

Abstract
The objective of the present research work was to prepare and evaluate sustained release subcutaneous (s.c.) nanoparticles of low molecular weight heparin (LMWH). The nanoparticles were prepared by water–in-oil in-water (w/o/w) emulsion and evaporation method using different grades of polylactide co-glycolide (50:50, 85:15), and different concentrations of polyvinyl alcohol (0.1%, 0.5%, 1%) aqueous solution as surfactant. The fabricated nanoparticles were evaluated for size, shape, zeta potential, encapsulation efficiency, in vitro drug release, and in vivo biological activity (anti-factor Xa activity) using the standard kit. The drug and excipient compatibility was analyzed by Fourier transform infrared spectroscopy (FTIR), differential scanning calorimetry (DSC) and X-ray diffraction (XRD) studies. The formation of nanoparticles was confirmed by scanning electron microscopy; nanoparticles were spherical in shape. The size of prepared nanoparticles was found between 195 nm and 251 nm. The encapsulation efficiency of the nanoparticles was found between 46% and 70%. In vitro drug, release was about 16–38% for 10 days. In vivo drug, release shows the sustained release of drug for 10 days in rats. FTIR studies indicated that there was no loss in chemical integrity of the drug upon fabrication into nanoparticles. DSC and XRD results demonstrated that the drug was changed from the crystalline form to the amorphous form in the formulation during the fabrication process. The results of this study revealed that the s.c. nanoparticles were suitable candidates for sustained delivery of LMWH.
Keywords: Anti-factor Xa activity, low molecular weight heparin, nanoparticles, polylactide co-glycolide, subcutaneous, sustained release
INTRODUCTION
Low molecular weight heparin (LMWH) is a glycosaminoglycan widely used for its anti-coagulant activity. Venous thromboembolism and unstable angina can be prevented or treated with LMWH.[1,2] Venous thromboembolism is the third most common cause of death among hospitalized patients. It is also used in the prevention of clots after implantation of a medical device.[1] Apart from anti-coagulant activity, it is also reported for its use in the therapy of rheumatoid arthritis, asthma, and cancer.[3,4,5] LMWH shows excellent anti-coagulant activity with less severe toxicities like thrombocytopenia and bleeding.[1] LMWH is water-soluble macromolecular drug, with a short physiological half-life. On oral administration, it shows very low bioavailability because of its size and negative charge and the poor permeation through the intestinal wall. To overcome this problem, it is administered via the parenteral route.[1] Formulations approved by Food and Drug Administration are lyophilized powders to be reconstituted into solutions. Repeated parenteral administration that is, intravenous (I.V.)/subcutaneous (s.c.) solutions and therapeutically monitored I.V. infusions are invasive and further, repeated administrations add insult to the existing invasiveness. This is a major problem with currently used therapy with LMWH. To improve the therapy with LMWH, several routes, and several delivery systems were previously investigated.[6] For instance, Lanke et al., investigated oral microspheres along with penetration enhancers to enhance oral bioavailability of LMWH.[7] In this study, the oral bioavailability of LMWH was enhanced by 21%. Bai et al., investigated dendrimers as carriers for pulmonary delivery of LMWH.[8] This formulation enhanced the bioavailability of LMWH by 40% and was more effective in preventing venous thromboembolism in a rodent model compared with its solution form administered subcutaneously. Similarly, several studies to improve the therapy with LMWH have been investigated. However, these formulations are in the investigational stages only. Parenteral sustained release dosage forms for LMWH was recently addressed in one study by Choubey et al.[9] This study demonstrated better benefits for parenteral sustained release dosage forms for LMWH. However, this is the only study, which addressed this issue so far. More investigations are necessary for clinical realization of such a strategy.
Development of parenteral sustained release dosage forms finds a solution to the problem with current LMWH therapy and this solution is clinically viable. The most convenient and the clinically viable route for sustained release LMWH is parenteral route. Intramuscular (i.m.), s.c., intraperitoneal (i.p.) and I.V. formulations belong to various parenteral formulations. Sustained release parenteral formulations can be administered via any of these aforementioned routes. Significant improvement in the therapy in the form of prolonged release via the parenteral route was achieved with heparin. Heparin implantable dosage forms such as stents, nanoparticles, liposomes are safe for administration in the body and resulted in sustained systemic release of heparin.[10,11,12] Similar observations can be extrapolated to LMWH, which is a similar molecular as that of heparin. Parenteral sustained release I.V. dosage forms such as microspheres, nanoparticles, liposomes upon administration are immediately engulfed by reticuloendothelial system (RES) in the liver and then slowly releases the drug within RES.[13,14] To overcome this problem, development of sustained release i.m., s.c., and i.p. nanoparticles are needed. However, precedence has been established with s.c route for LMWH. Presently marketed preparations of LMWH are administered via s.c. route and the dosage forms require frequent administration.[1] Thus, development of s.c. sustained release dosage forms offer a promising option for LMWH. Thus, the most convenient and the clinically viable route for LMWH nanoparticles is s.c. As per our knowledge, for LMWH, subcutaneously administered sustained release nanoparticles were not reported previously. Thus, the objective of this study was to develop s.c. sustained release nanoparticles of LWMH and further evaluate the formulation for various in vitro and in vivo properties.
MATERIALS AND METHODS
Materials
Low molecular weight heparin (Enoxaparin) was a gift sample from Gland Pharma Pvt. Ltd., (Hyderabad, India). Polylactide co-glycolide (PLGA) (50:50, 85:15) was purchased from PolySciTech, USA. Dichloromethane and polyvinyl alcohol (PVA) were purchased from SD Fine Chemicals Ltd. An ultraviolet (UV) - visible spectrophotometer from Thermo scientific was used. Qsonica probe sonicator, Cooling centrifuge Hittech MIKRO 220 R, Freeze dryer (Mini Lyodel) Chennai, India were used for formulation of Nanoparticles. A JSM-5200 scanning electron microscope (SEM), Japan, was used to study the surface morphology of nanoparticles. Zetasizer Nano ZS (Malvern Instruments, Malvern, United Kingdom) was used to measure the particle size and zeta potential of prepared nanoparticles. Differential scanning calorimeter from Shimadzu, Fourier transform infrared spectrophotometer (FTIR) from Perkin-Elmer and X-ray diffractometer, X-ray powder diffraction (XRPD) from PAN analytical were used. All other ingredients used in this study were of analytical grade.
Methods
Preparation of nanoparticles
The LMWH nanoparticles were prepared by the water–in-oil in-water (w/o/w) emulsion and evaporation method. Required quantity of drug was taken in 2 ml of distilled water. To this 10 ml of methylene chloride containing the polymer (PLGA 50:50/PLGA 85:15) was added and emulsified with a probe sonicator (Qsonica) for 30 s at 60 W. The resulting w/o emulsion was then poured in 40 ml of PVA (0.1%) solution and sonicated for 60 s at 60 W using probe sonicator resulting in the formation of w/o/w emulsion. This emulsion was kept on the magnetic stirrer (15 rpm) for 10 min to evaporate methylene chloride.[14] Nanoparticles were then isolated by centrifugation (Hittich, MIKRO 220R), at 18,000 rpm for 15 min. The supernatant was removed and assayed to find out entrapment efficiency. The nanoparticles were washed three times with deionized water and subjected to freeze drying (Lyophilizer, Mini–Lyodel, Delvac, Chennai, India).
Characterization of the nanoparticles
Size and surface analysis
The mean diameter of nanoparticles and their surface potential were evaluated with zeta sizer, which was regularly calibrated and validated. Surface morphology was evaluated using SEM. A concentrated aqueous suspension was spread over a slab and dried under vacuum. The sample was shadowed in a cathodic evaporator with gold layer 20 nm thick. Photographs were taken using a JSM-5200 Scanning Electron Microscope (Tokyo, Japan) operated at 20 kV.
Zeta potential
The zeta potential is used to measure the electric charge at the surface of the particles, indicating the physical stability of colloidal systems. The zeta potential was measured using a Zetasizer Nano ZS (Malvern Instruments, Malvern, United Kingdom). The equipment is maintenance-free system, which is regularly calibrated and validated. Samples were diluted with the respective original dispersion medium, which provides information regarding the thickness of the diffuse layer. Diluted nanosuspension was added to the sample cell (quartz cuvette) and was put into the sample holder unit, and zeta potential was measured.
Encapsulation efficiency
The amount of drug entrapped in nanoparticles was determined by turbidimetric assay by measuring the amount of nonentrapped drug in the supernatant recovered after centrifugation. Briefly, 1 ml of supernatant was taken. To this 1 ml of acetate buffer (1M, pH 5) followed by 4 ml of cetylpyridinium solution (0.1%) in NaCl 0.94% was added and assayed for drug release at 500 nm by UV spectrophotometer.[15]
In vitro drug release
An aliquot of 10 mg of nanoparticles was suspended in flask containing 3 ml of phosphate buffer saline (phosphate-buffered saline 0.011 M, NaCl 0.15 M, pH 7.4), and kept it for stirring (150 rpm) on magnetic stirrer. Entire sample was taken every 24 h up to 10 days and centrifuged for 15 min at 18,000 rpm, using cooling centrifuge (Hittich, MIKRO 220 R). To the pellet obtained after centrifugation, again 3 ml of phosphate buffer saline was added and kept for stirring for further readings. A volume of 1 ml of supernatant was taken, and LMWH was assayed.
In vivo drug release from low molecular weight heparin nanoparticles
The in vivo biological activity of LMWH was evaluated by measuring the anti-factor Xa activity with a chromogenic substrate using the standard kit (KRIBIOLISA™ Xa) from Krishgen Bio Systems, Mumbai, India. according to the method described by the supplier.
In vivo biological activity of LMWH was investigated in male Wistar rats. All the experiments were conducted according to the guidelines of CPCSEA. The study was approved by Animal Ethical Committee of Synapse Life Sciences, Warangal registered under CPCSEA, India (IAEC No. vcop/VI/2014/10/2). The animals were divided into 5 groups (n = 6) as given below. Based on the results of size and surface analysis, encapsulation efficiency, in vitro drug release four formulations (F5, F6, F11, F12) were selected for in vivo biological activity. LMWH was administered to the control group subcutaneously.
Group 1: S.c. administration of F5
Group 2: S.c. administration of F6
Group 3: S.c. administration of F11
Group 4: S.c. administration of F12
Group 5: Control group administered s.c drug solution.
Group 1 received 0.3 ml of F5 nanoparticular formulation. Group 2 received 0.3 ml F6 nanoparticular formulation. Group 3 received the 0.3 ml suspension of formulation F11, Group 4 received the 0.3 ml suspension of F12 formulation. All the rats were administered 4.5 mg/kg equivalent of the drug. After administration of formulations blood samples were collected at 6, 12, 18, 24 h and 2, 3, 4, 5, 6, 7, 8, 9, and 10 days, then plasma was collected and the concentration of drug of drug in plasma was quantified by anti-Xa assay method using standard kit (KRIBIOLISA™ Xa) according to the method described by the supplier. The peak plasma concentration (Cmax) and time to reach the maximum peak concentration (Tmax) and area under the curve (AUC) were obtained as described earlier.[16,17]
Selection and characterization of optimized formulation
Using size, zeta potential, encapsulation, in vitro and in vivo drug release, an optimum formulation that releases the drug for 10 days in vivo was selected. The optimized formulation was characterized for various properties as chemical integrity, thermal state and crystalline state using FTIR, differential scanning calorimetry (DSC) and PXRD, respectively, as described earlier.[18]
Statistics
All experiments were done six times and the data were expressed as mean ± standard deviation and Tukey's post-hoc test was done to analyze significance of difference between different groups using the statistical analysis software package SPSS (version 16.0, IBM, USA).
RESULTS AND DISCUSSION
Studies that addressed parenteral sustained release of LMWH are scanty. A recent study developed a pegylated enoxaparin for sustained s.c. delivery of LMWH. Choubey et al. developed polyethylene glycol-LMWH (P-ENX) conjugates for s.c. administration. P-ENX conjugate exhibited enhancement in anti Xa activity by three-fold as compared to free ENX, and it exhibited an increase in AUC by four-fold.[9] Thus, s.c. administration of this drug a novel approach for extended and enhanced activity of LMWH. Development of subcutaneously administered nanoparticular formulation is another similar attempt that can be investigated for LMWH. In recent years, nanoparticles have become very attractive for their applications in biology and medicine.[19] In this study, nanoparticles incorporating LMWH were prepared to develop a parenteral sustained release dosage form. The particular focus of the route of administration of these nanoparticles is subcutaneous. Studies on s.c. nanoparticles for sustained systemic release of the active appeared in the scientific literature. They have tremendous potential. S.c. nanoparticles for systemic delivery of antitubercular drugs, tetracycline have been previously reported.[20,21] One recent study reported s.c. etoposide-loaded solid lipid nanoparticles (ETPL) for local delivery in tumors.[22] A comparison of s.c., I.V., and i.p. routes has been mentioned to treat Daltons lymphoma in this mice model. The results indicated that s.c. route for nanoparticles was better compared with other two routes. S.c. administration also showed a significant reduction in drug uptake by organs of the reticuloendothelial system (i.e., lung, liver, and spleen), which resulted in longer circulation of ETPL nanoparticles. This route also had a relatively low tissue distribution, which can reduce the systemic side-effects of etoposide. Similar results we envision with s.c. administration of LMWH nanoparticles.
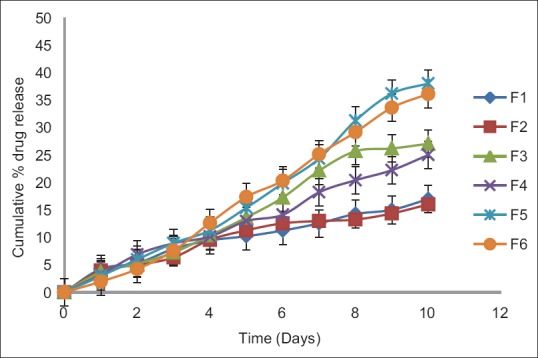
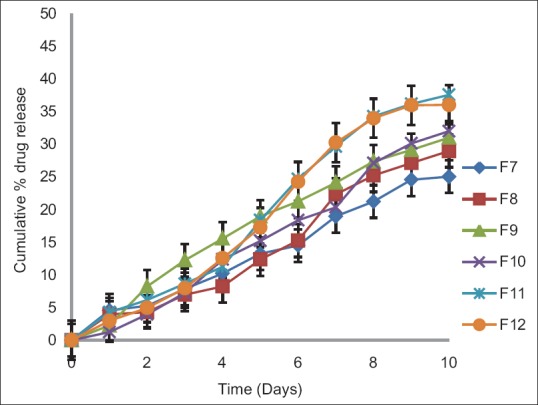
In this study, for the first time, we report s.c. nanoparticles for sustained systemic delivery of the LMWH. The main goals in the design of nanoparticles are to control the particle size, surface properties and release of the active drug.[19] In this study, we used PLGAs to prepare s.c. LMWH nanoparticles. PLGA is a biocompatible and biodegradable polymer approved by USFDA. Several commercial products utilize this polymer. We successfully fabricated LMWH nanoparticles using PLGA (50:50, 85:15) and different concentration PVA (0.1%, 0.5%, 1%) aqueous solution by employing water-in-oil in-water (w/o/w) emulsion and evaporation method. The compositions are shown in Table 1. The particle size of the all the nanoparticle formulations was found between 195 nm and 251 nm. The study of SEM was conducted to confirm the formation and surface morphology of nanoparticles. All the nanoparticles were spherical in shape. These are shown in Figure 1. Particle size of the particles decreased as the concentration of surfactant increased. This may due to low concentration of surfactant, which would fail to stabilizing the nanoparticles and thereby promoting aggregation and then enhancement in particle size. As the concentration of PLGA increased, particle size also increased. The zeta potential of all the particles was between − 22.1 mV and − 35.2 mV. Nanoparticles prepared using PLGA have invariably negative charge, and sometimes depends on the characteristics of the drug encapsulated. Particle size and zeta potential of all the nanoparticle formulations depicted in Table 2. The encapsulation efficiency of all the fabricated nanoparticles was found between 46% and 70%. As the concentration of polymer increases, there was an increase in encapsulation efficiency. The lowest encapsulation efficiencies were 46 ± 2 and 48 ± 1.56 found in F1, F6 formulations, which consist of lower amount of polymer concentration (150 mg) and lower surfactant concentration (0.1%). The in vitro drug release studies were performed for all the formulations. The results of drug release demonstrated sustained release of drug from all the formulations for 10 days [Figure 2]. Among the nanoparticles fabricated with PLGA 50:50 (F1–F6), F5 formulation demonstrated maximum drug release, which was about 38.02% in 10 days and the reason could be attributed to the surfactant concentration. As the concentration of surfactant increased, there was a decrease in particle size of an increase in surface area of the particles, which tends to increase the drug release from the nanoparticles. The same results were demonstrated with nanoparticles fabricated by using PLGA 85:15. F11 and F12 formulations show maximum drug release for 10 days about 37.56% and 36.01%, respectively. The release represented in Figure 3. From the various formulations we prepared, we selected four formulations (F5, F6, F11, F12) for in vivo biological activity based on the results of size and surface analysis, encapsulation efficiency, in vitro drug release. The selected four formulations were used for measurement of anti-factor Xa activity in rats. Software was used to calculate the pharmacokinetic parameters, and the results are summarized in Table 3. Plasma concentration versus time profile of selected formulations and pure drug solution is presented in Figure 4. For pure drug peak plasma concentration, Cmax was 12.5 μg/ml and Tma × 3 h, and AUC0-∞ was 37.84 μg/ml/h. Pharmacokinetic parameters of selected formulations and pure drug solutions are represented in Table 3. S.c. nanoparticles exhibited increased Cmax, AUC0-∞, and Tmax values when compared to pure drug solutions for 10 days. Among the four formulations (F5, F6, F11, F12) F6 formulation exhibited peak plasma concentration 9.5 μg/ml at 7th day (Tmax). F6 formulation exhibited a four-fold enhancement (37.485 vs. 158.484 μg/ml/h) in the area under the curve (AUC0-∞) From the in vitro and in vivo drug release data, as well as zeta potential and surface size, an optimum formulation (F6) was selected, and this was further characterized.
Table 1.
Composition of various nanoparticle formulations
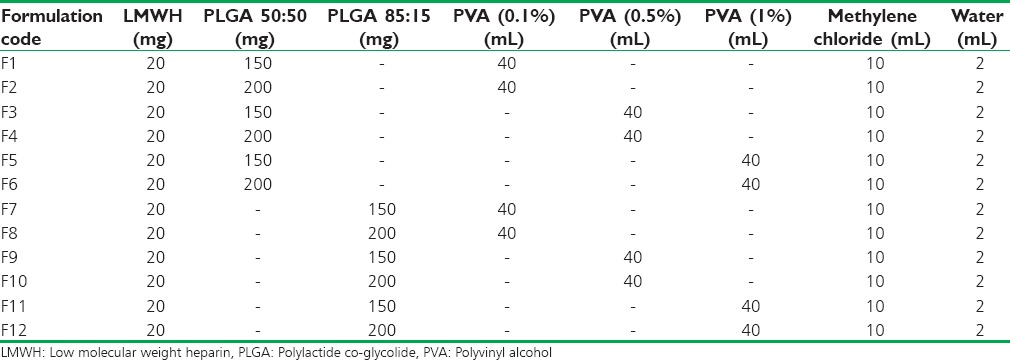
Figure 1.
In vitro drug release profiles of nanoparticle formulations containing low molecular weight heparin prepared using different concentrations of polylactide co-glycolide 50:50 and different concentrations of surfactant (0.1%, 0.5%, 1%)
Table 2.
Particle size, zeta potential and encapsulation efficiency of all the formulations
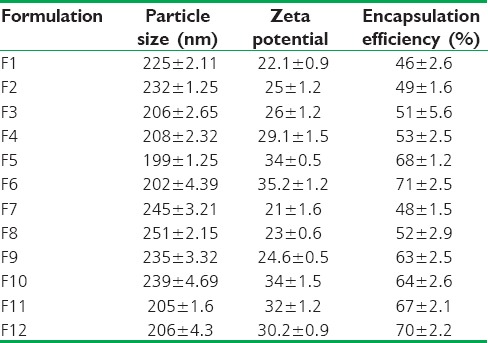
Figure 2.
In vitro drug release profiles of nanoparticle formulations containing low molecular weight heparin prepared using different concentrations of polylactide co-glycolide 85:15 (150, 200 mg) and different concentrations of surfactant (0.1%, 0.5%, 1%)
Figure 3.
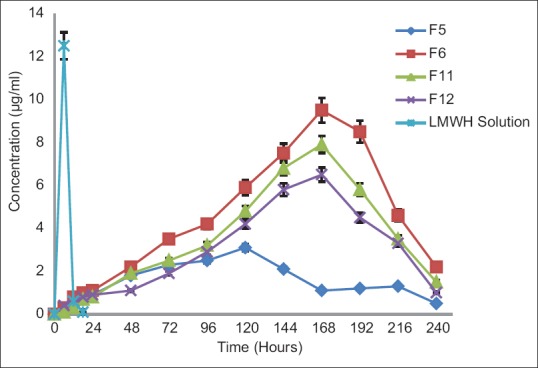
Plasma concentration-time profiles of selected formulations and low molecular weight heparin solution
Table 3.
Pharmacokinetic parameters of LMWH and selected formulations
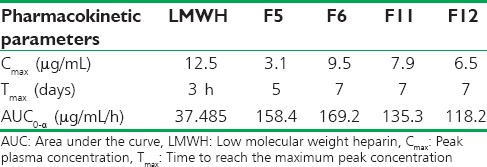
Figure 4.

Scanning electron microscope picture of low molecular weight heparin nanoparticles
Fourier transforms infrared spectroscopy spectra of drug, polymers, and optimized formulation was recorded. The presence of peaks at the expected range confirmed that the materials taken for the study are genuine, and there were no possible chemical interactions between drug and the excipients. XRPD results yielded interesting results. The drug was crystalline due to the sharp peaks demonstrated in XRPD. With the optimized nanoparticles, there was a small bump in the peak suggesting partial conversion of LMWH to less crystalline form and that may be due to the formation of amorphous form upon formulation. XRPD spectra are shown in Figure 5. The DSC of the pure drug and the optimized formulations is shown in Figure 6. There was a change in the thermograms of pure drug and the optimized formulation, suggesting a change in the physical state of the drug upon formulation. DSC of physical mixture was used as a control. As DSC of the physical mixture was very much similar to that of the optimized formulation, it can be inferred that there was a change in the solid state of the drug upon formulating into the nanoformulation.
Figure 5.
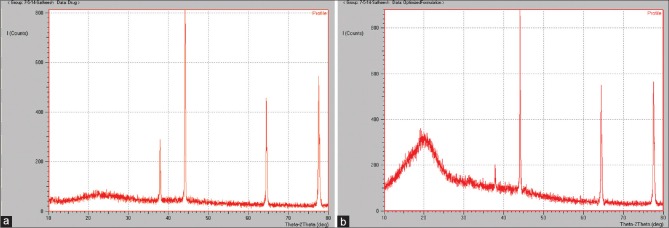
XRPD pattern of LMWH in the optimized formulation ((a) pure drug; (b) optimized formulation
Figure 6.

Differential scanning calorimetry thermograms of low molecular weight heparin in the optimized formulation ((a) pure drug; (b) optimized formulation; (c) physical mixture)
CONCLUSIONS
The results of this study revealed that s.c. nanoparticular formulation developed here will be a suitable form for sustained delivery of LMWH. Currently, LMWH is administered subcutaneously or intravenously twice or thrice a day in the form of solution. This could be conveniently avoided with the formulations we developed in our study. This is definitely a new observation and will have a tremendous impact for the therapy in all the diseases where LMWH is useful.
ACKNOWLEDGMENTS
The work is funded by Nanomission, DST Government of India. Dr. Jithan Aukunuru is the principal investigator of the project (SR/NM/NS-1118/2011). The authors would like to acknowledge the Management of Mother Teresa College of Pharmacy, Hyderabad for providing necessary facilities utilized in the conduction of the work. The authors would also like to acknowledge Synapse Life Sciences, Warangal for providing facilities utilized in conduction of animal studies and Osmania University for providing facilities utilized in conduction of analytical work.
Footnotes
Source of Support: Funded by Nanomission, DST Government of India
Conflict of Interest: Nil.
REFERENCES
- 1.Hirsh J, Warkentin TE, Shaughnessy SG, Anand SS, Halperin JL, Raschke R, et al. Heparin and low molecular weight heparin mechanisms of action, pharmacokinetics, dosing, monitoring, efficacy and safety. Chest. 2001;119:64S–94. doi: 10.1378/chest.119.1_suppl.64s. [DOI] [PubMed] [Google Scholar]
- 2.McRae S. Treatment options for venous thromboembolism: Lessons learnt from clinical trials. Thromb J. 2014;12:27. doi: 10.1186/s12959-014-0027-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hwang SR, Seo DH, Al-Hilal TA, Jeon OC, Kang JH, Kim SH, et al. Orally active desulfated low molecular weight heparin and deoxycholic acid conjugate, 6ODS-LHbD, suppresses neovascularization and bone destruction in arthritis. J Control Release. 2012;163:374–84. doi: 10.1016/j.jconrel.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 4.Patel B, Gupta N, Ahsan F. Low-molecular-weight heparin (LMWH)-loaded large porous PEG-PLGA particles for the treatment of asthma. J Aerosol Med Pulm Drug Deliv. 2014;27:12–20. doi: 10.1089/jamp.2013.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hornsby LB, Sutter D, Sexton K, Eckley M, Durhan S. A review of suggested anti-cancer benefits of warfarin, low-molecular weight heparin and aspirin. ClotCare Online Resource. [Last accessed on 2015 Jan 22]. Available from: http://www.clotcare.com/anticancer_benefits_warfarin_lmwh_aspirin.aspx .
- 6.Paliwal R, Paliwal SR, Agrawal GP, Vyas SP. Recent advances in search of oral heparin therapeutics. Med Res Rev. 2012;32:388–409. doi: 10.1002/med.20217. [DOI] [PubMed] [Google Scholar]
- 7.Lanke SS, Gayakwad SG, Strom JG, D'souza MJ. Oral delivery of low molecular weight heparin microspheres prepared using biodegradable polymer matrix system. J Microencapsul. 2009;26:493–500. doi: 10.1080/02652040802465719. [DOI] [PubMed] [Google Scholar]
- 8.Bai S, Thomas C, Ahsan F. Dendrimers as a carrier for pulmonary delivery of enoxaparin, a low-molecular weight heparin. J Pharm Sci. 2007;96:2090–106. doi: 10.1002/jps.20849. [DOI] [PubMed] [Google Scholar]
- 9.Choubey AK, Dora CP, Bhatt TD, Gill MS, Suresh S. Development and evaluation of PEGylated Enoxaparin: A novel approach for enhanced anti-Xa activity. Bioorg Chem. 2014;54:1–6. doi: 10.1016/j.bioorg.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Kim TD, Sakon M, Kawasaki T, Kambayashi J, Ohshiro T, Mori T. Studies on liposome-encapsulated heparin. Thromb Res. 1986;43:603–12. doi: 10.1016/0049-3848(86)90097-6. [DOI] [PubMed] [Google Scholar]
- 11.Köse GT, Arica MY, Hasirci V. Low-molecular-weight heparin-conjugated liposomes with improved stability and hemocompatibility. Drug Deliv. 1998;5:257–64. doi: 10.3109/10717549809065756. [DOI] [PubMed] [Google Scholar]
- 12.Kolandaivelu K, Swaminathan R, Gibson WJ, Kolachalama VB, Nguyen-Ehrenreich KL, Giddings VL, et al. Stent thrombogenicity early in high-risk interventional settings is driven by stent design and deployment and protected by polymer-drug coatings. Circulation. 2011;123:1400–9. doi: 10.1161/CIRCULATIONAHA.110.003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonepally CR, Gandey SJ, Bommineni K, Gottumukkala KM, Aukunuru J. Preparation, characterization and in vivo evaluation of sylibin nanoparticles for the treatment of Liver Fibrosis. Trop J Pharm Res. 2013;12:1–6. [Google Scholar]
- 14.Elthuri I, Bonepally CR, Kokkula S, Thadkapally R, Aukunuru J. Preparation and optimization of Kupffer cell targeted catechin loaded spherical particles. Turk J Pharm Sci. 2013;10:35–7. [Google Scholar]
- 15.Bajpai AK, Bhanu S. Dynamics of controlled release of heparin from swellable crosslinked starch microspheres. J Mater Sci Mater Med. 2007;18:1613–21. doi: 10.1007/s10856-007-3020-y. [DOI] [PubMed] [Google Scholar]
- 16.Sridhar A, Jithan AV, Malla Reddy V. Comparative pharmacokinetics of free and liposome-encapsulated catechin after intravenous and intraperitoneal administration. Int J Pharm Sci Nanotechnol. 2008;1:152–9. [Google Scholar]
- 17.Konatham S, Nyathani HK, Bonepally CR, Yannamaneni PK, Aukunuru J. Liposomal delivery of curcumin to liver. Turk J Pharm Sci. 2010;7:89–98. [Google Scholar]
- 18.Thadkala K, Nanam PK, Rambabu B, Sailu C, Aukunuru J. Preparation and characterization of amorphous ezetimibe nanosuspensions intended for enhancement of oral bioavailability. Int J Pharm Investig. 2014;4:131–7. doi: 10.4103/2230-973X.138344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sadat Tabatabaei Mirakabad F, Nejati-Koshki K, Akbarzadeh A, Yamchi MR, Milani M, Zarghami N, et al. PLGA-based nanoparticles as cancer drug delivery systems. Asian Pac J Cancer Prev. 2014;15:517–35. doi: 10.7314/apjcp.2014.15.2.517. [DOI] [PubMed] [Google Scholar]
- 20.Pandey R, Khuller GK. Subcutaneous nanoparticle-based antitubercular chemotherapy in an experimental model. J Antimicrob Chemother. 2004;54:266–8. doi: 10.1093/jac/dkh260. [DOI] [PubMed] [Google Scholar]
- 21.Xu XM, Wang YS, Chen RY, Feng CL, Yao F, Tong SS, et al. Formulation and pharmacokinetic evaluation of tetracycline-loaded solid lipid nanoparticles for subcutaneous injection in mice. Chem Pharm Bull (Tokyo) 2011;59:260–5. doi: 10.1248/cpb.59.260. [DOI] [PubMed] [Google Scholar]
- 22.Harivardhan Reddy L, Sharma RK, Chuttani K, Mishra AK, Murthy RS. Influence of administration route on tumor uptake and biodistribution of etoposide loaded solid lipid nanoparticles in Dalton's lymphoma tumor bearing mice. J Control Release. 2005;105:185–98. doi: 10.1016/j.jconrel.2005.02.028. [DOI] [PubMed] [Google Scholar]