

Abstract
A series of thiol-Michael and radical thiol-ene network polymers were successfully prepared from ester-free as well as ester-containing monomer formulations. Polymerization reaction rates, dynamic mechanical analysis, and solvent resistance experiments were performed and compared between compositions with varied ester loading. The incorporation of ester-free alkyl thiol, vinyl sulfone and allylic monomers significantly improved the mechanical properties when compared with commercial, mercaptopropionate-based thiol-ene or thiol-Michael networks. For polymers with no hydrolytically degradable esters, glass transition temperatures (Tg's) as high as 100 °C were achieved. Importantly, solvent resistance tests demonstrated enhanced stability of ester-free formulations over PETMP-based polymers, especially in concentrated basic solutions. Kinetic analysis showed that glassy step-growth polymers are readily formed at ambient conditions with conversions reaching 80% and higher.
Keywords: thiol-ene reaction, thiol-Michael reaction, hydrolytic degradation, DMA
Introduction
Over the past decade most of the research in the thiol-ene and thiol-Michael chemistry has been focused on the development of new types of polymeric materials and applications including surface modification1,2, formation of networks/hydrogels with unique properties3-5, polymer functionalization6,7, photolitography8,9, nano-, and microparticle synthesis10, 11, and dental composites12,13, to name a few. Given attributes such as high reaction efficiency at mild conditions, these processes have often been described as ‘click’ reactions. There are a number of comprehensive reviews on the topic covering a broad range of thiol-X reaction implementations.14-17
Thiol-ene and thiol-Michael crosslinking polymerizations proceed via a step-growth mechanism, generally to high functional group conversions. The step-growth mechanism results in lower shrinkage stress and uniform polymer networks because gelation occurs at later conversions as compared to conventional multi(meth)acrylate systems,18 which was the motivation for implementing thiol-enes into ternary thiol-ene-methcarylate dental resin mixtures.12, 19, 20
Often, to achieve the desired performance, such as in the above example of ternary dental formulations, thiol-ene reactions are used in tandem with other types of sequential, simultaneous or orthogonal reactions. Many examples point out the benefits of hybrid networks based on thiol-ene reactions in combination with thiol-yne21, 22, thiol-epoxy23, 24 and thiol-isocyanate25, 26 reactions, and also other chemistries involving e.g. siloxanes27 or benzoxazines28. It is not surprising that thiol-ene reactions dominate in many material science areas.
The term thiol-ene generally refers to the radical-mediated reaction of thiols with a wide range of unsaturated vinyl monomers (e.g., thiol-ene photopolymerizations). Radical thiol-ene photopolymerizations have the benefits of typical photopolymerization reactions; spatial and temporal control, rapid polymerization rates, easy to cure in mild reaction conditions, and the ability for solventless processing.29 In contrast, the thiol-Michael addition reaction proceeds via an anionic step-growth mechanism, and it is catalyzed by a base or nucleophile. In addition, it is not oxygen inhibited, and it can be implemented with a variety of monomers: methacrylates, vinyl sulfones, acrylamides, maleimides etc.16 As the thiols can react with many types of double bonds following either one of the two different mechanisms, thiol-X polymerizations exhibit great versatility in their chemistry.
For both of these reactions, multifunctional thiols are typically synthesized via esterification reactions from readily available precursors (multifunctional alcohols and acids such as mercaptopropionic or mercaptoacetic acid). Due to this facile process, most of the commercially available thiols are mercaptopropionates or thioglycolates.
Due to the ester content in their structures these monomers and the resulting polymers are readily hydrolyzed in either basic or acidic solutions. The hydrolysis process diminishes the materials' properties over time and ultimately leads to mechanical failure.30, 31 Further, the polarity of the ester group and its affinity to water increases water sorption. Swelling in water (or other aqueous solutions) decreases the mechanical performance by lowering the modulus, glass transition and strength. Often, neat thiol-ene network materials are soft materials, and only a few thiol-ene compositions exhibit glass transition temperatures that are significantly higher than ambient.
Recently, we showed that the rubbery nature of the thiol-Michael systems is not simply an artifact of the low crosslinking density of these materials or the high content of thioethers formed but it is also caused by the ester presence.32 Eliminating the ester moieties from the monomers, and by that from final network structure, leads to a considerable increase in the materials' Tg. We also postulated that ester free thiol-ene or thiol-Michael networks should exhibit improved hydrolytic degradation resistance. Reducing hydrolytic degradation is of great biological significance for thiol-ene materials since the leachable components may be eliminated and the overall biocompatibility improved.
In the present report we extended the scope of thioether forming reactions to ester-free radical thiol-ene polymerizations. Thiol-Michael as well as radical thiol-ene polymer networks containing no interchain ester moieties were assessed for curing rates, solvent resistance, and relevant mechanical properties. To achieve this aim, we synthesized an alkyl thiol i.e. tetra(2-mercaptoethyl)silane and used it in thiol-X reactions with divinyl sulfone (DVS), triallyl-1,3,5-triazine-2,4,6-(1H,3H,5H)-trione (TTT) and trimethylolpropane triacrylate (TMPTA). For comparison, we prepared and assessed analogue ester-based networks from a mercaptopropionate tetrathiol, namely pentaerythritol tetra(3-mercaptopropionate) (PETMP).
Experimental
Materials
Trimethylolpropane triacrylate (TMPTA), triethylamine (TEA) triallyl-1,3,5-triazine-2,4,6-(1H,3H,5H)-trione (TTT), 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) were purchased from Sigma-Aldrich. Divinyl sulfone (DVS) was purchased from Oakwood Chemicals. Irgacure 819 (Bis(2,4,6-trimethylbenzoyl)-phenylphosphineoxide–BPO) was obtained from BASF. Pentaerythritol tetra(3-mercaptopropionate) (PETMP) was donated by Bruno Bock. All chemicals were used as received. Tetra(2-mercaptoethyl)silane (SiTSH) was synthesized according to previously reported procedure.33-35 The structures of the thiol and vinyl monomers are depicted in Figure 1.
Fig. 1.
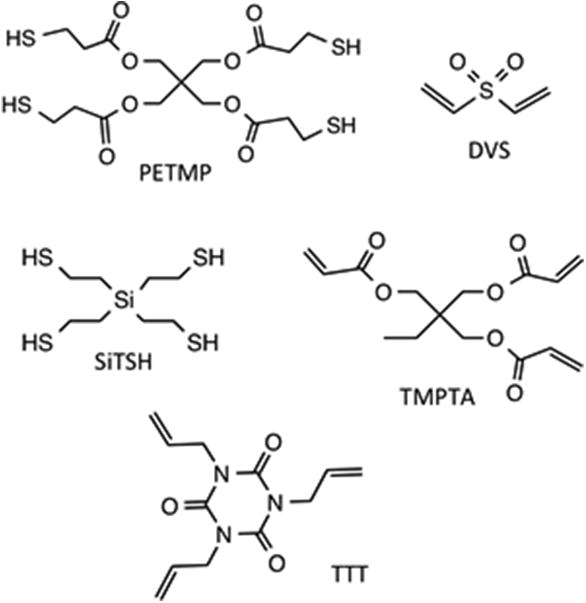
Chemical structures and abbreviations for the thiol and vinyl monomers utilized in this study.
Monomer Synthesis
The ester-free thiol, tetra(2-mercaptoethyl)silane (SiTSH) was synthesized by a two-step methodology which was previously reported in the literature.35 The synthesis starts with a radical thiol-ene step followed by an acetyl deprotection step. To a flask with tetravinyl silane (10.0 g, 73 mmol), thioacetic acid (28.8 g, 378 mmol) was added slowly in an ice-bath. After addition of azobisisobutylronitrile (AIBN) (0.776 g), the solution was stirred at 65 °C for 36 h. After the thiol-ene reaction was complete, the excess amount of thioacetic acid was distilled in vacuo at 65 °C. No further purification was performed on the crude product from the first step. Methanol (50 mL) and hydrochloric acid (20 mL) were added to a flask with the crude product from the first step. The solution was stirred at 60 °C for 12 h. After hydrolysis was completed the solvent was distilled and the product washed twice with 5 wt.% sodium bicarbonate solutions. Finally the crude product was purified by column chromatography (silica gel, DCM/Hexane 1:2) to give a colorless liquid. 1H-NMR (400 MHz, CDCl3, ppm): δ: 2.74 – 2.53 (m, 2H), 1.60 (t, 1H, J=6.9Hz), 1.18-1.00 (m, 2H).
Characterization
Fourier Transform Infrared Spectroscopy (Nicolet 6700 FT-IR) combined with a vertical light source was utilized to measure the real-time conversion during curing. The thiol-ene samples were cured in the FTIR chamber using an irradiation intensity of 50 mW/cm2 (400-500nm) at the surface of the sample which is monitored by a radiometer (model IL1400A equipped with a GaAsP detector and a quartz diffuser). To couple with various mechanical property measurements, near-IR was utilized to evaluate the functional group conversions in polymerizations of 1 mm (DMA, solvent resistance and kinetic analysis) and/or 0.2 mm (kinetic analysis only) thick samples sandwiched between glass slides separated by appropriate spacers. The first C=C overtone signal at 6160 cm-1 was monitored during all real-time kinetic runs. Also, the signal of the thiol group at 2560 cm-1 was monitored for some of the samples to confirm a stoichiometric pathway of thiol-Michael reaction. For thermal curing a heating stage was utilized which allows for real-time runs at predetermined temperature. Basically, the sample surrounding temperature was gradually increased in the chamber until the final temperature was achieved. The runs were recorded from the moment the samples were placed in the heating compartment. The final temperature was set to be 90 °C.
Solvent resistance tests were performed using disc-shaped specimens (n=3) of 1mm in thickness and 5mm in diameter. The curing was performed in the IR chamber according to the methodology described above. During the immersion tests the sample weight was recorded on a daily, then weekly, and then monthly basis over the maximum period of three months. 10 % NaOHaq and 10 % HClaq were chosen as the hydrolytic degradation media.
Dynamic mechanical analysis (DMA) is used to measure the viscoelastic properties of polymers (glass transition temperature, rubbery storage modulus, tan delta etc). A DMA Q800 (TA Instruments) and was utilized in these measurements. Sample specimens with 1×4×10 mm rectangular dimensions were tested in multifrequency strain mode by applying a sinusoidal stress of 1 Hz frequency with the temperature ramping at 3 °C/min. The Tg was determined as the maximum of the tan delta curve. The rubbery moduli were determined in the rubbery region at Tg + 30 °C. Tg half widths were taken as the half width of the tan delta peak at half maximum values.
The monomer and resin viscosities were measured on a TA instruments ARES rheometer. The liquids were placed between 20 mm glass plates with a gap spacing of 0.4 mm. The reported values were recorded at shear rate of 63 s-1.
Results and discussion
Resin Characterization
Pentaerythritoltetra(3-mercaptopropionate) (PETMP) is the most commonly used thiol ester derivative in many crosslinking thiol-ene or thiol-Michael reactions. Most other commercially available thiol monomers are either thioacetates or other thiopropionates. Their use does not allow for a control in the ester loading through variations in monomer structure or monomer selection. Here, we prepared six thiol-ene/thiol-Michael mixtures with varied ester content to assess the resin curing behavior and the resulting materials' mechanical properties and hydrolytic stability. For the latter, solvent resistance tests in 10 % acidic (HCl) and 10 % basic (NaOH) solutions over the period of three months were performed.
To enable systematic adjustment of the ester content, we synthesized an ester-free tetrathiol using tetravinyl silane and thioacetic acid as the starting materials in an adopted synthetic procedure from the work reported by Lundberg et al. and Suzuki et al.33, 34 The synthesized tetra(2-mercaptoethyl)silane is an alkyl thiol of relatively low molecular weight (272.2 g/mol) which is approximately half of the PETMP molecular weight. Importantly, it exhibits one order of magnitude lower viscosity than PETMP (Table 1) which should facilitate composite development and processing of mixtures prior to and during polymerization. As seen from Table 1, all formulations containing SiTSH exhibit significantly lower viscosities in pair-wise comparisons with PETMP.
Table 1.
The ambient temperature viscosity of the thiol monomers as well as the formulated thiol resin compositions. Standard deviations are included in the brackets.
| Monomer/Formulation | Viscosity (mPa·s) |
|---|---|
| SiTSH | 65 (1) |
| PETMP | 369 (1) |
| SiTSH/DVS | 8.7 (0.3) |
| PETMP/DVS | 30.2 (0.5) |
| SiTSH/TMPTA | 65 (2) |
| PETMP/TMPTA | 184 (1) |
| SiTSH/TTT | 96 (3) |
| PETMP/TTT | 332 (3) |
Reaction Rates and Conversion Analysis
PETMP and SITSH were both stoichiometrically reacted in thiol-Michael reactions with TMPTA and DVS as well as in radical thiol-ene reactions with TTT. Therefore, from six different monomer systems, two i.e. SITHS/DVS and SITSH/TTT contained no interchain ester linkages at all. On the other hand, the highest ester content exists for the PETMP/TMPTA system. Regardless of the reaction type, the polymeric samples were cured in pairs (and/or postcured) in the same way to enable unbiased data comparison. To prepare the thiol-Michael polymers, TEMPO (1wt%) was used as a base/nucleophile initiator and the reactions were carried out at 90°C. The use of a radical scavenger, i.e., TEMPO, to initiate anionic polymerization may seem counterintuitive, but the kinetic analysis confirms a stoichiometric reaction (Fig.2c) between thiols and double bonds. It has been reported that TEMPO abstracts hydrogen from protic species, and the thermal decay of TEMPO may involve hydroxylamine generation,36 which in this case would serve as the necessary base to accelerate the thiol-Michael reaction. Though it is used successfully here to initiate the thiol-Michael pathway, thermal initiation with TEMPO, in particular its detailed decomposition pathway and initiation mechanism, require more detailed investigation. The radical thiol-ene systems were cured with the visible light initiator BPO (1 wt%) at ambient conditions. The relevant kinetic plots for thermally and photochemically initiated polymerizations are depicted in Fig 2. Maximal reaction rates obtained between 10-40 % double bond conversions are summarized in the Supporting Information Table S1. Comparing the thiol and double bond consumption rate for the SiTSH/TMPTA system in Fig. 2c, it can be seen that the monomers react stoichiometrically when TEMPO is used and the reaction is initiated thermally. The maximal double bond consumption rate is one order of magnitude faster for the silane thiol/acrylate mixture, indicating higher reactivity of SiTSH when compared to PETMP (Table S1). The thiol-Michael reactions are even more rapid when DVS is used as the Michael acceptor (Fig. 2b, Table S1). It should be mentioned here that the silane thiol-divinyl sulfone reaction was far too rapid to yield a uniformly crosslinked polymer even when initiated with weak nucleophiles/bases such as triphenylphosphine and triethylamine. The rapidness of the SiTSH/DVS reaction was the reason for the eventual use of TEMPO. It is therefore possible that TEMPO acts as a very weak base, and the elevated temperature speeds up the polymerization. Supportive of such a conclusion, a slow reaction resulting in ultimate gelation over several hours occurred when the SiTSH/DVS TEMPO-containing mixture was shelf-stored at ambient temperature.
Fig. 2.
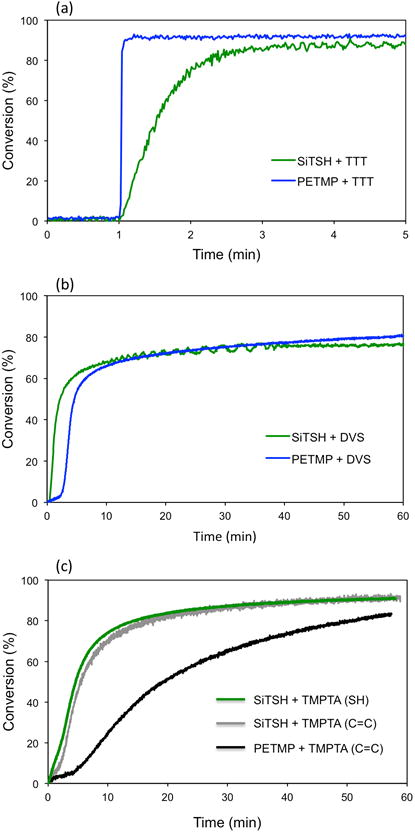
Real time kinetic profiles (C=C conversions) for the: (a) radical thiol-ene reaction between SiTSH and PETMP with TTT; (b) thiol-Michael reaction between SiTSH and PETMP with DVS and (c) thiol-Michael reaction between SiTSH and PETMP with TMPTA. The radical thiol-ene processes reach high conversions after 4 min of irradiation with visible light (400-500nm) at 50 mW/cm2. It is evident that the alkyl thiol reacts with lower initial rates. The thiol-Michael reaction is initiated thermally with 1 wt% TEMPO, and longer reaction times are required to reach high conversions.
Fairly high conversions were achieved in the tested thiol-Michael formulations which usually exceeded 80% after one hour in the heating compartment in the IR instrument. On the other hand the silane thiol, being an alkyl thiol, reacts with lower reaction rates in radical thiol-ene polymerization (0.072 mmol×s-1 as opposed to 0.386 mmol×s-1 for PETMP/TTT) but despite that it can be reacted to high conversions over relatively short time. Further, after thermal annealing of the samples at 100 °C for an additional hour, near-quantitative conversions were achieved (see Table 2).
Table 2.
Summary of the mechanical, and solvent resistance properties of the six tested systems. Maximal conversions after thermal annealing are also included. 10 % HClaq and 10 % NaOHaq were selected as the media for hydrolytic tests. +/- at each value stands for sample mass gain/loss. Brackets show standard deviations values.
| Resin | Properties | ||||
|---|---|---|---|---|---|
|
| |||||
| Ester groups (mol %) | Tg (°C) | Conv. (%) | wt% Loss/Gain (10 wt% HClaq) | wt% Loss/Gain (10wt% NaOHaq) | |
| SiTSH/TTT | 0 | 95 (6) | 84 (2) | +0.4 (0.1) | + 0.5 (0.1) |
| PETMP/TTT | 0.49 | 79 (1) | 93 (1) | +0.7 (0.1) | -8 (2) |
| SiTSH/TMPTA | 0.59 | 35 (3) | 99 (1) | +0.4 (0.1) | -11 (2) |
| PETMP/TMPTA | 0.91 | 21 (1) | 96 (1) | -13 (1) | Degraded after 7 days |
| SiTSH/DVS | 0 | 64 (8) | 99 (1) | +0.7 (0.2) | +0.7 (0.1) |
| PETMP/DVS | 0.55 | 44 (3) | 98 (1) | -4.0 (0.1) | -41 (4) |
Dynamic Mechanical Analysis
Viscoelastic behavior was analyzed by DMA on thermally postcured samples (Fig. 3). All formulations created fairly homogenous networks, as indicated by the narrow tan delta peak widths (Tg1/2width ∼ 20-30 °C). This outcome was expected as these are step-growth systems. Interestingly, the networks containing SiTSH achieved significantly higher glass transition temperatures and rubbery moduli (i.e., crosslinking density) compared to using PETMP. This result is explained by better crosslinking capability of the low molecular weight silane tetrathiol as well as the reductions in ester content in SiTSH-containing formulations. In our previous report we showed that incorporating sulfone groups in the network lead to better mechanical response of the thiol-Michael materials.32 Also, in this case the DVS-derived networks exhibit higher Tg's than the networks based on TMPTA. However, the highest glass transition temperatures were achieved for the two thiol-ene systems, most probably due to the highly crosslinked structure of these, and the rigid segments provided by the TTT monomer. It is worth noting that depending on the conversions, Tg's exceeding 100 °C are obtained for SiTSH/TTT composition and importantly, this performance was achieved without the use of excessively viscous resin mixtures. Some recent reports show improvements in mechanical properties, such as fracture toughness or modulus of elasticity, in thiol-ene materials devoid of ester functionalities. For example to achieve better flexural strength ester free multifunctional thiols derived from an array of bisphenol structures were used.37 Such materials showed also an improved performance in relation to PETMP/TTT after conditioning in water for 24 hours. Other reports described the use of ester-free thiol-ene materials as dental resins.38 Reduced shrinkage stress and again improved toughness were emphasized. Therefore, the silane thiol presented here seems to be a promising crosslinking candidate for coating, lithography and dental resin applications as it allows for the generation of glassy thiol-ene systems even when the curing is performed at ambient conditions. Its additional advantage, besides the absence of relatively “soft” and hydrophilic ester groups, is very low viscosity which should help to increase the filler loading in any composite material.
Fig. 3.
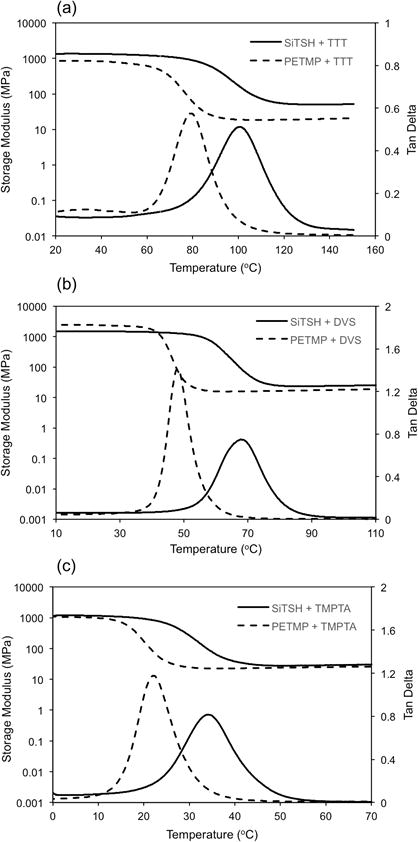
Storage modulus and loss tangent (tan delta) plots for (a) SiTSH/TTT and PETMP/TTT radical thiol-ene networks; (b) SiTSH/DVS and PETMP/DVS thiol-vinyl sulfone networks and (c) SiTSH/TMPTA and PETMP/TMPTE thiol-acrylate networks. It is evident from the curves that the higher ester content present in the network, the softer the material. The SiTSH-based polymers exhibit exclusively higher Tg's as compared to PETMP-based polymers.
Solvent Resistance Tests
To assess the solvent resistance and hydrolytic stability of the networks with varying ester levels, samples were immersed in acidic and basic solutions for 90 days (Table 2, Figs. S1 and S2). As summarized in Table 2 and also in Supporting Information Table S2 and S3, only ester-free thiol formulations exhibit less than 1% mass change regardless of the environment type. The higher the ester concentration in the network, the higher the mass loss that is observed, especially in basic solutions. The most ester-rich PETMP/TMPTA system degraded completely after less than one week in concentrated basic solvent, and has the highest mass loss in acidic conditions after 90 days. Further, it can be seen that lowering the ester content decelerates the hydrolysis rate as the differences in solvent uptake become less pronounced. Also apparent is the higher hydrophilicity of the DVS containing networks which absorb more water, facilitating faster degradation of the ester-containing networks. Detailed plots showing the specimen' mass change (Δm) over time can be found in the Supporting Information (Figs. S1 and S2 and Table S2 and S3). Additionally, we immersed the SiTSH/TTT composition in 20% boiling NaOH aqueous solution for one hour and afterwards tested the sample in DMA (Fig. 4). Comparing the samples before and after the NaOH treatment it is evident that there is no marked difference between the runs whatsoever. This behavior is therefore quite promising in all applications where the corrosive base effects have to be prevented. Decreased water uptake/swelling is also of significance for many other coating, lithography and dental resin applications.
Fig. 4.
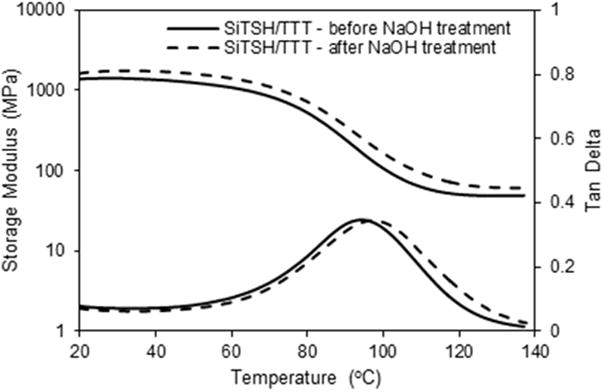
Dynamic mechanical analysis of the SiTSH/TTT ester-free system, before (solid line), and after (dashed line) base treatment (20 wt% NaOHaq) at boiling conditions for one hour. No obvious change in the mechanical response is detected before and after base treatment.
Conclusion
In our study we showed that even conventional crosslinking step-growth systems based on efficient thiol-X reactions can result in glassy materials when the ester functionalities are removed or significantly reduced in content. Uniform network polymers were readily prepared with impressive conversions, which is one of the attributes of step-growth systems that also holds in this case. More importantly, step-growth polymer networks devoid of ester groups are an excellent class of materials resistant to corrosive basic or acidic conditions. Ester-free thiol-ene networks were shown to withstand concentrated basic treatment for an extended amount of time without any detrimental effects observed in the thermo-mechanical properties.
Supplementary Material
Acknowledgments
The authors acknowledge the National Institute of Health (1U01DE023777-01) for providing funding for this research.
References
- 1.Harant AW, Khire VS, Thibodaux MS, Bowman CN. Macromolecules. 2006;39:1461–1466. [Google Scholar]
- 2.Sparks BJ, Hoff EFT, Xiong L, Goetz JT, Patton DL. ACS Appl Mater Interfaces. 2013;5:1881–1817. doi: 10.1021/am303165e. [DOI] [PubMed] [Google Scholar]
- 3.Gupta N, Lin BF, Campos LM, Dimitriou MD, Hikita ST, Treat ND, Tirrell MV, Clegg DO, Kramer EJ, Hawker CJ. Nature Chem. 2010;2:138–145. doi: 10.1038/nchem.478. [DOI] [PubMed] [Google Scholar]
- 4.Kade MJ, Burke DJ, Hawker CJ. J Polym Sci, Part A: Polym Chem. 2010;48:743–750. [Google Scholar]
- 5.Schreck KM, Leung D, Bowman CN. Macromolecules. 2011;44:7520–7529. doi: 10.1021/ma201695x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koo SP, Stamenović MM, Prasath RA, Inglis AJ, Du Prez FE, Barner-Kowollik C, Van Camp W, Junkers T. J Polym Sci, Part A: Polym Chem. 2010;48:1699–1713. [Google Scholar]
- 7.Connal LA, Kinnanae CR, Zelkin AN, Caruso F. Chem Mater. 2009;21:576–578. [Google Scholar]
- 8.Polizzotti BD, Fairbanks BD, Anseth KS. Biomacromolecules. 2008;9:1084–1087. doi: 10.1021/bm7012636. [DOI] [PubMed] [Google Scholar]
- 9.Besson E, Gue AM, Sudor J, Korri-Youssoufi H, Jaffrezic N, Tardy J. Langmuir. 2006;22:8346–8352. doi: 10.1021/la053303l. [DOI] [PubMed] [Google Scholar]
- 10.Amato DV, Amato DN, Flynt AS, Patton DL. Polym Chem. 2015 doi: 10.1039/C4PY01449A. [DOI] [Google Scholar]
- 11.Wang C, Podgórski M, Bowman CN. Mater Horiz. 2014;1:535–539. [Google Scholar]
- 12.Beigi S, Yeganeh H, Atai M. Dent Mater. 2013;29:777–787. doi: 10.1016/j.dental.2013.04.015. [DOI] [PubMed] [Google Scholar]
- 13.Carioscia JA, Lu H, Stansbury JW, Bowman CN. Dent Mater. 2005;12:1137–1143. doi: 10.1016/j.dental.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 14.Lowe AB. Polym Chem. 2014;5:4820–4870. [Google Scholar]
- 15.Hoyle CE, Bowman CN. Angew Chem Int Ed. 2010;49:1540–1573. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 16.Nair DP, Podgoŕski M, Chatani S, Gong T, Xi W, Fenoli CR, Bowman CN. Chem Mater. 2014;26:724–744. [Google Scholar]
- 17.Lowe AB. Polym Chem. 2010;1:17–36. [Google Scholar]
- 18.Kloxin CJ, Bowman CN. AIChE Journal. 2008;54:2775–2795. [Google Scholar]
- 19.Cramer NB, Couch CL, Schreck KM, Boulden JE, Wydra R, Stansbury JW, Bowman CN. Dent Mater. 2010;26:799–806. doi: 10.1016/j.dental.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boulden JE, Cramer NB, Schreck KM, Couch CL, Bracho-Troconis C, Stansbury JW, Bowman CN. Dent Mater. 2011;27:267–272. doi: 10.1016/j.dental.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prasath RA, Gokmen MT, Espeel P, Du Prez FE. Polym Chem. 2010;1:685–692. [Google Scholar]
- 22.Lovelady E, Kimmins SD, Wu J, Cameron NR. Polym Chem. 2011;2:559–562. [Google Scholar]
- 23.Flores M, Tomuta AM, Fernández-Francos X, Ramis X, Sangermano M, Serra A. Polymer. 2013;54:5473–5481. [Google Scholar]
- 24.Sangermano M, Roppolo I, Ortiz RA, Tovar AGN, Valdez AEG, Duarte MLB. Prog Org Coat. 2015;78:244–248. [Google Scholar]
- 25.Shin J, Matsushima H, Comer CM, Bowman CN, Hoyle CE. Chem Mater. 2010;22:2616–2625. [Google Scholar]
- 26.Li Q, Zhou H, Wicks DA, Hoyle Ch E. J Polym Sci, Part A: Polym Chem. 2007;45:5103–5111. [Google Scholar]
- 27.Cole MA, Bowman CN. J Polym Sci, Part A: Polym Chem. 2012;50:4325–4333. doi: 10.1002/pola.26245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narayanan J, Jungman MJ, Patton DL. React Funct Polym. 2012;72:799–806. [Google Scholar]
- 29.Cramer NB, Scott JP, Bowman CN. Macromolecules. 2002;35:5361–5365. [Google Scholar]
- 30.Ferracane JL. Dent Mater. 2006;22:211–222. doi: 10.1016/j.dental.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 31.Rydholm AE, Bowman CN, Anseth KS. Biomaterials. 2005;26:4495–4506. doi: 10.1016/j.biomaterials.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 32.Podgórski M, Chatani S, Bowman CN. Macromol Rapid Commun. 2014;35:1497–1502. doi: 10.1002/marc.201400260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lundberg P, Bruin A, Klijnstra JW, Nystrom AM, Johansson M, Malkoch M, Hult A. ACS Appl Mater Interfaces. 2010;2:903–912. doi: 10.1021/am900875g. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki, Higashihara T, Ando S, Ueda M. Macromolecules. 2012;45:3402–3408. [Google Scholar]
- 35.Podgórski M, Nair DP, Chatani S, Berg G, Bowman CN. ACS Appl Mater Interfaces. 2014;16:6111–6119. doi: 10.1021/am405371r. [DOI] [PubMed] [Google Scholar]
- 36.Ma Y, Loyns C, Price P, Chechik V. Org Biomol Chem. 2011;9:5573–5578. doi: 10.1039/c1ob05475a. [DOI] [PubMed] [Google Scholar]
- 37.Reinelt S, Tabatabai M, Fisher UK, Moszner N, Utterodt A, Ritter H. Beilstein J Org Chem. 2014;10:1733–1740. doi: 10.3762/bjoc.10.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reinelt S, Tabatabai M, Moszner N, Fisher UK, Utterodt A, Ritter H. Macromol Chem Phys. 2014;215:1415–1425. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.