

Abstract
Skeletal and cardiac muscles play key roles in the regulation of systemic energy homeostasis and display remarkable plasticity in their metabolic responses to caloric availability and physical activity. In this Perspective we discuss recent studies highlighting transcriptional mechanisms that govern systemic metabolism by striated muscles. We focus on the participation of the Mediator complex in this process, and suggest that tissue-specific regulation of Mediator subunits impacts metabolic homeostasis.
Introduction
In the 21st century, obesity and type II diabetes mellitus have become worldwide epidemics. The prevalence of these diseases continues to rise as technologic innovation has decreased reliance on physical activity for survival and allowed for easy access to high-calorie food. Metabolic syndrome precedes type II diabetes in many patients, and is characterized by abdominal obesity, hypertension, insulin resistance, and inflammation. Consequently, metabolic syndrome carries an increased risk of heart attack, stroke, and premature death (Malik et al., 2004). Skeletal muscle and the heart play central roles in metabolic syndrome and are regulators of total body mass and energy consumption (Rolfe and Brown, 1997). Excess triglycerides, free fatty acids, and glucose, coupled with physical inactivity, perturbs metabolism in skeletal and cardiac muscle. As striated muscles adapt to increased substrate availability, systemic metabolic homeostasis is altered, contributing to the onset of obesity and diabetes.
Obesity and diabetes evoke a characteristic cardiac phenotype known as “diabetic cardiomyopathy” (Hamby et al., 1974) withan underlying transcriptional basis associated with diminished cardiac function (Battiprolu et al., 2010). Transcriptional regulation of metabolic genes occurs through interactions of ligand-binding nuclear receptors (NRs), transcriptional coregulators, chromatin modifiers, and the Mediator complex among other factors (Burris et al., 2013; Finck and Kelly, 2006; Mouchiroud et al., 2014). Recent investigations have revealed that changes in metabolic gene transcription in heart and skeletal muscle, induced by muscle-specific manipulation of Mediator subunits, modulate systemic metabolic disease (Baskin et al., 2014b; Chen et al., 2010; Grueter et al., 2012; Lee et al., 2014). These studies imply that metabolic transcriptional adaptations in muscle are not only a consequence of metabolic disease, but also a potential disease modifier. Here we summarize the ways in which muscle transcription affects whole-body energy homeostasis, and review the tissue-specific roles of Mediator components in this process.
The Role of Muscle in Systemic Metabolic Homeostasis
Skeletal muscle comprises ~40% of total human body mass in a healthy-weight individual (Rolfe and Brown, 1997). Together, skeletal muscle and the heart account for almost 30% of resting energy consumption and nearly 100% of increased energy consumption during exercise (Gallagher et al., 1998). Skeletal muscle is heterogeneous and composed of slow and fast-twitch fiber types, which differ in the composition of contractile proteins, oxidative capacity, and substrate preference for ATP production. Slow-twitch fibers display low fatigability, high oxidative capacity, and a preference for fatty acids as substrate for ATP production. Fast-twitch fibers have a higher fatigability, higher strength of contraction, lower oxidative capacity, and a preference for glucose as a substrate for ATP production through anaerobic glycolysis (Bassel-Duby and Olson, 2006; Schiaffino and Reggiani, 2011). Thus, fiber type composition of skeletal muscle profoundly impacts systemic energy consumption (Figure 1).
Figure 1. The Role of Muscle Fiber Types in the Regulation of Systemic Metabolism.
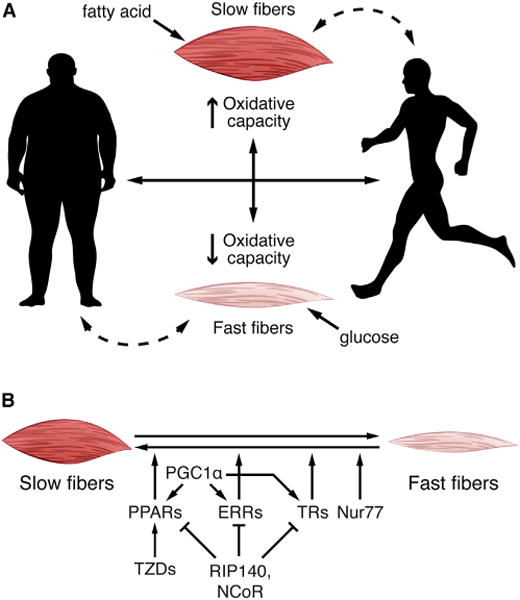
(A) Slow-twitch myofibers have a high oxidative capacity and prefer fatty acids as substrate for ATP production. Fast-twitch fibers have a lower oxidative capacity and prefer glucose. Muscle fiber type can be altered by external and internal factors. Exercise increases the number of slow-twitch fibers, thus enhancing fatty acid utilization, while obesity increases fast-twitch fibers and causes slow-twitch fibers to become insulin resistant. Muscle fiber type is transcriptionally regulated, ultimately impacting systemic metabolism.
(B) Nuclear receptors, such as PPARs, ERRs, TRs, and Nur77 activate transcription of genes involved in myofiber switching. PGC1α is a coregulator and acts with PPAR, ERR, and TR to drive the switch from fast to slow fibers. RIP140 and NCoR1 are corepresors for PPARs, ERRs, and TRs. TZDs, insulin sensitizing drugs, reactivate PPAR in the setting of diabetes and obesity, which can also lead to myofiber switching. PPAR, peroxisome proliferator-activated receptor; ERR, estrogen-related receptor; TR, thyroid hormone receptor; Nur77, orphan nuclear receptor NR4A1; PGC1α, peroxisome proliferator-activated receptor gamma coactivator 1 alpha; RIP140, corepressor receptor-interacting protein 140; NCoR, nuclear receptor corepressor; TZD, thiazolidinediones.
Endurance or aerobic exercise increases mechanical and metabolic demand on skeletal muscle, resulting in a switch from a fast-twitch to a slow-twitch fiber type (Figure 1A). Conversely, in obesity and diabetes, characterized by excess caloric intake without increased metabolic demand, a slow-to-fast fiber type switch occurs in muscle, which decreases oxidative capacity (Mootha et al., 2003). Insulin resistance, a hallmark of metabolic syndrome and diabetes, correlates with a higher composition of fast-twitch myofibers (Simoneau et al., 1995). Resistance training also impacts skeletal muscle metabolism by increasing muscle mass and enhancing the oxidative and glycolytic capacity of fast-twitch fibers (LeBrasseur et al., 2011). Diabetic patients on a regimen of resistance training have improved insulin sensitivity (Zanuso et al., 2010), and obese patients subjected to resistance training develop increased lean mass and a higher resting metabolic rate (Willis et al., 2012). Exercise impacts systemic glucose and lipid homeostasis and alters muscle fiber type composition, which is regulated at the level of transcription.
Transcriptional Regulation of Metabolic Genes in Striated Muscle
Transcriptional regulation of metabolic genes in muscle begins with tissue-extrinsic signals such as peptides, steroid hormones, and catecholamines, as well as tissue-intrinsic signals including mechanical stretch, intracellular calcium, and nutrient availability. Myocyte enhancer factor 2 (MEF2) is a downstream mediator of many of these signals and plays a critical role in muscle metabolism by stimulating the transcriptional activity of two key metabolic regulators: peroxisome proliferator-activated receptor alpha (PPARα) and peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC1α) (Czubryt et al., 2003). These factors, in turn, coordinate with others such as GLUT4 enhancer factor (GEF), cAMP response element-binding protein (CREB), and nuclear respiratory factor (NRF) to mediate transcription of genes involved in fatty acid and glucose metabolism (reviewed in Egan and Zierath, 2013).
During exercise, many signaling pathways are activated that ultimately lead to transcriptional changes in muscle. For example, increased intracellular calcium activates calcium/calmodulin-dependent protein kinase II (CaMKII), mechanical stretch activates mitogen-activated protein (MAP) and c-Jun N-terminal (JNK) kinases, and changes in NAD/NADH and AMP/ATP ratios activate sirtuins and AMP-activated protein kinase (AMPK), respectively (Egan and Zierath, 2013). As a result, PPARα and PGC1α transcriptional activity is increased, mitochondrial biogenesis is induced, and fatty acid oxidation is enhanced. Conversely, in metabolic syndrome, adiposity is increased as energy supply outpaces demand. Circulating glucose, triglycerides, and free fatty acids are increased, as well as inflammatory mediators, insulin, leptin, and other adipokines. Excess free fatty acids and inflammatory cytokines released from adipose tissue macrophages disrupt insulin signaling and lead to a decrease in heart and skeletal muscle glucose utilization (Malik et al., 2004). Transcriptional regulation of metabolic genes is essential for adaptation of striated muscle to physiological challenges, and is predominantly regulated by NRs.
Metabolic Control by NRs in Muscle
NRs are transcription factors that are activated by multiple ligands including lipids, steroids, retinoids, and hormones (Burris et al., 2013). Many NRs are expressed in skeletal muscle and heart and transcriptionally regulate various aspects of metabolism including mitochondrial biogenesis, substrate utilization, and fiber type switching (Fan et al., 2013; Huss and Kelly, 2004). NRs well studied in this regard are peroxisome proliferator-activated receptors (PPARs), estrogen-related receptors (ERRs), thyroid hormone receptors (TRs), and Nur77 (Figure 1B).
PPARs are critical regulators of metabolic genes in striated muscle (Madrazo and Kelly, 2008). PPARα is expressed in tissues with a high capacity for fatty acid catabolism, such as heart and skeletal muscle. When bound to long chain fatty acids or eicosanoids, PPARα activates transcription of genes required for fatty acid uptake and oxidation. The role of PPARα in muscle and heart has been revealed using genetic mouse models. Pparα-deficient mice develop myocardial lipotoxicity, and inhibition of fatty acid metabolism in these mice results in lethal hypoglycemia (Djouadi et al., 1998) (Table 1). PPARα activation also robustly induces fatty acid utilization in human muscle cells (Muoio et al., 2002). In transgenic mouse models of skeletal muscle (Finck et al., 2005) and cardiac-specific overexpression of PPARα (Finck et al., 2002), skeletal muscle and the heart become insulin resistant. In diabetic patients, insulin resistance enhances PPARα activation in the heart resulting in altered cardiac metabolism (Young et al., 2002), and PPARα activity is decreased in the failing human heart (Lopaschuk et al., 2010).
Table 1.
Mediator Subunits and Interacting Nuclear Receptors that Regulate Metabolism
| Mediator Subunit or NR | Target Tissue (Organism, Driver) | Phenotype | Reference |
|---|---|---|---|
| MED1 | skeletal muscle deletion (mouse, MCK-Cre) | enhanced insulin sensitivity, improved glucose tolerance on high-fat diet | Chen et al., 2010 |
| MED1 | liver deletion (mouse, Albumin-Cre) | reduced hepatic steatosis on high-fat diet | Jia et al., 2004 |
| MED12 | heart/muscle deletion (Drosophila, Mef2-Gal4) | increased susceptibility to obesity | Lee et al., 2014 |
| MED13 | heart/muscle deletion (Drosophila, Mef2-Gal4) | increased susceptibility to obesity | Lee et al., 2014 |
| MED13 | heart deletion (mouse, αMHC-Cre) | increased susceptibility to obesity | Grueter et al., 2012 |
| MED13 | heart transgenic (mouse, αMHC) | enhanced lipid metabolism and insulin sensitivity, leanness, decreased susceptibility to obesity | Grueter et al., 2012; Baskin et al., 2014b |
| MED15 | global knockdown (C. elegans, RNAi) | decreased fat storage | Yang et al., 2006 |
| MED23 | liver deletion (mouse, Albumin-Cre) | increased glucose and lipid metabolism and insulin responsiveness | Chu et al., 2014 |
| MED30 | global missense mutation (mouse) | mitochondrial dysfunction in heart and heart failure | Krebs et al., 2011 |
| CDK8 | liver knockdown (mouse, tail vein injection of adenovirus expressing shRNA against CDK8) | hepatic steatosis | Zhao et al., 2012 |
| PPARα | global deletion (mouse) | etoxomir-induced cardiac lipotoxicity, hypoglycemia, lethality | Djouadi et al., 1998 |
| PPARα | heart transgenic (mouse, αMHC) | cardiac insulin resistance, diabetic cardiomyopathy | Finck et al., 2002 |
| PPARα | skeletal muscle transgenic (mouse, MCK) | skeletal muscle insulin resistance | Finck et al., 2005 |
| PPARβ/δ | heart deletion (mouse, αMHC-Cre) | lipotoxic cardiomyopathy | Cheng et al., 2004 |
| PPARβ/δ | heart transgenic (mouse, αMHC) | increased cardiac glucose metabolism, hearts protected from ischemia-reperfusion injury | Burkart et al., 2007 |
| PPARβ/δ | skeletal muscle deletion (mouse, HSA-Cre) | increased number of fast muscle fibers with reduced oxidative capacity, insulin resistance, adiposity | Schuler et al., 2006 |
| PPARβ/δ | skeletal muscle transgenic (mouse, HSA) | increased number of slow muscle fibers, leanness, decreased susceptibility to obesity, increased exercise endurance | Wang et al., 2004 |
| PPARγ | heart transgenic (mouse, αMHC) | dilated cardiomyopathy, increased cardiac lipid and glycogen storage | Son et al., 2007 |
| PPARγ | skeletal muscle deletion (mouse, MCK-Cre) | glucose intolerant, insulin resistant, increased adiposity | Hevener et al., 2003; Norris et al., 2003; |
| PPARγ | muscle constitutively active transgenic (mouse, HSA) | decreased intramuscular lipid accumulation, decreased susceptibility to obesity and insulin resistance, increased number of slow muscle fibers | Amin et al., 2010 |
| TRα | global deletion of α1 (mouse) | decreased muscle weight, increased proportion of slow-twitch fibers | Yu et al., 2000 |
| TRα/β | global deletion of α1/β (mouse) | decreased body and muscle weight, increased proportion of slow-twitch fibers | Yu et al., 2000 |
| TRα | global knock-in mutation (mouse) | decreased body weight, heart size, blood pressure, cardiac glucose utilization | Esaki et al., 2004 |
| TRβ | global knock-in mutation (mouse) | increased cardiac glucose utilization | Esaki et al., 2004 |
| TRβ | heart dominant-negative β1 (mouse, αMHC) | cardiac dysfunction, reduced substrate flux through multiple pathways in the heart | Pazos-Moura et al., 2000; Hyyti et al., 2008; |
| ERRα | global deletion (mouse) | impaired cardiac metabolism, cardiac maladaptation to hemodynamic stress and ischemia | Huss et al., 2007 |
| ERRα | skeletal muscle deletion (mouse, MCK-Cre) | impaired muscle regeneration and recovery of mitochondrial function in muscle after injury | LaBarge et al., 2014 |
| ERRβ/γ | skeletal muscle deletion (mouse, HSA-Cre) | decreased exercise endurance | Gan et al., 2013 |
| ERRγ | global deletion (mouse) | lethal, decreased embryonic cardiac function, disruption of perinatal cardiometabolic switching | Alaynick et al., 2007 |
| ERRγ | skeletal muscle transgenic (mouse, HSA) | increased exercise endurance, resistance to diet-induced obesity | Narkar et al., 2011 |
| Nur77 | global deletion (mouse) | high-fat-diet-induced skeletal muscle insulin resistance, increased susceptibility to obesity | Chao et al., 2009 |
| Nur77 | skeletal muscle transgenic (mouse, MCK) | increased mitochondrial number and function in skeletal muscle, improved muscle performance | Chao et al., 2012 |
Summary of genetic models of Mediator subunits that display a metabolic phenotype, and genetic mouse models of nuclear receptors that affect striated muscle, some of which interact with Mediator subunits, as discussed in the text. MCK, muscle creatine kinase; αMHC, alpha myosin heavy chain; HSA, human skeletal muscle actin.
PPARβ/δ, like PPARα, is activated by fatty acids derived from triglycerides (Neels and Grimaldi, 2014). PPARβ/δ facilitates the formation of oxidative muscle fibers through PGC1α activation (Schuler et al., 2006), and its overexpression in skeletal muscle causes a lean phenotype, mimicking exercise training (Wang et al., 2004). While deletion of Pparβ/δ in cardiac muscle causes lipotoxicity and progressive heart failure (Cheng et al., 2004), its overexpression in the heart increases cardiac glucose metabolism and protects the heart from ischemia-reperfusion injury (Burkart et al., 2007) (Figure 1B). PPARβ/δ may be a beneficial therapeutic target to combat metabolic syndrome given the role of the PPARβ/δ-selective agonist GW501516 as a potential exercise mimetic, although side effects of PPARβ/δ activation in other tissues may still confound its therapeutic potential (Fan et al., 2013).
PPARγ is enriched in adipose tissue, but also regulates metabolism in striated muscle (Ahmadian et al., 2013). Global deletion of Pparγ causes lethality in mice, and deletion in the heart leads to cardiac hypertrophy and dysfunction, although the metabolic effects of Pparγ deletion in the heart have not been evaluated (Madrazo and Kelly, 2008). Cardiac overexpression of PPARγ causes dilated cardiomyopathy accompanied by increased cardiac lipid and glycogen storage (Son et al., 2007). Interestingly, a constitutively active form of PPARγ expressed in skeletal muscle and heart decreased intramuscular lipid accumulation, susceptibility to insulin resistance and obesity, and induced a shift to slow-twitch myofibers (Amin et al., 2010). Conversely, mice with the deletion of Pparγ in skeletal muscle developed insulin resistance and increased adiposity (Hevener et al., 2003; Norris et al., 2003) (Table 1). Mouse models of PPARγ are useful tools for understanding tissue-specific functions of PPARγ, particularly because PPARγ is a therapeutic target for the management of metabolic diseases (Burris et al., 2013).
TRs are NRs that activate and repress transcription. Thyroid hormones activate TRs, and have long been known to play an important role in metabolism (Tata, 2013). TRs regulate expression of myosin heavy chain isoforms, a determinant of striated muscle contractility and fiber type, and ultimately energy substrate preference (Mullur et al., 2014; Yu et al., 2000). Increased TR transcriptional activity in muscle is accompanied by a switch from fast- to slow-twitch fiber type (Salvatore et al., 2014) (Figure 1B). Genetic models of TRs have also implicated TR in cardiac metabolism and function (Esaki et al., 2004; Hyyti et al., 2008; Kahaly and Dillmann, 2005; Pazos-Moura et al., 2000) (Table 1). Indeed, decreased thyroid hormone activity has been correlated with worsening heart failure, but efforts to improve heart function with TR agonists have been unsuccessful and are associated with adverse side effects (Burris et al., 2013). To date most in vivo studies on TRs have utilized genetic models with global TR gene disruption; however, TR isoforms have unique expression patterns. Given their role in striated muscle function and systemic metabolism, it will be important for future studies to determine tissue-specific regulation and function of TR isoforms (Flamant and Gauthier, 2013).
Orphan NRs, such as ERRs and Nur77, have no known natural ligands. ERRα is abundantly expressed in tissues with high metabolic and oxidative capacity, and its transcriptional activity is highly dependent on coregulatory proteins, such as PGC1α (Huss et al., 2004; Villena and Kralli, 2008). ERRα induces expression of fatty acid metabolism genes in skeletal and cardiac muscle, but regulates expression of metabolic genes in other cell types as well (Giguère, 2008). Mouse gene deletion studies have revealed a role for ERRα in regulation of cardiac energy metabolism and adaptation to pressure overload (Huss et al., 2007; Huss and Kelly, 2004; Villena and Kralli, 2008). ERRα is also important in muscle regeneration and recovery of mitochondrial function in muscle after injury (LaBarge et al., 2014). Global deletion of Errγ is lethal due to impairment in perinatal cardiac function and cardiometabolic switching (Alaynick et al., 2007). Overexpression of ERRγ in skeletal muscle increases exercise endurance as a result of fast-to-slow muscle fiber type switching (Narkar et al., 2011), and recently, ERRs were shown to regulate muscle fiber type switching in cooperation with PPARs through a muscle microRNA network (Gan et al., 2013). The orphan NR Nur77 is also important in muscle fiber type switching; it regulates expression of metabolic genes in muscle (Pearen and Muscat, 2010). Nur77 knockout mice develop skeletal muscle insulin resistance in response to a high-fat diet (Chao et al., 2009), whereas overexpression of Nur77 in muscle increases oxidative metabolism (Chao et al., 2012). While additional NRs may be involved in muscle fiber type switching, those more thoroughly studied are shown in Figure 1B.
NR transcriptional activities are facilitated by transcriptional coactivators, such as PGC1α, which promotes histone acetylation, binding to the Mediator complex, and recruitment of RNA polymerase II (Pol II) to initiate transcription (Mouchiroud et al., 2014; Wallberg et al., 2003). PGC1α was originally identified in brown adipose tissue through its association with PPARγ. It functions in tissues with high oxidative capacity to regulate myofiber switching, mitochondrial biogenesis, and expression of fatty acid oxidation enzymes (Figure 1B) (reviewed in Chan and Arany, 2014; Finck and Kelly, 2006; Lin et al., 2005). Overexpression of PGC1α in skeletal muscle causes a switch from fast to slow muscle fiber type accompanied by resistance to fatigue (Lin et al., 2002), and overexpression of PGC1α in the heart enhances mitochondrial biogenesis (Lehman et al., 2000). Conversely, global deletion of Pgc1α decreases exercise capacity and cardiac function, and disrupts cardiac metabolism (Arany et al., 2005; Leone et al., 2005). Based on these and other studies, it is evident that PGC1α-mediated NR activity is an important regulator of striated muscle metabolism and function. Indeed, PGCα1 expression is decreased in muscles of insulinresistant diabetic patients, and this is associated with decreased expression of fatty acid oxidation genes (Chan and Arany, 2014; Lin et al., 2005).
NR corepressors (NCoRs) are also important regulators of transcription (Mouchiroud et al., 2014). The two most relevant in the context of muscle metabolism and myofiber switching are corepressor receptor-interacting protein 140 (RIP140) and NCoRs. RIP140 was originally identified as a repressor of ERR transcriptional activity, but it also inhibits PPAR and TR activity. Mice with global overexpression of RIP140 present with decreased mitochondrial gene expression, mitochondrial activity, cardiac hypertrophy, and early mortality (reviewed in Nautiyal et al., 2013). NCoR also represses transcriptional activity of PPARs, ERRs, and TRs. Consequently, deletion of Ncor in muscle increases mitochondrial function and causes a switch from fast- to slow-twitch muscle fiber type (reviewed in Mottis et al., 2013) (Figure 1B).
Given their robust induction of metabolic gene transcription, NRs are attractive drug targets for treatment of metabolic disease. Indeed, PPARα was identified as the target of the triglyceride-lowering fibric acid derivative medications fenofibrate and gemfibrozil, which are used to treat hypertriglyceridemia (Krey et al., 1997). Thiazolidinediones (TZDs), also known as glitazones, are a class of drugs used in the treatment of diabetes. TZDs are PPARγ agonists and are currently the most potent insulin-sensitizing drugs. They are, however, limited in their use by cardiovascular side effects including increased incidence of heart failure and myocardial infarction (Burris et al., 2013). Side effects of targeting NRs are most likely due to the pleiotropic nature of NRs themselves, but could also be due to drug promiscuity. In this regard, it will be critical to determine the tissue-specific roles of the Mediator complex in regulating transcription in response to modulation of various NRs.
The Mediator Complex
The interaction of NRs, transcriptional coactivators and corepressors, general transcription factors, and chromatin modification complexes (collectively referred to as the pre-initiation complex [PIC]) with RNA Pol II occurs through the Mediator complex. Mediator comprises a ubiquitous group of nuclear proteins that regulate gene transcription through multiple mechanisms. First discovered in yeast as an essential regulator of activatorinduced transcription (Flanagan et al., 1991), the Mediator complex has been isolated from mammalian cells in active and inactive forms. The core Mediator complex contains 26 subunits and consists of three domains: the head, body, and tail (Taatjes, 2010). The CDK8 kinase submodule, consisting of four subunits, acts as a transcriptional regulator of the core Mediator complex. The kinase submodule represses the Mediator complex by allosterically inhibiting RNA Pol II binding, but also activates TR-dependent transcription by promoting Pol II recruitment to TR target genes (Belakavadi and Fondell, 2010; Fondell, 2013) (Figure 2). The Mediator complex is essential for PIC formation, by interacting with all the components of the PIC to initiate recruitment of RNA Pol II, resulting in activation of transcription (Poss et al., 2013).
Figure 2. The Mediator Complex.
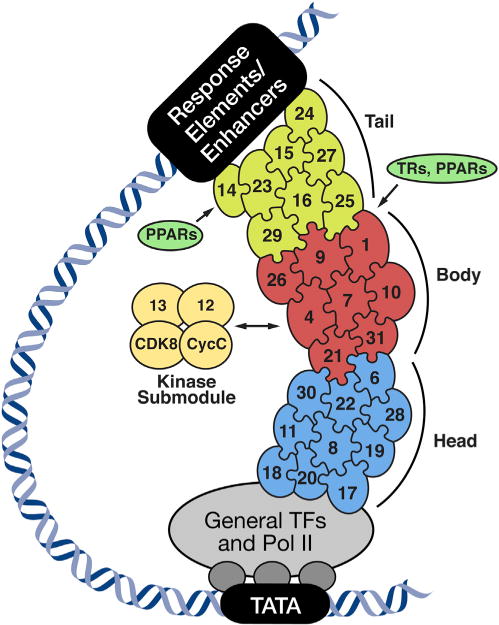
The transcriptionally active core Mediator complex consists of the head (blue), body (red), and tail (light green) domains, and the relative location of the subunits is depicted. The CDK8 kinase submodule (yellow) reversibly associates with the core complex and regulates Mediator transcriptional activity. The Mediator complex regulates transcription by interacting with nuclear receptors, general transcription factors (GTFs), RNA polymerase II (Pol II), and enhancers. TR, thyroid hormone receptor; PPARs, peroxisome proliferatoractivated receptors.
In human cells, the Mediator complex was first identified as a TR coactivator (Fondell et al., 1996). MED1 (also called TRAP220) was the first subunit identified and has since been shown to directly and functionally interact with nearly all NRs for which a ligand is known, including steroid and TRs, vitamin D and retinoic acid receptors, and PPARs (Chen and Roeder, 2011; Fondell, 2013). The Mediator requires an open chromatin conformation for binding and recruitment of RNA Pol II. In this regard, PGC1α has been shown to bind and recruit histone acetyltransferases, opening chromatin conformation, and facilitating transcription of MEF2 and PPAR regulated genes, likely through a Mediator-dependent mechanism (Puigserver et al., 1999). Given the significance of NRs in regulating metabolic gene expression, it is essential to determine the role of Mediator as it pertains to metabolic homeostasis.
Electron microscopy studies have demonstrated that the Mediator binds to transcription factor activation domains and subsequently changes conformation upon binding (Taatjes et al., 2002). The conformation adopted by the Mediator appears to be distinct for different transcription factors. Such a mechanism may enable the Mediator to differentially regulate transcription, depending upon transcription factor availability, possibly in a tissue-specific manner. There is preliminary evidence supporting this hypothesis in studies investigating the role of MED23 in different cell types (Balamotis et al., 2009). Because multiple signaling pathways converge on transcription factors, which are differentially regulated in a cell-specific manner, it is feasible that the ultimate effect on the Mediator complex and resulting gene expression is dependent on cell type and physiologic context. One could also imagine a scenario in which tissue-specific transcription factors are activated and bound to Mediator subunits, inducing a conformational change in Mediator and subsequently exposing other Mediator subunits for binding by additional transcriptional regulators.
Mediator-regulated transcription is not only controlled by signaling pathways and their downstream action on transcription factors, but also by posttranslational modifications (PTMs) on Mediator itself. Although these studies are still in their infancy, a number of phosphorylation sites have been identified on Mediator subunits (Miller et al., 2012). Interestingly, MED1 phosphorylation by extracellular signal-regulated kinase (ERK) leads to enhanced transcription of TR-regulated genes (Belakavadi et al., 2008). Additionally, MED13/MED13L are targeted for degradation through ubiquitination by SCF-Fbw7, which ultimately affects the transcriptional activity of the Mediator complex (Davis et al., 2013). It is likely that other PTMs regulate Mediator subunits, and of particular interest are those generated by metabolic pathways. For example, palmitoylation and/or O-linked beta-N-acetylglucosamine (O-GlcNAc) modification of Mediator subunits could enhance or inhibit interactions with other transcription factors and coactivators/corepressors (Ozcan et al., 2010). Acetylation, a more commonly studied metabolic PTM, not only regulates histone architecture and access to DNA, but also NR activity, and could also regulate Mediator activity (Lu and Thompson, 2012; Wang et al., 2011). While very little is known, it will be interesting to investigate the role of metabolically driven PTMs on Mediator activity, particularly as a regulator of tissue- and cell-specific transcriptional regulation. We suggest that metabolic PTMs and regulation of the Mediator complex are intimately tied to the metabolic demand of specific tissues.
In addition to being directly modified, Mediator components may also posttranslationally modify transcription factors. For example, the kinase CDK8, a component of the kinase submodule of the Mediator complex, was shown to phosphorylate the transcription factor sterol regulatory element binding protein (SREBP-1), leading to its ubiquitination and degradation, consequently decreasing lipogenesis (Zhao et al., 2012). PTMs of the Mediator complex, and modification of other transcriptional components by the Mediator, adds another layer of complexity to the regulation of transcription. These studies further support the hypothesis that Mediator components are regulated, and perhaps function, in a tissue-specific manner.
Metabolic Functions of Mediator Components in Muscle
The Mediator complex is required for normal cellular function, and genetic mutations in components of the Mediator complex have been implicated in a number of diseases. Several Mediator subunits are directly involved in the development or progression of cancer (Spaeth et al., 2011), and nearly every Mediator subunit is dysregulated in some type of cancer (Schiano et al., 2014a). Mediator subunits have also been implicated in neurodevelopmental disorders and cardiovascular defects and diseases (Grueter, 2013; Napoli et al., 2012; Schiano et al., 2014b). Additionally, MED1, MED23, MED25, MED15, MED12, and MED13 play key roles in regulating tissue and systemic metabolic homeostasis (Grueter, 2013; Jia et al., 2004; Napoli et al., 2012) (summarized in Table 1). The Mediator components that alter systemic metabolism in a muscle-specific manner are of particular interest (Figure 3).
Figure 3. Mediator Subunits Alter Systemic Metabolism in a Muscle-Specific Manner.
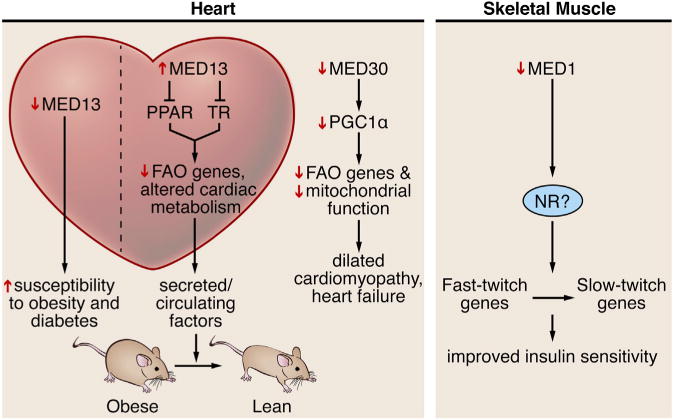
Perturbations of MED13 and MED30 in the heart alter metabolism. Overexpression of MED13 in the heart causes leanness, and loss of MED13 in the heart increases susceptibility of obesity and diabetes. Loss-of-function mutations in MED30 lead to altered substrate availability, heart failure, and death. Loss of MED1 in muscle causes a fast-to-slow muscle fiber type switch, which enhances insulin sensitivity and protects against obesity.
In metazoans, Mediator components are expressed ubiquitously throughout embryonic development and adulthood (Poss et al., 2013). Genetic deletion of Med1 (Zhu et al., 2000), Med24 (Ito et al., 2002), Cdk8 (Westerling et al., 2007), or MED12 (Hong et al., 2005) in mice results in embryonic or preimplantation lethality. However, mice with skeletal muscle-specific deletion of Med1 are viable. Female Med1 muscle knockout mice are resistant to high-fat-diet-induced obesity and have enhanced insulin sensitivity (Chen et al., 2010). These mice displayed an increase in mitochondrial gene expression and mitochondrial content of fast-twitch muscle, which also correlated with fast-to-slow myofiber switching. This could account for the improved insulin sensitivity in the setting of caloric excess. As mentioned above, patients with diabetes showed decreased oxidative capacity in muscle, which is suggestive of a slow- to fast-twitch fiber type switch. The inability of TR to activate transcription of fast-twitch genes in the absence of MED1 may account for a slow fiber type switch. Therefore, MED1 may function specifically in muscle to control whole-body metabolism through its influence on muscle fiber type composition. While the role of MED1 in diabetic patients is currently unknown, it will be of therapeutic interest to investigate the role of Mediator subunits in regulation of metabolic diseases.
Metabolic alterations in heart failure have been studied extensively and are likely transcriptionally regulated, perhaps through the Mediator complex (Lopaschuk et al., 2010). Flux through creatine kinase alters the phosphocreatine:ATP ratio, which leads to increased substrate demand. The heart meets this demand by increasing glucose utilization, ultimately shifting away from fatty acids as the main energy source (Doenst et al., 2013). Corresponding myosin switching occurs, with αMHC decreasing in abundance as βMHC increases, in addition to a reversion to a fetal metabolic gene profile (Razeghi et al., 2001). Underlying gene expression changes include downregulation of PGC1α-, PPARα-, and TR- regulated genes. Because NRs interact with Mediator to regulate transcription, components of the Mediator complex are likely involved in regulation of cardiac metabolism that occurs in heart failure, although this has not yet been investigated.
MED30 controls PGC1α-mediated expression of metabolic genes. Mice with a loss-of-function mutation in Med30 die from severe heart failure characterized by myocardial fibrosis, necrosis, and impaired contractility (Krebs et al., 2011). Heart failure progressed after weaning Med30 mutant mice due to mitochondrial dysfunction in the heart. Progressive downregulation of PGC1α and many oxidative phosphorylation and electron transport genes were observed post weaning. These changes suggested that altered substrate availability, which occurs with changes in diet post weaning (from a high- to a low-fat diet), could be responsible for altered gene expression and mitochondrial dysfunction. Indeed, Med30 mutant mice weaned onto a ketogenic diet, which is similar to breast milk in fat content, survived longer and showed partial restoration of mitochondrial gene expression (Krebs et al., 2011). While mutations in Med30 severely affect cardiac function and cardiac metabolism, Med30 mutant mice did not display a detectable skeletal muscle phenotype. These results suggest that MED30 and possibly other Mediator components could have tissue-specific roles. Alternatively, the presence of other tissue-specific nuclear proteins that interact with Mediator may be responsible for the cardiac phenotype in Med30 mutant mice.
MED13 is a component of the kinase submodule of the Mediator complex, which regulates Mediator activity. Studies in our laboratory identified MED13 as a regulator of systemic metabolism (Baskin et al., 2014b; Grueter et al., 2012). Overexpression of MED13 specifically in the heart led to a striking metabolic phenotype in which mice were resistant to obesity and maintained insulin sensitivity when placed on a high-fat diet (Grueter et al., 2012). In contrast, mice with cardiac-specific deletion of Med13 had increased susceptibility to obesity and diabetes (Grueter et al., 2012). We also demonstrated that Med13 deficiency in striated muscles results in a similar phenotype in Drosophila (Lee et al., 2014). However, the potential influence of MED13 in mammalian skeletal muscle on systemic energy homeostasis is currently under investigation.
MED13 is a target of microRNA-208a (miR-208a), a cardiac-specific microRNA that is critically important in myosin fiber type switching in response to cardiac stress (van Rooij et al., 2007). As in MED13 cardiac transgenic mice, wild-type (WT) mice treated with an inhibitor of miR-208a became lean and resistant to diet-induced obesity, and cardiac MED13 levels were increased (Grueter et al., 2012). Given the tissue-specific expression of some miRs, and the therapeutic potential of anti-miRs, MED13, and perhaps other Mediator components, are attractive anti-miR targets to combat obesity (Olson, 2014; Rottiers and Näär, 2012). However, much more still needs to be understood about the Mediator complex in order to unlock its therapeutic potential.
Other components of the Mediator complex have been implicated in the regulation of tissue-specific and systemic metabolism. CDK8 suppresses lipogenesis by phosphorylating and inhibiting SREBP-1 in the liver (Zhao et al., 2012). A separate study demonstrated that MED17 is required for liver X receptor (LXR)-dependent regulation of SREBP-1, thus activating hepatic lipogenesis (Kim et al., 2014). Yet another study demonstrated that MED15 is required for SREBP-1 activation of lipogenic target genes in nematodes (Yang et al., 2006). MED1, MED14, and MED23 are required for adipocyte development, and are ultimately involved in lipid metabolism as well (Chu et al., 2014; Zhang et al., 2013). Interestingly, the nuclear corepressor RIP140 interacts with CDK8 to regulate gene expression during fibroblast-adipocyte differentiation (Nautiyal et al., 2013). One function of the Mediator complex in muscle may be to inhibit transcription of PPAR-activated genes, which could result in release of molecular signals that modulate lipid metabolism in distant tissues, implicating Mediator in the suppression of diabetes and obesity. We consider it important to identify other Mediator subunits that modulate muscle and systemic metabolism, given the broad systemic metabolic effect of manipulation of Mediator components in only one tissue.
Mediator, Myokines, and the Control of Systemic Energy Homeostasis
The mechanism(s) whereby nuclear proteins in the heart and muscle can mediate metabolic changes in distant tissues is a clinically relevant and ongoing topic of investigation. It is likely that the Mediator plays a role in this process as evidenced by the influence of cardiac MED13 on leanness and resistance to diet-induced obesity (Grueter et al., 2012). We have recently demonstrated, using heterotypic parabiosis, that circulating/secreted factors control leanness in transgenic mice with cardiac-specific MED13 overexpression (TG) (Baskin et al., 2014b). WT mice from WT-TG heterotypic parabiotic pairs gained less weight over time than their WT-WT isotypic parabiotic controls. Furthermore, metabolic rates in liver and adipose tissue from WT mice subjected to heterotypic parabiosis were significantly elevated, to the same extent as in TG mice. Therefore, circulating factors within the systemic milieu of TG mice, perhaps secreted by the heart, are responsible for enhanced metabolic rate and leanness (Baskin et al., 2014b). We also identified wingless (wg) and armadillo as muscle-secreted proteins in Drosophila that were necessary and sufficient to suppress fat accumulation, and wg overexpression rescued the obese phenotype in Med13 knockout flies (Lee et al., 2014). These findings suggest that the heart and skeletal muscle produce signaling molecules that increase metabolic rate of distal tissues, and that this is regulated by Mediator.
There are other examples of secreted proteins that modulate systemic metabolism and facilitate inter-organ communication, and perhaps future studies will address the role of Mediator in secretion of these factors (Figure 4). The adipokine leptin is the most well-known adipose-derived hormone that regulates fat storage (Halaas et al., 1995). Adiponectin, a hormone released by adipose tissue, also acts on muscle to stimulate fatty acid oxidation, and acts on liver to suppress gluconeogenesis (Kadowaki et al., 2006). Other adipokines that affect systemic metabolism include adipsin, resistin, and secreted frizzled-related protein 5 (sfrp5) (Deng and Scherer, 2010). Insulin-like growth factor 1 (IGF-1) and fibroblast growth factor 21 (FGF-21) also regulate systemic metabolism and are secreted from adipose tissue and liver, thus acting as adipokines and hepatokines (Stefan and Häring, 2013).
Figure 4. Proposed Roles for Mediator in Regulating Secreted Factors and Systemic Metabolism.
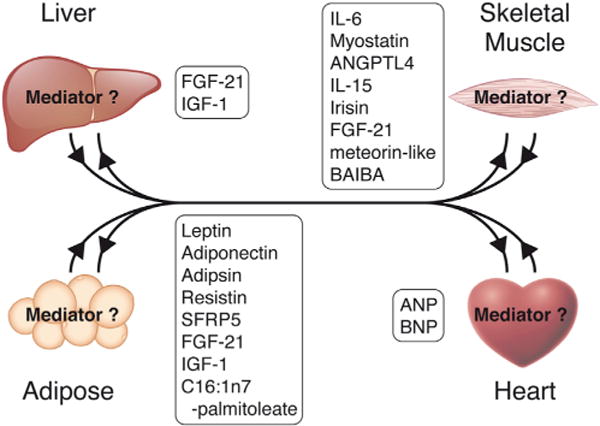
Secreted factors released from adipose tissue and liver regulate skeletal and cardiac muscle metabolism, and many factors are released from heart and muscle that exert feedback on these tissues. While the role of Mediator in regulating secreted factors has not yet been investigated, it is highly likely that Mediator is involved in this process. See text for discussion.
Similar to adipokines, myokines are factors secreted from muscle that act in an autocrine and/or paracrine manner (Pedersen and Febbraio, 2012; Weigert et al., 2014). Interleukin 6 (IL-6), released from muscle with exercise, was the first classified myokine (Pedersen et al., 2004). IL-6 has been investigated in many experimental settings and has tissue-specific effects (reviewed in Pal et al., 2014), although it seems to play a role in glucose and fatty acid metabolism in muscle and other organs acutely after exercise (Muñoz-Cánoves et al., 2013). Myostatin also regulates glucose metabolism, and myostatin-deficient mice are resistant to diet-induced obesity (Huang et al., 2011). Other myokines with roles in systemic metabolism include angiopoietin-like protein 4 (ANGPTL4) (Mattijssen and Kersten, 2012), IL-15, irisin, FGF-21 (reviewed in Pedersen and Febbraio, 2012), and meteorin-like protein (Rao et al., 2014). GDF11 is another important circulating factor that has beneficial effects on multiple tissues, and it has most recently been shown to act as a “rejuvenating” factor by improving muscle function in old mice (Sinha et al., 2014).
Cardiokines are factors secreted from the heart, although few have been identified to date (Doroudgar and Glembotski, 2011; Shimano et al., 2012). Unlike many of the known myokines, most cardiokines identified act in an autocrine manner to regulate cardiac function and response to stress. For example, follistatin-like 1 is antiapoptotic, and heat shock protein 20 (Hsp20) regulates angiogenesis in the heart. Thrombospondin 4 (TWEAK), mesencephalic astrocyte-derived neurotrophic factor (MANF), the fibroblast growth factors FGF-21 and FGF-23 regulate cardiac hypertrophy (Shimano et al., 2012), and FGF-21 and IGF-1 are cardiokines that act in an autocrine manner to regulate cardiac metabolism (Brahma et al., 2014; Kajstura et al., 2001). Perhaps the only cardiokines known to regulate systemic metabolism are atrial natriuretic peptide (ANP) and B-type/ventricular natriuretic peptide (BNP), which lead to browning of white adipose tissue (Bordicchia et al., 2012).
There are also examples of non-protein circulating factors that act on distant tissues. For example, mice lacking adipose fatty acid binding proteins FABP4 and FABP5 revealed a “lipokine” (C16:1n7-palmitoleate), secreted by adipocytes that acts on liver and skeletal muscle, rendering mice resistant to metabolic syndrome (Cao et al., 2008). Along these same lines, PGC1α was recently shown to regulate the metabolic myokine β-aminoisobutyric acid (BAIBA). Consequently, BAIBA induced a browning phenotype in white adipose tissue (Roberts et al., 2014). Additionally, metabolic substrates, products, and intermediary metabolites can act as “metabokines.” Indeed, altered substrate flux has been suggested to regulate systemic metabolism through cross-talk between heart and liver in the setting of hypertrophic cardiomyopathy (Baskin et al., 2014a; Magida and Leinwand, 2014). These discoveries are surely the tip of the iceberg in deciphering inter-organ communication driven by Mediator regulation of secreted factors, particularly the most recent class of factors dubbed “myometabokines” (Weigert et al., 2014).
Questions for the Future
Investigation of Mediator components in muscle tissue is a developing field that can potentially reveal novel molecules and pathways that regulate metabolism. Deletion or overexpression of Mediator components in heart and skeletal muscle modulates transcription of numerous genes and, in particular, NR-regulated genes. Many of these genes are also regulated in muscle in response to heart failure, obesity, and diabetes, indicating that transcriptional manipulation by the Mediator in muscle may possibly modulate systemic metabolism. Regulation of metabolic homeostasis by secreted factors may likely be controlled by Mediator in a tissue-specific manner as well. The interaction between Mediator components, muscle-specific transcription factors, NRs, coregulators, and other factors may explain the systemic effects of tissue-specific manipulation of ubiquitous Mediator components.
Our understanding of the Mediator complex has developed quite rapidly, including the identification of Mediator components, the structures of various subunits, and its general role in transcription. These studies are just the beginning as we translate what we know from in vitro studies to the role of the Mediator in specific tissues in vivo. Based on the studies we reviewed here, it is evident that the Mediator complex is an important regulator of metabolism and plays a significant role in striated muscle. We speculate that modulation of other Mediator components in muscle will likely affect systemic metabolism.
NRs are prominent regulators of metabolism and muscle fiber type, and recent evidence suggests that NRs interact with Mediator components to regulate transcription (Chen and Roeder, 2011; Fondell, 2013). NRs are regulated by a variety of physiological stimuli including exercise, and therefore it is logical to presume that the Mediator complex is regulated in a similar manner. There are no known instances of regulation of Mediator with exercise, and to date, only a few studies on Mediator in muscle have been reported (Figure 3). Future studies should focus on how the Mediator complex is regulated in striated muscle at baseline and in response to various physiological stimuli, such as exercise and metabolic and nutrient stress (e.g., fasting or different diets). It will also be of interest to determine the role of Mediator components using gain- and loss-of-function mouse models.
Of course, many questions remain unanswered regarding the regulation of the Mediator complex itself. MED13, a regulator of systemic metabolism, is inhibited by miR-208a, and perhaps other Mediator components follow suit. Indeed, in a screen for target genes, several miRs have been shown to regulate the expression of Mediator components in an embryonic stem cell line (Davis et al., 2012). Additionally, miR-146, miR-1, and miR-205 were shown to bind and regulate MED1 expression in spermatogonia, cancer cells, prostate cells, and trophoblasts, respectively (Hulf et al., 2013; Huszar and Payne, 2013; Jiang et al., 2014; Mouillet et al., 2010). Because MED1 seems to be regulated by multiple miRs, perhaps in a cell-specific manner, it will be important to determine the endogenous role of MED1 in these cell types. Furthermore, it will be interesting to extend these findings in vivo.
Finally, human mutations in Mediator subunits have been associated with developmental disorders, cardiovascular diseases, and cancer (Grueter, 2013; Napoli et al., 2012; Schiano et al., 2014b). Genetic mutations of Mediator components have not been directly linked to metabolic diseases; however, it is likely that genetic variants could disrupt Mediator-NR interactions causing adverse metabolic consequences. Perhaps genome-wide association studies (GWASs) can provide clues about possible genetic mutations in Mediator subunits underlying metabolic diseases. However, it may very well be the case that the interaction with NRs, which facilitate transcriptional activity of the Mediator, is most important in the context of metabolic regulation by the Mediator complex.
Acknowledgments
We apologize to our colleagues whose work could not be cited due to space limitations and our narrowly focused Perspective. We thank Dr. Angie L. Bookout for thought-provoking discussions and careful reading of the manuscript, and Jose Cabrera for the graphics. This work was supported by grants from the NIH (HL-077439, HL-111665, HL-093039, DK-099653, and U01-HL-100401), Foundation Leducq Networks of Excellence, Cancer Prevention & Research Institute of Texas, and the Robert A. Welch Foundation (grant 1-0025 to E.N.O.). K.K.B. was supported by a fellowship from the American Heart Association (14POST18320034), and B.R.W. was supported by NIH T32 grant.
References
- Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19:557–566. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alaynick WA, Kondo RP, Xie W, He W, Dufour CR, Downes M, Jonker JW, Giles W, Naviaux RK, Giguère V, Evans RM. ERRgamma directs and maintains the transition to oxidative metabolism in the postnatal heart. Cell Metab. 2007;6:13–24. doi: 10.1016/j.cmet.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Amin RH, Mathews ST, Camp HS, Ding L, Leff T. Selective activation of PPARgamma in skeletal muscle induces endogenous production of adiponectin and protects mice from diet-induced insulin resistance. Am J Physiol Endocrinol Metab. 2010;298:E28–E37. doi: 10.1152/ajpendo.00446.2009. [DOI] [PubMed] [Google Scholar]
- Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Balamotis MA, Pennella MA, Stevens JL, Wasylyk B, Belmont AS, Berk AJ. Complexity in transcription control at the activation domainmediator interface. Sci Signal. 2009;2:ra20. doi: 10.1126/scisignal.1164302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskin KK, Bookout AL, Olson EN. The heart-liver metabolic axis: defective communication exacerbates disease. EMBO Mol Med. 2014a;6:436–438. doi: 10.1002/emmm.201303800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskin KK, Grueter CE, Kusminski CM, Holland WL, Bookout AL, Satapati S, Kong YM, Burgess SC, Malloy CR, Scherer PE, et al. MED13-dependent signaling from the heart confers leanness by enhancing metabolism in adipose tissue and liver. EMBO Mol Med. 2014b;6:1610–1621. doi: 10.15252/emmm.201404218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassel-Duby R, Olson EN. Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem. 2006;75:19–37. doi: 10.1146/annurev.biochem.75.103004.142622. [DOI] [PubMed] [Google Scholar]
- Battiprolu PK, Gillette TG, Wang ZV, Lavandero S, Hill JA. Diabetic cardiomyopathy: mechanisms and therapeutic targets. Drug Discov Today Dis Mech. 2010;7:e135–e143. doi: 10.1016/j.ddmec.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belakavadi M, Fondell JD. Cyclin-dependent kinase 8 positively cooperates with Mediator to promote thyroid hormone receptor-dependent transcriptional activation. Mol Cell Biol. 2010;30:2437–2448. doi: 10.1128/MCB.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belakavadi M, Pandey PK, Vijayvargia R, Fondell JD. MED1 phosphorylation promotes its association with mediator: implications for nuclear receptor signaling. Mol Cell Biol. 2008;28:3932–3942. doi: 10.1128/MCB.02191-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordicchia M, Liu D, Amri EZ, Ailhaud G, Dessì-Fulgheri P, Zhang C, Takahashi N, Sarzani R, Collins S. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J Clin Invest. 2012;122:1022–1036. doi: 10.1172/JCI59701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahma MK, Adam RC, Pollak NM, Jaeger D, Zierler KA, Poecher N, Schreiber R, Romauch M, Moustafa T, Eder S, et al. Fgf21 is induced upon cardiac stress and alters cardiac lipid homeostasis. J Lipid Res. 2014;55:2229–2241. doi: 10.1194/jlr.M044784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkart EM, Sambandam N, Han X, Gross RW, Courtois M, Gierasch CM, Shoghi K, Welch MJ, Kelly DP. Nuclear receptors PPAR-beta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest. 2007;117:3930–3939. doi: 10.1172/JCI32578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris TP, Solt LA, Wang Y, Crumbley C, Banerjee S, Griffett K, Lundasen T, Hughes T, Kojetin DJ. Nuclear receptors and their selective pharmacologic modulators. Pharmacol Rev. 2013;65:710–778. doi: 10.1124/pr.112.006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Gerhold K, Mayers JR, Wiest MM, Watkins SM, Hotamisligil GS. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell. 2008;134:933–944. doi: 10.1016/j.cell.2008.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan MC, Arany Z. The many roles of PGC-1α in muscle–recent developments. Metabolism. 2014;63:441–451. doi: 10.1016/j.metabol.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LC, Wroblewski K, Zhang Z, Pei L, Vergnes L, Ilkayeva OR, Ding SY, Reue K, Watt MJ, Newgard CB, et al. Insulin resistance and altered systemic glucose metabolism in mice lacking Nur77. Diabetes. 2009;58:2788–2796. doi: 10.2337/db09-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LC, Wroblewski K, Ilkayeva OR, Stevens RD, Bain J, Meyer GA, Schenk S, Martinez L, Vergnes L, Narkar VA, et al. Skeletal muscle Nur77 expression enhances oxidative metabolism and substrate utilization. J Lipid Res. 2012;53:2610–2619. doi: 10.1194/jlr.M029355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Roeder RG. Mediator-dependent nuclear receptor function. Semin Cell Dev Biol. 2011;22:749–758. doi: 10.1016/j.semcdb.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Zhang X, Birsoy K, Roeder RG. A muscle-specific knockout implicates nuclear receptor coactivator MED1 in the regulation of glucose and energy metabolism. Proc Natl Acad Sci USA. 2010;107:10196–10201. doi: 10.1073/pnas.1005626107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N, Evans RM, Schneider MD, Brako FA, Xiao Y, et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med. 2004;10:1245–1250. doi: 10.1038/nm1116. [DOI] [PubMed] [Google Scholar]
- Chu Y, Gómez Rosso L, Huang P, Wang Z, Xu Y, Yao X, Bao M, Yan J, Song H, Wang G. Liver Med23 ablation improves glucose and lipid metabolism through modulating FOXO1 activity. Cell Res. 2014;24:1250–1265. doi: 10.1038/cr.2014.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czubryt MP, McAnally J, Fishman GI, Olson EN. Regulation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1 alpha) and mitochondrial function by MEF2 and HDAC5. Proc Natl Acad Sci USA. 2003;100:1711–1716. doi: 10.1073/pnas.0337639100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MP, Abreu-Goodger C, van Dongen S, Lu D, Tate PH, Bartonicek N, Kutter C, Liu P, Skarnes WC, Enright AJ, Dunham I. Large-scale identification of microRNA targets in murine Dgcr8-deficient embryonic stem cell lines. PLoS ONE. 2012;7:e41762. doi: 10.1371/journal.pone.0041762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MA, Larimore EA, Fissel BM, Swanger J, Taatjes DJ, Clurman BE. The SCF-Fbw7 ubiquitin ligase degrades MED13 and MED13L and regulates CDK8 module association with Mediator. Genes Dev. 2013;27:151–156. doi: 10.1101/gad.207720.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Scherer PE. Adipokines as novel biomarkers and regulators of the metabolic syndrome. Ann N Y Acad Sci. 2010;1212:E1–E19. doi: 10.1111/j.1749-6632.2010.05875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouadi F, Weinheimer CJ, Saffitz JE, Pitchford C, Bastin J, Gonzalez FJ, Kelly DP. A gender-related defect in lipid metabolism and glucose homeostasis in peroxisome proliferator-activated receptor alpha-deficient mice. J Clin Invest. 1998;102:1083–1091. doi: 10.1172/JCI3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. 2013;113:709–724. doi: 10.1161/CIRCRESAHA.113.300376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroudgar S, Glembotski CC. The cardiokine story unfolds: ischemic stress-induced protein secretion in the heart. Trends Mol Med. 2011;17:207–214. doi: 10.1016/j.molmed.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013;17:162–184. doi: 10.1016/j.cmet.2012.12.012. [DOI] [PubMed] [Google Scholar]
- Esaki T, Suzuki H, Cook M, Shimoji K, Cheng SY, Sokoloff L, Nunez J. Cardiac glucose utilization in mice with mutated alpha- and beta-thyroid hormone receptors. Am J Physiol Endocrinol Metab. 2004;287:E1149–E1153. doi: 10.1152/ajpendo.00078.2004. [DOI] [PubMed] [Google Scholar]
- Fan W, Atkins AR, Yu RT, Downes M, Evans RM. Road to exercise mimetics: targeting nuclear receptors in skeletal muscle. J Mol Endocrinol. 2013;51:T87–T100. doi: 10.1530/JME-13-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–130. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Bernal-Mizrachi C, Han DH, Coleman T, Sambandam N, LaRiviere LL, Holloszy JO, Semenkovich CF, Kelly DP. A potential link between muscle peroxisome proliferator- activated receptor-alpha signaling and obesity-related diabetes. Cell Metab. 2005;1:133–144. doi: 10.1016/j.cmet.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Flamant F, Gauthier K. Thyroid hormone receptors: the challenge of elucidating isotype-specific functions and cell-specific response. Biochim Biophys Acta. 2013;1830:3900–3907. doi: 10.1016/j.bbagen.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Flanagan PM, Kelleher RJ, 3rd, Sayre MH, Tschochner H, Kornberg RD. A mediator required for activation of RNA polymerase II transcription in vitro. Nature. 1991;350:436–438. doi: 10.1038/350436a0. [DOI] [PubMed] [Google Scholar]
- Fondell JD. The Mediator complex in thyroid hormone receptor action. Biochim Biophys Acta. 2013;1830:3867–3875. doi: 10.1016/j.bbagen.2012.02.012. [DOI] [PubMed] [Google Scholar]
- Fondell JD, Ge H, Roeder RG. Ligand induction of a transcriptionally active thyroid hormone receptor coactivator complex. Proc Natl Acad Sci USA. 1996;93:8329–8333. doi: 10.1073/pnas.93.16.8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher D, Belmonte D, Deurenberg P, Wang Z, Krasnow N, PiSunyer FX, Heymsfield SB. Organ-tissue mass measurement allows modeling of REE and metabolically active tissue mass. Am J Physiol. 1998;275:E249–E258. doi: 10.1152/ajpendo.1998.275.2.E249. [DOI] [PubMed] [Google Scholar]
- Gan Z, Rumsey J, Hazen BC, Lai L, Leone TC, Vega RB, Xie H, Conley KE, Auwerx J, Smith SR, et al. Nuclear receptor/microRNA circuitry links muscle fiber type to energy metabolism. J Clin Invest. 2013;123:2564–2575. doi: 10.1172/JCI67652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguère V. Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr Rev. 2008;29:677–696. doi: 10.1210/er.2008-0017. [DOI] [PubMed] [Google Scholar]
- Grueter CE. Mediator complex dependent regulationof cardiac development and disease. Genomics Proteomics Bioinformatics. 2013;11:151–157. doi: 10.1016/j.gpb.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueter CE, van Rooij E, Johnson BA, DeLeon SM, Sutherland LB, Qi X, Gautron L, Elmquist JK, Bassel-Duby R, Olson EN. A cardiac microRNA governs systemic energy homeostasis by regulation of MED13. Cell. 2012;149:671–683. doi: 10.1016/j.cell.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- Hamby RI, Zoneraich S, Sherman L. Diabetic cardiomyopathy. JAMA. 1974;229:1749–1754. [PubMed] [Google Scholar]
- Hevener AL, He W, Barak Y, Le J, Bandyopadhyay G, Olson P, Wilkes J, Evans RM, Olefsky J. Muscle-specific Pparg deletion causes insulin resistance. Nat Med. 2003;9:1491–1497. doi: 10.1038/nm956. [DOI] [PubMed] [Google Scholar]
- Hong SK, Haldin CE, Lawson ND, Weinstein BM, Dawid IB, Hukriede NA. The zebrafish kohtalo/trap230 gene is required for the development of the brain, neural crest, and pronephric kidney. Proc Natl Acad Sci USA. 2005;102:18473–18478. doi: 10.1073/pnas.0509457102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Chen X, Chen D. Myostatin: a novel insight into its role in metabolism, signal pathways, and expression regulation. Cell Signal. 2011;23:1441–1446. doi: 10.1016/j.cellsig.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Hulf T, Sibbritt T, Wiklund ED, Patterson K, Song JZ, Stirzaker C, Qu W, Nair S, Horvath LG, Armstrong NJ, et al. Epigenetic-induced repression of microRNA-205 is associated with MED1 activation and a poorer prognosis in localized prostate cancer. Oncogene. 2013;32:2891–2899. doi: 10.1038/onc.2012.300. [DOI] [PubMed] [Google Scholar]
- Huss JM, Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res. 2004;95:568–578. doi: 10.1161/01.RES.0000141774.29937.e3. [DOI] [PubMed] [Google Scholar]
- Huss JM, Torra IP, Staels B, Giguère V, Kelly DP. Estrogenrelated receptor alpha directs peroxisome proliferator-activated receptor alpha signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol Cell Biol. 2004;24:9079–9091. doi: 10.1128/MCB.24.20.9079-9091.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huss JM, Imahashi K, Dufour CR, Weinheimer CJ, Courtois M, Kovacs A, Giguére V, Murphy E, Kelly DP. The nuclear receptor ERRalpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 2007;6:25–37. doi: 10.1016/j.cmet.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Huszar JM, Payne CJ. MicroRNA 146 (Mir146) modulates spermatogonial differentiation by retinoic acid in mice. Biol Reprod. 2013;88:15. doi: 10.1095/biolreprod.112.103747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyyti OM, Olson AK, Ge M, Ning XH, Buroker NE, Chung Y, Jue T, Portman MA. Cardioselective dominant-negative thyroid hormone receptor (Delta337T) modulates myocardial metabolism and contractile efficiency. Am J Physiol Endocrinol Metab. 2008;295:E420–E427. doi: 10.1152/ajpendo.90329.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Okano HJ, Darnell RB, Roeder RG. The TRAP100 component of the TRAP/Mediator complex isessential in broad transcriptional events and development. EMBO J. 2002;21:3464–3475. doi: 10.1093/emboj/cdf348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Qi C, Kashireddi P, Surapureddi S, Zhu YJ, Rao MS, Le Roith D, Chambon P, Gonzalez FJ, Reddy JK. Transcription coactivator PBP, the peroxisome proliferator-activated receptor (PPAR)-binding protein, is required for PPARalpha-regulated gene expression in liver. J Biol Chem. 2004;279:24427–24434. doi: 10.1074/jbc.M402391200. [DOI] [PubMed] [Google Scholar]
- Jiang C, Chen H, Shao L, Wang Q. MicroRNA-1 functions as a potential tumor suppressor in osteosarcoma by targeting Med1 and Med31. Oncol Rep. 2014;32:1249–1256. doi: 10.3892/or.2014.3274. [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006;116:1784–1792. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahaly GJ, Dillmann WH. Thyroid hormone action in the heart. Endocr Rev. 2005;26:704–728. doi: 10.1210/er.2003-0033. [DOI] [PubMed] [Google Scholar]
- Kajstura J, Fiordaliso F, Andreoli AM, Li B, Chimenti S, Medow MS, Limana F, Nadal-Ginard B, Leri A, Anversa P. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes. 2001;50:1414–1424. doi: 10.2337/diabetes.50.6.1414. [DOI] [PubMed] [Google Scholar]
- Kim GH, Oh GS, Yoon J, Lee GG, Lee KU, Kim SW. Hepatic TRAP80 selectively regulates lipogenic activity of liver X receptor. J Clin Invest. 2014 doi: 10.1172/JCI73615. Published online December 1, 2014. http://dx.doi.org/10.1172/JCI73615. [DOI] [PMC free article] [PubMed]
- Krebs P, Fan W, Chen YH, Tobita K, Downes MR, Wood MR, Sun L, Li X, Xia Y, Ding N, et al. Lethal mitochondrial cardiomyopathy in a hypomorphic Med30 mouse mutant is ameliorated by ketogenic diet. Proc Natl Acad Sci USA. 2011;108:19678–19682. doi: 10.1073/pnas.1117835108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krey G, Braissant O, L’Horset F, Kalkhoven E, Perroud M, Parker MG, Wahli W. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivatordependent receptor ligand assay. Mol Endocrinol. 1997;11:779–791. doi: 10.1210/mend.11.6.0007. [DOI] [PubMed] [Google Scholar]
- LaBarge S, McDonald M, Smith-Powell L, Auwerx J, Huss JM. Estrogen-related receptor-alpha (ERRalpha) deficiency in skeletal muscle impairs regeneration in response to injury. FASEB J. 2014;28:1082–1097. doi: 10.1096/fj.13-229211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBrasseur NK, Walsh K, Arany Z. Metabolic benefits of resistance training and fast glycolytic skeletal muscle. Am J Physiol Endocrinol Metab. 2011;300:E3–E10. doi: 10.1152/ajpendo.00512.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Bassel-Duby R, Olson EN. Heart- and muscle-derived signaling system dependent on MED13 and Wingless controls obesity in Drosophila. Proc Natl Acad Sci USA. 2014;111:9491–9496. doi: 10.1073/pnas.1409427111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, et al. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepaticsteatosis. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM. Metabolic controlthrough the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab. 2012;16:9–17. doi: 10.1016/j.cmet.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrazo JA, Kelly DP. The PPAR trio: regulators of myocardial energy metabolism in health and disease. J Mol Cell Cardiol. 2008;44:968–975. doi: 10.1016/j.yjmcc.2008.03.021. [DOI] [PubMed] [Google Scholar]
- Magida JA, Leinwand LA. Metabolic crosstalk between the heart and liver impacts familial hypertrophic cardiomyopathy. EMBO Mol Med. 2014;6:482–495. doi: 10.1002/emmm.201302852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, Wong ND, Franklin SS, Kamath TV, L’Italien GJ, Pio JR, Williams GR. Impact of the metabolic syndrome on mortality from coronary heart disease, cardiovascular disease, and all causes in United States adults. Circulation. 2004;110:1245–1250. doi: 10.1161/01.CIR.0000140677.20606.0E. [DOI] [PubMed] [Google Scholar]
- Mattijssen F, Kersten S. Regulation of triglyceride metabolism by Angiopoietin-like proteins. Biochim Biophys Acta. 2012;1821:782–789. doi: 10.1016/j.bbalip.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Miller C, Matic I, Maier KC, Schwalb B, Roether S, Strässer K, Tresch A, Mann M, Cramer P. Mediator phosphorylation prevents stress response transcription during non-stress conditions. J Biol Chem. 2012;287:44017–44026. doi: 10.1074/jbc.M112.430140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Mottis A, Mouchiroud L, Auwerx J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 2013;27:819–835. doi: 10.1101/gad.214023.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchiroud L, Eichner LJ, Shaw RJ, Auwerx J. Transcriptional coregulators: fine-tuning metabolism. Cell Metab. 2014;20:26–40. doi: 10.1016/j.cmet.2014.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouillet JF, Chu T, Nelson DM, Mishima T, Sadovsky Y. MiR-205 silences MED1 in hypoxic primary human trophoblasts. FASEB J. 2010;24:2030–2039. doi: 10.1096/fj.09-149724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullur R, Liu YY, Brent GA. Thyroid hormone regulation of metabolism. Physiol Rev. 2014;94:355–382. doi: 10.1152/physrev.00030.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Cánoves P, Scheele C, Pedersen BK, Serrano AL. Interleukin-6 myokine signaling in skeletal muscle: a double-edged sword? FEBS J. 2013;280:4131–4148. doi: 10.1111/febs.12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muoio DM, Way JM, Tanner CJ, Winegar DA, Kliewer SA, Houmard JA, Kraus WE, Dohm GL. Peroxisome proliferator-activated receptor-alpha regulates fatty acid utilization in primary human skeletal muscle cells. Diabetes. 2002;51:901–909. doi: 10.2337/diabetes.51.4.901. [DOI] [PubMed] [Google Scholar]
- Napoli C, Sessa M, Infante T, Casamassimi A. Unraveling framework of the ancestral Mediator complex in human diseases. Biochimie. 2012;94:579–587. doi: 10.1016/j.biochi.2011.09.016. [DOI] [PubMed] [Google Scholar]
- Narkar VA, Fan W, Downes M, Yu RT, Jonker JW, Alaynick WA, Banayo E, Karunasiri MS, Lorca S, Evans RM. Exercise and PGC-1α-independent synchronization of type I muscle metabolism and vasculature by ERRγ. Cell Metab. 2011;13:283–293. doi: 10.1016/j.cmet.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nautiyal J, Christian M, Parker MG. Distinct functions for RIP140 in development, inflammation, and metabolism. Trends Endocrinol Metab. 2013;24:451–459. doi: 10.1016/j.tem.2013.05.001. [DOI] [PubMed] [Google Scholar]
- Neels JG, Grimaldi PA. Physiological functions of peroxisome proliferator-activated receptor β. Physiol Rev. 2014;94:795–858. doi: 10.1152/physrev.00027.2013. [DOI] [PubMed] [Google Scholar]
- Norris AW, Chen L, Fisher SJ, Szanto I, Ristow M, Jozsi AC, Hirshman MF, Rosen ED, Goodyear LJ, Gonzalez FJ, et al. Muscle-specific PPARgamma-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. J Clin Invest. 2003;112:608–618. doi: 10.1172/JCI17305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson EN. MicroRNAs as therapeutic targets and biomarkers of cardiovascular disease. Sci Transl Med. 2014;6:239ps233. doi: 10.1126/scitranslmed.3009008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan S, Andrali SS, Cantrell JE. Modulation of transcription factor function by O-GlcNAc modification. Biochim Biophys Acta. 2010;1799:353–364. doi: 10.1016/j.bbagrm.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal M, Febbraio MA, Whitham M. From cytokine to myokine: the emerging role of interleukin-6 in metabolic regulation. Immunol Cell Biol. 2014;92:331–339. doi: 10.1038/icb.2014.16. [DOI] [PubMed] [Google Scholar]
- Pazos-Moura C, Abel ED, Boers ME, Moura E, Hampton TG, Wang J, Morgan JP, Wondisford FE. Cardiac dysfunction caused by myocardium-specific expression of a mutant thyroid hormone receptor. Circ Res. 2000;86:700–706. doi: 10.1161/01.res.86.6.700. [DOI] [PubMed] [Google Scholar]
- Pearen MA, Muscat GE. Minireview: nuclear hormone receptor 4A signaling: implications for metabolic disease. Mol Endocrinol. 2010;24:1891–1903. doi: 10.1210/me.2010-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol. 2012;8:457–465. doi: 10.1038/nrendo.2012.49. [DOI] [PubMed] [Google Scholar]
- Pedersen BK, Steensberg A, Fischer C, Keller C, Keller P, Plomgaard P, Wolsk-Petersen E, Febbraio M. The metabolic role of IL-6 produced during exercise: is IL-6 an exercise factor? Proc Nutr Soc. 2004;63:263–267. doi: 10.1079/PNS2004338. [DOI] [PubMed] [Google Scholar]
- Poss ZC, Ebmeier CC, Taatjes DJ. The Mediator complex and transcription regulation. Crit Rev Biochem Mol Biol. 2013;48:575–608. doi: 10.3109/10409238.2013.840259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Adelmant G, Wu Z, Fan M, Xu J, O’Malley B, Spiegelman BM. Activation of PPARgamma coactivator-1 through transcription factor docking. Science. 1999;286:1368–1371. doi: 10.1126/science.286.5443.1368. [DOI] [PubMed] [Google Scholar]
- Rao RR, Long JZ, White JP, Svensson KJ, Lou J, Lokurkar I, Jedrychowski MP, Ruas JL, Wrann CD, Lo JC, et al. Meteorin-like is a hormone that regulates immune-adipose interactions to increase beige fat thermogenesis. Cell. 2014;157:1279–1291. doi: 10.1016/j.cell.2014.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H. Metabolic gene expression in fetal and failing human heart. Circulation. 2001;104:2923–2931. doi: 10.1161/hc4901.100526. [DOI] [PubMed] [Google Scholar]
- Roberts LD, Boström P, O’Sullivan JF, Schinzel RT, Lewis GD, Dejam A, Lee YK, Palma MJ, Calhoun S, Georgiadi A, et al. β-Aminoisobutyric acid induces browning of white fat and hepatic β-oxidation and is inversely correlated with cardiometabolic risk factors. Cell Metab. 2014;19:96–108. doi: 10.1016/j.cmet.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77:731–758. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- Rottiers V, Näär AM. MicroRNAs in metabolism and metabolic disorders. Nat Rev Mol Cell Biol. 2012;13:239–250. doi: 10.1038/nrm3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatore D, Simonides WS, Dentice M, Zavacki AM, Larsen PR. Thyroid hormones and skeletal muscle—new insights and potential implications. Nat Rev Endocrinol. 2014;10:206–214. doi: 10.1038/nrendo.2013.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiaffino S, Reggiani C. Fiber types in mammalian skeletal muscles. Physiol Rev. 2011;91:1447–1531. doi: 10.1152/physrev.00031.2010. [DOI] [PubMed] [Google Scholar]
- Schiano C, Casamassimi A, Rienzo M, de Nigris F, Sommese L, Napoli C. Involvement of Mediator complex in malignancy. Biochim Biophys Acta. 2014a;1845:66–83. doi: 10.1016/j.bbcan.2013.12.001. [DOI] [PubMed] [Google Scholar]
- Schiano C, Casamassimi A, Vietri MT, Rienzo M, Napoli C. The roles of mediator complex in cardiovascular diseases. Biochim Biophys Acta. 2014b;1839:444–451. doi: 10.1016/j.bbagrm.2014.04.012. [DOI] [PubMed] [Google Scholar]
- Schuler M, Ali F, Chambon C, Duteil D, Bornert JM, Tardivel A, Desvergne B, Wahli W, Chambon P, Metzger D. PGC1 alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006;4:407–414. doi: 10.1016/j.cmet.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Shimano M, Ouchi N, Walsh K. Cardiokines: recent progress in elucidating the cardiac secretome. Circulation. 2012;126:e327–e332. doi: 10.1161/CIRCULATIONAHA.112.150656. [DOI] [PubMed] [Google Scholar]
- Simoneau JA, Colberg SR, Thaete FL, Kelley DE. Skeletal muscle glycolytic and oxidative enzyme capacities are determinants of insulin sensitivity and muscle composition in obese women. FASEB J. 1995;9:273–278. [PubMed] [Google Scholar]
- Sinha M, Jang YC, Oh J, Khong D, Wu EY, Manohar R, Miller C, Regalado SG, Loffredo FS, Pancoast JR, et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science. 2014;344:649–652. doi: 10.1126/science.1251152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son NH, Park TS, Yamashita H, Yokoyama M, Huggins LA, Okajima K, Homma S, Szabolcs MJ, Huang LS, Goldberg IJ. Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J Clin Invest. 2007;117:2791–2801. doi: 10.1172/JCI30335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaeth JM, Kim NH, Boyer TG. Mediator and human disease. Semin Cell Dev Biol. 2011;22:776–787. doi: 10.1016/j.semcdb.2011.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan N, Häring HU. The role of hepatokines in metabolism. Nat Rev Endocrinol. 2013;9:144–152. doi: 10.1038/nrendo.2012.258. [DOI] [PubMed] [Google Scholar]
- Taatjes DJ. The human Mediator complex: a versatile, genome-wide regulator of transcription. Trends Biochem Sci. 2010;35:315–322. doi: 10.1016/j.tibs.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taatjes DJ, Näär AM, Andel F, 3rd, Nogales E, Tjian R. Structure, function, and activator-induced conformations of the CRSP coactivator. Science. 2002;295:1058–1062. doi: 10.1126/science.1065249. [DOI] [PubMed] [Google Scholar]
- Tata JR. The road to nuclear receptors of thyroid hormone. Biochim Biophys Acta. 2013;1830:3860–3866. doi: 10.1016/j.bbagen.2012.02.017. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- Villena JA, Kralli A. ERRalpha: a metabolic function for the oldest orphan. Trends Endocrinol Metab. 2008;19:269–276. doi: 10.1016/j.tem.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallberg AE, Yamamura S, Malik S, Spiegelman BM, Roeder RG. Coordination of p300-mediated chromatin remodeling and TRAP/mediator function through coactivator PGC-1alpha. Mol Cell. 2003;12:1137–1149. doi: 10.1016/s1097-2765(03)00391-5. [DOI] [PubMed] [Google Scholar]
- Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, Ham J, Kang H, Evans RM. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004;2:e294. doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Tian L, Popov VM, Pestell RG. Acetylation and nuclear receptor action. J Steroid Biochem Mol Biol. 2011;123:91–100. doi: 10.1016/j.jsbmb.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigert C, Lehmann R, Hartwig S, Lehr S. The secretome of the working human skeletal muscle—a promising opportunity to combat the metabolic disaster? Proteomics Clin Appl. 2014;8:5–18. doi: 10.1002/prca.201300094. [DOI] [PubMed] [Google Scholar]
- Westerling T, Kuuluvainen E, Mäkelä TP. Cdk8 is essential for preimplantation mouse development. Mol Cell Biol. 2007;27:6177–6182. doi: 10.1128/MCB.01302-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis LH, Slentz CA, Bateman LA, Shields AT, Piner LW, Bales CW, Houmard JA, Kraus WE. Effects of aerobic and/or resistance training on body mass and fat mass in overweight or obese adults. J Appl Physiol. 2012;113:1831–1837. doi: 10.1152/japplphysiol.01370.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Vought BW, Satterlee JS, Walker AK, Jim Sun ZY, Watts JL, DeBeaumont R, Saito RM, Hyberts SG, Yang S, et al. An ARC/Mediator subunit required for SREBP control of cholesterol and lipid homeostasis. Nature. 2006;442:700–704. doi: 10.1038/nature04942. [DOI] [PubMed] [Google Scholar]
- Young ME, McNulty P, Taegtmeyer H. Adaptation and maladaptation of the heart in diabetes: Part II: potential mechanisms. Circulation. 2002;105:1861–1870. doi: 10.1161/01.cir.0000012467.61045.87. [DOI] [PubMed] [Google Scholar]
- Yu F, Göthe S, Wikström L, Forrest D, Vennström B, Larsson L. Effects of thyroid hormone receptor gene disruption on myosin isoform expression in mouse skeletal muscles. Am J Physiol Regul Integr Comp Physiol. 2000;278:R1545–R1554. doi: 10.1152/ajpregu.2000.278.6.R1545. [DOI] [PubMed] [Google Scholar]
- Zanuso S, Jimenez A, Pugliese G, Corigliano G, Balducci S. Exercise for the managementof type 2diabetes:a reviewof the evidence. Acta Diabetol. 2010;47:15–22. doi: 10.1007/s00592-009-0126-3. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiaoli Zhao X, Yang F. The Mediator complex and lipid metabolism. J Biochem Pharmacol Res. 2013;1:51–55. [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Feng D, Wang Q, Abdulla A, Xie XJ, Zhou J, Sun Y, Yang ES, Liu LP, Vaitheesvaran B, et al. Regulation of lipogenesis by cyclin-dependent kinase 8-mediated control of SREBP-1. J Clin Invest. 2012;122:2417–2427. doi: 10.1172/JCI61462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YJ, Qi C, Jia YZ, Nye JS, Rao MS, Reddy JK. Deletion of PBP/PPARBP, the gene for nuclear receptor coactivator peroxisome proliferator-activated receptor-binding protein, results in embryonic lethality. J Biol Chem. 2000;275:14779–14782. doi: 10.1074/jbc.C000121200. [DOI] [PubMed] [Google Scholar]