

Abstract
Objective
To assess the activity of non-lysosomal proteolytic systems in skeletal and cardiac muscle during burn induced hypermetabolism in rats.
Methods
Rats underwent 30% TBSA scald burn or sham injury and were observed for up to 42 days. Body weights and resting energy expenditures (REE) were determined weekly. Skeletal (soleus/pectoral) muscle and hearts were harvested on days 0 (=control), 7, 14, 21 and 42 post burn. Calpain, caspase-1, -3/7, -6, -8, -9 and proteasome peptidase activities were measured in tissue extracts.
Results
Hypermetabolism developed within 3 weeks after burns, as documented by increased REEs and decreased body weights on post burn days 21–42 (p<0.05 vs. control). Calpain activities did not show significant alterations. Pan caspase activities increased by time and were significantly elevated in skeletal and cardiac muscle extracts during hypermetabolism. Whereas increases in caspase-1, caspase-8 and caspase-9 activities were predominantly responsible for elevated pan caspase activities in skeletal muscle, increases in caspase-6 activities dominated in the heart. Proteasome peptidase activities in skeletal muscle extracts were not significantly altered. Proteasome peptidase activities in heart extracts increased time dependently and were significantly increased during burn induced hypermetabolism.
Conclusions
Activation of caspase cascades during burn induced hypermetabolism constitutes a uniform response in skeletal and cardiac muscle and may contribute to enhanced metabolic protein turnover. Activation of myocardial proteasome activities may reflect persistent cardiac stress. Further exploration of caspase cascades and the proteasome as therapeutic targets to influence long term consequences of burn induced hypermetabolism appears justified.
Keywords: Caspase, calpain, proteasome, resting energy expenditure, muscle catabolism
INTRODUCTION
Severe burn injury induces hypermetabolism, which persists for months after complete wound closure (1–5). Depending on the degree of the burn injury, resting energy expenditure (REE) increases up to 180% of normal and can remain elevated for more than two years after injury (2, 6–8). Burn induced hypermetabolism results in muscle catabolism, which is associated with adverse outcomes and prolonged recovery periods (1, 2, 9). The molecular mechanisms that lead to and perpetuate burn induced hypermetabolism and muscle catabolism, however, are complex and not well understood (1). Anabolic agents such as oxandrolone, human growth hormone and insulin-like growth factor-1 have been used with some success in pediatric burn patients and in animal models of burn injury in an attempt to reverse muscle catabolism (10–15). Nonetheless, the mechanisms through which these therapeutic interventions attenuate the deleterious effects of hypermetabolism and muscle catabolism remain to be determined.
Studies on muscle protein turnover showed that protein degradation and synthesis are increased in burned patients (16). Because increases in protein degradation exceeded increases in protein synthesis, enhanced protein degradation appears as the primary effector of muscle catabolism (16). Whereas the regulation of major proteolytic systems in human muscle after burns is unknown, the majority of previous studies on the regulation of proteolysis in muscle after burns in animals have focused on the acute post burn period (17–23). Information on the temporal pattern of muscle protease activities after burns is scarce, and the activities of important proteolytic systems in muscle during established burn induced hypermetabolism are unknown. The understanding of the enzyme systems that may participate in the protein degradation process, however, is essential to develop improved strategies that attenuate the whole body protein loss and improve outcomes from severe burn injuries. Thus, it was the aim of the present study to assess the activities of major proteolytic systems in skeletal and cardiac muscle during burn induced hypermetabolism in rats.
The major proteolytic systems in muscle are comprised of the lysosomal system and the non-lysosomal calpain, caspase and proteasome systems. Lysosomal proteolysis, however, is unlikely to be responsible for a significant proportion of metabolic protein turnover in muscle under normal conditions and after burn injury (17, 18, 24–26). Thus, we focused our studies on non-lysosomal proteolytic systems and measured REE along with caspase, calpain and proteasome peptidase activities in skeletal and cardiac muscle extracts obtained from animals after 30% total body surface area (TBSA) dorsal scald burn throughout a six week observation period.
MATERIALS AND METHODS
Animal protocol
All procedures were performed according to NIH Guidelines for Use of Laboratory Animals and approved by the Loyola IACUC. Male Sprague Dawley rats (330 – 380 g body weight, Harlan, Indianapolis, IN) were anesthetized with ketamine 100 mg/kg i.p., 2.5% isoflurane inhalational, shaved and placed into a template that exposes a dorsal body area corresponding to 30% TBSA. A full thickness burn was then induced by immersion of the dorsal skin into boiling water for 25 seconds (27). We did not observe any evidence for thermal injury of the muscles adjacent to the dorsal burn injury on any day after burn during routine necropsy. Sham animals were anesthetized, shaved and their dorsal surface immersed in tepid water. Animals were then resuscitated with Lactated Ringers solution i.p. as per the Parkland formula (4 mL/kg/percent burn) over the first 24 h after burn injury. Throughout the observation period animals had free access to standard rat chow and water. Food consumption was quantified on days 1–3, 7, 14, 21 and 28 post burn. On days 7, 14, 21 and 42 after burn injury, animals (n=5/time point) were anesthetized (ketamine 100 mg/kg, xylazine 10 mg/kg) and cardioplegic arrest was achieved by injection of 10 mL of 4°C University of Wisconson solution (UW, Duramed) into the right ventricle (28, 29). The heart, ventral pectoral and soleus muscle were rapidly excised, snap frozen in liquid nitrogen and stored at −70°C until further processing, as described (28, 29). Tissues from sham control animals served as controls (= day 0).
Resting energy expenditure (REE)
REE was measured using indirect calorimetry, as described (14). Respiratory gas exchange was measured in an open-circuit respirometer (Columbus Instruments, OH). Food was withdrawn from morning until mid-afternoon (6 h) before measuring REE. Rats were placed in a plexiglas metabolic chamber (4 L capacity). Air inlets and outlets contained columns of calcium sulfate to dry both inlet and expired air. Airflow rate was monitored continuously for 10 min for 6 cycles (total of 60 min) and oxygen consumption and CO2 production were calculated by multiplying the rate of airflow by changes in O2 and CO2 concentrations of air entering and exiting the chamber. From these values, the differences in O2 intake (DO2), CO2 (DCO2) output and REE were calculated using the Oxymax software (Columbus Instruments, OH). REE was measured at baseline (day 0), on day 7 after burn or sham injury and weekly for 5 weeks thereafter.
Tissue extract preparation
Snap frozen tissues were homogenized in 1/10 phosphate buffered saline, pH 7.4 (1:5 weight/volume), centrifuged (16,600xg, 4°C, 30min) and supernatants (=extracts) aliquoted, as described (29, 30). Protein concentrations in the tissue extracts were determined using the DC protein assay (Bio-Rad, Hercules, CA). All measurements in tissue extracts were standardized to total protein content.
ATP assay
Tissues were homogenized in 1% TCA, centrifuged (2000xg, 10min, 4°C) and supernatants collected. Supernatants were assayed for ATP using a bioluminescence assay (Invitrogen), as described (29, 31).
Proteasome peptidase activity
Proteasome peptidase activities (=total peptidase activity minus activity in the presence of the specific proteasome inhibitor epoxomicin) in tissue extracts were measured employing the chymotryptic-like (CT-L) fluorogenic peptide substrate N-Suc-LLVY-7-amino-4-methylcoumarin (Enzo Life Sciences, Farmingdale, NY), as described (28–30). Incubation mixtures contained 50 μg of extract protein, 200 μM of the peptide substrate and ATP at the actual tissue concentration. All enzyme assays were performed immediately after preparation of the tissue extracts to prevent proteasome inactivation by freeze-thawing. Enzyme time progression curves showed linearity for 40 min.
Calpain peptidase activity
Calpain activity in tissue extracts (total activity minus activity in the presence of calpain inhibitor) was measured utilizing the BioVision Calpain Activity Fluorometric Assay Kit (Milpitas CA) as per manufacturer’s instructions. In brief, 50 μg of tissue extract protein in a volume of 85 μL, 10 μL of 10 x reaction buffer and 5 μL of fluorogenic calpain substrate (Ac-LLY-AFC) were incubated in the dark at 37°C in the presence and absence of 1 μL of the calpain inhibitor Z-LLY-FMK. Positive controls contained 1 μL of active calpain. After incubation, the fluorescence signal was read at λemission/excitation 485/528 nm in a Synergy 2 microplate reader (Biotek). Enzyme time progression curves showed linearity for 60 min. Calpain activities were calculated as relative fluorescence units per min and mg of extract protein.
Caspase peptidase activity
Caspase activities in tissue extracts were measured utilizing the SensoLyte AFC Caspase Sampler Fluorimetric Kit (Freemont, CA) as per manufacturer’s instructions. In brief, incubation mixtures contained 50 μg of extract protein, 0.5 mM caspase substrates (caspase-1 - Ac-YYVAD-7-amino-4-trifluoromethyl-coumarin (AFC), caspase-2 - Ac-VDVAD-AFC, caspase-3/7 - Ac-DEVD-AFC, caspase-6 - Ac-VEID-AFC, caspase-8 - Ac-IETD-AFC, caspase-9 - Ac-LEHD-AFC), and 10 mM DTT in caspase assay buffer (SensoLyte AFC Caspase Sampler Fluorimetric Kit). Mixtures were placed in a black clear bottom microplate and the fluorescence signals (λexcitation/emission 380/508 nm) were measured every 5 min for 1 hour at room temperature in a Synergy 2 microplate reader (Biotek). Enzyme time progression curves showed linearity for 60 min. Enzyme activities were calculated as relative fluorescence units per min and mg of extract protein and pan caspase activity was expressed as the sum of all individual caspase activities measured.
Statistical analyses
Data are described as mean ± SEM. Two-way analyses of variance (ANOVA) with Bonferroni post-tests were used for comparisons of REEs and body weights. Enzyme activities at the various time points after burns were analyzed by one-way ANOVA with Dunnett’s post-tests to compare each time point with the normal enzyme activity measured at day 0 (=control) and with post-tests for linear trend to evaluate time dependent changes. A two-tailed p < 0.05 was considered significant. Statistical analyses were calculated using GraphPad Prism 5 (GraphPad Software Inc.) software.
RESULTS
Resting energy expenditures (REEs) and body weights
All animals recovered from the burn injury and showed normal feeding and drinking behavior within seven days. After seven days, there were no differences in food consumption between rats after burn or sham injury (24–26 g/day for all animals, p>0.05; Fig. 1A). None of the animals developed wound infection after burns. REEs and body weights of animals after burn and sham injury are shown in Fig. 1B/C. REE after burns increased from 2.6 ± 0.1 kcal/h on day 0 to 3.1 ± 0.13 kcal/h on day 28 and remained unchanged thereafter (day 35: 3.1 ± 0.1 kcal/h; day 42: 3.1 ± 0.15 kcal/h). REE remained constant throughout the observation period in sham control animals (2.3 ± 0.1 – 2.6 ± 0.1 kcal/h). As compared with REEs of sham control animals, REEs were significantly elevated after burn injury on days 21 – 42.
Fig. 1.

Food consumption (g/day) (A), resting energy expenditures (REE, kcal/h) (B) and body weights (g) (C) after 30%TBSA burn. Grey squares: burn. Open circles: sham control. N=5–6 per group, mean ± SEM. *: p<0.05 vs. sham control.
Body weights of sham control animals increased from 355 ± 7 g on day 0 to 380 ± 9 g on day 42. In animals after burn injuries, body weights decreased from 360 ± 4 g on day 0 to 334 ± 7 g on day 21 and remained between 339 ± 5 – 343 ± 4 g on days 28 – 42. Compared with that of sham injured animals, body weights of animals after burn injury were significantly lower on days 21 – 42.
Calpain activities
There were no significant differences between calpain peptidase activities in control skeletal or cardiac muscle extracts and extracts from tissues obtained on any day after burns throughout the observation period (Fig. 2A–C, light grey bars). Furthermore, comparison of calpain activities in extracts from tissues obtained at time points when REE was significantly increased (post burn days 21 and 42 = burn induced hypermetabolism (BHM)) with those measured at day 0 and after burns before REE increased (days 7 and 14 = pre BHM) did not reveal statistically significant differences (Fig. 2A–C, dark grey bars).
Fig. 2.
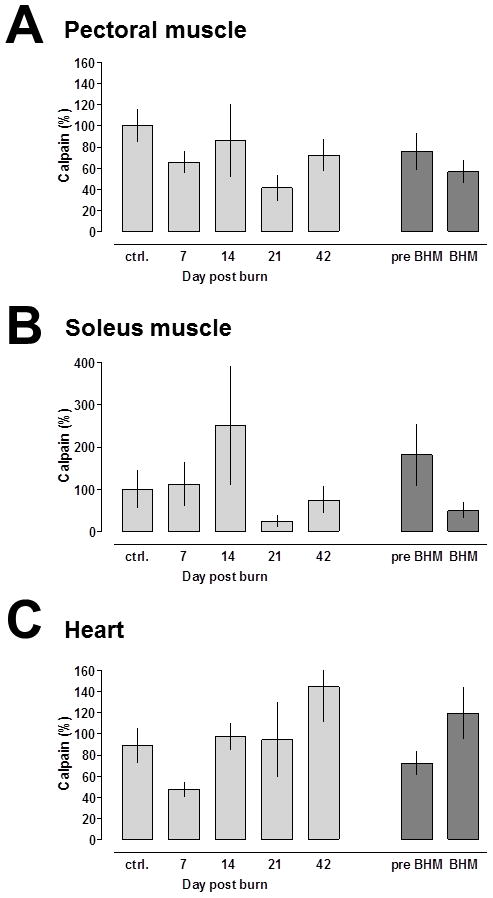
Calpain activities after 30% TBSA burn. Enzyme activities are expressed as % of uninjured control (day 0); mean ± SEM. Light grey bars: Calpain activities at the individual time points. N = 5/time point. Dark grey bars: Calpain activities before (pre-, post burn days 7 and 14) and during burn induced hypermetabolism (BHM; post burn days 21 and 42). A. Pectoral muscle. B. Soleus muscle. C. Cardiac muscle.
Caspase activities
Pan caspase peptidase activities in pectoral muscle extracts increased time dependently after burns. Maximal pan caspase activities reached 525 ± 173% of control (day 0) on post burn day 21 and declined to 250 ± 83% of control on post burn day 42 (Fig. 3A, light grey bars). As compared with pan caspase activities in pectoral muscle extracts on day 0 and pre BHM (days 7 and 14), pan caspase activities were significantly increased during BHM (days 21 and 42) (Fig 3A, dark grey bars).
Fig. 3.
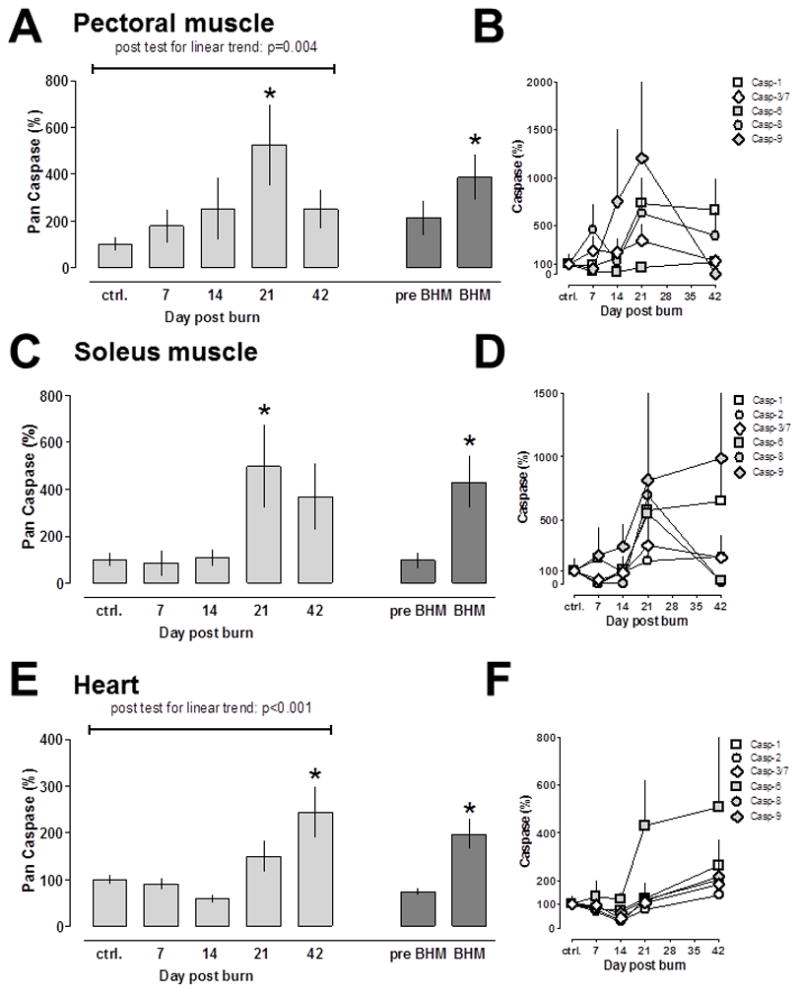
Caspase activities after 30% TBSA burn. Enzyme activities are expressed as % of uninjured control (day 0); mean ± SEM. N = 5/time point. A/C/E: Pan caspase activities. Light grey bars: Pan caspase activities at the individual time points. Dark grey bars: Pan caspase activities before (pre-, post burn days 7 and 14) and during burn induced hypermetabolism (BHM; post burn days 21 and 42). *: p<0.05 vs. day 0 (control). B/D/F: Individual caspase activities, as shown in the graph. There was no detectable caspase-2 activity in pectoral muscle extracts. A/B: Pectoral muscle. C/D: Soleus muscle. E/F: Cardiac muscle.
Whereas we did not observe incremental increases in pan caspase activities in soleus muscle extracts on post burn days 7 and 14, pan caspase activities were increased to 497 ± 117% of control on post burn day 21 and decreased slightly to 370 ± 140% of control on post burn day 42 (Fig. 3C, light grey bars). Comparison of pan caspase activities in soleus muscle extracts between day 0, pre BHM and BHM showed significantly increased pan caspase activities during BHM (Fig. 3C, dark grey bars).
Pan caspase activities increased time dependently in heart extracts (Fig. 3E, light grey bars). In contrast to skeletal muscle extracts, peak pan caspase activities in cardiac extracts were detectable on post burn day 42 (244 ± 55% of control). As observed in skeletal muscle extracts, pan caspase activities were also significantly increased in heart extracts obtained during BHM (200 ± 32% of control), as compared to control and cardiac extracts obtained pre BHM (Fig. 3E, dark grey bars).
The individual caspase activities measured in skeletal and cardiac extracts during the post burn observation period are shown in Fig. 3B/D/F. The increase in pan caspase activities during BHM in pectoral muscle extracts was a result of increased caspase-1, caspase-8 and caspase-9 peptidase activities (Fig. 3B) and of increased caspase-1, caspase-6, caspase-8 and caspase-9 activities in soleus muscle, respectively (Fig. 3D). Whereas caspase-1 and caspase-8 peptidase activities showed sustained increases during BHM in pectoral muscle extracts, increases in caspase-1 and caspase-9 activities were sustained during BHM in soleus muscle extracts. Unlike that in skeletal muscle extracts, the increase in pan caspase activities during BHM in heart extracts was dominated by a sustained increase in caspase-6 activities (Fig. 3F). Nevertheless, caspase-1, caspase-8 and caspase-9 peptidase activities also increased to more than 200% of control on post burn day 42 in heart extracts (Fig. 3F).
Proteasome activities
As compared with ATP levels in skeletal and cardiac extracts on day 0, ATP levels were unchanged throughout the post burn period (data not shown). Thus, we measured proteasome peptidase activities at a concentration of 2 mM ATP in skeletal muscle extracts to reflect the physiological tissue ATP concentration, and at 5 mM ATP in cardiac extracts, respectively (32–36). In pectoral and soleus muscle extracts, proteasome peptidase activities were not significantly altered on any day after burns (Fig. 4A/B). In contrast, proteasome peptidase activities in cardiac extracts increased time dependently to 143 ± 17% on post burn day 42 (Fig. 4C, light grey bars). Cardiac proteasome peptidase activities were significantly higher during BHM (141 ± 9% of control), when compared with control and the pre BHM period (114 ± 8% of control).
Fig. 4.
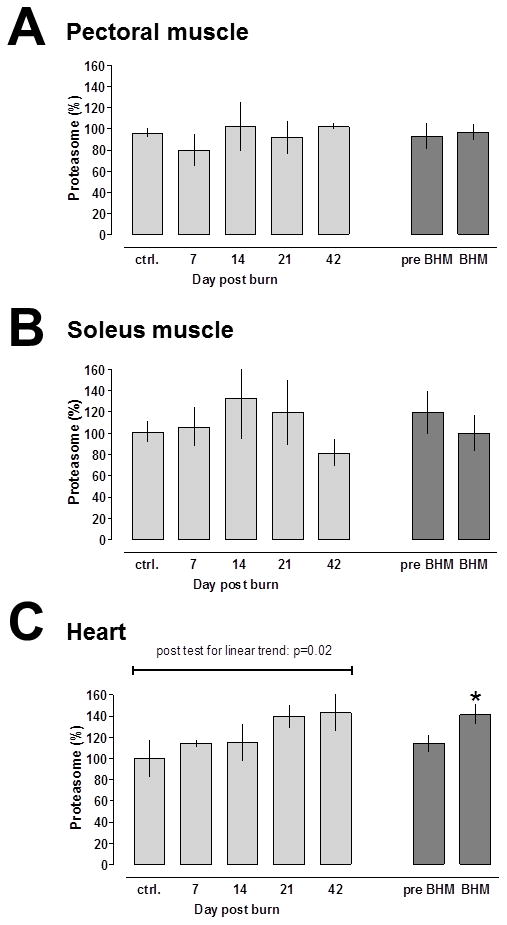
Proteasome activities after 30% TBSA burn. Enzyme activities are expressed as % of uninjured control (day 0); mean ± SEM. Light grey bars: Proteasome activities at the individual time points. N = 5/time point. Dark grey bars: Proteasome activities before (pre-, post burn days 7 and 14) and during burn induced hypermetabolism (BHM; post burn days 21 and 42). A. Pectoral muscle. B. Soleus muscle. C. Cardiac muscle. *: p<0.05 vs. day 0 (control).
DISCUSSION
In the present study, we provide an initial assessment of calpain, caspase and proteasome activities in skeletal and cardiac muscle during burn induced hypermetabolism in rats. Because the heart, pectoral and soleus muscles were not exposed to thermal injury, the observed changes in enzyme activities can be attributed to the systemic whole body response following burns. Our data suggest that activation of the muscle caspase system coincides with burn induced hypermetabolism. Whereas we could not provide evidence for enhanced calpain activities in skeletal and cardiac muscle after burns, activation of proteasome peptidase activities during burn induced hypermetabolism was limited to cardiac muscle.
The observed changes in REE after burn injury in rats are in good agreement with the expected changes after burns in humans. On average, REE increased to 127.3 % of normal between days 21–42 after 30% TBSA in rats. Based on the previously described linear relationship between REE and %TBSA burn in patients, REEs after a similar burn injury in humans would be expected at 129.1 % (37, 38). The hallmark of the post-burn hypermetabolic response in patients is increased energy expenditure with profound whole body protein loss. Because muscle accounts for over 50% of body cell dry weight and for the majority of body protein, muscle catabolism dominates this response (1, 39). Thus, the observed increase in REE in combination with the parallel loss of body weight of the animals in our model documents that relevant burn induced hypermetabolism developed within 21 days after injury and persisted throughout the observation period.
Previous animal studies suggested that muscle proteasome activities, and possibly calpain activites, are elevated 24 – 72 hours after burn injury (17, 19, 23, 40). Information on the activities these enzymes in muscle throughout longer time periods after burns has as yet not been reported.
As ATP has direct regulatory effects on proteasome function, we first determined tissue ATP levels after burns to be able to measure proteasome activities at the actual tissue ATP concentration (41). The observation that ATP tissue levels were not affected after burn injury is consistent with previous nuclear magnetic resonance spectroscopy measurements of post burn muscle ATP levels (42). We found that proteasome and calpain activities were not significantly altered in skeletal muscle after burns when measured at day 7 post burn and weekly thereafter. This observation suggests that the previously described activation of these enzyme systems may occur transiently during the acute inflammatory response to burn injury (17, 19, 23, 40). Furthermore, our data argue against the assumption that globally enhanced calpain or proteasome enzyme activities in skeletal muscle are responsible for the enhanced protein turnover and protein loss that results in hypermetabolism and muscle catabolism after burns.
Skeletal muscle caspase activities have previously been studied for up to 14 days after burn injury in rats and mice (20, 21, 43). Whereas caspase-3 peptidase activities in abdominal muscle continuously increased during seven day post burn observation periods (21, 43), transient increases in caspase-3, caspase-8 and caspase-9 activities with peak activities at post burn day four have been reported in tibialis anterior muscle during a 14-day observation period after burns (20). Caspases exist in latent forms in cells; caspase activation occurs through autoproteolytic processing and subsequent activation of other downstream latent caspases by activated caspases, leading to activation of the caspase cascades (44). Thus, in the present study we utilized pan caspase activities as a global indicator of the activation status of this highly regulated enzyme system. Our findings that pan caspase activities increase within three weeks after burns in skeletal muscle and remain elevated during burn induced hypermetabolism are consistent with previous observations in abdominal muscle during shorter time periods (21, 43). As caspases play essential roles during induction and execution of apoptosis, our data further support the notion that muscle apoptosis contributes to the development and perpetuation of burn induced hypermetabolism and muscle wasting (20–22, 43, 44). In addition, caspases can cleave hundreds of cellular proteins, including actin (26, 45, 46). Caspase cleavage of proteins is thought to generate peptide fragments carrying C- or N-terminal PEST motifs (sequences rich in Pro, Glu, Asp, Ser and Thr), which can function as initiation sites for proteasomal degradation (45–47). Therefore, it appears possible that increased caspase activities in muscle during burn induced hypermetabolism lead to an increase in cellular substrates for proteasomal degradation, which in turn would result in enhanced metabolic protein turnover. As such, enhanced proteasomal degradation of proteins would be a result of increased cellular substrate concentrations and not detectable in in vitro enzyme activity assays with test substrates.
Our observation that the increases in caspase activities in skeletal and cardiac muscle were differentially regulated and that proteasome activation was only detectable in cardiac muscle is in agreement with previous observations on post burn muscle caspase activities and the differences of individual muscles to respond with changes in protein turnover (20, 21, 23, 43, 48).
Whereas myocardial proteasome and calpain activities after burns have not been studied previously, activation of myocardial caspase cascades and induction of apoptosis has been described in short term burn models (49–51). The findings of the present study suggest that activation of caspase cascades is a uniform response during burn induced hypermetabolism in skeletal and cardiac muscle. Furthermore, post burn hypermetabolism is accompanied by cardiac stress, as manifested by long term increases in heart rate, cardiac output and myocardial oxygen consumption (52). As activation of the proteasome occurs during adaptation of the heart to hemodynamic overload (53), the observation that proteasome activities were elevated during post burn hypermetabolism likely corresponds to sustained cardiac stress and implies activation of molecular pathways that can lead to cardiac hypertrophy and failure (53–55).
In the present study, we have utilized crude tissue extracts for enzyme activity measurements. The enzyme activities, therefore, reflect the activities of the sum of enzymes released from muscle and non-muscle cell populations that were present in the tissue biopsies. As the tissue extracts, however, were prepared from uninjured muscles remote from the dorsal burn injury, confounding effects of enzymes released from small amounts of local non-muscle cells or infiltrating leukocytes appear negligible.
Our study is limited by the small sample size at each of the multiple time points in combination with a considerable variability of some of the enzyme activities measurements, in particular calpain. Based on the observed variability in the present study, we calculate that our sample size provided a power of 0.8 to detect a minimal difference of 80% between groups on a two tailed p<0.05 level.
Another limitation of the present study is that it does not address the mechanisms leading to the observed changes in caspase and proteasome activities. Various mechanisms, however, have been associated with protease activation and muscle catabolism, such as inflammation, acidosis, abnormal insulin signaling or glucocorticoids (56–58). Although these mechanisms may have also contributed to the observed changes in our model, further studies are required to define their relative contribution after burns.
In conclusion, our findings suggest that activation of the caspase system during development and perpetuation of burn induced hypermetabolism constitutes a uniform response in skeletal and cardiac muscle, which may, at least partially, contribute to enhanced metabolic protein turnover. Activation of proteasome activities in the heart after burns likely reflects persistent cardiac stress. Our data justify further exploration of caspase cascades and the proteasome as therapeutic targets to reduce detrimental long term consequences of burn induced hypermetabolism.
Acknowledgments
This research was made possible a grant that was awarded and administered by the U.S. Army Medical Research & Materiel Command (USAMRMC) and the Telemedicine & Advanced Technology Research Center (TATRC), at Fort Detrick, MD, under Contract Number W81XWH1110559. The views, opinions and/or findings contained in this research are those of the author(s) and do not necessarily reflect the views of the Department of Defense and should not be construed as an official DoD/Army position, policy or decision unless so designated by other documentation. No official endorsement should be made. This work was also supported in part by National Institutes of Health Grant NIHT32GM008750 and the Dr. Ralph and Marian Falk Medical Research Trust.
References
- 1.Pereira C, Murphy K, Jeschke M, et al. Post burn muscle wasting and the effects of treatments. Int J Biochem Cell Biol. 2005;37(10):1948–1961. doi: 10.1016/j.biocel.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 2.Hart DW, Wolf SE, Chinkes DL, et al. Determinants of skeletal muscle catabolism after severe burn. Ann Surg. 2000;232(4):455–465. doi: 10.1097/00000658-200010000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia de Lorenzo y Mateos A, Ortiz Leyba C, Sanchez SM. Guidelines for specialized nutritional and metabolic support in the critically-ill patient: update. Consensus SEMICYUC-SENPE: critically-ill burnt patient. Nutr Hosp. 2011;26 (Suppl 2):59–62. doi: 10.1590/S0212-16112011000800013. [DOI] [PubMed] [Google Scholar]
- 4.Williams FN, Herndon DN, Jeschke MG. The hypermetabolic response to burn injury and interventions to modify this response. Clin Plast Surg. 2009;36(4):583–596. doi: 10.1016/j.cps.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Porter C, Hurren NM, Herndon DN, et al. Whole body and skeletal muscle protein turnover in recovery from burns. Int J Burns Trauma. 2013;3(1):9–17. [PMC free article] [PubMed] [Google Scholar]
- 6.Matsuda T, Clark N, Hariyani GD, et al. The effect of burn wound size on resting energy expenditure. J Trauma. 1987;27(2):115–118. doi: 10.1097/00005373-198702000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Jeschke MG, Gauglitz GG, Kulp GA, et al. Long-term persistance of the pathophysiologic response to severe burn injury. PLoS One. 2011;6(7):e21245. doi: 10.1371/journal.pone.0021245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilmore DW, Aulick LH, Mason AD, et al. Influence of the burn wound on local and systemic responses to injury. Ann Surg. 1977;186(4):444–458. doi: 10.1097/00000658-197710000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porter C, Herndon DN, Sidossis LS, et al. The impact of severe burns on skeletal muscle mitochondrial function. Burns. 2013;39(6):1039–1047. doi: 10.1016/j.burns.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tuvdendorj D, Chinkes DL, Zhang XJ, et al. Long-term oxandrolone treatment increases muscle protein net deposition via improving amino acid utilization in pediatric patients 6 months after burn injury. Surgery. 2011;149(5):645–653. doi: 10.1016/j.surg.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hart DW, Wolf SE, Ramzy PI, et al. Anabolic effects of oxandrolone after severe burn. Ann Surg. 2001;233(4):556–564. doi: 10.1097/00000658-200104000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suman OE, Mlcak RP, Herndon DN. Effects of exogenous growth hormone on resting pulmonary function in children with thermal injury. J Burn Care Rehabil. 2004;25(3):287–293. doi: 10.1097/01.bcr.0000124792.22931.d7. [DOI] [PubMed] [Google Scholar]
- 13.Herndon DN, Ramzy PI, DebRoy MA, et al. Muscle protein catabolism after severe burn: effects of IGF-1/IGFBP-3 treatment. Ann Surg. 1999;229(5):713–720. doi: 10.1097/00000658-199905000-00014. discussion 720–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyer NA, Barrow RE, Herndon DN. Combined insulin-like growth factor-1 and growth hormone improves weight loss and wound healing in burned rats. J Trauma. 1996;41(6):1008–1012. doi: 10.1097/00005373-199612000-00011. [DOI] [PubMed] [Google Scholar]
- 15.Strock LL, Singh H, Abdullah A, et al. The effect of insulin-like growth factor I on postburn hypermetabolism. Surgery. 1990;108(2):161–164. [PubMed] [Google Scholar]
- 16.Biolo G, Fleming RY, Maggi SP, et al. Inverse regulation of protein turnover and amino acid transport in skeletal muscle of hypercatabolic patients. J Clin Endocrinol Metab. 2002;87(7):3378–3384. doi: 10.1210/jcem.87.7.8699. [DOI] [PubMed] [Google Scholar]
- 17.Fang CH, Tiao G, James H, et al. Burn injury stimulates multiple proteolytic pathways in skeletal muscle, including the ubiquitin-energy-dependent pathway. J Am Coll Surg. 1995;180(2):161–170. [PubMed] [Google Scholar]
- 18.Fang CH, Li BG, Wray CJ, et al. Insulin-like growth factor-I inhibits lysosomal and proteasome-dependent proteolysis in skeletal muscle after burn injury. J Burn Care Rehabil. 2002;23(5):318–325. doi: 10.1097/00004630-200209000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Fang CH, Li BG, Fischer DR, et al. Burn injury upregulates the activity and gene expression of the 20 S proteasome in rat skeletal muscle. Clin Sci. 2000;99(3):181–187. [PubMed] [Google Scholar]
- 20.Duan H, Chai J, Sheng Z, et al. Effect of burn injury on apoptosis and expression of apoptosis-related genes/proteins in skeletal muscles of rats. Apoptosis. 2009;14(1):52–65. doi: 10.1007/s10495-008-0277-7. [DOI] [PubMed] [Google Scholar]
- 21.Yasuhara S, Perez ME, Kanakubo E, et al. Skeletal muscle apoptosis after burns is associated with activation of proapoptotic signals. Am J Physiol Endocrinol Metab. 2000;279(5):E1114–1121. doi: 10.1152/ajpendo.2000.279.5.E1114. [DOI] [PubMed] [Google Scholar]
- 22.Yasuhara S, Kanakubo E, Perez ME, et al. The 1999 Moyer award. Burn injury induces skeletal muscle apoptosis and the activation of caspase pathways in rats. J Burn Care Rehabil. 1999;20(6):462–470. doi: 10.1097/00004630-199920060-00007. [DOI] [PubMed] [Google Scholar]
- 23.Chai J, Wu Y, Sheng Z. The relationship between skeletal muscle proteolysis and ubiquitin-proteasome proteolytic pathway in burned rats. Burns. 2002;28(6):527–533. doi: 10.1016/s0305-4179(02)00049-9. [DOI] [PubMed] [Google Scholar]
- 24.Lowell BB, Ruderman NB, Goodman MN. Evidence that lysosomes are not involved in the degradation of myofibrillar proteins in rat skeletal muscle. Biochem J. 1986;234(1):237–240. doi: 10.1042/bj2340237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wildenthal K, Wakeland JR, Ord JM, et al. Interference with lysosomal proteolysis fails to reduce cardiac myosin degradation. Biochem Biophys Res Commun. 1980;96(2):793–798. doi: 10.1016/0006-291x(80)91424-2. [DOI] [PubMed] [Google Scholar]
- 26.Goll DE, Neti G, Mares SW, et al. Myofibrillar protein turnover: the proteasome and the calpains. J Anim Sci. 2008;86(14 Suppl):E19–35. doi: 10.2527/jas.2007-0395. [DOI] [PubMed] [Google Scholar]
- 27.Walker HL, Mason AD., Jr A standard animal burn. J Trauma. 1968;8(6):1049–1051. doi: 10.1097/00005373-196811000-00006. [DOI] [PubMed] [Google Scholar]
- 28.Baker TA, Geng Q, Romero J, et al. Prolongation of myocardial viability by proteasome inhibition during hypothermic organ preservation. Biochem Biophys Res Commun. 2010;401(4):548–553. doi: 10.1016/j.bbrc.2010.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geng Q, Romero J, Saini V, et al. A subset of 26S proteasomes is activated at critically low ATP concentrations and contributes to myocardial injury during cold ischemia. Biochem Biophys Res Commun. 2009;390(4):1136–1141. doi: 10.1016/j.bbrc.2009.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Majetschak M, Patel MB, Sorell LT, et al. Cardiac proteasome dysfunction during cold ischemic storage and reperfusion in a murine heart transplantation model. Biochem Biophys Res Commun. 2008;365(4):882–888. doi: 10.1016/j.bbrc.2007.11.092. [DOI] [PubMed] [Google Scholar]
- 31.Majetschak M, Sorell LT. Immunological methods to quantify and characterize proteasome complexes: development and application. J Immunol Methods. 2008;334(1–2):91–103. doi: 10.1016/j.jim.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 32.Gribble FM, Loussouarn G, Tucker SJ, et al. A novel method for measurement of submembrane ATP concentration. J Biol Chem. 2000;275(39):30046–30049. doi: 10.1074/jbc.M001010200. [DOI] [PubMed] [Google Scholar]
- 33.Ando T, Imamura H, Suzuki R, et al. Visualization and measurement of ATP levels in living cells replicating hepatitis C virus genome RNA. PLoS Pathog. 2012;8(3):e1002561. doi: 10.1371/journal.ppat.1002561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beer M, Seyfarth T, Sandstede J, et al. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol. 2002;40(7):1267–1274. doi: 10.1016/s0735-1097(02)02160-5. [DOI] [PubMed] [Google Scholar]
- 35.Bottomley PA, Atalar E, Weiss RG. Human cardiac high-energy phosphate metabolite concentrations by 1D-resolved NMR spectroscopy. Magn Reson Med. 1996;35(5):664–670. doi: 10.1002/mrm.1910350507. [DOI] [PubMed] [Google Scholar]
- 36.Kostler H, Landschutz W, Koeppe S, et al. Age and gender dependence of human cardiac phosphorus metabolites determined by SLOOP 31P MR spectroscopy. Magn Reson Med. 2006;56(4):907–911. doi: 10.1002/mrm.21027. [DOI] [PubMed] [Google Scholar]
- 37.Jones LK. Resting energy expenditure in patients with thermal injuries. JPEN J Parenter Enteral Nutr. 1993;17(1):94–96. doi: 10.1177/014860719301700194. [DOI] [PubMed] [Google Scholar]
- 38.Carlson DE, Cioffi WG, Jr, Mason AD, Jr, et al. Resting energy expenditure in patients with thermal injuries. Surg Gynecol Obstet. 1992;174(4):270–276. [PubMed] [Google Scholar]
- 39.Majetschak M, Waydhas C. Infection, bacteremia, sepsis and the sepsis syndrome- metabolic alterations, hypermetabolism, and cellular alterations. In: Baue AE, Faist E, Fry DE, editors. Multiple organ failure: pathophysiology, prevention, and therapy. Springer; New York: 2000. pp. 101–107. [Google Scholar]
- 40.Tan Y, Peng X, Wang F, et al. Effects of tumor necrosis factor-alpha on the 26S proteasome and 19S regulator in skeletal muscle of severely scalded mice. J Burn Care Res. 2006;27(2):226–233. doi: 10.1097/01.BCR.0000203378.85736.38. [DOI] [PubMed] [Google Scholar]
- 41.Majetschak M. Regulation of the proteasome by ATP: Implications for ischemic myocardial injury and donor heart preservation. Am J Physiol Heart Circ Physiol. 2013;305(3):H267–278. doi: 10.1152/ajpheart.00206.2012. [DOI] [PubMed] [Google Scholar]
- 42.Sprague DB, Gadian DG, Williams SR, et al. Intracellular metabolites in rat muscle following trauma: a 31P and 1H nuclear magnetic resonance study. J R Soc Med. 1987;80(8):495–498. doi: 10.1177/014107688708000813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee HY, Kaneki M, Andreas J, et al. Novel mitochondria-targeted antioxidant peptide ameliorates burn-induced apoptosis and endoplasmic reticulum stress in the skeletal muscle of mice. Shock. 2011;36(6):580–585. doi: 10.1097/SHK.0b013e3182366872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan TJ, Han LH, Cong RS, et al. Caspase family proteases and apoptosis. Acta Biochim Biophys Sin (Shanghai) 2005;37(11):719–727. doi: 10.1111/j.1745-7270.2005.00108.x. [DOI] [PubMed] [Google Scholar]
- 45.Du J, Wang X, Miereles C, et al. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest. 2004;113(1):115–123. doi: 10.1172/JCI200418330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belizario JE, Alves J, Garay-Malpartida M, et al. Coupling caspase cleavage and proteasomal degradation of proteins carrying PEST motif. Curr Protein Pept Sci. 2008;9(3):210–220. doi: 10.2174/138920308784534023. [DOI] [PubMed] [Google Scholar]
- 47.Rogers S, Wells R, Rechsteiner M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science. 1986;234(4774):364–368. doi: 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- 48.Fang CH, Li BG, Tiao G, et al. The molecular regulation of protein breakdown following burn injury is different in fast- and slow-twitch skeletal muscle. Int J Mol Med. 1998;1(1):163–169. doi: 10.3892/ijmm.1.1.163. [DOI] [PubMed] [Google Scholar]
- 49.Cao W, Xie YH, Li XQ, et al. Burn-induced apoptosis of cardiomyocytes is survivin dependent and regulated by PI3K/Akt, p38 MAPK and ERK pathways. Basic Res Cardiol. 2011;106(6):1207–1220. doi: 10.1007/s00395-011-0199-3. [DOI] [PubMed] [Google Scholar]
- 50.Zhang JP, Ying X, Liang WY, et al. Apoptosis in cardiac myocytes during the early stage after severe burn. J Trauma. 2008;65(2):401–408. doi: 10.1097/TA.0b013e31817cf732. [DOI] [PubMed] [Google Scholar]
- 51.Carlson DL, Horton JW. Cardiac molecular signaling after burn trauma. J Burn Care Res. 2006;27(5):669–675. doi: 10.1097/01.BCR.0000237955.28090.41. [DOI] [PubMed] [Google Scholar]
- 52.Williams FN, Herndon DN, Suman OE, et al. Changes in cardiac physiology after severe burn injury. J Burn Care Res. 2011;32(2):269–274. doi: 10.1097/BCR.0b013e31820aafcf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Depre C, Wang Q, Yan L, et al. Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation. 2006;114(17):1821–1828. doi: 10.1161/CIRCULATIONAHA.106.637827. [DOI] [PubMed] [Google Scholar]
- 54.Warren SA, Briggs LE, Zeng H, et al. Myosin light chain phosphorylation is critical for adaptation to cardiac stress. Circulation. 2012;126(22):2575–2588. doi: 10.1161/CIRCULATIONAHA.112.116202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma Y, Chen Y, Yang Y, et al. Proteasome inhibition attenuates heart failure during the late stages of pressure overload through alterations in collagen expression. Biochem Pharmacol. 2013;85(2):223–233. doi: 10.1016/j.bcp.2012.10.025. [DOI] [PubMed] [Google Scholar]
- 56.Mitch WE, Price SR. Mechanisms activating proteolysis to cause muscle atrophy in catabolic conditions. J Ren Nutr. 2003;13(2):149–152. doi: 10.1053/jren.2003.50019. [DOI] [PubMed] [Google Scholar]
- 57.McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5(4):a008656. doi: 10.1101/cshperspect.a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bailey JL. Insulin resistance and muscle metabolism in chronic kidney disease. ISRN Endocrinol. 2013;2013:329606. doi: 10.1155/2013/329606. Epub 2013 Feb 3. [DOI] [PMC free article] [PubMed] [Google Scholar]