

Abstract
Objectives
In the light of increasing drug resistance in Staphylococcus aureus, bacteriophage endolysins [peptidoglycan hydrolases (PGHs)] have been suggested as promising antimicrobial agents. The aim of this study was to determine the antimicrobial activity of nine enzymes representing unique homology groups within a diverse class of staphylococcal PGHs.
Methods
PGHs were recombinantly expressed, purified and tested for staphylolytic activity in multiple in vitro assays (zymogram, turbidity reduction assay and plate lysis) and against a comprehensive set of strains (S. aureus and CoNS). PGH cut sites in the staphylococcal peptidoglycan were determined by biochemical assays (Park–Johnson and Ghuysen procedures) and MS analysis. The enzymes were tested for their ability to eradicate static S. aureus biofilms and compared for their efficacy against systemic MRSA infection in a mouse model.
Results
Despite similar modular architectures and unexpectedly conserved cleavage sites in the peptidoglycan (conferred by evolutionarily divergent catalytic domains), the enzymes displayed varying degrees of in vitro lytic activity against numerous staphylococcal strains, including cell surface mutants and drug-resistant strains, and proved effective against static biofilms. In a mouse model of systemic MRSA infection, six PGHs provided 100% protection from death, with animals being free of clinical signs at the end of the experiment.
Conclusions
Our results corroborate the high potential of PGHs for treatment of S. aureus infections and reveal unique antimicrobial and biochemical properties of the different enzymes, suggesting a high diversity of potential applications despite highly conserved peptidoglycan target sites.
Keywords: peptidoglycan hydrolase, antimicrobial, biofilm, antibiotic resistance
Introduction
Staphylococcus aureus is a Gram-positive bacterial pathogen responsible for mortality and morbidity in both humans and animals worldwide. It is an important foodborne pathogen and a major cause of nosocomial infections, generating excessive healthcare costs and associated with a wide array of diseases.1 Besides its role as a human pathogen, S. aureus has high impact on the dairy industry (annual loss of ∼$2 billion in the USA) as the major causative agent of bovine mastitis.2 S. aureus is also capable of producing biofilms,3 which are a persistent problem in medicine, food safety and agriculture.4,5 In both human clinics and agricultural settings, MDR S. aureus strains are becoming increasingly prevalent.6,7 MRSA accounts for up to 60% of hospital- and community-acquired S. aureus infections.8,9 For this reason, alternative therapeutics for treatment of S. aureus infections are urgently needed.
Endolysins are bacteriophage-encoded peptidoglycan (PG) hydrolases (PGHs) that are produced inside an infected host cell at the end of the phage lytic cycle to enzymatically degrade the bacterial PG ‘from within’, resulting in cell lysis and liberation of progeny phages. In Gram-positive bacteria, which lack an outer membrane, lysis can also be achieved ‘from without’, by exogenously applying (recombinant) endolysins. Therefore, these enzymes have attracted increasing attention as potential antimicrobials in recent years.10,11 Development of resistance to endolysins is believed to be rare compared with antibiotics because of their target cell specificity, their highly conserved targets in the PG,10 and because their application ‘from without’ avoids many possible resistance mechanisms.12 While various studies have unsuccessfully attempted to generate endolysin-resistant strains,10 resistance against non-endolysin PGHs, such as lysostaphin,13 has been reported. This bacteriocin targets the weakly conserved pentaglycine cross-bridge,14 modification of which has been shown to cause lysostaphin resistance in several strains.15 Endolysins from a Gram-positive background show a modular design, consisting of enzymatically active domains (EADs) and cell wall binding domains (CBDs).16 CBDs confer specificity by recognizing certain cell wall ligands, whereas EADs catalyse PG degradation. The EADs can be classified with regard to their cleavage sites in the PG, with glycosidases, amidases and endopeptidases cutting within the sugar strand, between sugar and peptide moieties, and within the peptide portion, respectively. Endolysin architectures found in public databases are diverse regarding the number and arrangement of individual domains.4
The largest subgroup of staphylococcal PGHs feature C-terminal SH3b CBDs17 and, in most cases, two different EADs [N-terminal cysteine, histidine-dependent amidohydrolase/peptidase (CHAP) and centrally located N-acetylmuramoyl-l-alanine amidase], as predicted by bioinformatics.4,18 PG cut sites have been determined only for the endolysins of the phages K (LysK) and phi11 (phi11). Both cleave the MurNAc-l-Ala amide bond (amidase) and the d-Ala-Gly bond between the stem peptide and the pentaglycine bridge (CHAP).19,20 SH3b-bearing staphylococcal PGHs fall into five homology groups that show >90% within-group and <50% between-group sequence identity, and six ‘stand-alone’ proteins (it should be noted that the SH3b domain corresponds to the region of highest sequence conservation between the different groups).18 Some of these highly diverse enzymes have been partially characterized.19–25 However, comparison of results from different laboratories is hampered by the multitude of methods and assay conditions used.11
Here, we comparatively characterized nine different recombinant PGHs representing all of the homology groups mentioned above and four stand-alone proteins. This analysis included determination of (i) lytic activity by multiple in vitro assays and against various staphylococcal strains; (ii) cleavage sites in the PG; (iii) anti-biofilm activity; and (iv) therapeutic potential using a mouse model of systemic MRSA infection.
Materials and methods
Constructs, plasmids and strains
The nucleotide sequences encoding the endolysins of the staphylococcal phages phi80α (80α; GenBank accession number ABF71642.1), phi11 (phi11; YP_500516.1), K (LysK; YP_024461), phiP68 (P68; NP_817332.1) and Twort (Twort; AAX92311) were kindly provided by G. Christie,26 R. Jayaswal,27 P. Ross,28 U. Bläsi29 and M. Loessner,25 respectively. The phage 2638A endolysin (2638A; AAX90995) gene was amplified from S. aureus 2854 (HER 1283, Laval University, Quebec, Canada).22 Sequences of the Staphylococcus haemolyticus prophage phiSH2 (phiSH2; BAE05642.1)23 and the Staphylococcus warneri phage phiWMY (WMY; BAD83402)24 endolysins were synthesized (GeneArt, Regensburg, Germany). The sequence coding for mature lysostaphin (P10547) was a gift from David Kerr.30 All sequences were inserted into NdeI and XhoI restriction sites of pET21a (EMD Biosciences, San Diego, CA) using standard molecular techniques, thereby adding 6 × His-tag coding sequences to the 3′ end of each gene (yielding C-terminally 6 × His-tagged proteins). Alternatively, N-terminally His-tagged versions were created by inserting the coding sequences into BamHI and SalI sites of pQE30 (Qiagen, Hilden, Germany). Unless stated otherwise, the C-terminally 6 × His-tagged constructs were used for experiments in this study. Escherichia coli BL21 (DE3) or XL1-Blue MRF′ (for pQE30-based constructs) was used for overexpression of recombinant proteins and was grown in LB medium at 37°C with 150 mg/L ampicillin for plasmid selection (+30 mg/L tetracycline in the case of XL1-Blue MRF′). Staphylococcal strains used in this study are listed in Table 1. They were grown in tryptic soy broth (TSB) or brain heart infusion (BHI) at 37°C, supplemented with suitable antibiotics if required.
Table 1.
Activity of purified PGHs against multiple staphylococcal strains as determined by plate lysis assay
| Straina | Sourceb | 80α | Phi11 | LysK | P68 | Lysostaphin | 2638Ac |
Twort | PhiSH2 | WMY | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| day 1 | day 3+ | ||||||||||
| Newman | 1 | ++d | ++ | ++ | + | +++ | − | − | ++ | − | (+) |
| 305 (Newbould) | 2 | ++ | ++ | ++ | ++ | +++ | − | − | ++ | + | ++ |
| Newman smr | 1 | ++ | ++ | ++ | ++ | +++ | − | − | ++ | (+) | + |
| Newman ΔtagO | 1 | +++ | +++ | ++ | ++ | +++ | +++ | +++ | ++ | + | +++ |
| Newman Δica | 1 | ++ | ++ | ++ | + | +++ | − | − | ++ | − | (+) |
| Newman ΔdltA | 1 | ++ | ++ | ++ | + | +++ | − | − | ++ | − | + |
| Newman ΔsrtA | 1 | ++ | ++ | ++ | + | +++ | − | − | + | − | + |
| MN8 | 3 | ++ | ++ | ++ | + | ++ | − | ++ | + | (+) | + |
| MN8 Δica | 3 | ++ | ++ | ++ | + | ++ | − | ++ | ++ | − | + |
| MN8 ΔsarA | 3 | ++ | ++ | ++ | + | ++ | (+) | +++ | ++ | − | + |
| ALC 1342 | 4 | ++ | + | ++ | + | ++ | − | − | ++ | (+) | (+) |
| ANG 133 | 5 | ++ | ++ | + | + | ++e | − | − | + | − | (+) |
| ANG 144 | 5 | (+) | (+) | + | (+) | ++e | − | − | + | − | − |
| SA113 | 6 | ++ | ++ | ++ | + | +++ | + | ++ | ++ | − | ++ |
| SA113 ΔtagO | 6 | ++ | +++ | ++ | ++ | +++ | +++ | +++ | ++ | − | +++ |
| SA113 ΔdltA | 6 | ++ | ++ | ++ | ++ | +++ | + | ++ | ++ | + | ++ |
| Reynolds (CP–) | 1 | ++ | ++ | ++ | + | +++ | − | − | ++ | (+) | + |
| Reynolds (CP5) | 1 | ++ | ++ | ++ | ++ | +++ | − | − | ++ | (+) | ++ |
| Reynolds (CP8) | 1 | ++ | ++ | ++ | ++ | +++ | − | − | ++ | (+) | + |
| NRS 382 (MRSA) | 7 | ++ | + | ++ | + | ++ | − | − | + | (+) | (+) |
| NRS 383 (MRSA) | 7 | ++ | ++ | ++ | + | +++ | (+) | ++ | ++ | + | + |
| NRS 384 (MRSA) | 7 | ++ | ++ | ++ | + | +++ | − | (+) | + | (+) | + |
| NRS 385 (MRSA) | 7 | ++ | ++ | ++ | + | +++ | − | − | ++ | (+) | + |
| SA001 | 8 | ++ | ++ | ++ | ++ | +++ | − | − | ++ | (+) | ++ |
| SA002 | 8 | ++ | ++ | ++ | ++ | +++ | − | + | ++ | (+) | + |
| SA003 | 8 | ++ | ++ | ++ | ++ | +++ | − | − | ++ | (+) | + |
| SA009 | 8 | ++ | ++ | ++ | ++ | +++ | − | − | ++ | + | ++ |
| SA019 | 8 | ++ | ++ | ++ | ++ | +++ | − | + | ++ | (+) | (+) |
| SA020 | 8 | ++ | ++ | ++ | ++ | +++ | − | + | ++ | + | + |
| SA021 | 8 | ++ | + | ++ | + | ++ | − | (+) | + | (+) | (+) |
| SA026 | 8 | ++ | ++ | ++ | ++ | ++ | − | + | ++ | (+) | + |
| SA028 | 8 | ++ | + | ++ | + | ++ | − | − | + | (+) | (+) |
| SA029 | 8 | ++ | ++ | ++ | ++ | ++ | (+) | + | ++ | (+) | ++ |
| SA031 | 8 | ++ | ++ | ++ | + | ++ | − | − | ++ | (+) | + |
| SA033 | 8 | ++ | ++ | ++ | + | ++ | − | + | ++ | (+) | ++ |
| SA047 | 8 | ++ | ++ | ++ | + | ++ | − | (+) | ++ | (+) | + |
| SA048 | 8 | ++ | ++ | ++ | + | ++ | − | + | ++ | (+) | + |
| SA049 | 8 | ++ | ++ | ++ | + | ++ | − | + | ++ | (+) | ++ |
| S. chromogenes | 9 | +++ | +++ | +++ | +++ | +++ | + | ++ | ++ | + | ++ |
| S. epidermidis | 9 | ++ | (+) | ++ | + | ++ | − | + | ++ | + | − |
| S. hyicus | 9 | ++ | + | +++ | + | +++ | − | − | ++ | + | + |
| S. simulans | 9 | ++ | + | ++ | + | ++ | − | (+) | ++ | + | + |
| S. warneri | 9 | ++ | − | ++ | + | ++ | − | − | ++ | + | − |
| S. xylosus | 9 | +++ | +++ | +++ | ++ | +++ | − | − | ++ | + | ++ |
aThe species was S. aureus unless stated otherwise.
bStrains were obtained from: 1Jean C. Lee, Channing Laboratory, Brigham and Women's Hospital, Boston, MA, USA; 2ATCC 29740; 3Gerald B. Pier, Channing Laboratory, Brigham and Women's Hospital, Boston, MA, USA; 4Ambrose L. Cheung, Dartmouth Medical School, Hanover, NH, USA; 5Olaf Schneewind, Department of Microbiology, University of Chicago, Chicago, IL, USA; 6Andreas Peschel, Medical Microbiology and Hygiene Department, University of Tübingen, Tübingen, Germany; 7Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA), Chantilly, VA, USA; 8Yasunori Tanji, Tokyo Institute of Technology, Yokohama, Japan (bovine mastitis isolate); and 9Max Paape, ARS, USDA, Beltsville, MD, USA (bovine mastitis isolate).
cFor 2638A, lysis zones were evaluated after overnight incubation at 37°C (day 1), then further incubated at room temperature and evaluated again after 3 days (day 3+).
dEnzyme amounts causing a lysis zone: +++, 1 pmol; ++, 10 pmol; +, 100 pmol; (+) faint lysis zone observed at 100 pmol; −, no lysis zone.
eANG133 and ANG144 showed 4-fold reduced susceptibility to lysostaphin in MIC assays compared with the WT Newman strain.15
Protein expression and purification
Expression and purification of recombinant 6 × His-tagged PGHs was performed essentially as previously described,31 with the following modifications. Induced E. coli cultures were harvested, resuspended in 10 mL of lysis buffer (50 mM NaH2PO4/300 mM NaCl/10 mM imidazole/30% glycerol, pH 8.0) per 1 L of culture and sonicated on ice for 5 min (1 s pulses separated by 1 s rests). After removal of debris by centrifugation (9000 g for 30 min), 6 × His-tagged proteins were purified from the cleared supernatant by immobilized metal ion affinity chromatography using nickel-NTA Superflow resin (Qiagen, Valencia, CA, USA). When proteins were prepared for in vivo experiments, purification columns were washed with 25 column volumes (CV) of lysis buffer supplemented with 0.1% Triton X-114 for removal of endotoxin,32 40 CV of lysis buffer and 15 CV of wash buffer (50 mM NaH2PO4/300 mM NaCl/20 mM imidazole/30% glycerol, pH 8.0). For all other experiments, the Triton X-114 wash was omitted and columns were washed with 25 CV of lysis buffer and 15 CV of wash buffer instead. Target proteins were eluted with elution buffer (50 mM NaH2PO4/300 mM NaCl/250 mM imidazole/30% glycerol, pH 8.0) in 500 μL fractions. Fractions with high protein concentrations were combined and, in the case of in vivo experiments, dialysed against dialysis buffer [50 mM NaH2PO4/300 mM NaCl/10% glycerol (20% in the case of P68), pH 7.5]. Proteins were filtered (0.2 μM), concentrations were measured spectrophotometrically using a NanoDrop ND-1000 (NanoDrop Technologies, Wilmington, DE, USA) and purity was determined by SDS–PAGE. In the case of C-terminally 6 × His-tagged 2638A (for which purification yields a double band), the concentration was determined based on the estimation that the full-length endolysin accounts for 50% of the total protein amount (Figure 1). Representative random samples were evaluated for endotoxin content using the Limulus amoebocyte lysate assay (LAL QCL-1000, Lonza, Walkersville, MD, USA) and shown to be <5 EU/mg.
Figure 1.

Modular organization, purification and staphylolytic activity of nine different staphylococcal SH3b domain-containing PGHs. (a) Schematic representation of nine PGHs representing different homology groups reported previously.18 EADs are depicted as hatched and white/dotted bars and SH3b CBDs as black bars. The scale indicates amino acid (AA) positions. M23, M23 endopeptidase domain; SH3b, bacterial SH3 domain (CBD). All proteins harbour a C-terminal 6 × His-tag (not represented). (b) SDS–PAGE of 6 × His-tagged PGHs purified by immobilized metal ion affinity chromatography; 5 μg of protein was loaded in each lane. Expected molecular weights: 80α, 54.9 kDa; phi11, 55.1 kDa; LysK, 55.8 kDa; P68, 29.6 kDa; lysostaphin, 28.2 kDa; 2638A, 56.6 kDa; Twort, 54.3 kDa; phiSH2, 57.4 kDa; and WMY, 55.0 kDa. Note that C-terminally 6 × His-tagged 2638A purification yielded a double band (for an explanation see the text). (c) Zymogram with the enzymes shown in (b) and S. aureus Newman cells embedded in the gel. The same amounts of protein as in SDS–PAGE were loaded. (d) Specific activity of the nine PGHs against S. aureus Newman determined by turbidity reduction assays performed with 0.2 μM enzyme in buffer containing 200 mM NaCl. Error bars indicate standard deviations from three independent experiments.
SDS–PAGE and zymogram
SDS–PAGE and zymogram analyses of purified staphylococcal PGHs were carried out as described previously,33 using 15% or 12% polyacrylamide gels and Kaleidoscope Precision Plus Protein Standard (Bio-Rad). For the zymogram, a 300 mL culture volume equivalent of live mid-log phase (OD600 = 0.4–0.6) cells of S. aureus strain Newman was embedded in the gel during polymerization. After electrophoresis, zymograms were washed extensively with water to remove SDS and enable re-folding of proteins, followed by incubation in 10 mM Tris/150 mM NaCl, pH 8.0, for 30 min and then 10 mM Tris/300 mM NaCl, pH 8.0, for another 30 min.
Turbidity reduction assay
Turbidity reduction assays for quantification of enzymatic activity were performed in a 96-well plate format essentially as described by Becker et al.,19 using frozen but viable cells of S. aureus Newman as substrate. In brief, equimolar concentrations (0.2 μM) of PGH were added to bacterial suspensions adjusted to an OD600 of 1.0 in 10 mM Tris/200 mM NaCl, pH 7.5, and the OD was measured at 20 s intervals for 30 min. The steepest slopes of the resulting lysis curves were used for calculation of specific lytic activities, which were expressed as ΔOD600 min–1 μM–1. In order to determine the effect of ionic strength on lytic activity, assays were carried out as described above with varying NaCl concentrations, ranging from 0 to 600 mM. All assays were performed in triplicate.
Plate lysis assay
For plate lysis assays, the Staphylococcus strains listed in Table 1 were grown to mid-log phase, diluted to an OD600 of 0.1 using PBS and 3 mL of the suspension was spread on gridded tryptic soy agar (TSA) plates. Excess liquid was removed and the plates were air dried for 15 min in a laminar flow hood. Equimolar amounts (100, 10 and 1 pmol) of PGHs in 10 μL volumes were spotted onto the plates; the plates were air dried for 10 min and then incubated overnight at 37°C. Cleared spots within the resulting bacterial lawn indicating cell lysis were scored within 24 h. Plate lysis assays were performed in duplicate.
Biochemical assays for PGH cut site analysis
Preparation of crude cell walls from S. aureus strain SA113 was performed as published earlier.34 Free amino groups in the peptidoglycan were acetylated with acetic anhydride.35 Pre-acetylated cell walls were washed three times with water and stored at –20°C for endolysin activity assays. Cell wall digestion was performed in 20 mM NaH2PO4/200 mM NaCl, pH 7.4, in a total volume of 100 μL containing cell walls (400 μg) and enzyme (10 μg of P68, 2638A, 80α, Twort, WMY or mutanolysin). Mixtures were incubated at 30°C for 16 h and finally boiled at 100°C for 10 min to stop reactions.
For reducing sugar analysis, digests were assayed by a modified Park–Johnson method.36,37 A 10 μL aliquot of digested PG was mixed with 90 μL of water, 200 μL of 0.05% (w/v) potassium ferricyanide (in water) and 200 μL of 0.53% (w/v) sodium carbonate/0.065% (w/v) potassium cyanide (in water) and heated at 100°C for 15 min. Reactions were cooled and 1 mL of ferric iron solution [0.15% (w/v) ferric ammonium sulphate, 0.1% (w/v) SDS in 0.025 M sulphuric acid] was added. After 10 min of incubation, absorbance was measured at 690 nm. Undigested cell walls were used as control. The content of reducing groups was calculated using a calibration curve of glucose.
The liberation of free amino acids was determined by a modified Ghuysen procedure using 1-fluoro-2,4-dinitrobenzene (FDNB).38 Enzymes (10 μg) were added to each cell wall digest and the mixture was incubated for 6 h at 37°C for complete cell wall degradation. After inactivation at 100°C for 15 min, 15 μL of 10% K2B7O4 and 15 μL of FDNB solution (0.1 M in ethanol) were added to the sample. The mixture was incubated at 65°C for 45 min in the dark. After hydrolysis with 4 M HCl (800 μL, 12 h at 95°C), the DNP amino acid derivatives were measured at 365 nm. l-Alanine was used as the standard for calculations. All experiments were performed in triplicate.
MS analysis of digested S. aureus cell walls
For determination of PGH cut sites within the staphylococcal PG, cell walls were prepared and digested with the respective enzymes (the N-terminally 6 × His-tagged version was used in the case of 2638A) and purified, and the resulting fragments were analysed by MS essentially as described before.19,39 In brief, cell walls of strain Newman or SA113 ΔtagO were digested at 37°C for 2 h with the respective enzymes, filtered through 5000 MW cut-off Vivaspin 500 U (Sartorius North America Inc., Bohemia, NY, USA) and PG fragments were desalted using Sep-Pak C18 cartridges (Water Corporation, Milford, MA, USA). The eluted fragments (in 50% methanol) were dried in a SpeedVac, dissolved in deionized water and analysed by positive-ion electrospray ionization MS performed on a Micromass Q-TOF2 device (Micromass Ltd, Manchester, UK). For exact determination of endopeptidase cut sites, double digests with full-length parental enzymes and a truncated version of LysK containing only the CHAP domain19 were performed. For digests with P68, a double digest was performed (with both P68 and an N-terminal truncation of 2638A comprising only the amidase and SH3b domains22) in order to see whether the added amidase activity was able to reproduce the ions created with the dual lytic domain endolysins. Fragments were filtered as described above and desalted using C18 ZipTips (Millipore, Zug, Switzerland), from which they were eluted with 10 μL of 50 : 50: 0.01 (v/v/v) CH3OH : H2O : HCOH (pH ∼2). NanoESI–MS analysis of the samples was performed on a Q-TOF Ultima API mass spectrometer (Micromass, UK). The solutions were infused through a fused silica capillary [internal diameter (ID) 75 μm] at a flow rate of 0.50 μL/min. Electrospray Pico Tips (ID 30 μm) were obtained from New Objective (Woburn, MA). Mass spectra were acquired by scanning an m/z range from 50 to 3000 with a scan duration of 1 s and an interscan delay of 0.1 s. Spray voltage was set to 2.1 kV, cone voltage to 35V, RF lens 1 energy to 50 V and collision to 10 eV.
Biofilm reduction assay
A method modified from previous studies40,41 was used to test the effect of PGHs on staphylococcal biofilms in a static 96-well plate (polystyrene; SPL Life Sciences, Korea)-based model. S. aureus SA113 was grown in TSB + 0.25% d(+)-glucose at 37°C overnight, diluted 1/200 in the same fresh medium, aliquoted in a 96-well plate (200 μL per well) and further incubated for 24 h for biofilm growth. Biofilms were washed twice with PBS. Serial dilutions of PGHs in elution buffer (70 μL per well) were added and the plate was incubated at 37°C for 2.5 h. Buffer alone served as control. After washing twice with PBS, biofilms were stained with 100 μL of 0.4% crystal violet for 15 min at room temperature, followed by washing three times with PBS. The stain was dissolved in 33% acetic acid in water and the OD590 of each well diluted 1/10 in water was measured spectrophotometrically. Similar experiments were carried out in parallel using N-terminally His-tagged versions of the enzymes and PBS for preparation of enzyme dilutions. All experiments were performed in triplicate.
Mouse model for evaluation of PGHs against MRSA in vivo
Female BALB/c mice aged 4–6 weeks (weight range 22–24 g, Harlan Laboratories, Frederick, MD, USA) were used in biosafety level 2 facilities in accordance with the University of Maryland institutional animal care and use committee (College Park, MD, USA) regulations and national animal care guidelines. Briefly, MRSA strain NRS382 was grown overnight in BHI medium at 37°C. The culture was then diluted 1 : 100 and grown to OD600 = 0.3–0.4, centrifuged and resuspended in BHI supplemented with 5% mucin (Sigma-Aldrich, St Louis, MO, USA) for the mouse experiment, according to a previously reported protocol.42 The mouse model was designed to be an LD90 model, i.e. to yield lethality in 90% of control mice at the 48 h post-infection endpoint. To this end, ∼4 × 107 cfu bacteria in suspension (in a volume of 0.2 mL) were injected intraperitoneally. Actual inoculum titres were derived by plating serial dilutions of each inoculum on BHI agar plates.
To determine the in vivo efficacy of PGHs, 30 min post-infection, infected mice were divided into groups (10 mice per group) and injected intraperitoneally with either PGHs in dialysis buffer (200 μg/mouse in 200 μL) or PBS or dialysis buffer as controls (200 μL/mouse). The antibiotics vancomycin and oxacillin (Sigma-Aldrich), prepared in distilled H2O, were used as additional controls. Antibiotics were administered subcutaneously (vancomycin, 375 μg/mouse; oxacillin, 1250 μg/mouse) 30 min post-infection. The administration route and diluent most appropriate for each therapeutic was chosen for these experiments. While subcutaneous administration and use of water as diluent is recommended for antibiotics, a phosphate buffer (dialysis buffer) was used for diluting PGHs to avoid possible aggregation of the proteins in water, and intraperitoneal administration of PGHs was chosen since it is well documented in the literature.10 Oxacillin served as negative control for the subcutaneous route of administration.
The survival rate for each experimental group was monitored every 4 h up to 48 h post-bacterial challenge. The data were statistically analysed using Kaplan–Meier survival curves and Fisher's exact test. In addition, a septicaemia score index described by Biswas et al.43 was used to evaluate the health condition of MRSA-infected mice at intervals of 4 h for up to 48 h (Figure 5c).
Figure 5.

Efficacy of PGHs and antibiotics in a mouse model of systemic MRSA infection. (a) Survival of mice infected intraperitoneally with MRSA and subjected to different treatment regimens. Treatment groups listed on the right and corresponding survival curves are displayed in matching colours. Numbers of animals in each group (N) are given in parentheses. Antibiotics are shown in italics and PGHs are underlined. Differences in survival rates between PBS-treated control mice and animals treated with 80α, phi11, LysK, lysostaphin, 2638A, WMY or vancomycin were statistically significant (P < 0.02; Fisher's exact test). (b) Average septicaemia scores of mice recorded for 48 h following infection. Scores for animals in different treatment groups listed on the right (displayed in matching colours) correspond to the matrix shown in (c). (c) Composite matrix of septicaemia rating the disease state of infected animals characterized by defined clinical signs.43
To detect bacteria in the bloodstream, mice surviving until the end of the experiment (48 h post-infection) were euthanized and 100 μL blood samples were taken, mixed with 900 μL of PBS and then serially diluted and plated on BHI agar plates.
Results
Staphylococcal PGHs from five different homology groups kill S. aureus in vitro
Nine staphylococcal PGHs (Figure 1a) were expressed in E. coli with C-terminal 6 × His-tags and purified by nickel affinity chromatography, yielding preparations of >95% purity as demonstrated by SDS–PAGE (Figure 1b). Purification of 2638A yielded a shadow band at 36.3 kDa in addition to the expected 56.6 kDa band, this has been shown to be due to a secondary translational start site within the 2638A coding sequence22 that was not present when the enzyme was purified with an N-terminal 6 × His-tag (Figure S1, available as Supplementary data at JAC Online). Zymogram analysis revealed staphylolytic activity of all purified enzymes, as apparent from cleared zones of lysis within the opaque gel containing S. aureus Newman cells (Figure 1c). While intensities of clearing zones differed between enzymes (with phiSH2 and 80α yielding barely visible and lysostaphin and P68 the most intense bands), the zymograms demonstrated that the major bands corresponding to the target proteins accounted for ∼98% of the activity within the preparations, whereas minor contaminating bands (as visible on the SDS gel) contributed little or no activity. Lytic activity of the nine PGHs was further analysed by two quantitative PGH activity assays, turbidity reduction and plate lysis. Despite their mostly conserved modular architecture, the enzymes displayed vastly different degrees of specific activity [ΔOD600 min–1μM–1 ranging from <0.1 (phiSH2) to 1.3 (LysK)] against live bacteria in suspension (Figure 1d). Also, optimum salt concentrations differed among the enzymes. While some PGHs (phi11, 2638A, WMY) exhibited pronounced optima at 100 mM NaCl, others (LysK, phiSH2) were most active at higher ionic strength (300–400 mM NaCl) (Figure 2 and Figure S2). It is of note that lysostaphin, a well-characterized potent anti-staphylococcal protein, showed only moderate activity in turbidity reduction assays compared with other PGHs. In contrast, lysostaphin was clearly the most active enzyme in the plate lysis assay (Table 1 and Figure S3A). When tested against the Newman strain, it caused a lysis zone at the lowest concentration used (1 pmol per 10 μL spot), whereas at least 10 pmol/10 μL was required for other PGHs.
Figure 2.

Comparison of lytic activity of nine PGHs against live S. aureus cells at varying ionic strength. Specific activity was determined by turbidity reduction assays using 0.2 μM enzyme against S. aureus Newman cells suspended in Tris buffer, pH 7.5, with NaCl concentrations ranging from 0 to 600 mM. All assays were performed in triplicate. For graphs of individual enzymes with error bars, see Figure S2.
PGH activity against multiple S. aureus strains and CoNS
To further elucidate their therapeutic potential, the lytic activity of the nine PGHs was determined in plate lysis assays against a comprehensive collection of staphylococcal strains, including strains relevant to clinical and agricultural settings, such as MRSA strains, bovine mastitis isolates (S. aureus strains 305 and SA001–SA04944) and CoNS. Furthermore, various mutant strains with altered surface structures were tested, including mutants lacking wall teichoic acids (ΔtagO45) or devoid of d-alanine in the teichoic acids (ΔdltA46); mutants lacking the ability to produce poly-N-acetyl-glucosamine, a surface polysaccharide often associated with biofilm formation (Δica47); an srtA mutant of strain Newman (lacking cell wall-associated proteins48); strains with mutations in sarA, a global regulator of virulence factors, resulting in increased protease secretion (MN8 ΔsarA, ALC134247); strains with reduced susceptibility to lysostaphin (4-fold higher MIC compared with the WT Newman strain), including a femAB mutant having mono- instead of pentaglycine bridges (ANG133) and a lyrA mutant with as yet uncharacterized cell wall alterations (ANG144);15 and isogenic mutants expressing different capsular polysaccharides or no capsule [Reynolds (CP5), Reynolds (CP8) and Reynolds (CP−)49]. With the exception of phiSH2 and 2638A, most PGHs exhibited strong antimicrobial activity against all or the majority of the strains (Table 1). As previously reported,22 2638A produced ill-defined lysis zones that increased in diameter and intensity when incubated for extended periods of time (Figure S3B). Most PGHs showed enhanced activity against tagO mutants in both Newman and SA113 genetic backgrounds (Figure S3A). By contrast, reduced activity was observed for all enzymes against ANG144 compared with the Newman WT, whereas the femAB mutation (ANG133) showed reduced susceptibility to lysostaphin but only minor changes in susceptibility to the other PGHs.
Dual lytic domain endolysins have conserved PG cut sites
Bioinformatic analysis of the dual lytic domain endolysins (80α, phi11, LysK, 2638A, Twort, phiSH2 and WMY) predicted similar modular architectures (except for 2638A, which harbours an M23 instead of a CHAP domain) (Figure 1a), suggesting similar ontology and thus shared cleavage mechanisms. The absence of reducing groups in the Park–Johnson chemical assay36 performed with 80α, P68, 2638A, Twort and WMY on purified S. aureus PG indicated that none of the enzymes yielded detectable glycosidase activity, as opposed to the muramidase mutanolysin50 (Table S1). In a modified Ghuysen assay, which detects endopeptidase or amidase activity, mutanolysin yielded only low (<0.1 mM) concentrations of free amino groups, whereas P68 (harbouring one catalytic domain) and the dual lytic domain endolysins produced ∼0.5 and 1 mM of free amino groups, respectively (Table S1). When S. aureus cell walls digested with 80α, Twort or WMY were subjected to electron spray ionization MS (ESI–MS), a dominant peak at m/z = 702 was observed. This peak was identical to the one detected after LysK digestion (Figure 3a), corresponding to the fragment Ala2-iGln-Lys-Gly5 produced by simultaneous action of a MurNAc-l-Ala amidase and a d-Ala-Gly endopeptidase19 (Figure 3c). Surprisingly, digestion with 2638A (N-terminally 6 × His-tagged) yielded the same 702 peak (Figure 3b). By contrast, phiSH2 produced two larger double-charged ions (m/z = 684 and 693), which presumably correspond to the fragment Ala4-iGln2-Lys2-Gly10 (with and without a water molecule) in addition to the (weaker) 702 peak, suggesting incomplete digest by this relatively weak enzyme (Figure 1d). P68 digestion yielded only background signal, as expected from this single lytic domain enzyme, which presumably produces fragments comprising complete sugar strands; such strands would be too large to be detected in this MS analysis (data not shown). To further define the cleavage sites of the endopeptidase domains, double digests were performed with each endolysin and the previously reported CHAP-K deletion construct harbouring just the LysK CHAP domain (d-Ala-Gly activity).19 In all spectra, the 702 peak was dominant (except for P68) and no additional fragments compared with the single digests were detected, confirming identical cut sites for all of the dual lytic domain endolysins (Figure 3b). P68/CHAP-K double digestion again produced no dominant peaks, whereas after double digestion with P68 and a deletion construct harbouring just the 2638A amidase domain22 the 702 peak was detected in addition to the larger 684 fragment (product of incomplete CHAP digestion), further confirming that the P68 CHAP domain exhibits d-Ala-Gly activity.
Figure 3.

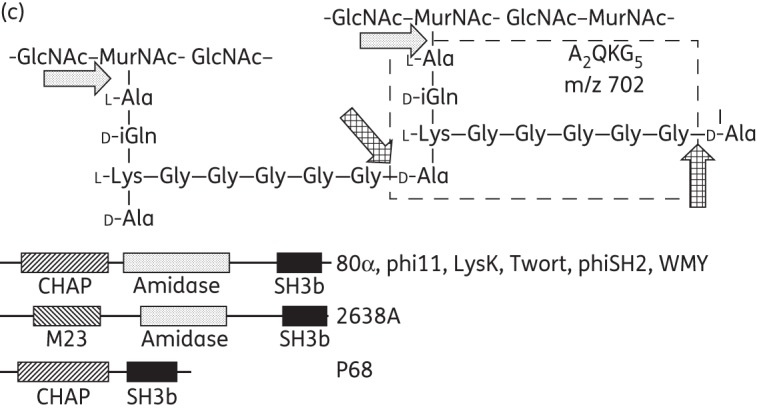
ESI–MS analysis of PGH cleavage sites. (a) ESI–MS spectra (m/z range 400–1000) of S. aureus Newman PG digested with four different dual lytic domain endolysins. All spectra contain an identical dominant peak at m/z = 702. (b) Mass spectra (m/z range 650–750) of S. aureus SA113 ΔtagO PG after single digestion with full-length PGHs or double digestion with two different enzymes. Data were normalized to the highest peak (=100%) within the shown m/z range. Note that the numerous peaks in the P68 + CHAP-K spectrum merely represent amplified background noise. (c) Portion of the S. aureus PG (top) with dotted and cross-hatched arrows indicating cleavage sites of the amidase and CHAP/M23 endopeptidase domains, respectively (cross-hatched arrows indicate that both the M23 domain of 2638A and the CHAP domains of the other seven endolysins exhibit d-Ala-Gly endopeptidase activity). Domains are depicted with corresponding patterns in the schematics (bottom). Simultaneous digestion with both catalytic domains yields the boxed fragment (A2QKG5) of m/z = 702, corresponding to the dominant peak in (a).
PGHs degrade staphylococcal biofilms
In view of the significance of biofilms for medicine, agriculture and food safety, the nine PGHs were compared for their potential to degrade S. aureus SA113 biofilms cultured for 24 h in polystyrene microtitre plates. Using this method, biofilms were formed primarily at the bottoms of the wells, whereas the amount of biofilm attached to the side walls was negligible. In a first series of experiments with C-terminally 6 × His-tagged PGHs in elution buffer, most enzymes degraded the biofilms in a concentration-dependent manner (Figure 4). Whereas LysK and lysostaphin were effective at concentrations as low as 40–80 nM, phi11, P68 and 2638A exhibited no measurable or only weak effects under these conditions. In similar experiments with N-terminally 6 × His-tagged proteins in PBS, phi11, 2638A and P68 markedly degraded the biofilms at 160 nM, 310 nM and 2.5 μM, respectively, whereas LysK, lysostaphin, Twort and phiSH2 were up to one order of magnitude less effective under these alternative conditions (Figure S4).
Figure 4.

Biofilm-disrupting activity of staphylococcal PGHs. Biofilms of S. aureus SA113 were grown in polystyrene 96-well plates for 24 h at 37°C. After washing and incubation for 2.5 h at 37°C with various concentrations of C-terminally His-tagged PGHs in elution buffer or elution buffer only as a control, wells were stained with crystal violet.
PGHs protect mice from systemic MRSA infection
To compare the in vivo efficacy of the nine PGHs and the antibiotics oxacillin and vancomycin, BALB/c mice were inoculated intraperitoneally with 4 × 107 cfu of the MRSA strain NRS382 (a USA100 isolate) together with mucin as an immunosuppressant and treated after 30 min with PGHs (intraperitoneally; 200 μg/mouse), antibiotics or buffer. Whereas only 25%–30% of the mice treated with PBS, dialysis buffer or oxacillin survived after 48 h, 80α, phi11, LysK, lysostaphin, 2638A, WMY and vancomycin protected 100% of the animals from death (Figure 5a). Twort- or phiSH2-treated mice showed less protection from systemic infection (50%–60% survivors) and treatment with P68 (administered in 20% instead of 10% glycerol and at ∼120 μg per mouse due to low solubility) resulted in the least protection, with no survivors at the end of the experiment. Average septicaemia scores determined over the period of 48 h were in good agreement with the survival rates (Figure 5b; for scores of individual animals, see Figure S5). While the health status of mice treated with buffers, oxacillin or P68 rapidly deteriorated, reaching a moribund disease state after 20 h, animals treated with 80α, phi11, LysK, lysostaphin, 2638A, WMY or vancomycin only showed temporary slight illness and completely recovered by the end of the study. In the cases of Twort and phiSH2, surviving mice developed clinical signs of slight to moderate illness (Figure S5). When surviving animals (after 48 h) were examined for presence of S. aureus in the bloodstream, mice with bacteraemia were identified in the buffer, antibiotics, phi11, 2638A, Twort and phiSH2 treatment groups. No bacteria were detected in the blood of animals treated with 80α, LysK, lysostaphin or WMY (Table 2).
Table 2.
Recovery of MRSA from murine blood 48 h post-infection and after different treatments
| Treatment | Number of mice testeda | Percentage of tested mice with bacteraemiaa | Average MRSA concentration in the blood (log10 cfu/mL)b |
|---|---|---|---|
| PBS | 3 | 67% | 2.48 ± 0.31 |
| Dialysis buffer | 3 | 33% | 2.55 ± 0.71 |
| Oxacillin | 3 | 33% | 2.61 ± 0.81 |
| Vancomycin | 10 | 40% | 2.18 ± 0.11 |
| 80α | 10 | 0% | <2 |
| Phi11 | 10 | 10% | 2.05 ± 0.06 |
| LysK | 10 | 0% | <2 |
| P68 | 0 | NA | NA |
| Lysostaphin | 10 | 0% | <2 |
| 2638A | 10 | 20% | 2.10 ± 0.08 |
| Twort | 5 | 60% | 2.51 ± 0.30 |
| PhiSH2 | 6 | 83% | 2.64 ± 0.44 |
| WMY | 10 | 0% | <2 |
NA, not applicable.
aOnly blood from mice surviving after 48 h was tested.
bAverage concentrations are displayed as geometric means ± SEM. The detection limit in this experiment was 2 log10 cfu/mL. For calculating means, individual mice with MRSA concentrations below the detection limit were factored in with 2 log10 cfu/mL. For treatments where no MRSA was detected in any of the tested mice, average concentrations are displayed as <2 log10 cfu/mL.
Discussion
This is the first comparative study analysing enzymatic properties and evaluating the antimicrobial/therapeutic potential of a diverse collection of SH3b domain-containing staphylococcal PGHs. In the past, poor expression and/or purification of some full-length endolysins included in this study28,29 had hampered their testing as antimicrobials and made it necessary to develop strategies for enhancement of solubility.29,51 Under the conditions employed here, all nine PGHs were readily expressed and yielded preparations of high purity. However, solubility of P68 rapidly decreased with decreasing glycerol concentrations, reducing the putative advantages conferred by this candidate therapeutic for systemic administration, as demonstrated by the in vivo results presented here. It should also be noted that the co-purification of two enzymatically active (full-length and truncated) versions of C-terminally 6 × His-tagged 2638A impedes exact specific activity determinations of the full-length protein.22
Overall, the PGHs examined in this study exhibited lytic activity against S. aureus in three different in vitro activity assays: zymogram, turbidity reduction and plate lysis. It is apparent that the three assays did not always yield consistent results on a quantitative level, an inconsistency previously reported and discussed.11,52 It has also been proposed that small proteins and those with low cell wall affinity exhibit higher activity in assays that rely on diffusion through a semi-solid matrix,53 which is consistent with the finding that P68 and lysostaphin produced the most intense lysis zones in the zymogram (Figure 1c).
Whereas lysostaphin is a potent and well-characterized staphylolytic agent with the ability to lyse staphylococcal species other than S. aureus54 (Table 1), its application as a single therapeutic for treatment of staphylococcal infections is not supported due to its documented susceptibility to bacterial resistance development. However, the use of lysostaphin in combination with other PGHs or as part of a chimeric PGH construct such as PRF-11955 could circumvent this limitation. Resistance development may explain why the majority of the SH3b domain-containing staphylococcal phage endolysins have adopted a dual-EAD architecture (two simultaneous mutations within a highly conserved PG region would likely be required to evoke resistance). Protein truncation analyses suggest that in the man-made scenario of ‘lysis from without’, activity is usually ascribed to primarily one of the two catalytic domains (mostly the CHAP domain).19,22,23 Nonetheless, the results of our cut site determination suggest that both lytic domains are functional and that their maintenance confers an evolutionary advantage on the phage. This advantage might also include a synergistic effect of both domains acting simultaneously, e.g. with one EAD increasing accessibility to the cut site for the second catalytic domain.11 Particularly noteworthy and surprising (given the diverse amino acid sequences and highly variable activities in functional assays) is the finding that both the conserved CHAP domains and the unrelated M23 domain of 2638A target the same PG bonds, providing a prime example for convergent evolution among phage endolysins. This finding provides the strongest support for the notion that this combination of PGH activities (MurNAc-l-Ala amidase and d-Ala-Gly endopeptidase) allows the most effective destabilization of the cell wall in the context of progeny phage particle release and raises the question of whether, over many millennia of evolution, the phages have identified the cut sites least susceptible to modification by the host, thus ensuring longevity of the phage clade. The fact that the resistance-prone lysostaphin harbours an M23 endopeptidase with different (glycyl-glycine) activity supports this hypothesis.
Our plate lysis susceptibility tests represent the first time that such an extensive collection of PGHs have been tested against a panel of known cell wall mutants and revealed that this class of enzymes is relatively unaffected by these surface structure alterations. Increased susceptibility of the ΔtagO mutants can possibly be explained by better accessibility of the PG substrate due to the absence of teichoic acids in the cell walls of these strains and/or the diminished cross-linking of PG from the ΔtagO mutants.45,56 It should be mentioned in this context that the glycine-rich inter-peptide bridges in the PG have been suggested to be the targets for the SH3b CBDs of staphylococcal PGHs (not the teichoic acids) and this has been demonstrated for lysostaphin's SH3b domain.11,57 It is important to note that the lyrA mutant ANG144 showed reduced susceptibility not only to lysostaphin but also to all endolysins. In contrast to the femAB mutant ANG133, which features gross alterations of the pentaglycine bridge responsible for lysostaphin tolerance, reduced susceptibility of ANG144 has been attributed mostly to other, as yet unknown alterations of the cell envelope.15 A theoretically plausible resistance mechanism for all cell wall-lytic agents acting from without is the modification of secondary cell wall structures, creating steric hindrance and thereby reducing access to binding or cut sites. Our results with the ANG144 mutant demonstrate for the first time that development of tolerance against endolysins is possible, albeit likely at a lower frequency than for lysostaphin or antibiotics that act inside the bacterial cell. Activity of the endolysins against both coagulase-positive and coagulase-negative strains (Table 1) was not unexpected, since the endolysin PG cleavage sites are conserved throughout the genus Staphylococcus,14 and the SH3b domains of several staphylococcal endolysins have been shown to recognize strains from various staphylococcal species.11
The effectiveness of individual PGH constructs in treating systemic infections caused by various bacterial pathogens has been demonstrated in several studies (reviewed in Schmelcher et al.11) and the results of our in vivo experiments with nine enzymes corroborate these findings. Six out of nine PGHs tested showed efficacy similar to vancomycin when administered as a single dose (Figure 5) and four PGHs were able to prevent bacteraemia, whereas MRSA was recovered from the blood of vancomycin-, oxacillin- and buffer-treated animals (Table 2). It is of note that all negative control animals (PBS-, dialysis buffer- and oxacillin-treated) developed bacteraemia and that these three different treatments yielded almost identical results in terms of survival rates and average septicaemia scores, suggesting that the choice of different administration routes and diluents did not adversely affect the outcome of the experiment. Given the high amino acid sequence diversity of the nine PGHs (<50% identical) and the diverse results they yielded in the various in vitro assays described here, it was not surprising that some of the enzymes were less effective than others at protecting mice from systemic MRSA infection. While the inefficacy of P68 can likely be attributed to its low solubility, the low efficacy of phiSH2 is in good agreement with the low activity of this enzyme in various in vitro assays (Figures 1 and 2 and Table 1). The capability of PGHs investigated here and in previous studies40,58 to degrade staphylococcal biofilms is certainly encouraging, since biofilms are estimated to be involved in >60% of bacterial infections in humans59 and significantly contribute to the resistance of pathogens to conventional treatment regimens.4,60 However, the results obtained here using a static biofilm model need to be followed up by studies employing more sophisticated models.
In conclusion, this work represents the most comprehensive comparison to date of: (i) staphylolytic antimicrobial activity; (ii) enzymatic specificity; (iii) anti-biofilm activity; and (iv) therapeutic potential, featuring a diverse group of SH3b domain-containing PGH enzymes, including eight bacteriophage endolysins and lysostaphin. Together with previous findings regarding synergistic action, safety, near-species specificity, low immunogenicity and low frequency of resistant strain formation associated with phage endolysins,10,11,61 these results support the promise of these enzymes as antimicrobials to combat bacterial infection/contamination by either CoNS or S. aureus, including drug-susceptible and MDR strains. With regard to immunogenicity, several studies have found that although antibodies are produced against systemically administered endolysins, they do not inactivate the PGHs in vivo. No adverse side effects have been reported after systemic endolysin application in animal models (reviewed by Fischetti10 and Schmelcher et al.11). One concern associated with systemic endolysin administration is the possible release of pro-inflammatory cellular debris, such as peptidoglycan fragments and teichoic and lipoteichoic acids.10 Increased concentrations of pro-inflammatory cytokines were reported in mice treated by continuous intravenous infusion of the streptococcal phage endolysin Cpl-1, whereas reduced cytokine concentrations compared with untreated animals were detected when the same enzyme was administered at 12 h intervals.62,63 This difference was explained by the possibly higher degree of cell wall fragmentation in the case of continuous infusion,10 which suggests that repeated as opposed to continuous application of endolysins may represent a more appropriate treatment regimen when aiming to minimize problems associated with the release of cellular debris. It is worth noting that our group of nine enzymes is actually representative of multiple subgroups, totalling ∼60 SH3b-containing enzymes with >90% identity with at least one of the nine lysins tested.18 The highly conserved targets resulting from such a diverse set of protein sequences allow individual optimizations (e.g. the unique plate lysis results for 2638A, the high optimum ionic strength for phiSH2 activity, the high efficacy of LysK and lysostaphin against biofilms under certain buffer conditions, and the high activity of lysostaphin and P68 in semi-solid matrices such as zymograms) and suggest that each enzyme (or subgroup) may be best suited for a particular application among the diverse antimicrobial needs of food safety, agriculture, biotechnology and healthcare.
Funding
This work was supported by the National Institutes of Health (grant number 1RO1AI075077–01A1 to D. M. D. and J. C. L.), the National Research Initiative (grant number 2007-35204-18395 to D.M.D.) and United States State Department funds supporting a US-Pakistani (to D. M. D.) and US-Russian collaboration (to D. M. D.).
Transparency declarations
M. S. has a patent pending (U.S. Serial No. 13/605,200, filed 6 September 2012). J. C. L. received support from the National Institutes of Health for travel to the annual Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) meeting. S. C. B. has a patent issued (U.S. Patent No. 8,568,714). D. M. D. has two patents issued (U.S. Patent No. 8,012,730; U.S. Patent No. 8,361,772) and four patents pending (Published Patent application 201000214450 RCe, filed 5 March 2012; U.S. Serial No. 13/495,536, filed 13 June 2012; U.S. Serial No. 13/605,200, filed 6 September 2012; and U.S. Serial No. 13/298,966, filed 17 November 2012). All other authors: none to declare.
Disclaimer
Mentioning of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the United States Department of Agriculture. The USDA is an equal opportunity provider and employer.
Supplementary data
Acknowledgements
We thank Udo Bläsi, Ambrose Cheung, Gail Christie, Radheshyam Jayaswal, David Kerr, Andreas Peschel, Gerald Pier, Paul Ross, Olaf Schneewind and Yasunori Tanji for the gift of bacterial strains or plasmids. The NARSA isolates were obtained through the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) Program: NRS382, NRS383, NRS384 and NRS385 supported under NIAID/ NIH Contract No. HHSN272200700055C.
References
- 1.Götz F, Bannerman T, Schleifer KH. The genera Staphylococcus and Macrococcus. In: Falkow S, Rosenberg E, Schleifer KH, et al., eds. The Prokaryotes. New York: Springer, 2006; 5–75. [Google Scholar]
- 2.Sordillo LM, Streicher KL. Mammary gland immunity and mastitis susceptibility. J Mammary Gland Biol Neoplasia 2002; 7: 135–46. [DOI] [PubMed] [Google Scholar]
- 3.Götz F. Staphylococcus and biofilms. Mol Microbiol 2002; 43: 1367–78. [DOI] [PubMed] [Google Scholar]
- 4.Nelson DC, Schmelcher M, Rodriguez-Rubio L, et al. Endolysins as antimicrobials. Adv Virus Res 2012; 83: 299–365. [DOI] [PubMed] [Google Scholar]
- 5.Fox LK, Zadoks RN, Gaskins CT. Biofilm production by Staphylococcus aureus associated with intramammary infection. Vet Microbiol 2005; 107: 295–9. [DOI] [PubMed] [Google Scholar]
- 6.Klevens RM, Morrison MA, Nadle J, et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007; 298: 1763–71. [DOI] [PubMed] [Google Scholar]
- 7.De Oliveira AP, Watts JL, Salmon SA, et al. Antimicrobial susceptibility of Staphylococcus aureus isolated from bovine mastitis in Europe and the United States. J Dairy Sci 2000; 83: 855–62. [DOI] [PubMed] [Google Scholar]
- 8.Klevens RM, Edwards JR, Tenover FC, et al. Changes in the epidemiology of methicillin-resistant Staphylococcus aureus in intensive care units in US hospitals, 1992–2003. Clin Infect Dis 2006; 42: 389–91. [DOI] [PubMed] [Google Scholar]
- 9.Moran GJ, Amii RN, Abrahamian FM, et al. Methicillin-resistant Staphylococcus aureus in community-acquired skin infections. Emerg Infect Dis 2005; 11: 928–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischetti VA. Bacteriophage endolysins: a novel anti-infective to control Gram-positive pathogens. Int J Med Microbiol 2010; 300: 357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmelcher M, Donovan DM, Loessner MJ. Bacteriophage endolysins as novel antimicrobials. Future Microbiol 2012; 7: 1147–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spratt BG. Resistance to antibiotics mediated by target alterations. Science 1994; 264: 388–93. [DOI] [PubMed] [Google Scholar]
- 13.Schindler CA, Schuhardt VT. Lysostaphin: a new bacteriolytic agent for the Staphylococcus. Proc Natl Acad Sci USA 1964; 51: 414–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schleifer KH, Kandler O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol Rev 1972; 36: 407–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gründling A, Missiakas DM, Schneewind O. Staphylococcus aureus mutants with increased lysostaphin resistance. J Bacteriol 2006; 188: 6286–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loessner MJ. Bacteriophage endolysins—current state of research and applications. Curr Opin Microbiol 2005; 8: 480–7. [DOI] [PubMed] [Google Scholar]
- 17.Whisstock JC, Lesk AM. SH3 domains in prokaryotes. Trends Biochem Sci 1999; 24: 132–3. [DOI] [PubMed] [Google Scholar]
- 18.Becker SC, Foster-Frey J, Stodola AJ, et al. Differentially conserved staphylococcal SH3b_5 cell wall binding domains confer increased staphylolytic and streptolytic activity to a streptococcal prophage endolysin domain. Gene 2009; 443: 32–41. [DOI] [PubMed] [Google Scholar]
- 19.Becker SC, Dong S, Baker JR, et al. LysK CHAP endopeptidase domain is required for lysis of live staphylococcal cells. FEMS Microbiol Lett 2009; 294: 52–60. [DOI] [PubMed] [Google Scholar]
- 20.Navarre WW, Ton-That H, Faull KF, et al. Multiple enzymatic activities of the murein hydrolase from staphylococcal phage φ11. Identification of a d-alanyl-glycine endopeptidase activity. J Biol Chem 1999; 274: 15847–56. [DOI] [PubMed] [Google Scholar]
- 21.Schindler CA, Schuhardt VT. Purification and properties of lysostaphin—a lytic agent for Staphylococcus aureus. Biochim Biophys Acta 1965; 97: 242–50. [DOI] [PubMed] [Google Scholar]
- 22.Abaev I, Foster-Frey J, Korobova O, et al. Staphylococcal phage 2638A endolysin is lytic for Staphylococcus aureus and harbors an inter-lytic-domain secondary translational start site. Appl Microbiol Biotechnol 2013; 97: 3449–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmelcher M, Korobova O, Schischkova N, et al. Staphylococcus haemolyticus prophage PhiSH2 endolysin relies on cysteine, histidine-dependent amidohydrolases/peptidases activity for lysis ‘from without’. J Biotechnol 2012; 162: 289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yokoi KJ, Kawahigashi N, Uchida M, et al. The two-component cell lysis genes holWMY and lysWMY of the Staphylococcus warneri M phage varphiWMY: cloning, sequencing, expression, and mutational analysis in Escherichia coli. Gene 2005; 351: 97–108. [DOI] [PubMed] [Google Scholar]
- 25.Loessner MJ, Gaeng S, Wendlinger G, et al. The two-component lysis system of Staphylococcus aureus bacteriophage Twort: a large TTG-start holin and an associated amidase endolysin. FEMS Microbiol Lett 1998; 162: 265–74. [DOI] [PubMed] [Google Scholar]
- 26.Christie GE, Matthews AM, King DG, et al. The complete genomes of Staphylococcus aureus bacteriophages 80 and 80α—implications for the specificity of SaPI mobilization. Virology 2010; 407: 381–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jayaswal RK, Lee YI, Wilkinson BJ. Cloning and expression of a Staphylococcus aureus gene encoding a peptidoglycan hydrolase activity. J Bacteriol 1990; 172: 5783–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Flaherty S, Coffey A, Meaney W, et al. The recombinant phage lysin LysK has a broad spectrum of lytic activity against clinically relevant staphylococci, including methicillin-resistant Staphylococcus aureus. J Bacteriol 2005; 187: 7161–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manoharadas S, Witte A, Bläsi U. Antimicrobial activity of a chimeric enzybiotic towards Staphylococcus aureus. J Biotechnol 2009; 139: 118–23. [DOI] [PubMed] [Google Scholar]
- 30.Kerr DE, Plaut K, Bramley AJ, et al. Lysostaphin expression in mammary glands confers protection against staphylococcal infection in transgenic mice. Nat Biotechnol 2001; 19: 66–70. [DOI] [PubMed] [Google Scholar]
- 31.Donovan DM, Foster-Frey J. LambdaSa2 prophage endolysin requires Cpl-7-binding domains and amidase-5 domain for antimicrobial lysis of streptococci. FEMS Microbiol Lett 2008; 287: 22–33. [DOI] [PubMed] [Google Scholar]
- 32.Reichelt P, Schwarz C, Donzeau M. Single step protocol to purify recombinant proteins with low endotoxin contents. Protein Expr Purif 2006; 46: 483–8. [DOI] [PubMed] [Google Scholar]
- 33.Donovan DM, Dong S, Garrett W, et al. Peptidoglycan hydrolase fusions maintain their parental specificities. Appl Environ Microbiol 2006; 72: 2988–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eugster MR, Haug MC, Huwiler SG, et al. The cell wall binding domain of Listeria bacteriophage endolysin PlyP35 recognizes terminal GlcNAc residues in cell wall teichoic acid. Mol Microbiol 2011; 81: 1419–32. [DOI] [PubMed] [Google Scholar]
- 35.Riordan JF, Vallee BL. Acetylation. In: Hirs CHW, ed. Methods in Enzymology. New York: Academic Press, 1967; 565–70. [Google Scholar]
- 36.Park JT, Johnson MJ. A submicrodetermination of glucose. J Biol Chem 1949; 181: 149–51. [PubMed] [Google Scholar]
- 37.Spiro RG. Analysis of sugars found in glycoproteins. In: Neufeld EF, Ginsburg V, eds. Methods in Enzymology. New York: Academic Press, 1966; 3–26. [Google Scholar]
- 38.Ghuysen JM, Tipper DJ, Strominger JL. Enzymes that degrade bacterial cell walls. Methods Enzymol 1966; 8: 685–99. [Google Scholar]
- 39.Pritchard DG, Dong S, Baker JR, et al. The bifunctional peptidoglycan lysin of Streptococcus agalactieae bacteriophage B30. Microbiology 2004; 150: 2079–87. [DOI] [PubMed] [Google Scholar]
- 40.Sass P, Bierbaum G. Lytic activity of recombinant bacteriophage phi11 and phi12 endolysins on whole cells and biofilms of Staphylococcus aureus. Appl Environ Microbiol 2007; 73: 347–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Christensen GD, Simpson WA, Younger JJ, et al. Adherence of coagulase-negative staphylococci to plastic tissue culture plates: a quantitative model for the adherence of staphylococci to medical devices. J Clin Microbiol 1985; 22: 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Daniel A, Euler C, Collin M, et al. Synergism between a novel chimeric lysin and oxacillin protects against infection by methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 2010; 54: 1603–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biswas B, Adhya S, Washart P, et al. Bacteriophage therapy rescues mice bacteremic from a clinical isolate of vancomycin-resistant Enterococcus faecium. Infect Immun 2002; 70: 204–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Synnott AJ, Kuang Y, Kurimoto M, et al. Isolation from sewage influent and characterization of novel Staphylococcus aureus bacteriophages with wide host ranges and potent lytic capabilities. Appl Environ Microbiol 2009; 75: 4483–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weidenmaier C, Kokai-Kun JF, Kristian SA, et al. Role of teichoic acids in Staphylococcus aureus nasal colonization, a major risk factor in nosocomial infections. Nat Med 2004; 10: 243–5. [DOI] [PubMed] [Google Scholar]
- 46.Peschel A, Vuong C, Otto M, et al. The d-alanine residues of Staphylococcus aureus teichoic acids alter the susceptibility to vancomycin and the activity of autolytic enzymes. Antimicrob Agents Chemother 2000; 44: 2845–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kropec A, Maira-Litran T, Jefferson KK, et al. Poly-N-acetylglucosamine production in Staphylococcus aureus is essential for virulence in murine models of systemic infection. Infect Immun 2005; 73: 6868–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weidenmaier C, Kokai-Kun JF, Kulauzovic E, et al. Differential roles of sortase-anchored surface proteins and wall teichoic acid in Staphylococcus aureus nasal colonization. Int J Med Microbiol 2008; 298: 505–13. [DOI] [PubMed] [Google Scholar]
- 49.Watts A, Ke D, Wang Q, et al. Staphylococcus aureus strains that express serotype 5 or serotype 8 capsular polysaccharides differ in virulence. Infect Immun 2005; 73: 3502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yokogawa K, Kawata S, Nishimura S, et al. Mutanolysin, bacteriolytic agent for cariogenic streptococci: partial purification and properties. Antimicrob Agents Chemother 1974; 6: 156–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fenton M, Casey PG, Hill C, et al. The truncated phage lysin CHAP(k) eliminates Staphylococcus aureus in the nares of mice. Bioeng Bugs 2010; 1: 404–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kusuma C, Kokai-Kun J. Comparison of four methods for determining lysostaphin susceptibility of various strains of Staphylococcus aureus. Antimicrob Agents Chemother 2005; 49: 3256–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gaeng S, Scherer S, Neve H, et al. Gene cloning and expression and secretion of Listeria monocytogenes bacteriophage-lytic enzymes in Lactococcus lactis. Appl Environ Microbiol 2000; 66: 2951–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cisani G, Varaldo PE, Grazi G, et al. High-level potentiation of lysostaphin anti-staphyloccocal activity by lysozyme. Antimicrob Agents Chemother 1982; 21: 531–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Idelevich EA, von Eiff C, Friedrich AW, et al. In vitro activity against Staphylococcus aureus of a novel antimicrobial agent, PRF-119, a recombinant chimeric bacteriophage endolysin. Antimicrob Agents Chemother 2011; 55: 4416–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Atilano ML, Pereira PM, Yates J, et al. Teichoic acids are temporal and spatial regulators of peptidoglycan cross-linking in Staphylococcus aureus. Proc Natl Acad Sci USA 2010; 107: 18991–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gründling A, Schneewind O. Cross-linked peptidoglycan mediates lysostaphin binding to the cell wall envelope of Staphylococcus aureus. J Bacteriol 2006; 188: 2463–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Son JS, Lee SJ, Jun SY, et al. Antibacterial and biofilm removal activity of a Podoviridae Staphylococcus aureus bacteriophage SAP-2 and a derived recombinant cell-wall-degrading enzyme. Appl Microbiol Biotechnol 2010; 86: 1439–49. [DOI] [PubMed] [Google Scholar]
- 59.Lewis K. Riddle of biofilm resistance. Antimicrob Agents Chemother 2001; 45: 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abee T, Kovacs AT, Kuipers OP, et al. Biofilm formation and dispersal in Gram-positive bacteria. Curr Opin Biotechnol 2011; 22: 172–9. [DOI] [PubMed] [Google Scholar]
- 61.Borysowski J, Weber-Dabrowska B, Gorski A. Bacteriophage endolysins as a novel class of antibacterial agents. Exp Biol Med (Maywood) 2006; 231: 366–77. [DOI] [PubMed] [Google Scholar]
- 62.Entenza JM, Loeffler JM, Grandgirard D, et al. Therapeutic effects of bacteriophage Cpl-1 lysin against Streptococcus pneumoniae endocarditis in rats. Antimicrob Agents Chemother 2005; 49: 4789–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Witzenrath M, Schmeck B, Doehn JM, et al. Systemic use of the endolysin Cpl-1 rescues mice with fatal pneumococcal pneumonia. Crit Care Med 2009; 37: 642–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.