, MA1-048; Millipore) or anti-GAD67 antibody (mouse, 1:1000 dilution, MAB5406; Millipore) or TH antibody (mouse, 1:50,000 dilution, MAB5280; Millipore) in blocking solution. After washing, sections were incubated for 2 h in blocking solution with the fluorescently labeled secondary antibodies: Alexa Fluor 488-labeled donkey anti-rabbit (1:200 dilution, A-10042; Invitrogen) and Alexa Fluor 568-labeled donkey anti-mouse (1:2000 dilution, S-11249; Invitrogen). Sections were then washed, mounted on chrome-alum gelatin-coated slides, air-dried, and coverslipped with Vectashield fluorescent mounting medium.
All of the fluorescent images were captured using a CCD camera (CoolSNAP; Photometrics) attached to a Zeiss Axioskop 2 microscope. Images for the colocalization of Fos and NeuN/GAD67/TH/CaMKII were captured at 200× magnification. The number of Fos-labeled and double-labeled cells from the NAc of one section per rat were counted using iVision 4.0.15 for Macintosh (BioVision Technologies).
The results are expressed as mean ± SEM. We analyzed the data by multifactorial ANOVA, considering the between-subjects factors vapor (exposed to the alcohol vapor, not exposed to the alcohol vapor), treatment (nicotine and saline), and sessions as the repeated measure.
Results
Escalation of alcohol self-administration
Figure 1 shows the number of reinforcements over 16 alcohol self-administration sessions of animals exposed (A) or not exposed (B) to the alcohol vapor and administered nicotine (0.8 mg/kg, s.c.) or 0.9% saline (1.0 ml/kg, s.c.) and animals that were administered nicotine (0.8 mg/kg, s.c.) and exposed or not exposed to the alcohol vapor (C). The ANOVA indicated a significant interaction between the three factors (Vapor/Air, Saline/Nicotine, and sessions, F(15,375) = 4.76, p < 0.05). Therefore, we performed separate repeated-measures ANOVAs for each experimental condition: one with animals exposed to only vapor, one with animals exposed to air, and one with animals that were administered nicotine (0.8 mg/kg, s.c.) and exposed or not exposed to alcohol vapor. For the animals that were exposed to alcohol vapor, the ANOVA revealed significant effects of treatment (F(1,15) = 54.96, p < 0.05) and sessions (F(1,15) = 23.06, p < 0.05), with a significant interaction between factors (F(1,15) = 4.19, p < 0.05). A further analysis (F test) showed a significant difference among groups and among sessions from session numbers 9–12 (p < 0.05). The Vapor-Nicotine group exhibited an acceleration of escalation compared with the Vapor-Saline group (p < 0.05). These results indicate that the combination of nicotine administration with alcohol vapor exposure accelerated the development of the escalation of alcohol intake in dependent rats.
Figure 1.
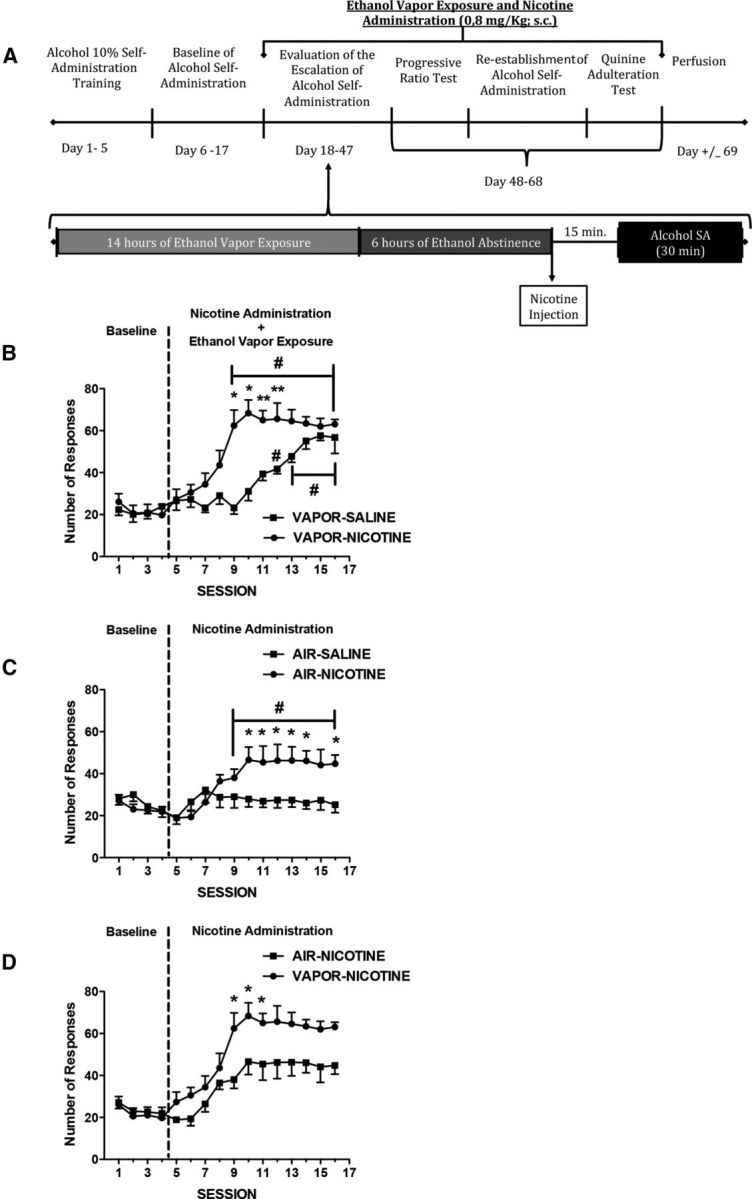
Escalation of alcohol self-administration. A, Timeline of the experiment. Dependent and nondependent rats were injected with saline or nicotine (0.8 mg/kg, s.c.) once per day, 15 min before each operant session after 6–8 h of withdrawal from alcohol vapor. The data represent the mean ± SEM number of active lever presses for 10% w/v alcohol in dependent (B) and nondependent (C) rats treated with saline (n = 8–9; black circles) or nicotine (n = 8–9; black squares) and dependent (n = 9; black squares) and nondependent (n = 9; black circles) rats treated with nicotine (0.8 mg/kg, s.c.; D). *p < 0.05, compared with saline group; **p < 0.001, compared with saline group; #p < 0.05, compared with baseline.
The ANOVA of the animals that were not exposed to alcohol vapor revealed significant effects of treatment (F(1,15) = 2.51, p < 0.05) and sessions (F(1,15) = 4.04, p < 0.05) and a significant interaction between factors (F(1,15) = 3.06, p < 0.05). A further analysis (F test) revealed a significant difference between groups in sessions 10–14 and 16 (p < 0.05). The Air-Nicotine group showed a significant difference relative to the last baseline session starting in session 9, but no difference from the Air-Saline group was observed (p < 0.05). These results indicate that nicotine administration enhanced alcohol self-administration in nondependent rats.
For the animals that received daily nicotine administration (0.8 mg/kg, s.c.) and exposed or not exposed to alcohol vapor, the ANOVA revealed significant effects of treatment (F(1,15) = 6.22, p < 0.05) and sessions (F(1,15) = 17.16, p < 0.05), with a significant interaction between factors (F(1,15) = 1.97, p < 0.05). A further analysis (F test) showed a significant difference between groups in sessions 9–11 (p < 0.05). These results indicate that rats that were coexposed to alcohol and nicotine exhibited higher escalation of alcohol intake than rats that were only exposed to nicotine.
To evaluate the role of nicotine receptors in the facilitating effects of nicotine in the escalation of alcohol self-administration, rats that were exposed to nicotine and alcohol were treated daily with mecamylamine (1.0 mg/kg, s.c.) or 0.9% saline (1.0 ml/kg, s.c.) 45 min before each nicotine injection. A two-way repeated-measures ANOVA revealed an effect of sessions (F(15,14) = 30.44, p < 0.05) and an interaction between treatment and sessions (F(15,210) = 2.85, p < 0.05), demonstrating that the behavioral effect of nicotine depends on the activation of nicotinic receptors, and mecamylamine blocks the facilitating effect of nicotine on alcohol self-administration (Fig. 2).
Figure 2.

Mecamylamine blocks the escalation of alcohol self-administration. Dependent rats were injected with mecamylamine (1 mg/kg, i.p.) or saline 45 min before each nicotine (0.8 mg/kg, s.c.) injection once per day, 15 min before each operant session after 6–8 h of withdrawal from alcohol vapor. The data represent the mean ± SEM number of active lever presses for 10% w/v alcohol in dependent rats treated with nicotine + saline (n = 7; black circles) or nicotine + mecamylamine (n = 7; black squares). *p < 0.05, compared with nicotine + saline group; #p < 0.05, compared with baseline.
Progressive ratio
Figure 3 depicts the average number of reinforcements and last ratio achieved during the PR sessions in rats that received 0.9% saline (1.0 ml/kg, s.c.) or nicotine (0.8 mg/kg, s.c.) and exposed (Vapor) or not exposed (Air) to alcohol vapor. For the number of reinforcements, the two-way ANOVA revealed significant effects of vapor (F(1,31) = 52.62, p < 0.05) and treatment (F(1,31) = 11.05, p < 0.05) but no interaction between factors (F(1,31) = 1.53, p > 0.05). The Newman–Keuls test indicated that nicotine administration enhanced the number of reinforcements in both groups compared with the saline group, and the vapor-nicotine group exhibited a higher number of reinforcements compared with the air-nicotine group (p < 0.05). For the last ratio achieved, the two-way ANOVA revealed significant effects of vapor (F(1,31) = 35.88, p < 0.05) and treatment (F(1,31) = 7.20, p < 0.05) but no interaction between factors (F(1,31) = 0.77, p > 0.05). The Newman–Keuls test indicated that nicotine administration enhanced the last ratio achieved in both groups compared with the saline group, and the vapor-nicotine group exhibited a higher number of reinforcements compared with the air-nicotine group (p < 0.05). These results indicate that nicotine administration increased the motivation for alcohol drinking and that the facilitating effect of nicotine on alcohol drinking was increased in alcohol-dependent rats.
Figure 3.

Mean (±SEM) reinforcement or last ratio achieved for alcohol during the PR sessions. Alcohol-dependent and nondependent Wistar rats responded for alcohol at 6–8 h of withdrawal from alcohol vapor and 15 min after an injection of saline (n = 8–9; white bars) or nicotine (n = 8–9; black bars). #p < 0.05, significant difference between air and vapor in nicotine-treated rats; *p < 0.05, significant difference from respective saline group.
Quinine adulteration test
Figure 4 depicts the average number of reinforcements during the quinine adulteration tests in rats that were administered 0.9% saline (1.0 ml/kg, s.c.) or nicotine (0.8 mg/kg, s.c.) and exposed (A) or not exposed (B) to alcohol vapor. Student's t test revealed significant differences in baseline between the saline and nicotine groups only for animals that were exposed to alcohol vapor (t(16) = 3.5, p < 0.05).
Figure 4.
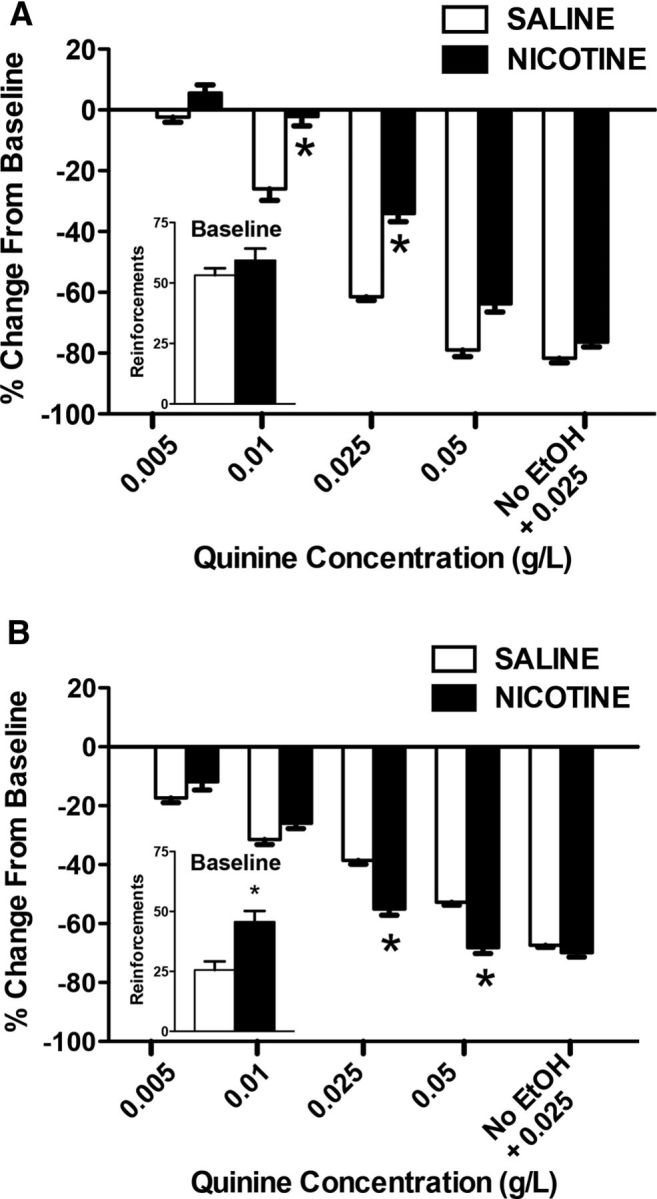
Compulsive-like drinking (i.e., persistent alcohol drinking despite the aversive bitter taste of quinine added to the alcohol solution). The data represent the percentage change from baseline (i.e., lever presses for alcohol alone before adulteration with quinine) in vapor-exposed (A) and air-control (B) rats that were treated with saline (n = 7–9; white bars) or nicotine (n = 7–9; black bars). *p < 0.05, significant difference between saline and nicotine.
We first performed a control test to evaluate taste differences in response to quinine between groups, in which rats were given a quinine solution (0.025 g/L) without alcohol. Vapor-exposed rats showed a greater reduction of quinine drinking (without alcohol) compared with air-exposed rats (−81.7 vs −67.3%, respectively; p < 0.05). Therefore, we performed two separate within-subjects analyses for the air-exposed and vapor-exposed groups to avoid the inherent bias of the between-subjects analysis. We analyzed the data using ANOVA, considering the factors vapor (alcohol vs air), treatment (nicotine vs saline), and quinine concentration as the repeated measure. The analysis showed a significant interaction among the three factors (F(1,155) = 5.36, p < 0.05). The statistical analysis was followed by two separate ANOVAs (dependent and nondependent rats).
For the animals that were exposed to alcohol vapor, the ANOVA revealed significant effects of treatment (F(1,16) = 15.43, p < 0.05) and quinine concentration (F(5,80) = 135.39, p < 0.05), with a significant interaction between factors (F(5,80) = 3.52, p < 0.05). These results indicate that rats that were exposed to alcohol vapor and treated with nicotine showed higher resistance to quinine at the 0.01 and 0.025 g/L doses (p < 0.05). Chronic treatment with nicotine and alcohol vapor exposure increased alcohol drinking despite the aversive bitter taste of quinine, suggesting increased compulsive alcohol intake.
For the animals that were not exposed to alcohol vapor, the ANOVA revealed significant effects of treatment (F(1,16) = 30.17, p < 0.05) and quinine concentration (F(5,80) = 51.97, p < 0.05), with a significant interaction between factors (F(5,80) = 6.06, p < 0.05). These results indicate that nicotine had an opposite effect on compulsive drinking in animals that were not exposed to alcohol vapor. The animals that received nicotine administration exhibited a higher reduction of intake of alcohol that was adulterated with quinine (0.025 and 0.05 g/L) compared with the saline group, demonstrating that the development of compulsive alcohol intake caused by nicotine depends on alcohol vapor exposure, and nicotine administration only increases compulsive drinking in alcohol-dependent animals.
Characterization of neuronal ensembles recruited by nicotine
We then investigated the neuronal ensembles that are recruited by nicotine-induced compulsive alcohol drinking in dependent rats. We evaluated the number of Fos-positive neurons in key brain regions that are involved in addiction (PFC, NAc, amygdala, BNST, and VTA) 60 min after the last session of escalated alcohol self-administration.
Prefrontal cortex
Figure 5 depicts the number of Fos-positive neurons per millimeter squared in the dmPFC, vmPFC, and OFC. Each graph shows the mean ± SEM (n = 6–8 animals per group). In the mPFC, the combination of nicotine treatment and alcohol vapor exposure increased the number of Fos-positive neurons only in the dmPFC compared with the other groups (vapor: F(1,28) = 0.86, p > 0.05; treatment: F(2,28) = 7.69, p < 0.05; interaction: F(2,28) = 4.13, p < 0.05). No difference was observed between groups in the vmPFC and OFC.
Figure 5.
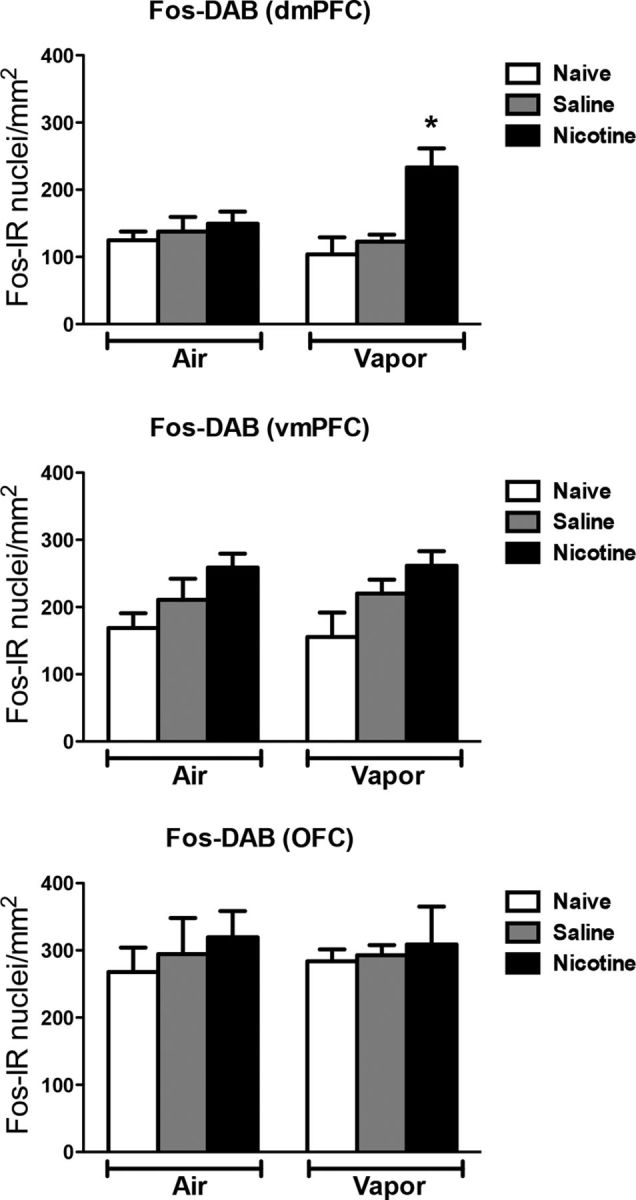
Nicotine accelerates the escalation of alcohol self-administration, associated with Fos induction in the dmPFC. Top, Number of Fos-immunoreactive nuclei per millimeter squared in the dmPFC. Middle, Number of Fos-immunoreactive nuclei per millimeter squared in the vmPFC. Bottom, Number of Fos-immunoreactive nuclei per millimeter squared in the OFC. The data are represented as mean ± SEM; *p < 0.05, different from the other groups; n = 6–8 per group.
Nucleus accumbens
Figure 6 depicts the number of Fos-positive neurons per millimeter squared in the NAc-Core and NAc-Shell. Each graph shows the mean ± SEM (n = 6–8 animals per group). The combination of nicotine treatment and alcohol vapor exposure increased the number of Fos-positive neurons only in the NAc-Core but not in the NAc-Shell (vapor: F(1,23) = 2.03, p > 0.05; treatment: F(2,23) = 4.16, p < 0.05; interaction: F(2,23) = 4.42, p < 0.05).
Figure 6.

Nicotine speeds up the escalation of alcohol self-administration, associated with Fos induction in the nucleus accumbens core. Top, Number of Fos-immunoreactive nuclei per millimeter squared in the NAc-Core. Bottom, Number of Fos-immunoreactive nuclei per millimeter squared in the NAc-Shell. *p < 0.05, different from the other groups; n = 6–8 per group.
Amygdala
Figure 7 depicts the number of Fos-positive neurons per millimeter squared in the BLA and CeA. Each graph shows the mean ± SEM (n = 6–8 animals per group). The combination of nicotine treatment and alcohol vapor exposure selectively increased the number of Fos-positive neurons in the CeA (vapor: F(1,28) = 0.03, p > 0.05; treatment: F(2,28) = 11.85, p < 0.05; interaction: F(2,28) = 2.63, p < 0.05). In contrast, nicotine administration increased the number of Fos-positive neurons in the BLA in both the vapor and air groups compared with the other groups (vapor: F(1,28) = 0.56, p > 0.05; treatment: F(2,28) = 12.94, p < 0.05; interaction: F(2,26) = 1.01, p > 0.05).
Figure 7.

Nicotine accelerates the escalation of alcohol self-administration, associated with Fos induction in the central amygdala, and nicotine treatment induces Fos in the basolateral amygdala. Top, Number of Fos-immunoreactive nuclei per millimeter squared in the CeA. Bottom, Number of Fos-immunoreactive nuclei per millimeter squared in the BLA. *p < 0.05, different from the other groups; n = 6–8 per group.
Bed nucleus of the stria terminalis
Figure 8 depicts the number of Fos-positive neurons per millimeter squared in different regions of the BNST. Each graph shows the mean ± SEM (n = 6–8 animals per group). In the BNST, the combination of nicotine treatment and alcohol vapor exposure increased the number of Fos-positive neurons in all subregions, including the juxtacapsular (vapor: F(1,28) = 25.70, p < 0.05; treatment: F(2,28) = 12.15, p < 0.05; interaction: F(2,28) = 9.24, p < 0.05), posterolateral (vapor: F(1,28) = 18.82, p < 0.05; treatment: F(2,28) = 19.52, p < 0.05; interaction: F(2,28) = 13.03, p < 0.05), and ventrolateral (vapor: F(1,28) = 25.58, p < 0.05; treatment: F(2,28) = 25.59, p < 0.05; interaction: F(2,28) = 11.47, p < 0.05), compared with the other groups.
Figure 8.
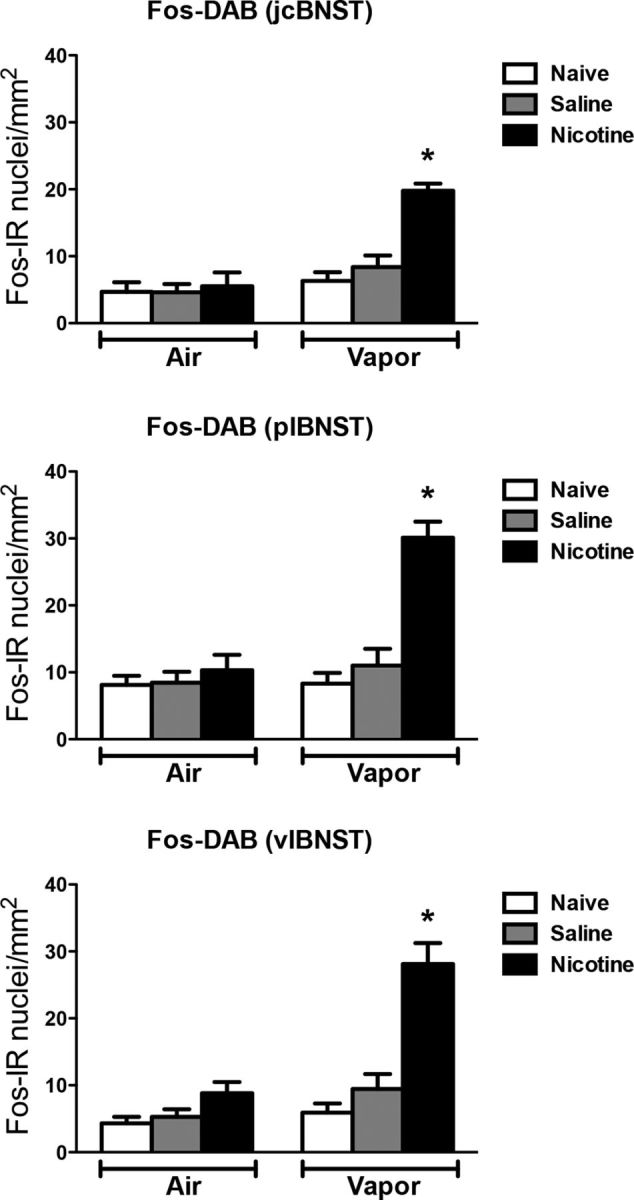
Nicotine accelerates the escalation of alcohol self-administration, associated with Fos induction in the BNST. Top, Number of Fos-immunoreactive nuclei per millimeter squared in the juxtacapsular bed nucleus of the stria terminalis (jcBNST). Middle, Number of Fos-immunoreactive nuclei per millimeter squared in the posterolateral bed nucleus of the stria terminalis (plBNST). Bottom, Number of Fos-immunoreactive nuclei per millimeter squared in the ventrolateral bed nucleus of the stria terminalis (vlBNST). *p < 0.05, different from the other groups; n = 6–8 per group.
Ventral tegmental area
Figure 9 depicts the number of Fos-positive neurons per millimeter squared in the aVTA and pVTA. Each graph shows the mean ± SEM (n = 6–8 animals per group). In the VTA, the combination of nicotine treatment and alcohol vapor exposure increased the number of Fos-positive neurons only in the posterior region compared with the other groups (vapor: F(1,28) = 7.66, p < 0.05; treatment: F(2,28) = 58.92, p < 0.05; interaction: F(2,28) = 38.21, p < 0.05). No difference was observed among the groups in the anterior region.
Figure 9.

Nicotine accelerates the escalation of alcohol self-administration, associated with Fos induction in the pVTA. Top, Number of Fos-immunoreactive nuclei per millimeter squared in the aVTA. Bottom, Number of Fos-immunoreactive nuclei per millimeter squared in the pVTA. *p < 0.05, different from the other groups; n = 6–8 per group.
Effect of mecamylamine on Fos expression
We then evaluated Fos expression in the dmPFC after alcohol self-administration in nicotine-treated rats that were given mecamylamine or saline pretreatment (for behavior; Fig. 2) and naive rats. The dmPFC was chosen because it exhibited the most robust effect of nicotine among all of the structures analyzed. Mecamylamine blocked the increase in the number of Fos neurons caused by pretreatment with nicotine (one-way ANOVA: F(2,18) = 12.61, p < 0.05, n = 7 animals per group, Fig. 10).
Figure 10.

Mecamylamine blocks the facilitating effect of nicotine on the escalation of alcohol self-administration, associated with a reduction of Fos expression in the dorsal medial prefrontal cortex. *p < 0.05, different from the other groups; n = 7 per group.
Characterization of Fos-expressing neurons using double-labeling immunohistochemistry
We used double-labeling immunohistochemistry to determine the cellular phenotype of Fos-expressing neurons in sections obtained from a subset of the brains used in our experiment (four rats from each group). Table 1 depicts the percentage of double labeling for Fos and the general neuronal marker NeuN. Only a small proportion of neurons was activated (Fig. 11). In the dmPFC, Fos was expressed in 7.6 ± 1.4%, 1.6 ± 0.1%, 1.7 ± 0.1%, and 2.2 ± 0.2% of all neurons after the last exposure to alcohol self-administration in the vapor-nicotine, vapor-saline, air-nicotine, and air-saline groups, respectively, and 1.9 ± 0.2% and 1.8 ± 0.1% of all neurons after saline injection in the vapor-control and air-control groups, respectively. In the NAc-Core, Fos was expressed in 7.4 ± 0.9%, 3.0 ± 0.5%, 3.9 ± 0.2%, and 3.5 ± 0.4% of all neurons after the last exposure to alcohol self-administration in the vapor-nicotine, vapor-saline, air-nicotine, and air-saline groups, respectively, and 2.5 ± 0.5% and 2.6 ± 0.1% of all neurons after the saline injection in the vapor-control and air-control groups, respectively. In the BNST, Fos was expressed in 4.1 ± 0.1%, 2.3 ± 0.2%, 2.5 ± 0.2%, and 1.4 ± 0.1% of all neurons after the last exposure to alcohol self-administration in the vapor-nicotine, vapor-saline, air-nicotine, and air-saline groups, respectively, and 1.0 ± 0.1% and 0.7 ± 0.1% of all neurons after the saline injection in the vapor-control and air-control groups, respectively. In the CeA, Fos was expressed in 3.5 ± 0.6%, 0.9 ± 0.2%, 1.1 ± 0.2%, and 1.4 ± 1.1% of all neurons after the last exposure to alcohol self-administration in the vapor-nicotine, vapor-saline, air-nicotine, and air-saline groups, respectively, and 0.3 ± 0.3% and 1.4 ± 0.6% of all neurons after the saline injection in the vapor-control and nonvapor-control groups, respectively. In the BLA, Fos was expressed in 6.2 ± 0.6%, 1.5 ± 0.4%, 1.7 ± 0.1%, and 1.9 ± 0.2% of all neurons after the last exposure to alcohol self-administration in the vapor-nicotine, vapor-saline, air-nicotine, and air-saline groups, respectively, and 1.1 ± 0.1% and 2.1 ± 0.2% of all neurons after the saline injection in the vapor-control and air-control groups, respectively. In the pVTA, Fos was expressed in 13.25 ± 2.5%, 7.7 ± 1.0%, 13.5 ± 2.0%, and 6.1 ± 0.1% of all neurons after the last exposure to alcohol self-administration in the vapor-nicotine, vapor-saline, air-nicotine, and air-saline groups, respectively, and 4.6 ± 0.2% and 4.6 ± 0.7% of all neurons after the saline injection in the vapor-control and air-control groups, respectively.
Table 1.
Percentage of Fos + NeuN double-labeled nuclei per millimeter squared in the dmPFC, NAc-Core, BNST, CeA, BLA, and pVTA associated with nicotine-induced acceleration of the escalation of alcohol self-administration
| Group | dmPFC (%) | Ac-Core (%) | BNST (%) | CeA (%) | BLA (%) | pVTA (%) |
|---|---|---|---|---|---|---|
| Air-Naive | 1.8 ± 0.1 | 2.6 ± 0.1 | 0.7 ± 0.1 | 1.4 ± 0.6 | 2.1 ± 0.2 | 4.6 ± 0.7 |
| Air-Saline | 2.2 ± 0.2 | 3.5 ± 0.4 | 1.4 ± 0.1 | 1.4 ± 1.1 | 1.9 ± 0.2 | 6.1 ± 0.1 |
| Air-Nicotine | 1.7 ± 0.1 | 3.9 ± 0.2 | 2.5 ± 0.2 | 1.1 ± 0.2 | 1.7 ± 0.1 | 6.7 ± 1.2 |
| Vapor-Naive | 1.9 ± 0.2 | 2.5 ± 0.5 | 1.0 ± 0.1 | 0.3 ± 0.3 | 1.1 ± 0.1 | 4.6 ± 0.2 |
| Vapor-Saline | 1.6 ± 0.1 | 3.0 ± 0.5 | 2.3 ± 0.0.2 | 0.9 ± 0.2 | 1.5 ± 0.4 | 7.7 ± 1.0 |
| Vapor-Nicotine | 7.6 ± 1.4* | 7.4 ± 0.9* | 4.1 ± 0.1* | 3.5 ± 0.6* | 6.2 ± 0.6* | 13.25 ± 2.5 |
Two-way ANOVA revealed a significant interaction between the vapor and drug factors (F(2,12) = 8.11, p < 0.05).
*p < 0.05, compared with other groups in the same brain area.
Figure 11.

Representative image of double-labeled immunohistochemistry detecting Fos and NeuN associated with nicotine-induced acceleration of the escalation of alcohol self-administration. A, Red-labeled nuclei indicate expression of the general neuronal nuclei marker NeuN. B, Green-labeled nuclei indicate Fos expression. C, Merged image indicating Fos + NeuN double-labeled nuclei in yellow. Scale bar, 50 μm. n = 4 per group.
Finally, we assessed the cellular phenotypes of these Fos-expressing neurons (Tables 2–5, Fig. 12). In the dmPFC, nicotine increased the percentage of Fos+/CaMKII neurons, indicating the preferential recruitment of glutamatergic neurons. The remaining Fos-expressing neurons were composed of GABAergic neurons (colabeled with GAD67) and another population of unidentified neurons since the total percentage does not add up to 100% (Table 2). In the NAc-Core, the majority of Fos-expressing neurons were colabeled with GAD67, indicating that they were GABAergic neurons. The phenotype of the remaining neurons was unknown (Table 3). In the BNST, a minority of Fos-expressing neurons were colabeled with GAD67, indicating that they were GABAergic neurons, whereas the phenotype of the remaining neurons was unknown (Table 4). In the pVTA, the majority of Fos-expressing neurons were colabeled with GAD67, indicating that they were GABAergic neurons. The remaining Fos-expressing neurons were composed of dopaminergic neurons (colabeled with TH) and other unknown neurons (Table 5).
Table 2.
Specific cell types of Fos-expressing neuronal ensembles in the dmPFC that were activated in the nicotine-induced acceleration of the escalation of alcohol self-administration
| Group | CaMKII | GAD67 |
|---|---|---|
| Air-Naive | 21.6 ± 7.1 | 52.8 ± 8.2 |
| Air-Saline | 20.6 ± 3.7 | 36.4 ± 6.2 |
| Air-Nicotine | 27.7 ± 7.7 | 27.2 ± 5.6 |
| Vapor-Naive | 5.9 ± 5.7 | 30.37 ± 7.5 |
| Vapor-Saline | 20.28 ± 2.0 | 30.62 ± 6.6 |
| Vapor-Nicotine | 54.58 ± 1.55* | 27.86 ± 3.6 |
These data are represented as the mean ± SEM of double-labeled neurons as a percentage of all Fos-labeled neurons. The total percentage does not necessarily add up to 100%, possibly due to incomplete labeling with cell-type markers. For CaMKII, the two-way ANOVA revealed a significant interaction between the vapor and drug factors (F(1,12) = 8.33, p < 0.05).
*p < 0.05 compared with the other groups in the same brain area.
Table 3.
Specific cell types of Fos-expressing neuronal ensembles in the NAc-Core that were activated in the nicotine-induced acceleration of the escalation of alcohol self-administration
| Group | GAD67 |
|---|---|
| Air-Naive | 77.7 ± 10.9 |
| Air-Saline | 46.29 ± 9.1 |
| Air-Nicotine | 49.3 ± 6.8 |
| Vapor-Naive | 50.1 ± 20.5 |
| Vapor-Saline | 36.1 ± 15 |
| Vapor-Nicotine | 71.1 ± 6.9 |
These data are represented as the mean ± SEM of double-labeled neurons as a percentage of all Fos-labeled neurons. The total percentage does not necessarily add up to 100%, possibly due to incomplete labeling with cell-type markers.
Table 4.
Specific cell types of Fos-expressing neuronal ensembles in the BSNST that were activated in the nicotine-induced acceleration of the escalation of alcohol self-administration
| Group | GAD67 |
|---|---|
| Air-Naive | 11.0 ± 1.1 |
| Air-Saline | 27.5 ± 18.7 |
| Air-Nicotine | 29.8 ± 9.9 |
| Vapor-Naive | 8.5 ± 8.2 |
| Vapor-Saline | 20.8 ± 6.36 |
| Vapor-Nicotine | 17.1 ± 3.2 |
These data are represented as the mean ± SEM of double-labeled neurons as a percentage of all Fos-labeled neurons. The total percentage does not necessarily add up to 100%, possibly due to incomplete labeling with cell-type markers.
Table 5.
Specific cell types of Fos-expressing neuronal ensembles in the pVTA that were activated in the nicotine-induced acceleration of the escalation of alcohol self-administration
| Group | GAD67 | TH |
|---|---|---|
| Air-Naive | 17.4 ± 3.9 | 18.4 ± 1.7 |
| Air-Saline | 23.8 ± 10.4 | 18.4 ± 10.2 |
| Air-Nicotine | 35.7 ± 16.1 | 26.8 ± 4.6 |
| Vapor-Naive | 8.6 ± 8.1 | 2.2 ± 4.6 |
| Vapor-Saline | 41.6 ± 20.4 | 19.32 ± 3.4 |
| Vapor-Nicotine | 62.5 ± 6.2 | 36.4 ± 3.1 |
These data are represented as the mean ± SEM of double-labeled neurons as a percentage of all Fos-labeled neurons. The total percentage does not necessarily add up to 100%, possibly due to incomplete labeling with cell-type markers.
Figure 12.

Representative image of double-labeled immunohistochemistry detecting Fos and CaMKII (A), Fos and TH (B), and Fos and GAD67 (C) associated with nicotine-induced acceleration of the escalation of alcohol self-administration. A, Red-labeled nuclei indicate expression of the general neuronal nuclei marker NeuN. B, Green-labeled nuclei indicate Fos expression. Merged images indicate nuclei in green. Scale bar, 100 μm. n = 4 per group.
Discussion
Here, we show that chronic treatment with nicotine (0.8 mg/kg, daily) dramatically accelerated the escalation of alcohol drinking, increased PR responding, and increased compulsive alcohol drinking despite adverse consequences. We further demonstrated that the facilitating effect of nicotine on compulsive alcohol drinking was associated with the recruitment of a widespread but selective set of neuronal ensembles in the dmPFC, CeA, NAc-Core, BNST, and pVTA but not vmPFC, NAc-Shell, OFC, BLA, or aVTA. The blockade of nicotinic receptors prevented both the recruitment of the nicotine neuronal ensembles in the dmPFC and the faster escalation of alcohol drinking. These results demonstrate that nicotine is a critical factor in the development of alcohol dependence that dysregulates a set of interconnected neuronal ensembles throughout the brain, including elements of the extended amygdala (CeA and BNST), dorsal corticostriatal module (dmPFC-NAc-Core), and mesocorticolimbic dopaminergic/GABAergic system (pVTA).
Previous studies have investigated the effect of nicotine on alcohol drinking in nondependent rats, but these reports had conflicting results. Some studies demonstrated an increase in alcohol drinking (Blomqvist et al., 1996; Lê et al., 2000, 2014; Abreu-Villaça et al., 2013; Britt and Bonci, 2013) and relapse after nicotine administration (Lê et al., 2003), whereas other studies showed a decrease in alcohol intake (Sharpe and Samson, 2002). Our results showed increased FR and PR responding in nondependent rats after chronic nicotine treatment, which is consistent with previous studies showing increased alcohol intake after nicotine administration (Blomqvist et al., 1996; Lê et al., 2000, 2014). Moreover, our results may explain the conflicting results observed in previous studies with nondependent rats. Indeed, we observed both an increase in responding under FR and PR schedules and a decrease in responding when alcohol drinking was associated with adverse consequences (quinine), suggesting that nicotine may decrease alcohol intake in nondependent rats under specific conditions.
The present results with dependent rats demonstrate that nicotine not only accelerates the development of alcohol dependence (faster escalation of alcohol intake) but also increases PR responding and responding despite adverse consequences. Although in our study the dependent rats that were treated with saline were more sensitive to the bitter taste of quinine (without alcohol) than nondependent rats, dependent rats that received nicotine administration showed higher resistance to quinine than nondependent rats, confirming the results in the FR and PR tests. The early escalation of alcohol intake observed in rats that were exposed to alcohol and nicotine is consistent with clinical studies showing the development of severe alcohol dependence in patients who use tobacco (Batel et al., 1995).
We recently hypothesized that drug and alcohol addiction may result from the dysregulation of different putative modules, including the incentive salience (VTA and NAc), negative affect (CeA and BNST), and loss of control modules (PFC and OFC; George and Koob, 2010). A possible mechanism for the facilitation of the escalation of alcohol drinking with chronic nicotine is the recruitment, by nicotine, of specific neuronal ensembles within one or more of these modules. Indeed, we found that the combination of nicotine and alcohol robustly increased the number of Fos-positive neurons in the dmPFC, NAc-Core, CeA, BNST, and pVTA in dependent rats. This increase in Fos-positive neurons was not observed in the OFC, NAc-Shell, or aVTA, demonstrating the neuronal specificity of these ensembles in dependent rats. Although an increase in Fos-positive neurons was also observed in the BLA, it was also observed in nondependent rats that were exposed to nicotine, ruling out a specific role in alcohol dependence. These results demonstrate that the combination of nicotine and alcohol produced specific dysregulation of discrete regions of the cognitive (dmPFC and NAc-Core), negative affect (CeA-BNST), and incentive salience (pVTA) modules in dependent rats. While at first glance it appears surprising that there is no increase in Fos in the CeA and PFC in rats exposed to vapor compared with air (without nicotine), it is actually consistent with our previous findings (George et al., 2012) that increases in Fos in the CeA and PFC are only observed during alcohol withdrawal, and not when withdrawal is followed by access to alcohol self-administration, as in the present report.
The causal role between activation of the dmPFC, NAc-Core, CeA, BNST, and pVTA in drug and alcohol seeking and taking has been largely demonstrated previously using pharmacological and optogenetic techniques (Fuchs et al., 2007; Linsenbardt and Boehm, 2009; Ding et al., 2012; Vendruscolo et al., 2012; Seif et al., 2013; Gilpin et al., 2014). For instance, activation of the CeA leads to negative emotional states during withdrawal and promotes excessive nicotine and alcohol intake (George et al., 2007; Gilpin et al., 2008; Koob, 2009; Koob and Volkow, 2010), and chronic nicotine enhances the activity of the extended amygdala (Picciotto and Kenny, 2013). Studies have also demonstrated that increased dopamine release in the pVTA-dmPFC and pVTA-NAc-Core pathways potentiates incentive salience and the motivation and compulsion to drink alcohol in rats (Koob and Volkow, 2010). Moreover, nicotine administration enhances dopamine release in the NAc (Di Chiara, 2000) while blockade of nicotinic receptors in the NAc prevents alcohol-induced increases in dopamine release and decreases alcohol intake in rats (Blomqvist et al., 1993, 1997; Ericson et al., 1998; Smith et al., 1999).
Evidence suggests that alcohol acts on specific elements of the ventral forebrain, such as the NAc and amygdala, to produce its acute positive reinforcing effects (Koob, 2003). The extended amygdala, composed of the CeA, BNST, and a transition area in the NAc-Shell, has been hypothesized to be the region for neuroadaptive changes in this reward circuit that provides the motivation for excessive drinking characterizing dependence (Koob, 2003; Koob and Le Moal, 2006). Studies have demonstrated that nicotine can also act in these same brain regions (Barik and Wonnacott, 2009; Picciotto and Kenny, 2013; Picciotto and Mineur, 2014) and alter the rewarding and aversive properties of other drugs (Wang et al., 2011; Li et al., 2014; Szabo et al., 2014).
The neuronal phenotype analysis demonstrated that the increased Fos expression in these brain regions in the alcohol-nicotine group is related to the activation of a small percentage of neurons (∼3–13%). Similarly, other authors have shown that only a small percentage of neurons within a specific brain region is responsible for the expression of behaviors related to drug addiction (Koya et al., 2009, 2012; Bossert et al., 2011; Fanous et al., 2012). Future experiments will be necessary to test whether inactivation of these small percentages of cells will prevent nicotine-induced compulsive alcohol drinking.
We further explored the phenotype of the neurons recruited in these different neuronal ensembles and found remarkable selectivity in the activation of glutamatergic versus GABAergic neurons in different regions. Glutamatergic neurons were specifically recruited in the dmPFC, while GABA-ergic represented the majority of neurons in the NAc-Core, CeA, and pVTA. Very few studies have sought to determine the phenotype of the specific population of neurons that are activated during alcohol withdrawal. The present results that showed specific dysfunction of the dmPFC and CeA but not vmPFC-NAc-Shell is consistent with our previous work, showing that withdrawal from alcohol binge drinking in rats is associated with a functional disconnection between the dmPFC and CeA but not between the vmPFC and NAc-Shell (George et al., 2012).
The specific recruitment of glutamatergic neurons in the dmPFC suggests a key role for cortical pyramidal neurons in the effects of nicotine on the escalation of alcohol self-administration. The glutamatergic system has been implicated in the reinforcing effects of alcohol (Rao and Sari, 2012), and chronic alcohol intake has been shown to be associated with the upregulation of NMDA receptors (Sanna et al., 1993; Snell et al., 1996; Siggins et al., 2003). Moreover, alcohol withdrawal increased extracellular glutamate levels in the striatum (Rossetti and Carboni, 1995; Siggins et al., 2003). Importantly, glutamatergic neuron projections from the dmPFC to NAc (Kalivas, 2009) and CeA (George et al., 2012) have been linked to the development of addiction and alcohol binge drinking, in particular. Thus, future investigations should target the glutamatergic system, which seems to play an important role in the development of alcohol dependence.
Finally, we found that the antagonism of nicotine receptors blocked both the facilitating effect of nicotine on the escalation of alcohol self-administration and the recruitment of a nicotine neuronal ensemble. These results demonstrate that activation of nicotinic receptors controls the escalation of alcohol self-administration and suggest that individual differences at the nicotinic receptor level may underlie individual vulnerability to alcoholism. Indeed, it has been suggested that alcohol and nicotine have a common pathway involving α4β2 nicotinic receptors (Sharma et al., 2014a, b). Furthermore, varenicline, a partial agonist of α4β2 receptors, reduces alcohol (Feduccia et al., 2014) and nicotine (George et al., 2011) intake in rats.
Our results demonstrate that nicotine is a critical factor in the development of alcohol dependence that dysregulates a set of interconnected neuronal ensembles involved in incentive salience, negative emotional states, and loss of control. Targeting specifically these neuronal ensembles may provide new opportunities for the treatment of comorbid tobacco and alcohol dependence.
Footnotes
This research was supported by National Institutes of Health (NIH) grants (AA018914, AA020608, AA008459, AA006420, AA022977, and DA023597); National Institute on Drug Abuse, Intramural Research Program, NIH; and Coordination for the Improvement of Higher Education Personnel (CAPES) PDEE fellowship 0093/11-4 (R.M.L.).
The authors declare no competing financial interests.
References
- Abreu-Villaça Y, Cavina CC, Ribeiro-Carvalho A, Correa-Santos M, Naiff VF, Filgueiras CC, Manhães AC. Combined exposure to tobacco smoke and ethanol during adolescence leads to short- and long-term modulation of anxiety-like behavior. Drug Alcohol Depend. 2013;133:52–60. doi: 10.1016/j.drugalcdep.2013.05.033. [DOI] [PubMed] [Google Scholar]
- Alén F, Gomez R, González-Cuevas G, Navarro M, López-Moreno JA. Nicotine causes opposite effects on alcohol intake: evidence in an animal experimental model of abstinence and relapse from alcohol. Nicotine Tob Res. 2009;11:1304–1311. doi: 10.1093/ntr/ntp139. [DOI] [PubMed] [Google Scholar]
- Barik J, Wonnacott S. Molecular and cellular mechanisms of action of nicotine in the CNS. Handb Exp Pharmacol. 2009:173–207. doi: 10.1007/978-3-540-69248-5_7. [DOI] [PubMed] [Google Scholar]
- Barrett SP, Darredeau C, Pihl RO. Patterns of simultaneous polysubstance use in drug using university students. Hum Psychopharmacol. 2006;21:255–263. doi: 10.1002/hup.766. [DOI] [PubMed] [Google Scholar]
- Batel P, Pessione F, Maître C, Rueff B. Relationship between alcohol and tobacco dependencies among alcoholics who smoke. Addiction. 1995;90:977–980. doi: 10.1111/j.1360-0443.1995.tb03507.x. [DOI] [PubMed] [Google Scholar]
- Blomqvist O, Engel JA, Nissbrandt H, Söderpalm B. The mesolimbic dopamine-activating properties of ethanol are antagonized by mecamylamine. Eur J Pharmacol. 1993;249:207–213. doi: 10.1016/0014-2999(93)90434-J. [DOI] [PubMed] [Google Scholar]
- Blomqvist O, Ericson M, Johnson DH, Engel JA, Söderpalm B. Voluntary ethanol intake in the rat: effects of nicotinic acetylcholine receptor blockade or subchronic nicotine treatment. Eur J Pharmacol. 1996;314:257–267. doi: 10.1016/S0014-2999(96)00583-3. [DOI] [PubMed] [Google Scholar]
- Blomqvist O, Ericson M, Engel JA, Söderpalm B. Accumbal dopamine overflow after ethanol: localization of the antagonizing effect of mecamylamine. Eur J Pharmacol. 1997;334:149–156. doi: 10.1016/S0014-2999(97)01220-X. [DOI] [PubMed] [Google Scholar]
- Bossert JM, Stern AL, Theberge FR, Cifani C, Koya E, Hope BT, Shaham Y. Ventral medial prefrontal cortex neuronal ensembles mediate context-induced relapse to heroin. Nat Neurosci. 2011;14:420–422. doi: 10.1038/nn.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt JP, Bonci A. Alcohol and tobacco: how smoking may promote excessive drinking. Neuron. 2013;79:406–407. doi: 10.1016/j.neuron.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz FC, Koya E, Guez-Barber DH, Bossert JM, Lupica CR, Shaham Y, Hope BT. New technologies for examining the role of neuronal ensembles in drug addiction and fear. Nat Rev Neurosci. 2013;14:743–754. doi: 10.1038/nrn3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz FC, Babin KR, Leao RM, Goldart EM, Bossert JM, Shaham Y, Hope BT. Role of nucleus accumbens shell neuronal ensembles in context-induced reinstatement of cocaine-seeking. J Neurosci. 2014;34:7437–7446. doi: 10.1523/JNEUROSCI.0238-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G. Role of dopamine in the behavioural actions of nicotine related to addiction. Eur J Pharmacol. 2000;393:295–314. doi: 10.1016/S0014-2999(00)00122-9. [DOI] [PubMed] [Google Scholar]
- DiFranza JR, Guerrera MP. Alcoholism and smoking. J Stud Alcohol. 1990;51:130–135. doi: 10.15288/jsa.1990.51.130. [DOI] [PubMed] [Google Scholar]
- Ding ZM, Katner SN, Rodd ZA, Truitt W, Hauser SR, Deehan GA, Jr, Engleman EA, McBride WJ. Repeated exposure of the posterior ventral tegmental area to nicotine increases the sensitivity of local dopamine neurons to the stimulating effects of ethanol. Alcohol. 2012;46:217–223. doi: 10.1016/j.alcohol.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon WM, Thomas AM, Ostroumov A, Dong Y, Dani JA. Potential substrates for nicotine and alcohol interactions: a focus on the mesocorticolimbic dopamine system. Biochem Pharmacol. 2013a;86:1181–1193. doi: 10.1016/j.bcp.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon WM, Dong Y, Ostroumov A, Thomas AM, Zhang TA, Dani JA. Nicotine decreases ethanol-induced dopamine signaling and increases self-administration via stress hormones. Neuron. 2013b;79:530–540. doi: 10.1016/j.neuron.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S, Guerrero M, Ghoneim OM, Roberts E, Koob GF. Evidence that vasopressin V1b receptors mediate the transition to excessive drinking in ethanol-dependent rats. Addict Biol. 2012;17:76–85. doi: 10.1111/j.1369-1600.2010.00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericson M, Blomqvist O, Engel JA, Söderpalm B. Voluntary ethanol intake in the rat and the associated accumbal dopamine overflow are blocked by ventral tegmental mecamylamine. Eur J Pharmacol. 1998;358:189–196. doi: 10.1016/S0014-2999(98)00602-5. [DOI] [PubMed] [Google Scholar]
- Fanous S, Goldart EM, Theberge FR, Bossert JM, Shaham Y, Hope BT. Role of orbitofrontal cortex neuronal ensembles in the expression of incubation of heroin craving. J Neurosci. 2012;32:11600–11609. doi: 10.1523/JNEUROSCI.1914-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feduccia AA, Simms JA, Mill D, Yi HY, Bartlett SE. Varenicline decreases ethanol intake and increases dopamine release via neuronal nicotinic acetylcholine receptors in the nucleus accumbens. Br J Pharmacol. 2014;171:3420–3431. doi: 10.1111/bph.12690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs RA, Eaddy JL, Su ZI, Bell GH. Interactions of the basolateral amygdala with the dorsal hippocampus and dorsomedial prefrontal cortex regulate drug context-induced reinstatement of cocaine-seeking in rats. Eur J Neurosci. 2007;26:487–498. doi: 10.1111/j.1460-9568.2007.05674.x. [DOI] [PubMed] [Google Scholar]
- George O, Koob GF. Individual differences in prefrontal cortex function and the transition from drug use to drug dependence. Neurosci Biobehav Rev. 2010;35:232–247. doi: 10.1016/j.neubiorev.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George O, Ghozland S, Azar MR, Cottone P, Zorrilla EP, Parsons LH, O'Dell LE, Richardson HN, Koob GF. CRF-CRF1 system activation mediates withdrawal-induced increases in nicotine self-administration in nicotine-dependent rats. Proc Natl Acad Sci U S A. 2007;104:17198–17203. doi: 10.1073/pnas.0707585104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George O, Lloyd A, Carroll FI, Damaj MI, Koob GF. Varenicline blocks nicotine intake in rats with extended access to nicotine self-administration. Psychopharmacology. 2011;213:715–722. doi: 10.1007/s00213-010-2024-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George O, Le Moal M, Koob GF. Allostasis and addiction: role of the dopamine and corticotropin-releasing factor systems. Physiol Behav. 2012;106:58–64. doi: 10.1016/j.physbeh.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Misra K, Koob GF. Neuropeptide Y in the central nucleus of the amygdala suppresses dependence-induced increases in alcohol drinking. Pharmacol Biochem Behav. 2008;90:475–480. doi: 10.1016/j.pbb.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Smith AD, Cole M, Weiss F, Koob GF, Richardson HN. Operant behavior and alcohol levels in blood and brain of alcohol-dependent rats. Alcohol Clin Exp Res. 2009;33:2113–2123. doi: 10.1111/j.1530-0277.2009.01051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Roberto M, Koob GF, Schweitzer P. Kappa opioid receptor activation decreases inhibitory transmission and antagonizes alcohol effects in rat central amygdala. Neuropharmacology. 2014;77:294–302. doi: 10.1016/j.neuropharm.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison EL, McKee SA. Young adult non-daily smokers: patterns of alcohol and cigarette use. Addict Behav. 2008;33:668–674. doi: 10.1016/j.addbeh.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser SR, Getachew B, Oster SM, Dhaher R, Ding ZM, Bell RL, McBride WJ, Rodd ZA. Nicotine modulates alcohol-seeking and relapse by alcohol-preferring (P) rats in a time-dependent manner. Alcohol Clin Exp Res. 2012;36:43–54. doi: 10.1111/j.1530-0277.2011.01579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M, Egli M, Crabbe JC, Becker HC. Acute withdrawal, protracted abstinence and negative affect in alcoholism: are they linked? Addict Biol. 2010;15:169–184. doi: 10.1111/j.1369-1600.2009.00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- Koob GF. Alcoholism: allostasis and beyond. Alcohol Clin Exp Res. 2003;27:232–243. doi: 10.1097/01.ALC.0000057122.36127.C2. [DOI] [PubMed] [Google Scholar]
- Koob GF. Neurobiological substrates for the dark side of compulsivity in addiction. Neuropharmacology. 2009;56(Suppl 1):18–31. doi: 10.1016/j.neuropharm.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug abuse: hedonic homeostatic dysregulation. Science. 1997;278:52–58. doi: 10.1126/science.278.5335.52. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nat Neurosci. 2005;8:1442–1444. doi: 10.1038/nn1105-1442. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Neurobiology of addiction. London: Academic; 2006. [Google Scholar]
- Koob GF, Le Moal M. Review. Neurobiological mechanisms for opponent motivational processes in addiction. Philos Trans R Soc Lond B Biol Sci. 2008a;363:3113–3123. doi: 10.1098/rstb.2008.0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Addiction and the brain antireward system. Annu Rev Psychol. 2008b;59:29–53. doi: 10.1146/annurev.psych.59.103006.093548. [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouri EM, McCarthy EM, Faust AH, Lukas SE. Pretreatment with transdermal nicotine enhances some of ethanol's acute effects in men. Drug Alcohol Depend. 2004;75:55–65. doi: 10.1016/j.drugalcdep.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Koya E, Golden SA, Harvey BK, Guez-Barber DH, Berkow A, Simmons DE, Bossert JM, Nair SG, Uejima JL, Marin MT, Mitchell TB, Farquhar D, Ghosh SC, Mattson BJ, Hope BT. Targeted disruption of cocaine-activated nucleus accumbens neurons prevents context-specific sensitization. Nat Neurosci. 2009;12:1069–1073. doi: 10.1038/nn.2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koya E, Cruz FC, Ator R, Golden SA, Hoffman AF, Lupica CR, Hope BT. Silent synapses in selectively activated nucleus accumbens neurons following cocaine sensitization. Nat Neurosci. 2012;15:1556–1562. doi: 10.1038/nn.3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lê AD, Corrigall WA, Harding JW, Juzytsch W, Li TK. Involvement of nicotinic receptors in alcohol self-administration. Alcohol Clin Exp Res. 2000;24:155–163. doi: 10.1111/j.1530-0277.2000.tb04585.x. [DOI] [PubMed] [Google Scholar]
- Lê AD, Wang A, Harding S, Juzytsch W, Shaham Y. Nicotine increases alcohol self-administration and reinstates alcohol seeking in rats. Psychopharmacology. 2003;168:216–221. doi: 10.1007/s00213-002-1330-9. [DOI] [PubMed] [Google Scholar]
- Lê AD, Lo S, Harding S, Juzytsch W, Marinelli PW, Funk D. Co-administration of intravenous nicotine and oral alcohol in rats. Psychopharmacology. 2010;208:475–486. doi: 10.1007/s00213-009-1746-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lê AD, Funk D, Lo S, Coen K. Operant self-administration of alcohol and nicotine in a preclinical model of co-abuse. Psychopharmacology. 2014;231:4019–4029. doi: 10.1007/s00213-014-3541-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Bu Q, Chen B, Shao X, Hu Z, Deng P, Lv L, Deng Y, Zhu R, Li Y, Zhang B, Hou J, Du C, Zhao Q, Fu D, Zhao Y, Cen X. Mechanisms of metabonomic for a gateway drug: nicotine priming enhances behavioral response to cocaine with modification in energy metabolism and neurotransmitter level. PLoS One. 2014;9:e87040. doi: 10.1371/journal.pone.0087040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsenbardt DN, Boehm SL., 2nd Agonism of the endocannabinoid system modulates binge-like alcohol intake in male C57BL/6J mice: involvement of the posterior ventral tegmental area. Neuroscience. 2009;164:424–434. doi: 10.1016/j.neuroscience.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson BJ, Koya E, Simmons DE, Mitchell TB, Berkow A, Crombag HS, Hope BT. Context-specific sensitization of cocaine-induced locomotor activity and associated neuronal ensembles in rat nucleus accumbens. Eur J Neurosci. 2008;27:202–212. doi: 10.1111/j.1460-9568.2007.05984.x. [DOI] [PubMed] [Google Scholar]
- O'Dell LE, Roberts AJ, Smith RT, Koob GF. Enhanced alcohol self-administration after intermittent versus continuous alcohol vapor exposure. Alcohol Clin Exp Res. 2004;28:1676–1682. doi: 10.1097/01.ALC.0000145781.11923.4E. [DOI] [PubMed] [Google Scholar]
- Olausson P, Ericson M, Löf E, Engel JA, Söderpalm B. Nicotine-induced behavioral disinhibition and ethanol preference correlate after repeated nicotine treatment. Eur J Pharmacol. 2001;417:117–123. doi: 10.1016/S0014-2999(01)00903-7. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 5. Amsterdam: Elsevier Academic; 2005. [Google Scholar]
- Picciotto MR, Kenny PJ. Molecular mechanisms underlying behaviors related to nicotine addiction. Cold Spring Harb Perspect Med. 2013;3:a012112. doi: 10.1101/cshperspect.a012112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Mineur YS. Molecules and circuits involved in nicotine addiction: the many faces of smoking. Neuropharmacology. 2014;76:545–553. doi: 10.1016/j.neuropharm.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao PS, Sari Y. Glutamate transporter 1: target for the treatment of alcohol dependence. Curr Med Chem. 2012;19:5148–5156. doi: 10.2174/092986712803530511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimondini R, Sommer W, Heilig M. A temporal threshold for induction of persistent alcohol preference: behavioral evidence in a rat model of intermittent intoxication. J Stud Alcohol. 2003;64:445–449. doi: 10.15288/jsa.2003.64.445. [DOI] [PubMed] [Google Scholar]
- Roberts AJ, Heyser CJ, Cole M, Griffin P, Koob GF. Excessive ethanol drinking following a history of dependence: animal model of allostasis. Neuropsychopharmacology. 2000;22:581–594. doi: 10.1016/S0893-133X(99)00167-0. [DOI] [PubMed] [Google Scholar]
- Root DH, Fabbricatore AT, Ma S, Barker DJ, West MO. Rapid phasic activity of ventral pallidal neurons during cocaine self-administration. Synapse. 2010;64:704–713. doi: 10.1002/syn.20792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetti ZL, Carboni S. Ethanol withdrawal is associated with increased extracellular glutamate in the rat striatum. Eur J Pharmacol. 1995;283:177–183. doi: 10.1016/0014-2999(95)00344-K. [DOI] [PubMed] [Google Scholar]
- Sanna E, Serra M, Cossu A, Colombo G, Follesa P, Cuccheddu T, Concas A, Biggio G. Chronic ethanol intoxication induces differential effects on GABAA and NMDA receptor function in the rat brain. Alcohol Clin Exp Res. 1993;17:115–123. doi: 10.1111/j.1530-0277.1993.tb00735.x. [DOI] [PubMed] [Google Scholar]
- Schulteis G, Markou A, Cole M, Koob GF. Decreased brain reward produced by ethanol withdrawal. Proc Natl Acad Sci U S A. 1995;92:5880–5884. doi: 10.1073/pnas.92.13.5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seif T, Chang SJ, Simms JA, Gibb SL, Dadgar J, Chen BT, Harvey BK, Ron D, Messing RO, Bonci A, Hopf FW. Cortical activation of accumbens hyperpolarization-active NMDARs mediates aversion-resistant alcohol intake. Nat Neurosci. 2013;16:1094–1100. doi: 10.1038/nn.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Sahota P, Thakkar MM. Nicotine administration in the cholinergic basal forebrain increases alcohol consumption in C57BL/6J mice. Alcohol Clin Exp Res. 2014a;38:1315–1320. doi: 10.1111/acer.12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma R, Dumontier S, DeRoode D, Sahota P, Thakkar MM. Nicotine infusion in the wake-promoting basal forebrain enhances alcohol-induced activation of nucleus accumbens. Alcohol Clin Exp Res. 2014b;38:2590–2596. doi: 10.1111/acer.12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe AL, Samson HH. Repeated nicotine injections decrease operant ethanol self-administration. Alcohol. 2002;28:1–7. doi: 10.1016/S0741-8329(02)00238-0. [DOI] [PubMed] [Google Scholar]
- Siggins GR, Martin G, Roberto M, Nie Z, Madamba S, De Lecea L. Glutamatergic transmission in opiate and alcohol dependence. Ann N Y Acad Sci. 2003;1003:196–211. doi: 10.1196/annals.1300.012. [DOI] [PubMed] [Google Scholar]
- Smith BR, Horan JT, Gaskin S, Amit Z. Exposure to nicotine enhances acquisition of ethanol drinking by laboratory rats in a limited access paradigm. Psychopharmacology. 1999;142:408–412. doi: 10.1007/s002130050906. [DOI] [PubMed] [Google Scholar]
- Snell LD, Nunley KR, Lickteig RL, Browning MD, Tabakoff B, Hoffman PL. Regional and subunit specific changes in NMDA receptor mRNA and immunoreactivity in mouse brain following chronic ethanol ingestion. Brain Res Mol Brain Res. 1996;40:71–78. doi: 10.1016/0169-328X(96)00038-1. [DOI] [PubMed] [Google Scholar]
- Sommer WH, Rimondini R, Marquitz M, Lidström J, Siems WE, Bader M, Heilig M. Plasticity and impact of the central renin-angiotensin system during development of ethanol dependence. J Mol Med. 2007;85:1089–1097. doi: 10.1007/s00109-007-0255-5. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Hölter SM. Long-term alcohol self-administration with repeated alcohol deprivation phases: an animal model of alcoholism? Alcohol Alcohol. 1999;34:231–243. doi: 10.1093/alcalc/34.2.231. [DOI] [PubMed] [Google Scholar]
- Szabo ST, Fowler JC, Froeliger B, Lee TH. Time-dependent changes in nicotine behavioral responsivity during early withdrawal from chronic cocaine administration and attenuation of cocaine sensitization by mecamylamine. Behav Brain Res. 2014;262:42–46. doi: 10.1016/j.bbr.2013.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez GR, Roberts AJ, Chan K, Davis H, Brennan M, Zorrilla EP, Koob GF. Increased ethanol self-administration and anxiety-like behavior during acute ethanol withdrawal and protracted abstinence: regulation by corticotropin-releasing factor. Alcohol Clin Exp Res. 2002;26:1494–1501. doi: 10.1111/j.1530-0277.2002.tb02448.x. [DOI] [PubMed] [Google Scholar]
- Vendruscolo LF, Barbier E, Schlosburg JE, Misra KK, Whitfield TW, Jr, Logrip ML, Rivier C, Repunte-Canonigo V, Zorrilla EP, Sanna PP, Heilig M, Koob GF. Corticosteroid-dependent plasticity mediates compulsive alcohol drinking in rats. J Neurosci. 2012;32:7563–7571. doi: 10.1523/JNEUROSCI.0069-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Yuan W, Li MD. Genes and pathways co-associated with the exposure to multiple drugs of abuse, including alcohol, amphetamine/methamphetamine, cocaine, marijuana, morphine, and/or nicotine: a review of proteomics analyses. Mol Neurobiol. 2011;44:269–286. doi: 10.1007/s12035-011-8202-4. [DOI] [PubMed] [Google Scholar]