

Abstract
Microbial populations associated with microbial enhanced oil recovery (MEOR) and their abundance in the Xinjiang Luliang water-flooding petroleum reservoir were investigated using 16S rRNA, nitrate reductases, dissimilatory sulfate reductase, and methyl coenzyme-M reductase-encoded genes to provide ecological information for the potential application of MEOR. 16S rRNA gene miseq sequencing revealed that this reservoir harbored large amounts of taxa, including 155 bacterial and 7 archeal genera. Among them, Arcobacter, Halomonas, Marinobacterium, Marinobacter, Sphingomonas, Rhodococcus, Pseudomonas, Dietzia, Ochrobactrum, Hyphomonas, Acinetobacter, and Shewanella were dominant, and have the potential to grow using hydrocarbons as carbon sources. Metabolic gene clone libraries indicated that the nitrate-reducing bacteria (NRB) mainly belonged to Pseudomonas, Azospirillum, Bradyrhizobium, Thauera, Magnetospirillum, Sinorhizobium, Azoarcus, and Rhodobacter; the sulfate-reducing bacteria (SRB) were Desulfarculus, Desulfomonile, Desulfosarcina, Desulfotignum, Desulfacinum, Desulfatibacillum, Desulfatibacillum, Desulfomicrobium, and Desulfovibrio; while the methanogens were archaea and belonged to Methanomethylovorans, Methanosaeta, Methanococcus, Methanolobus, and Methanobacterium. Real-time quantitative PCR analysis indicated that the number of bacterial 16S rRNA reached 106 copies/mL, while the metabolic genes of NRB, SRB, and methanogens reached 104 copies/mL. These results show that the Luliang reservoir has abundant microbial populations associated with oil recovery, suggesting that the reservoir has potential for MEOR.
Keywords: 16S rRNA, MEOR, microbial community, miseq, QPCR
Introduction
With an increasing global energy demand and the depletion of oil reserves, water- and chemical-flooding and microbial enhanced oil recovery (MEOR) are currently studied intensively (Youssef et al. 2009; Wackett 2012). In particular, MEOR is considered to be the most economically feasible because of its low energy consumption, low environmental impact, and cost-effectiveness (Youssef et al. 2009; Simpson et al. 2011). This technique uses reservoir microorganisms and their metabolites to reduce crude oil viscosity, enhance permeability of reservoirs, and selectively plug large pore paths to improve oil recovery.
Complex ecosystems comprising various types of microorganisms are present in petroleum reservoirs. Since Bastin et al. 1926 first isolated sulfate-reducing bacteria (SRB) from production water in 1926, culture-dependent and -independent methodologies, in particular, 16S rRNA-based molecular identification methods, have revealed diverse microorganisms inhabiting petroleum reservoirs (Kumaraswamy et al. 2011; Al-Bahry et al. 2013; Lenchi et al. 2013; Okoro et al. 2014). Among them, hydrocarbon-degrading bacteria (HDB), nitrate-reducing bacteria (NRB), SRB, and methanogens are the important populations of reservoir ecosystems, and have critical roles in the microbial enhancing of the oil recovery process (Youssef et al. 2009). The majority of HDB can produce biosurfactants when growing with hydrocarbon as the carbon source. These biosurfactants improve oil emulsification and lower interfacial tension between the oil and water phase, which further improves oil fluidity in oil-bearing reservoirs. Reducing interfacial tensions and decreasing oil viscosity are important mechanisms involved in MEOR. NRB and SRB are common inhabitants of the oil field ecosystem. The increase of H2S (production of SRB) is associated with the corrosion of pipelines, platform structures, and other equipment; increases refining costs of oil and gas; plugs reservoirs by the accumulation of sulfides minerals; and increases health risks because of the toxicity of H2S. Recently, the stimulation of NRB by the addition of nitrate, nitrite, or nitrate/molybdate mixtures has been used to inhibit SRB propagation by out-competing the growth of SRB (Bodtker et al. 2008; Gao et al. 2014). As the terminal process of the microbial metabolism chain, methanogens reflect the ecological integrality and metabolic activity of a reservoir ecosystem. In addition, methanogens metabolize hydrogen and CO2, acetate, methylamines, and dimethylsulfides with the concurrent production of methane that increases reservoir pressure and decreases oil viscosity.
In this study, 16S rRNA gene miseq sequencing, nitrate reductase, dissimilatory sulfite reductase, and methyl coenzyme-M reductase-encoded gene (napA, dsrB, and mcrA) clone libraries were performed to investigate the microbial communities and the distribution of NRB, SRB, and methanogens in the Luliang water-flooding petroleum reservoir in the XinJiang Oilfield. This reservoir will improve oil recovery by stimulating reservoir microorganisms. Therefore, the primary objective of the study was to provide ecological information on microbial populations and the biological control potential for SRB. This study also provides us the opportunity to evaluate the performance of 16S rRNA gene miseq sequencing for the analysis of reservoir microbial community by the metabolic gene clone libraries and gene quantification.
Materials and Methods
Sample collection and DNA extraction
Samples of injected water and produced water (oil-water mixture) were collected from the wellhead of injection and the production wells of a mesothermic water-flooding reservoir located in the Xinjiang Luliang Oil Field by PetroChina field personnel (Fig.1). This field block is a homogeneous sandstone reservoir with an average permeability of 522 × 10−3 μm2, and has been water-flooded since 2001. The water samples were collected randomly on October 2012, from sampling valves located on the production wellhead. Approximately 25 L of each sample were immediately sealed to avoid contamination and oxygen intrusion. The bottles were then transported to the laboratory as soon as possible for further analysis (7 days later). Microbial cells were collected from a 5 L water sample by centrifugation at 4°C for 15 min at 10,000g in a high-speed centrifuge (Beckman, CA 92821, USA). The genomic DNA was extracted as described by Li et al. (2014). To collect as much of the microbial genomes as possible, the collected cells were resuspended with a TE buffer, and then lysed using a mini bead-beater (BioSpec, Bartlesville, OK 74005, USA) at 4°C and 200 rpm for 1 min at room temperature with 0.1 mm glass beads. DNA was extracted from the suspension solution using an AxyPrep™ Bacterial Genomic DNA Miniprep Kit (Axygen Biosciences, Tewksbury, MA 01876, USA) according to the manufacturer's instructions and then stored at −80°C for subsequent study.
Figure 1.
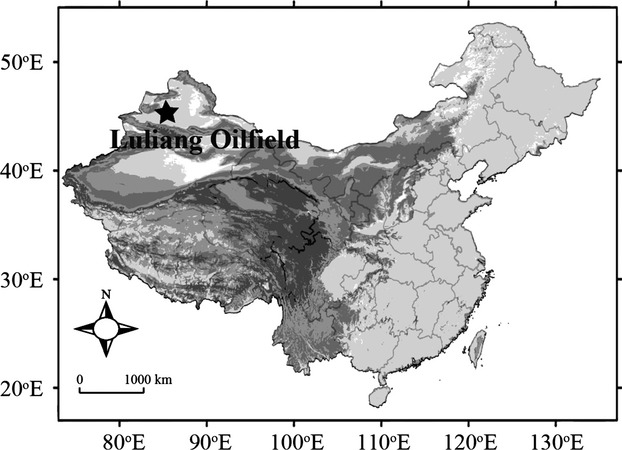
The geographical position of Luliang oilfield that is located in northwest of China. In this oilfield, injected water samples (Lu3064 and Lu3084) and produced water samples (Lu3065 and Lu3096) were collected on October 2012.
Real-time quantitative PCR analysis of microbial abundance
Evaluation of microbial community abundance by quantitative PCR (QPCR) was performed using 16S rRNA and napA, dsrB, and mcrA genes as molecular markers. Reactions were performed using the FastStart Universal SYBR Green Master PCR mix (Roche Applied Science, Mannheim, Germany) in a Bio-Rad iQ5 Sequence detection system Bio-Rad, CA 92821, USA. QPCR of bacterial 16S rRNA genes were performed with the primer set 8F (5′-AGA GTT TGA T(CT)(AC) TGG CTC-3′) and 338R (5′-GCT GCC TCC CGT AGG AGT-3′) as described by Schulz et al. (2010) and Li et al. (2013). QPCR of napA, dsrB, and mcrA were performed with the primer sets described in the Clone library construction of napA, dsrB, and mcrA genes. Plasmids containing the target genes were used as standards. The plasmid DNA concentration was determined on a Nanodrop spectrophotometer (Thermo Fisher Scientific, Wilmington, DE). The copy number of the target genes in the initial standard was calculated directly from the concentration of the extracted plasmid DNA. Gene copy numbers in unknown samples were determined based on standard curves constructed from 10-fold serial dilutions of the standard. Amplification efficiencies were calculated from the slope of standard curves. The specificity of PCR amplification was determined using the melting curve.
Miseq sequencing of partial 16S rRNA genes and sequence analysis
The V4 region of 16S rRNA gene (300–350 bp) was amplified with broadly conserved primer set 515f (GTG CCA GCM GCC GCG GTAA) and 806r (GGA CTA CHV GGG TWT CTA AT). The primer set was reported to be able to yield optimal community clustering with sequences of this length (Caporaso et al. 2011). PCR reactions were performed following the protocol described in Caporaso et al. Amplicon sequencing was conducted on an Illumina MiSeq platform at Novogene co., Beijing, China. Pairs of reads from the original DNA fragments were merged using FLASH (Magoc and Salzberg 2011). Sequences were then analyzed using the QIIME (Caporaso et al. 2010) and UPARSE pipeline (Edgar 2013). First, the reads were filtered using QIIME quality filters with default parameters. Then, a UPARSE pipeline was used to pick operational taxonomic units (OTUs) at 97% similarity. The resulting representative sequence set was aligned and given a taxonomic classification using RDP (Wang et al. 2007). The microbial distribution in the water samples was visualized using R package based on community composition information at taxonomic levels.
Clone library construction of napA, dsrB, and mcrA genes
Primer set napAf1 (5′-C TGG ACI ATG GGY TTI AAC CA-3′) and napAr1 (5′-CC TTC YTT YTC IAC CCA CAT-3′) were used to amplify napA gene (490 bp) (Feng et al. 2011). DSRp2060F (5′-CAA CAT CGT YCA YAC CCA GGG-3′) and DSR4R (5′-GTG TAG CAG TTA CCG CA-3′) were used to amplify dsrB gene (390 bp) (Geets et al. 2006). Primer set mcrAF (5′-GGT GGT GTM GGD TTC ACM CAR TA-3′) and mcrAR (5′-CGT TCA TBG CGT AGT TVG GRT AGT-3′) were used for the amplification of mcrA genes (450 bp) (Steinberg and Regan 2008). The PCR reaction mixtures and conditions are described in the supporting information. The purified PCR products were cloned into Escherichia coli using pEasy-T1 clone vector according to the manufacturer's instructions. The sequences of inserted PCR products were determined with an automated ABI 3730 DNA sequencer using M13 universal sequencing primers.
The retrieved napA, dsrB, and mcrA genes nucleotide sequences were truncated to exclude primers and vector sequences using the FinchTV 1.4.0 program (Wang et al. 2010), and were then translated into protein sequences using the “transeq” algorithm of the EMBOSS program. Deduced protein sequences were compared with sequences in the NCBI Gene Bank database, and were grouped into OTUs based on species taxa. Distance-based evolutionary trees were constructed using the neighbor-joining method with 1000 bootstrap replicates with MEGA 4 (Tamura et al. 2007).
Sequence accession numbers
The 16S rRNA genes reads were deposited in the National Center for Biotechnology Information (BioProject ID: PRJNA252404, http://www.ncbi.nlm.nih.gov/bioproject/252404). The sequences of dsrB genes were deposited in the GenBank database under accession numbers KC466037 to KC466050; the sequences of napA genes were deposited under accession numbers KC466065 to KC466079; and the sequences of mcrA genes were deposited under accession numbers KC466051 to KC466064.
Results
Physicochemical characteristics of the Luliang reservoir
The Luliang water-flooding reservoir is located in the Xinjiang Oil Field, northwest China. This block has been water-flooded since 2001, with an average water content of 80.3%. The formation temperature is ∽42°C. The average porosity is 29.9%, with an average permeability of 522 × 10−3 μm2. The physicochemical characteristics of the injected (Lu3064 and Lu3084) and produced (Lu3065 and Lu3096) water samples indicate that the concentrations of sodium, potassium, calcium, magnesium, and manganese are suitable for microbial growth (Table1). The nitrate and phosphate levels were lack for microbial growth. Sulfate (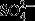 ) concentrations were between 4.9 and 116.2 mg/L, indicating that sulfate reduction could occur in this reservoir (Bodtker et al. 2008).
) concentrations were between 4.9 and 116.2 mg/L, indicating that sulfate reduction could occur in this reservoir (Bodtker et al. 2008).
Table 1.
Chemical properties of water samples obtained from Luliang reservoir
| Environmental characteristic | Lu3064 | Lu3084 | Lu3065 | Lu3096 | Average |
|---|---|---|---|---|---|
| Well type | Injection well | Production well | – | ||
| Water cut, % | – | – | 82% | 85% | – |
| pH | 5.5–6.0 | 5.5–6.0 | 5.5–6.0 | 5.5–6.0 | 5.5–6.0 |
| Salinity | 12478 | 10635 | 10177 | 9214 | 10700 |
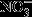 , mg/L , mg/L |
<0.1 | <0.1 | <0.1 | <0.1 | <0.1 |
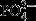 , mg/L , mg/L |
<0.1 | <0.1 | <0.1 | <0.1 | <0.1 |
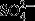 , mg/L , mg/L |
4.9 | 116.2 | 14.0 | 23.1 | 39.55 |
| Cl−, mg/L | 6640 | 5640 | 5050 | 4600 | 5482.5 |
| Na+, mg/L | 5460 | 4460 | 4759 | 4516 | 4798.5 |
| K+, mg/L | 56.8 | 64.9 | 44.1 | 49.3 | 53.8 |
| Ca2+, mg/L | 284.7 | 332.1 | 281.9 | 181.6 | 299.6 |
| Mg2+, mg/L | 31.6 | 21.7 | 27.8 | 26.03 | 26.8 |
Detection limit is 0.1 mg/L.
Quantification of microbial communities
Total bacteria, NRB, SRB, and methanogens were estimated based on the quantification of bacterial 16S rRNA, napA, dsrB, and mcrA genes using QPCR methods. The copy numbers of 16S rRNA, napA, dsrB, and mcrA genes in the injection and production water samples ranged from 1.86 × 106 to 5.62 × 106 copies/mL, 3.87 × 104 to 6.15 × 104 copies/mL, 1.88 × 104 to 4.02 × 104 copies/mL, and 1.88 × 104 to 2.72 × 104 copies/mL, respectively (Fig.2). Assuming that a bacteria contained one copy of the metabolic functional genes (Schulz et al. 2010; Li et al. 2013) and 3.6 copies of 16S rRNA genes per cell genome (Harms et al. 2003), the total bacterial density was calculated as 5.17 × 105 to 1.56 × 106 cells/L. The number of NRB, SRB, and methanogens reached 104 cells/L, which suggests that the ratio of these populations to total bacteria ranged from 39.4‰ to −74.9‰, 12.0‰ to 77.8‰, and 17.4‰ to 36.4‰, respectively.
Figure 2.
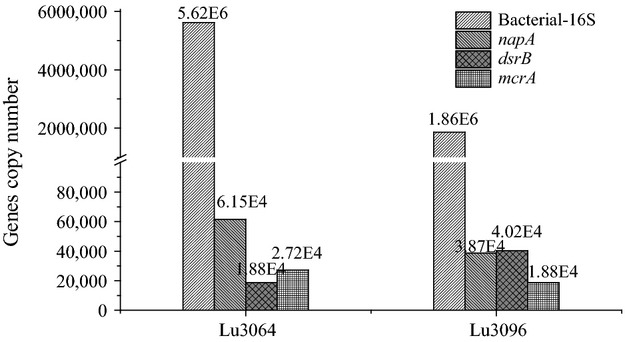
The copy numbers of 16S rRNA, napA, dsrB, and mcrA genes in the injected and produced water samples obtained from Luliang reservoir.
Statistical analysis of 16S rRNA miseq sequencing and the metabolic gene clone library
A total of 16,568 to 115,661 high-quality 16S rRNA gene sequences were retrieved from the four injected and produced water samples (Table2). The numbers of OTUs in each injected and produced water sample ranged in size from 1085 to 1515 (Table2). In combination with the community composition and relative abundance, the number of bacterial and archaeal sequences was calculated, with bacterial sequences in each injected and produced water samples ranging in size from 16554 to 115205, whereas only 13–455 archaeal sequences were obtained (Fig. S1). A total of 40–50 metabolic gene sequences were retrieved from the napA, dsrB, and mcrA gene clone libraries, with 4–11 OTUs per sample (Table2).
Table 2.
Statistical analysis of 16S rRNA miseq sequencing and metabolic genes clone libraries
| Library | Lu3084 | Lu3065 | Lu3064 | Lu3096 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 16S | 16S | 16S | napA | dsrB | mcrA | 16S | napA | dsrB | mcrA | |
| Sequences | 16568 | 19692 | 115661 | 45 | 42 | 40 | 48400 | 40 | 50 | 41 |
| OTUs | 1238 | 1197 | 1515 | 11 | 8 | 7 | 1085 | 4 | 6 | 7 |
| Shannon | 7.07 | 7.11 | 6.73 | – | – | – | 6.70 | – | – | – |
| Coverage, % | 99.9 | 99.9 | 99.9 | 88.1 | 98% | 95% | 99.8 | 97.5% | 96% | 97.6% |
OTU, operational taxonomic units.
Phylogenetic analysis of bacterial 16S rRNA genes
The classification and phylogenetic analysis indicated that all the bacterial sequences fell within 38 phyla (Fig. S2). The phylum Proteobacteria, Bacteroidetes, Chloroflexi, and Firmicutes predominated, representing 77.94–93.89% of the bacterial communities in the water samples (Fig. S2). The remaining bacterial sequences were mainly assigned to the phylum Cyanobacteria, Actinobacteria, Thermotogae, Planctomycetes, Chlamydiae, Spirochetes, Synergistetes, and candidate division WPS-2 (Fig. S2). At class level, 84.5–94.8% of bacterial sequences were assigned to Gammaproteobacteria, Alphaproteobacteria, Betaproteobacteria, Bacteroidia, Epsilonproteobacteria, Anaerolineae, Deltaproteobacteria, Sphingobacteriia, Bacilli, Actinobacteria, Thermotogae, and Phycisphaerae (Fig.3). A total of 155 genera were observed, accounting for 42.4–53.5% of the total bacterial communities in the water samples (Table S1).
Figure 3.
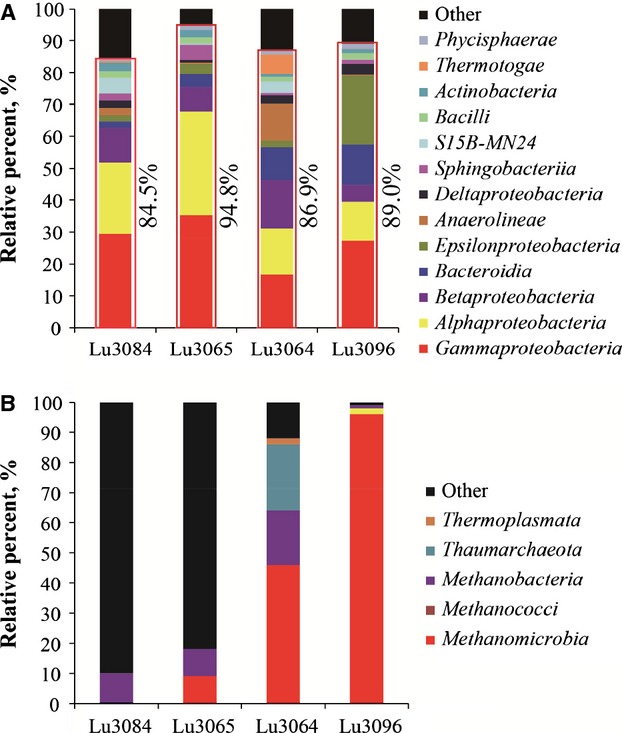
The relative proportion of (A) bacterial and (B) archaeal phylogenetic groups at class level in the injected (Lu3064 and Lu3084) and produced (Lu3065 and Lu3096) water samples obtained from Luliang reservoir.
Proteobacteria were mainly detected in this reservoir, with a relative percent of 16.6–35.3% in each water sample. Among them, Gammaproteobacteria were most dominant, with a relative percent of 16.6–35.3% in each water sample (Fig.3). Among them, Marinobacter, Pseudomonas, Acinetobacter, Halomonas, and Shewanella were most numerous (Fig.4). The remaining genera were closely related to Alishewanella, Pseudidiomarina, Trabulsiella, Enhydrobacter, Methylophaga, and Pseudoxanthomonas. Alphaproteobacteria, accounting for 16.6–35.3% of each community, were the second most common bacteria in the reservoir with 22 frequently detected genera (Figs.3 and 4). Among them, Agrobacterium, Rhodobacter, Rhodospirillum, and Azospirillum were dominant. Betaproteobacteria made up 5.3–15.3% of the reservoir bacterial communities (Fig.3). The most frequently sequenced genera were Achromobacter, Acidovorax, Aquabacterium, Hylemonella, Rubrivivax, Azovibrio, and Thauera (Fig.4). Epsilonproteobacteria accounted for 1.9–21.6% of the bacterial communities in the reservoir (Fig.3), with Arcobacter, Sulfurospirillum, and Sulfurimonas most frequently detected (Fig.4). Deltaproteobacteria accounted for 0.87–3.28% of the bacterial communities (Fig.3) and the dominant genera were Desulfomicrobium and Desulfovibrio, as well as Syntrophobacterales (Fig.4). Actinobacteriae accounted for 0.96–2.58% of the bacterial communities in the reservoir (Fig.3) and the dominant genera were Dietzia, Rhodococcus, Mycobacterium, Corynebacterium, and Propionibacterium (Fig.4).
Figure 4.
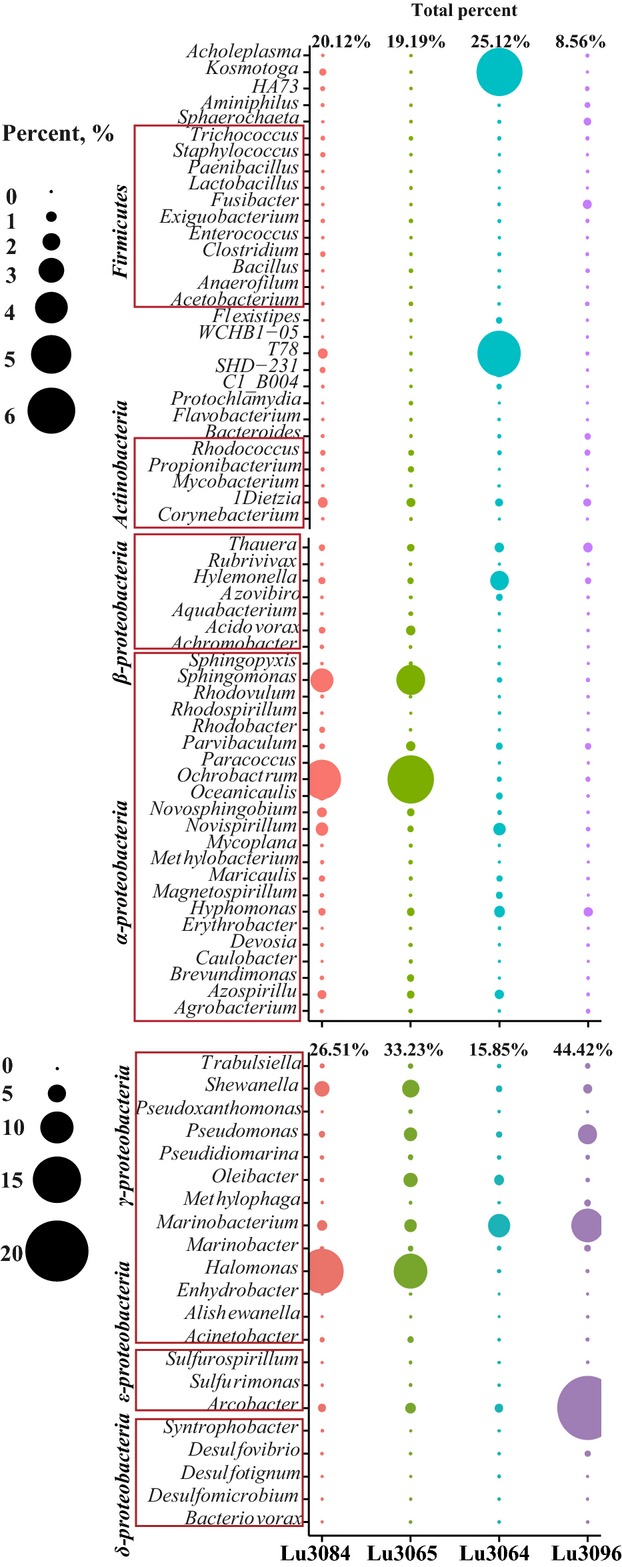
The dominant bacterial genera and their relative abundance in the injected (Lu3064 and Lu3084) and produced (Lu3065 and Lu3096) water samples obtained from Luliang reservoir. These genera were mainly affiliated with the phylum of Proteobacteria, Actinobateria, and Firmicutes.
Bacteroidia and Bacilli were also detected in the reservoir, with relative abundances of 1.89–12.7% and 1.46–2.11%, respectively (Fig.3). In the Bacteroidia class, most sequences could not be identified at the genus level with at least 80% confidence. Only genus Bacteroides was detected, with a relative abundance of 0.09–0.45% (Fig.4). In the Bacilli class, the dominant genera were Bacillus, Paenibacillus, Staphylococcus, Exiguobacterium, Trichococcus, Enterococcus, and Lactobacillus (Fig.4).
Phylogenetic analysis of archaeal 16S rRNA genes
Only 13 and 17 archaeal 16S rRNA gene sequences were obtained from Lu3084 and Lu3065 water samples, while a total of 455 and 389 archaeal sequences were retrieved from Lu3064 and Lu3096 water samples (Fig. S1), respectively. The classification and phylogenetic analysis indicated that all the archaeal sequences retrieved from Lu3064 and Lu3096 water samples fell within the Crenarchaeota and Euryarchaeota phyla. The classes Thaumarchaeota, Methanobacteria, Methanococci, and Methanomicrobia accounted for 86% and 99% of the archaeal communities in Lu3064 and Lu3096 water samples, respectively. The dominant genera were Methanobacterium, Methanothermobacter, Methanococcus, Methanocalculus, Methanomethylovorans, Methanolobus, and Nitrosopumilus (Table S3).
Phylogenetic analysis of napA, dsrB, and mcrA genes
The napA sequences retrieved from this reservoir were mainly assigned to Azospirillum sp., Magnetospirillum magnetotacticum, Rhodobacter, Sphaeroides, Bradyrhizobium sp., Sinorhizobium fredii, Azoarcus sp., Thauera sp., Candidatus Accumulibacter, phosphatis clade IIA, Bordetella petrii, Laribacter hongkongensis, Pseudomonas stutzeri, and Pseudomonas aeruginosa (Table3 and Fig.5A). The retrieved dsrB sequences were assigned to Desulfarculus baarsii, Desulfomonile tiedjei, Desulfacinum infernum, Desulfosarcina sp., Desulfobulbus sp., Desulfotignum balticum, Desulfatibacillum alkenivorans, Desulfovibrio alkalitolerans, Desulfovibrio aminophilus, and Desulfovibrio vulgaris (Table3 and Fig.5B). The retrieved mcrA sequences were assigned to Methanococcus maripaludis, Methanococcus vannielii, Methanothermococcus thermolithotrophicus, Methanosaeta thermophila, Methanolobus psychrophilus, Methanolobus tindarius, Methanolobus vulcani, uncultured Methanomethylovorans sp., uncultured Methanosarcinales archaeon, Methanobacterium formicicum, and Methanobacterium thermoautotrophicum (Table3 and Fig.5C).
Table 3.
NRB, SRB, and methanogens detected in metabolic genes clone libraries
| Metabolic function | Order | Family | Closest species (obtained from NCBI) |
|---|---|---|---|
| Nitrate-reducing bacteria | Rhodospirillales | Rhodospirillaceae | Azospirillum sp. B510 |
| Magnetospirillum magnetotacticum | |||
| Rhodobacterales | Rhodobacteraceae | Rhodobacter sphaeroides f. sp | |
| Rhizobiales | Bradyrhizobiaceae | Bradyrhizobium sp. | |
| Rhizobiaceae | 1Sinorhizobium fredii | ||
| Rhodocyclales | Rhodocyclaceae | 1Azoarcus sp. KH32C | |
| Thauera sp. MZ1T | |||
| 1Candidatus Accumulibacter | |||
| 1phosphatis clade IIA | |||
| Burkholderiales | Alcaligenaceae | 1Bordetella petrii | |
| Neisseriales | Neisseriaceae | 1Laribacter hongkongensis | |
| Pseudomonadales | Pseudomonadaceae | Pseudomonas stutzeri | |
| Pseudomonas aeruginosa | |||
| Sulfate-reducing bacteria | Desulfarculales | Desulfarculaceae | 1Desulfarculus baarsii |
| Syntrophobacterales | Syntrophaceae | 1Desulfomonile tiedjei | |
| 1Desulfacinum infernum | |||
| Desulfobacterales | Desulfobacteraceae | 1Desulfosarcina sp | |
| Desulfobulbaceae | Uncultured Desulfobulbus sp. | ||
| Desulfotignum balticum | |||
| Desulfobacteraceae | 1Desulfatibacillum alkenivorans | ||
| Desulfovibrionales | Desulfovibrionaceae | Desulfovibrio alkalitolerans | |
| Desulfovibrio aminophilus | |||
| Desulfovibrio vulgaris RCH1 | |||
| Methanogens | Methanococcales | Methanococcaceae | Methanococcus maripaludis |
| Methanococcus vannielii | |||
| 1Methanothermococcus thermolithotrophicus | |||
| Methanosarcinales | Methanosaetaceae | 1Methanosaeta thermophila | |
| Methanosarcinaceae | Methanolobus psychrophilus | ||
| Methanolubus tindarius | |||
| Methanolobus vulcani | |||
| Methanomethylovorans sp. | |||
| Methanosarcinales archaeon | |||
| Methanobacteriales | Methanobacteriaceae | Methanobacterium formicicum | |
| Methanobacterium thermoautotrophicum | |||
| Methanobacterium sp. SA-12 |
Represents microbial populations that only detected by metabolic genes clone libraries. The remaining microbial populations were detected by both of metabolic genes clone libraries and 16S rRNA gene miseq sequencing.
Figure 5.

Phylogenetic tree of napA (A), dsrB (B) and mcrA (C) protein sequences detected in the injected and produced water samples obtained from Luliang reservoir. Distance-based evolutionary trees were constructed by the neighboring-joining method with 1000 bootstrap replicates. The scale bar represents 0.05 inferred substitutions per nucleotide position. Percentages of bootstrap support are indicated at the branch points. The nucleotide sequence accession numbers and clones of each OTU were presented in brackets. OTU, operational taxonomic units.
Discussion
We investigated the microbial communities and the distribution of NRB, SRB, and methanogens in the Luliang water-flooding petroleum reservoir in the XinJiang Oil Field. The results provide ecological information on the microbial composition and the biological control potential for SRB during the stimulation of reservoir microorganisms to enhance oil recovery. The results indicate that the reservoir harbors diverse microbial populations. Based on 16s rRNA miseq sequencing, a total of 38 bacterial phyla and 155 genera were observed in the reservoir. In contrast with previous research (Tang et al. 2012; Lenchi et al. 2013; Okoro et al. 2014), this is the first study to show that so many microbial populations inhabit an oil reservoir. Statistical analysis of the 16S rRNA miseq sequencing indicated that 16,568 to 115,661 high-quality 16S rRNA gene sequences were retrieved from the water samples. The sequencing depth was ∽3- to 20-fold for 454 pyrosequences (assuming 5000 sequences per library), whereas it was 50–400 fold for the 16S rRNA gene clone library (assuming 300 clone per library). Miseq sequencing provides an opportunity to investigate the microbial community with an unprecedented level of detail. However, the current sequencing depth is still limited, in particular, for methanogens. In the present study, the bacterial and archaeal V4 region of 16S rRNA gene was simultaneously amplified with primer set 515f and 806r for miseq sequencing. Up to 16,554–115,205 bacterial sequences were obtained per sample, whereas only 13–455 archaeal sequences were obtained. The QPCR analysis indicated that the NRB, SRB, and methanogens only accounted for 39.4–74.9‰, 12.0–77.8‰, and 17.4–36.4‰ of the total bacteria, respectively. Therefore, in theory, only 12–77.8 NRB, SRB, or methanogens could be detected in the 16S rRNA gene library with 1000 sequences, suggesting the need for deeper sequencing for the detection of rare microbial species. This illustrates that even when water samples were obtained from the same production well (M17-10) at the same time and with the same sampling method, SRB were only detected in the Tang et al. (2012), but not by Wang et al. (2012).
Based on the RDP's FunGene library (Table S2) (http://fungene.cme.msu.edu//index.spr) (Cole et al. 2009), the bacterial populations with alkane monooxygenase gene (alk) were collected and categorized. In Gammaproteobacteria, Marinobacter, Pseudomonas, Acinetobacter, Halomonas, and Shewanella dominated, and have been reported to be able to degrade hydrocarbons. Furthermore, Marinobacterium, Pseudomonas, and Acinetobacter have also been described as halophilic oil-utilizing and rhamnolipids-producing bacteria (Abdel-Mawgoud et al. 2010; Satpute et al. 2010). In Alphaproteobacteria, the abundant species of Brevundimonas, Ochrobactrum, Hyphomonas, Paracoccus, and Sphingomonas have been reported to be able to degrade hydrocarbons. Among Betaproteobacteria, Thauera was reported to be able to reduce nitrate as well as degrade aromatic compounds (Song et al. 2000; Tang et al. 2012). Among Actinobacteriae, the dominant genera of Dietzia, Rhodococcus, and Mycobacterium are able to degrade hydrocarbons or produce biosurfactants (Cole et al. 2009; Wang et al. 2011; Xia et al. 2011). These microorganisms and the produced biosurfactants play important roles in enhancing oil recovery.
Alphaproteobacteria were the second most common bacteria in this reservoir. Among them, Agrobacterium, Rhodobacter, Rhodospirillum, and Azospirillum are closely related to denitrification. Within the Betaproteobacteria, Thauera has been reported to be able to reduce nitrate and degrade aromatic compounds (Song et al. 2000; Tang et al. 2012). Among Epsilonproteobacteria, most Sulfurospirillum and Arcobacter are associated with the cycling, oxidization, and reduction of sulfur and nitrogen (Tang et al. 2012). The dominant Deltaproteobacteria were affiliated with iron and sulfate reducers of Desulfomicrobium and Desulfovibrio, and with syntrophic bacteria of the Syntrophobacterales (Fig.4). The syntrophic bacteria of order Syntrophobacterales may convert propionate and butyrate to methanogenic substrates (Schmidt et al. 2014). The dominant methanogens were Methanobacterium, Methanothermobacter, Methanococcus, Methanocalculus, Methanomethylovorans, and Methanolobus (Table S3). Among them, Methanomethylovorans and Methanolobus are methyltrophic methanogens, while Methanothermobacter, Methanobacterium, Methanococcus, and Methanocalculus are CO2-reducing methanogens.
Metabolic gene clone libraries and QPCR analysis demonstrate that NRB, SRB, and methanogens were ubiquitous in the reservoir. The NRB mainly belonged to Pseudomonas, Azospirillum, Bradyrhizobium, Thauera, Magnetospirillum, Sinorhizobium, Azoarcus, and Rhodobacter. The SRB were Desulfarculus, Desulfomonile, Desulfosarcina, Desulfotignum, Desulfacinum, Desulfatibacillum, Desulfatibacillum, Desulfomicrobium, and Desulfovibrio. As reported in previous research (Wang et al. 2012; Zhao et al. 2012), the majority of the archaea identified in the reservoir were methanogens, including methyltrophic, acetoclastic, and CO2-reducing Methanomethylovorans, Methanosaeta, Methanococcus, Methanolobus, and Methanobacterium (Liu and Whitman 2008).
It is noteworthy that oil reservoirs have low redox potential and therefore harbor abundant anaerobic and facultative microorganisms. However, abundant aerobic microorganisms were also detected in the production well, and anaerobic microorganisms were also detected in the injected water. These microbial populations included Pseudomonas, Sphingomonas, Ochrobactrum, Dietzia, Arcobacter, Halomonas, Marinobacterium, and methanogens. This phenomenon may be closely related to the microbial populations in the injected water passing through reservoir strata and reaching production wells, while the produced water was collected and injected into the reservoir.
In summary, 16S rRNA gene miseq sequencing, metabolic gene clone libraries, and QPCR analysis indicate that abundant microbial populations, including HDB, NRB, SRB, and methanogens, are ubiquitous in the Luliang water-flooding reservoir. These results also suggest that this reservoir has potential for MEOR and biological control of SRB propagation by stimulating NRB.
Acknowledgments
This study was supported by the National High Technology Research and Development Program of China (grant no. 2009AA063502, 2013AA064402), the National Natural Science Foundation of China (grant no. 41373074 and 31100400), and the Chinese Postdoctoral Science Foundation (grant no. 2014M561175).
Conflict of Interest
The authors declare that obviously there is no conflict of interest regarding the publication of this article.
Supporting Information
The number of bacterial and archaeal sequences obtained from the injection and production water samples by 16S rRNA miseq sequencing.
Figure S2. The relative proportion of bacterial populations at phylum level in the injection and production water samples obtained from Luliang reservoir.
Table S1. The 155 bacterial genera detected in Luliang reservoir by 16S rRNA miseq sequencing.
Table S2. The bacterial populations with alkane monooxygenase gene (alk) obtained from RDP's FunGene.
Table S3. The archaeal genera detected in Luliang reservoir by 16S rRNA miseq sequencing.
References
- Abdel-Mawgoud AM, Lepine F. Deziel E. Rhamnolipids: diversity of structures, microbial origins and roles. Appl. Microbiol. Biotechnol. 2010;86:1323–1336. doi: 10.1007/s00253-010-2498-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Bahry SN, Elshafie AE, Al-Wahaibi YM, Al-Bemani AS, Joshi SJ, Al-Maaini RA, et al. Microbial consortia in oman oil fields: a possible use in enhanced oil recovery. J. Microbiol. Biotechnol. 2013;23:106–117. doi: 10.4014/jmb.1204.04021. [DOI] [PubMed] [Google Scholar]
- Bastin ES, Greer FE, Merritt CA. Moulton G. The presence of sulphate reducing bacteria in oil field waters. Science. 1926;63:21–24. doi: 10.1126/science.63.1618.21. [DOI] [PubMed] [Google Scholar]
- Bodtker G, Thorstenson T, Lillebo BL, Thorbjornsen BE, Ulvoen RH, Sunde E, et al. The effect of long-term nitrate treatment on SRB activity, corrosion rate and bacterial community composition in offshore water injection systems. J. Ind. Microbiol. Biotechnol. 2008;35:1625–1636. doi: 10.1007/s10295-008-0406-x. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl Acad. Sci. USA. 2011;108(Suppl 1):4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods. 2013;10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- Feng WW, Liu JF, Gu JD. Mu BZ. Nitrate-reducing community in production water of three oil reservoirs and their responses to different carbon sources revealed by nitrate-reductase encoding gene (napA) Int. Biodeterior. Biodegradation. 2011;65:1081–1086. [Google Scholar]
- Gao PK, Li GQ, Zhao LX, Dai XC, Tian HM, Dai LB, et al. Dynamic processes of indigenous microorganisms from a low-temperature petroleum reservoir during nutrient stimulation. J. Biosci. Bioeng. 2014;117:215–221. doi: 10.1016/j.jbiosc.2013.07.009. [DOI] [PubMed] [Google Scholar]
- Geets J, Borrernans B, Diels L, Springael D, Vangronsveld J, van der Lelie D, et al. DsrB gene-based DGGE for community and diversity surveys of sulfate-reducing bacteria. J. Microbiol. Methods. 2006;66:194–205. doi: 10.1016/j.mimet.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Harms G, Layton AC, Dionisi HM, Gregory IR, Garrett VM, Hawkins SA, et al. Real-time PCR quantification of nitrifying bacteria in a municipal wastewater treatment plant. Environ. Sci. Technol. 2003;37:343–351. doi: 10.1021/es0257164. [DOI] [PubMed] [Google Scholar]
- Kumaraswamy R, Ebert S, Gray MR, Fedorak PM. Foght JM. Molecular- and cultivation-based analyses of microbial communities in oil field water and in microcosms amended with nitrate to control H2S production. Appl. Microbiol. Biotechnol. 2011;89:2027–2038. doi: 10.1007/s00253-010-2974-8. [DOI] [PubMed] [Google Scholar]
- Lenchi N, İnceoğlu Ö, Kebbouche-Gana S, Gana ML, Llirós M, Servais P, et al. Diversity of microbial communities in production and injection waters of algerian oilfields revealed by 16S rRNA Gene Amplicon 454 Pyrosequencing. PLoS One. 2013;8:e66588. doi: 10.1371/journal.pone.0066588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wang XL, Mu BZ, Gu JD, Liu YD, Lin KF, et al. Molecular detection, quantification and distribution of alkane-degrading bacteria in production water from low temperature oilfields. Int. Biodeterior. Biodegradation. 2013;76:49–57. [Google Scholar]
- Li GQ, Gao PK, Wu YQ, Tian HM, Dai XC, Wang YS, et al. Microbial Abundance and community composition influence production performance in a low-temperature petroleum reservoir. Environ. Sci. Technol. 2014;48:5336–5344. doi: 10.1021/es500239w. [DOI] [PubMed] [Google Scholar]
- Liu YC. Whitman WB. Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann. N. Y. Acad. Sci. 2008;1125:171–189. doi: 10.1196/annals.1419.019. [DOI] [PubMed] [Google Scholar]
- Magoc T. Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okoro C, Smith S, Chiejina L, Lumactud R, An D, Park HS, et al. Comparison of microbial communities involved in souring and corrosion in offshore and onshore oil production facilities in Nigeria. J. Ind. Microbiol. Biotechnol. 2014;41:665–678. doi: 10.1007/s10295-014-1401-z. [DOI] [PubMed] [Google Scholar]
- Satpute SK, Banat IM, Dhakephalkar PK, Banpurkar AG. Chopade BA. Biosurfactants, bioemulsifiers and exopolysaccharides from marine microorganisms. Biotechnol. Adv. 2010;28:436–450. doi: 10.1016/j.biotechadv.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Schmidt O, Horn MA, Kolb S. Drake HL. Temperature impacts differentially on the methanogenic food web of cellulose-supplemented peatland soil. Environ. Microbiol. 2014 doi: 10.1111/1462-2920.12507. , and DOI: 10.1111/1462-2920.12507. [DOI] [PubMed] [Google Scholar]
- Schulz S, Perez-De-Mora A, Engel M, Munch JC. Schloter M. A comparative study of most probable number (MPN)-PCR vs. real-time-PCR for the measurement of abundance and assessment of diversity of alkB homologous genes in soil. J. Microbiol. Methods. 2010;80:295–298. doi: 10.1016/j.mimet.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Simpson DR, Natraj NR, McInerney MJ. Duncan KE. Biosurfactant-producing bacillus are present in produced brines from Oklahoma oil reservoirs with a wide range of salinities. Appl. Microbiol. Biotechnol. 2011;91:1083–1093. doi: 10.1007/s00253-011-3326-z. [DOI] [PubMed] [Google Scholar]
- Song B, Palleroni NJ. Häggblom MM. Isolation and characterization of diverse halobenzoate-degrading denitrifying bacteria from soils and sediments. Appl. Environ. Microbiol. 2000;66:3446–3453. doi: 10.1128/aem.66.8.3446-3453.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg LM. Regan JM. Phylogenetic comparison of the methanogenic communities from an acidic, oligotrophic fen and an anaerobic digester treating municipal wastewater sludge. Appl. Environ. Microbiol. 2008;74:6663–6671. doi: 10.1128/AEM.00553-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M. Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Tang YQ, Li Y, Zhao JY, Chi CQ, Huang LX, Dong HP, et al. Microbial communities in long-term, water-flooded petroleum reservoirs with different in situ temperatures in the Huabei Oilfield, China. PLoS One. 2012;7:e33535. doi: 10.1371/journal.pone.0033535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wackett LP. Petroleum microbiology. Microb. Biotechnol. 2012;5:579–580. [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM. Cole JR. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LP, Wang WP, Lai QL. Shao ZZ. Gene diversity of CYP153A and AlkB alkane hydroxylases in oil-degrading bacteria isolated from the Atlantic Ocean. Environ. Microbiol. 2010;12:1230–1242. doi: 10.1111/j.1462-2920.2010.02165.x. [DOI] [PubMed] [Google Scholar]
- Wang XB, Chi CQ, Nie Y, Tang YQ, Tan Y, Wu G, et al. Degradation of petroleum hydrocarbons (C6–C40) and crude oil by a novel Dietzia strain. Bioresour. Technol. 2011;102:7755–7761. doi: 10.1016/j.biortech.2011.06.009. [DOI] [PubMed] [Google Scholar]
- Wang LY, Duan RY, Liu JF, Yang SZ, Gu JD. Mu BZ. Molecular analysis of the microbial community structures in water-flooding petroleum reservoirs with different temperatures. Biogeosciences. 2012;9:5177–5203. [Google Scholar]
- Xia WJ, Dong HP, Yu L. Yu DF. Comparative study of biosurfactant produced by microorganisms isolated from formation water of petroleum reservoir. Colloid Surf. A. 2011;392:124–130. [Google Scholar]
- Youssef N, Elshahed MS. McInerney MJ. Microbial processes in oil fields: culprits, problems, and opportunities. Adv. Appl. Microbiol. 2009;66:141–251. doi: 10.1016/S0065-2164(08)00806-X. [DOI] [PubMed] [Google Scholar]
- Zhao LX, Ma T, Gao ML, Gao PK, Cao MN, Zhu XD, et al. Characterization of microbial diversity and community in water flooding oil reservoirs in China. World J. Microb. Biotechnol. 2012;28:3039–3052. doi: 10.1007/s11274-012-1114-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The number of bacterial and archaeal sequences obtained from the injection and production water samples by 16S rRNA miseq sequencing.
Figure S2. The relative proportion of bacterial populations at phylum level in the injection and production water samples obtained from Luliang reservoir.
Table S1. The 155 bacterial genera detected in Luliang reservoir by 16S rRNA miseq sequencing.
Table S2. The bacterial populations with alkane monooxygenase gene (alk) obtained from RDP's FunGene.
Table S3. The archaeal genera detected in Luliang reservoir by 16S rRNA miseq sequencing.