

Abstract
We have synthesized a panel of bivalent S-sialoside analogues, with modifications at the 4 position, as inhibitors of influenza virus. These first generation compounds show IC50 values ranging from low micromolar to high nanomolar in enzyme inhibition and plaque reduction assays with two intact viruses, Influenza H1N1 (A/California/07/2009) and H3N2 (A/Hongkong/8/68).
Keywords: Influenza virus, sialic acid, glycoconjugates, inhibitors, bifunctional
Influenza is an opportunistic pathogen that causes severe respiratory illnesses. The virus accounts for millions of infections worldwide each year, leading to significant morbidity and mortality. Senior citizens (over 65 years of age), young children, and individuals with underlying health problems are at increased risk for infection and subsequent secondary illnesses like pneumonia. 1 Along with vaccines, Oseltamivir®, Zanamivir®, and Premavir® (Figure 1B–D) are among the frontline drugs used to fight the infection. 2 These FDA approved transition state analogs inhibit the activity of the viral surface enzyme, Neuraminidase (NA), from cleaving the residual N-acetyl neuraminic acid (or sialic acid, Figure 1A) present on the infected host cell, consequently arresting the virus progeny from escaping the cell. However, some strains which include emerging, highly virulent strains that can potentially cause pandemics, have started to exhibit resistance to some of these inhibitors. In one recent surveillance study, 100% of all patients had a resistant strain to Oseltamivir®,3 and another study identified a strain that was resistant to both, Zanamivir® and Oseltamivir®.4 These studies emphasize the need for vigilance and continued development of novel drugs. Recently, a new class of mechanism based anti-viral compounds against NA has been reported to show broad spectrum anti-viral activity against all strains in vitro and in animal models. 5, 6(Figure 1E, F) Unlike transition state analogs, these compounds are similar to natural substrate, with modifications at the 3 and 4 positions, which enhance the binding activity. All of these recent reports are based on the natural substrate (sialic acid) with structure based drug design leading to increased and highly specific inhibition.
Figure 1.

Structures of N-acetyl Neuraminic Acid (sialic acid) analogs. A. Sialic acid with the numbering scheme. B. Oseltamivir ® C. Zanamivir®, D. Premavir® E-F. Flourinated analogs of Sialic acid. G. Fluorogenic substrate of sialic acid for obtaining IC50 values
A slightly different strategy of developing inhibitors follows Nature’s method of using multivalency that target Hemagluttinin (HA), a surface glycoprotein that binds to cell surface sialic acid to facilitate viral entry and NA. HA and NA are excellent targets for inhibition, because labeling experiments have shown that an individual viral particle has approximately 200–300 copies of trimeric HA and 50–100 copies of tetrameric NA, leading to over 800 binding sites per virion.7, 8 Indeed, mucins, endogenous sialylated proteins released by respiratory epithelial cells, capture viral particles by virtue of their multiple sialic acid residues which bind to HA and NA, and flush them away by the natural process of sneezing and coughing.9–11 A similar approach using glycopolymers and glycodendrimers with pendant sialic acids have been generated to capture the virus. It has been demonstrated that increase in the valency of the sialic acids increases the inhibitory effect significantly, from the micromolar IC50 value of a mono/di/tri saccharide, to the micromolar/submicromolar range. 12–14 These reports have focused on the architecture of the scaffolds, leaving the sialic acid unit unmodified.
In this report, we have increased the intrinsic binding affinity of a single sialic acid unit by introducing an amine/guanidine group at the 4 position of sialic acid. The basic amine/guanidine group fits perfectly into the binding pocket of viral NA as it interacts with the trio of amino acids present in the binding pocket. 2, 15 We introduced sulfur at the anomeric center, which provides additional advantages. First, replacing the O-sialoside with an S-sialoside makes the ligand more robust as we and others have demonstrated that S-sialosides are not readily cleaved by the virus. 16, 17 Second, the thiol group reacts with triflates and/or bromides present on multivalent scaffolds readily, thereby yielding rapid access to multivalent molecules. The combination of NA resistant sialosides combined with the multivalent presentation provides easy access to a new class of influenza virus inhibitors. Using this approach, we constructed a panel of bivalent compounds, (Schemes 1 and 2) that are similar to the natural substrate and evaluated their inhibitory activities with two viral NAs, N1 and N2, and intact viral strains. These first generation compounds show micromolar to nanomolar inhibition and could be further elaborated into potential therapies for influenza.
Scheme 1.

Synthesis of SA and SG. Reagents and Conditions: a) HCl (g), LiCl, CH3CN, 6 days; b) KSAc, TBAB (Tetrabutylammonium bisulfate), CH2Cl2/H2O, overnight, 50% over 2 steps; c) TosOC6H12SAc, DEA, DMF, 65%; d) PPh3, THF/H2O, 12h; e) (Boc)2O, TEA, THF, 60% over 2 steps f) MeS-(C=NBoc)NHBoc, HgCl2, TEA, CH2Cl2, 80% over 2 steps; g) MeOH/NaOH (aq), 1 h; h) TFA/DCM (1:1). 90% over two steps for SA and SG, respectively.
Scheme 2.

Synthesis of bivalent analogs of S-Sialosides. Reagents and Conditions: a) DEA, DMF; 76–85%; b) i) NaOH/CH3OH (aq), ii) H2/Pd(OH)2; 90–92% % over 2 steps; c) i) H2/Pd(OH)2, ii)MeS-(C=NBoc)NHBoc, HgCl2, TEA, CH2Cl2. 85–89% % over 2 steps; d) NaOH/CH3OH (aq), 1 h; i) TFA/DCM (1:1), 80–85% over 2 steps.
The design and synthesis of the compounds is shown in Scheme 1. Starting with the known azido compound, 1,18 hydrogen chloride was added across the double bond to produce the β-chloride 2, followed by SN2 type replacement of the chloride by a thioacetate to introduce the sulfur moiety at the anomeric center in good yield. The alpha conformation in 3 was confirmed by 1H and 13C NMR spectroscopies. Reaction of 3 with a hydrophobic six carbon spacer in the presence of diethylamine yielded 4. Next, the azide group of 4 was reduced using triphenylphospine to yield the amine 5, which was subsequently protected with a t-butylcarbonyl group or reacted with a suitable protected guanidinium group to yield 6 and 7, respectively. Removal of ester moieties was conducted under basic hydrolysis conditions, followed by removal of the acid sensitive t-butylcarbonyl groups using trifluoroacetic acid to yield analogs of sialic acid with an amine (SA) or guanidine group (SG) at the four position. While not the focus of this report, we incorporated a thiol group at the terminus of the alkyl spacer in SA and SG as it provides facile conjugation to surfaces for capture of the virus or to a scaffold/protein to produce multivalent inhibitors.
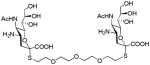
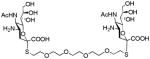
We used a series of activated homobifunctional hydrophobic and hydrophilic linkers to produce the dimeric compounds, shown in Scheme 2. Different spacers were used because the bivalent molecules can interact with viral NA in three ways. Briefly, the bivalent molecules can crosslink adjacent NAs on the same tetramer or crosslink two NAs from two different NA tetramers on the same viral particle or crosslink NA’s from two viral particles. The minimal distance between the active sites of the NAs in all three possibilities are ~ 16 Å, 30 Å, 50 Å, respectively.19 Therefore, using a series of activated hydrophilic and hydrophobic spacers, we generated a panel of gemini compounds, 8–12 in decent yield. The synthesis of the activated spacers for coupling are presented in the supplementary data. The azides in 8–12 were deprotected as described previously to yield five bivalent S-sialoside analogs with an amine at the 4 position. (C6-SA, C12-SA, TetraEG-SA, PentaEG-SA and HexaEG-SA). We also introduced a guanidine group at the 4 position as described for the synthesis of the monomeric SG to yield five bivalent S-sialoside analogs. (C6-SG, C12-SG, TetraEG-SG, PentaEG-SG and HexaEG-SG). All intermediates and final compounds were characterized by NMR and mass spectroscopies (Supplementary data)
Next, we performed NA inhibition studies using commercially available 2′-(4- Methylumbelliferyl)-α-D-N-acetylneuraminic acid (MUNANA) (Figure 1G) as the substrate. First, we used soluble viral NA as the enzymes before testing intact viruses. Since there is an initial binding component in the inhibitory mechanism, we premixed the compounds with NA and followed the cleavage of the substrate over two hours to obtain the IC50s using the linear slope between 0–35 min. The IC50 values with three independent experiments for each compound are given in Table 1 and the selective raw data is presented in the supplementary data. The commercial drugs, Zanamivir® and Ostelimivir® are included as controls. As expected, sialic acid exhibits millimolar inhibition and the antivirals exhibit nanomolar inhibitory values. Introduction of an amine at the 4 position decreases the IC50 by 1000 fold to micromolar levels for both influenza viral enzymes, N1 and N2; the IC50 values for SA range from 60–180 μM. The bivalent sialosides (C6-SA, C12-SA, TetraEG-SA, PentaEG-SA and HexaEG-SA) all exhibit higher inhibition activity, approximately four to five fold increase in inhibition, especially for the N1 enzyme, when compared to the monovalent compound. There were exceptions, the compounds with the longer spacers (PentaEG-SA and HexaEG-SA), were not that efficacious compared to the monomeric compounds. With the N2 enzyme, a similar trend was observed, with the bivalent compound exhibiting a two fold higher inhibition compared to the monomeric amine containing compound and the compounds with longer spacers not as effective. Introduction of the guanidine group led to increased inhibition with IC50 values ranging from 0.2–25 μM for SG, C6-SG, C12-SG, TetraEG-SG, PentaEG-SG and HexaEG-SG. The IC50 values for the bivalent compounds, irrespective of the spacers, are similar to SG, which is the monomeric compound and the values for both enzymes are similar, in the submicromolar range. This is because the soluble NAs are not membrane bound and are present as a mixture of monomers, dimers, trimers and some tetramers. To summarize this subsection, these experiments confirm that the compounds with amine at the 4 position (SA, C6-SA, C12-SA, TetraEG-SA, PentaEG-SA and HexaEG-SA) are better inhibitors than the natural receptor The compounds with guanidine at the 4 position (SG, C6-SG, C12-SG, TetraEG-SG, PentaEG-SG and HexaEG-SG) are better inhibitors than compounds with amine at the 4 position. We also confirmed that both classes of NA, NA1 with the more open binding pocket and a flexible loop that closes upon introduction of sialic acid or an inhibitor and NA2, which has a preset binding pocket, 20 are inhibited very well by the synthetic compounds.
Table 1.
IC50 values for the inhibition of soluble NAs and intact virus by the gemini sialoside analogs. The influenza strains used in these studies were Influenza H1N1 (A/California/07/2009) and H3N2 (A/Hongkong/8/68).
| Name | Structure | Virus | NA from Virus | ||
|---|---|---|---|---|---|
| H3N2 (μM) | H1N1 (μM) | H5N1 (μM) | H3N2 (μM) | ||
| SA |
|
64±14 | 56±6 | 127±29 | 178±20 |
| C6-SA |
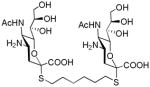
|
11.6±3.5 | 9.9±1.8 | 20±5 | 32±5 |
| C12-SA |
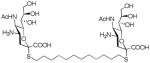
|
15±9 | 11±1 | 31±7 | 40±1 |
| TetraEG-SA |
|
11±5 | 9.8±2.4 | 28±3 | 39±14 |
| PentaEG-SA |
|
21±3 | 36±4 | 64±14 | 178±20 |
| HexaEG-SA |
|
33±15 | 45±1 | 85±12 | 216±56 |
| SG |
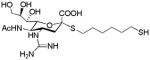
|
0.55±0.02 | 0.8±0.1 | 0.1±0.02 | 1±0.1 |
| C6-SG |
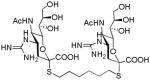
|
0.25±0.05 | 0.26±0.03 | 0.2±0.02 | 4.2±1.2 |
| C12-SG |
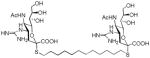
|
0.5±0.3 | 0.8±0.06 | 0.73±0.03 | 12±2 |
| TetraEG-SG |
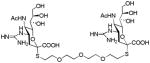
|
1.78±1.3 | 2.4±0.4 | 2.5±0.1 | 10±1 |
| PentaEG-SA |
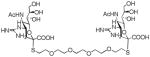
|
1±0.6 | 1.2±0.2 | 1.1±0.1 | 10±2 |
| HexaEG-SA |
|
0.5±0.2 | 0.9±0.1 | 0.57±0.06 | 11±2 |
| Zanamivir® |
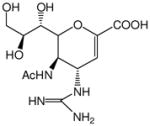
|
~0.001 | ~0.01 | ~0.04 | ~0.04 |
| Oseltamivir ® |
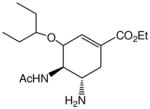
|
~0.008 | ~0.01 | ~0.05 | ~0.05 |
Since the compounds inhibited soluble NAs, we performed the same assay repeated with two intact viruses, Influenza H1N1 (A/California/07/2009) and H3N2 (A/Hongkong/8/68) to ensure that the compounds inhibit the transmembrane NAs present on the viruses. A similar trend as was observed for soluble NA. SA, the compound with an amine at the four position exhibits an IC50 value of 60 μM for both strains. The bivalent molecules, C6-SA, C12-SA, TetraEG-SA, PentaEG-SA and HexaEG-SA exhibit a 5 fold decrease in the IC50s compared to the monomer to ~ 10 μM. The lone exception is the compound with the longest oligoethylene glycol spacer, which did not have a similar effect as the other bivalent molecules, presumably because the spacer is too flexible to exhibit a bidentate effect. In the case of the guanidine containing compounds, the monomer SG has an IC50 value of ~ 1 μM for both strains, which is a 50 fold decrease compared to SA. The bivalent compounds, SG, C6-SG, C12-SG, TetraEG-SG, PentaEG-SG and HexaEG-SG all show significant decreases in the IC50 values, ranging from 250 nm to 2.4 μM for both viruses, with C6-SG the best inhibitor of all compounds. However, since the values for all bivalent compounds are similar, it is not clear if the compounds bind to two NA’s on the same virion or different virions. What is clear is that our hypothesis of increasing the intrinsic binding affinity of an individual sialoside analog coupled with bivalency decreases the IC50 values from the millimolar range by 10,000 fold to the nanomolar range.
We were also interested in testing the efficacy of these compounds to inhibit the virus in cell based systems. Towards that end, we performed a plaque assay for all the compounds. The virus was introduced to the cells for 30 minutes, followed by addition of the compounds to arrest the spread of the viruses. IC50 values based on 50% decrease in plaque size are given in Table 2. We were pleased to observe a similar trend for all compounds as was seen with the cell free system. Briefly, SA, the compound with an amine at the 4 position exhibited inhibition in the 10–1000 μM range for both, H1N1 and H3N2 strains, and the dimers C6-SA, C12-SA, TetraEG-SA, PentaEG-SA and HexaEG-SA, exhibiting a 10 fold decrease in the IC50 values (range from 1–100 μM) in comparison to SA. SG, the compounds with a guanidine at the 4 position, exhibited a 10 fold decrease in the IC50 values (1–10 μM) compared to SA. The bivalent derivatives, C6- SG, C12-SG, TetraEG-SG, PentaEG-SG and HexaEG-SG, all exhibiting lower IC50 values (10–1000 nM) than SG.
Table 2.
IC50 values of influenza strains by the gemini sialosides based on 50% decrease in plaque size. The strains used in these studies were Influenza H1N1 (A/California/07/2009) and H3N2 (A/Hongkong/8/68).
| Name | Structure | Virus | |
|---|---|---|---|
| H3N2 (μM) | H1N1 (μM) | ||
| SA |
|
10–100 | >100 |
| C6-SA |
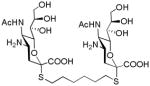
|
1–10 | 100 |
| C12-SA |
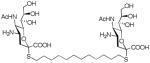
|
1–10 | 100 |
| TetraEG-SA |
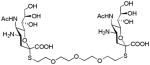
|
1–10 | 100 |
| PentaEG-SA |
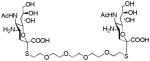
|
1–10 | 100 |
| HexaEG-SA |
|
1–10 | 100 |
| SG |
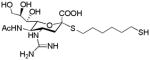
|
1–10 | 10 |
| C6-SG |
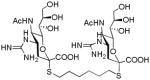
|
0.01–0.1 | 10 |
| C12-SG |
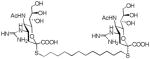
|
0.1 | 1.0 |
| TetraEG-SG |
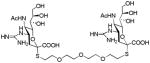
|
0.1 | 1.0 |
| PentaEG-SG |
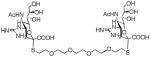
|
.01–0.1 | 1.0 |
| HexaEG-SG |
|
0.1–1.0 | 1.0 |
| Zanamivir® |
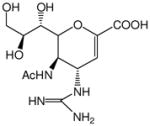
|
0.001 | 0.01 |
| Oseltamivir ® |
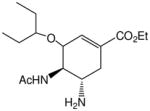
|
0.001–0.01 | 0.001–0.01 |
To summarize, we have combined robustness of S-sialosides, increased intrinsic affinity of a single unit and used bivalency to produce a panel of compounds that inhibit two different strains of influenza virus. The IC50 values range from the low micromolar to high nanomolar. We have also identified the appropriate length of the spacer for attachment to an appropriate scaffold, longer spacers such as a penta or hexa ethylene glycol do not seem appropriate for effective inhibition. Finally, we note that these compounds are not as efficacious as Zanamivir® or Oseltamivir®. However, we are currently making structural modifications, e.g. introducing a fluorine at the 3 position and tethering the molecules polymeric/dendrimeric scaffold. We anticipate that these modifications will enhance the IC50 values to produce highly potent inhibitors. These results will be reported soon.
Supplementary Material
Acknowledgments
We are grateful for NIH-NIAID (R01-AI089450) for funding. The viruses were obtained through BEI Resources, NIAID, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Fiore AE, Shay DK, Broder K, Iskander JK, Uyeki TM, Mootrey G, Bresee JS, Cox NJ. MMWR Recomm Rep. 2009;58:1. [PubMed] [Google Scholar]
- 2.von Itzstein M. Nat Rev Drug Discov. 2007;6:967. doi: 10.1038/nrd2400. [DOI] [PubMed] [Google Scholar]
- 3.Mai-Phuong HV, Hang Nle K, Phuong NT, Maile Q. Western Pac Surveill Response J. 2013;4:25. [Google Scholar]
- 4.Hay AJ, Hayden FG. Lancet. 2013;381:2230. doi: 10.1016/S0140-6736(13)61209-X. [DOI] [PubMed] [Google Scholar]
- 5.Vavricka CJ, Liu Y, Kiyota H, Sriwilaijaroen N, Qi J, Tanaka K, Wu Y, Li Q, Li Y, Yan J, Suzuki Y, Gao GF. Nat Commun. 2013;4:1491. doi: 10.1038/ncomms2487. [DOI] [PubMed] [Google Scholar]
- 6.Kim JH, Resende R, Wennekes T, Chen HM, Bance N, Buchini S, Watts AG, Pilling P, Streltsov VA, Petric M, Liggins R, Barrett S, McKimm-Breschkin JL, Niikura M, Withers SG. Science. 2013;340:71. doi: 10.1126/science.1232552. [DOI] [PubMed] [Google Scholar]
- 7.Murti KG, Webster RG. Virology. 1986;149:36. doi: 10.1016/0042-6822(86)90084-x. [DOI] [PubMed] [Google Scholar]
- 8.Amano H, Uemoto H, Kuroda K, Hosaka Y. J Gen Virol. 1992;73(Pt 8):1969. doi: 10.1099/0022-1317-73-8-1969. [DOI] [PubMed] [Google Scholar]
- 9.White MR, Helmerhorst EJ, Ligtenberg A, Karpel M, Tecle T, Siqueira WL, Oppenheim FG, Hartshorn KL. Oral Microbiol Immunol. 2009;24:18. doi: 10.1111/j.1399-302X.2008.00468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White MR, Crouch E, van Eijk M, Hartshorn M, Pemberton L, Tornoe I, Holmskov U, Hartshorn KL. Am J Physiol Lung Cell Mol Physiol. 2005;288:L831. doi: 10.1152/ajplung.00365.2004. [DOI] [PubMed] [Google Scholar]
- 11.Lieleg O, Lieleg C, Bloom J, Buck CB, Ribbeck K. Biomacromolecules. 2012;13:1724. doi: 10.1021/bm3001292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hidari KI, Murata T, Yoshida K, Takahashi Y, Minamijima YH, Miwa Y, Adachi S, Ogata M, Usui T, Suzuki Y, Suzuki T. Glycobiology. 2008;18:779. doi: 10.1093/glycob/cwn067. [DOI] [PubMed] [Google Scholar]
- 13.Sakamoto J, Koyama T, Miyamoto D, Yingsakmongkon S, Hidari KI, Jampangern W, Suzuki T, Suzuki Y, Esumi Y, Nakamura T, Hatano K, Terunuma D, Matsuoka K. Bioorg Med Chem. 2009;17:5451. doi: 10.1016/j.bmc.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 14.Sakamoto J, Koyama T, Miyamoto D, Yingsakmongkon S, Hidari KI, Jampangern W, Suzuki T, Suzuki Y, Esumi Y, Hatano K, Terunuma D, Matsuoka K. Bioorg Med Chem Lett. 2007;17:717. doi: 10.1016/j.bmcl.2006.10.085. [DOI] [PubMed] [Google Scholar]
- 15.Von Itzstein M, YW, Kok GB, Pegg MS, Dyason JC, Jin B, Van Phan T, Synthe ML, White HF, Oliver SW, Coleman PG, Varghese VJ, Cameron JM, Penn CR. Nature. 1993;363:418. doi: 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- 16.Wilson JC, Kiefel MJ, Angus DI, von Itzstein M. Org Lett. 1999;1:443. doi: 10.1021/ol990652w. [DOI] [PubMed] [Google Scholar]
- 17.Kale RR, Mukundan H, Price DN, Harris JF, Lewallen DM, Swanson BI, Schmidt JG, Iyer SS. J Am Chem Soc. 2008;130:8169. doi: 10.1021/ja800842v. [DOI] [PubMed] [Google Scholar]
- 18.Chandler M, Bamford MJ, Conroy R, Lamont B, Patel B, Patel VK, Steeples IP, Storer R, Weir NG, Wright M, Williamson C. Journal of the Chemical Society. Perkin Transactions. 1995;1:1173. [Google Scholar]
- 19.Macdonald SJF, Watson KG, Cameron R, Chalmers DK, Demaine DA, Fenton RJ, Gower D, Hamblin JN, Hamilton S, Hart GJ, Inglis GGA, Jin B, Jones HT, McConnell DB, Mason AM, Nguyen V, Owens IJ, Parry N, Reece PA, Shanahan SE, Smith D, Wu WY, Tucker SP. Antimicrobial Agents and Chemotherapy. 2004;48:4542. doi: 10.1128/AAC.48.12.4542-4549.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Russell RJ, Haire LF, Stevens DJ, Collins PJ, Lin YP, Blackburn GM, Hay AJ, Gamblin SJ, Skehel JJ. Nature. 2006;443:45. doi: 10.1038/nature05114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.