

Abstract
Mitochondria function as the core energy providers in the brain and symptoms of neurodegenerative diseases are often attributed to their dysregulation. Assessing mitochondrial function is classically performed in isolated mitochondria; however, this process requires significant isolation time, demand for abundant tissue and disruption of the cooperative mitochondrial reticulum, all of which reduce reliability when attempting to assess in vivo mitochondrial bioenergetics. Here we introduce a method that advances the assessment of mitochondrial respiration in the brain by permeabilizing existing brain tissue to grant direct access to the mitochondrial reticulum in situ. The permeabilized brain preparation allows for instant analysis of mitochondrial function with unaltered mitochondrial morphology using significantly small sample sizes (∼2 mg), which permits the analysis of mitochondrial function in multiple subregions within a single mouse brain. Here this technique was applied to assess regional variation in brain mitochondrial function with acute ischaemia–reperfusion injuries and to determine the role of reactive oxygen species in exacerbating dysfunction through the application of a transgenic mouse model overexpressing catalase within mitochondria. Through creating accessibility to small regions for the investigation of mitochondrial function, the permeabilized brain preparation enhances the capacity for examining regional differences in mitochondrial regulation within the brain, as the majority of genetic models used for unique approaches exist in the mouse model.
Key points
Mitochondrial function in the brain is traditionally assessed through analysing respiration in isolated mitochondria, a technique that possesses significant tissue and time requirements while also disrupting the cooperative mitochondrial reticulum.
We permeabilized brain tissue in situ to permit analysis of mitochondrial respiration with the native mitochondrial morphology intact, removing the need for isolation time and minimizing tissue requirements to ∼2 mg wet weight.
The permeabilized brain technique was validated against the traditional method of isolated mitochondria and was then further applied to assess regional variation in the mouse brain with ischaemia–reperfusion injuries.
A transgenic mouse model overexpressing catalase within mitochondria was applied to show the contribution of mitochondrial reactive oxygen species to ischaemia–reperfusion injuries in different brain regions.
This technique enhances the accessibility of addressing physiological questions in small brain regions and in applying transgenic mouse models to assess mechanisms regulating mitochondrial function in health and disease.
Introduction
In the brain, mitochondria regulate the provision of energy for maintaining electrochemical homeostasis and synaptic function, while additionally influencing calcium sequestration, neurite outgrowth, and the polar establishment and development of neural cells (Mattson & Partin, 1999; Li et al. 2004; MacAskill et al. 2010). Dysregulation of the mitochondria has been implicated in contributing towards many neurological and psychiatric disorders, decreased neural plasticity and to the poor prognosis resulting from ischaemia–reperfusion injuries (Fiskum et al. 1999; Manji et al. 2012; Sanderson et al. 2013). As such, the establishment of a methodology to rapidly assess mitochondrial function in the brain is necessary to determine potential mechanistic links between alterations in mitochondrial physiology and neurological disorders.
Most of our knowledge on the function of brain mitochondria stems from studies using isolated non-synaptosomal mitochondria, which are occasionally pooled with treated synaptosomal mitochondria for functional analysis. This approach offers several drawbacks, as in addition to the significant amount of time needed to isolate mitochondria (Sims & Anderson, 2008), this technique also possesses substantial tissue requirements that often demand the use of an entire rodent brain to obtain sufficient yield, or the pooling of multiple brains when assessing regional variation. Further, the properties of mitochondria differ between brain regions, neural and glial mitochondrial populations, as well as between subpopulations of mitochondria within synaptosomes and axons (Dubinsky, 2009). As many mitochondria are damaged and lost during mechanical disruption, the proportions of the varying recovered mitochondrial populations from neurons and glia contributing to function are unknown. Therefore, it is possible that in cases of a cell-type-specific bias in the progression of a pathology, analysing function in isolated mitochondria may result in the preferential yield of one mitochondrial population over another, altering the interpretation of the dysfunctional phenotype. Finally, mitochondria work cooperatively as dynamic reticular networks capable of fission, translocation and fusion for the purpose of sharing substrates, proteins (Liu et al. 2009), DNA (Legros et al. 2004) and membrane potential (Amchenkova et al. 1988), all which influence mitochondrial function (Picard et al. 2011). Therefore, assessing function following isolation is also limited in that mechanical disruption distorts the reticular structure (Picard et al. 2011), which not only alters the cooperative nature by which mitochondria interact, but probably either selects mitochondria with similar properties following differential centrifugation, or fails to maintain those properties altogether after considerable isolation time. Owing to these limitations, the interpretation of the results obtained through the analysis of isolated mitochondria may not accurately reflect the metabolic environment in vivo.
Considering changes in mitochondrial behaviour occur rapidly, the ability to analyse mitochondrial function immediately following removal from the brain is ideal for determining acute changes in bioenergetic regulation. These collective considerations prompted us to establish an in situ mitochondrial preparation for the brain that forgoes the need to disrupt the native mitochondrial environment. In permeabilizing freshly extracted brain tissue, we have removed the time requirement for isolation and minimized sample size to create the opportunity to analyse rapidly substrate-specific changes in multiple regions within a single mouse brain. Here we validated the efficacy of this preparation through assessing the functional responses to the presence of mitochondrial substrates and inhibitors and confirmed the morphological integrity of the mitochondrial reticulum through transmission electron microscopy. We then applied this technique to investigate the effects of acute ischaemia–reperfusion injuries on mitochondrial function in multiple brain regions within the mouse and validated the improved reliability of the permeabilized brain method against the existing method done in isolated mitochondria. Finally, we then applied this method to investigate a protective strategy in preventing metabolic dysfunction following ischaemia–reperfusion injuries by employing the use of a transgenic mouse line. These experiments demonstrate a technique that advances the applicability of assessing mitochondrial function in small brain regions when examining disease and treatment.
Methods
Ethical approval
C57BL/6 mice were bred on site at the University of Guelph (Guelph, ON, Canada). All procedures in this study were approved by the University of Guelph Animal Care Committee.
Permeabilizing brain tissue for respiration
High-resolution respirometry was performed on obtained brain samples using an Oroboros Oxygraph-2K (Oroboros Instruments, Innsbruck, Austria). Experiments were run at 37°C in MiR05 respiration medium (0.5 mm EGTA, 3 mm MgCl2, 60 mm potassium lactobionate, 10 mm KH2PO4, 20 mm Hepes, 110 mm sucrose, 1 g l−1 bovine serum albumin) with constant stirring at 750 rpm.
All experiments were initially performed in the cortex and confirmed in other brain regions. Following removal from the brain, samples were quickly dissected, weighed and placed into the respirometer. Optimal sample size was determined through varying sample size and examining the consistency of respiration values and examining for the presence of diffusion limitations. Samples were allowed ∼10 min for residual substrates to deplete before permeabilization or substrate addition to avoid drift during experiments, where periods >10 min were not additionally beneficial.
In determining the optimal saponin concentration for permeabilization of the plasma membrane, saponin treatment prior to respirometry resulted in insufficient permeabilization, determined by increased respiration from subsequent saponin additions during analysis. Therefore we tested saponin concentrations within the respirometer at concentrations of 0, 20, 40, 50, 60 and 80 μg ml−1 in the presence of 10 mm glutamate, 5 mm malate, and 5 mm ADP, and ran additional saponin titrations in the presence of 10 mm succinate, 5 mm ADP, and 0.5 μm rotenone, to approach permeabilization with multiple substrates with different membrane transport properties. Finally, mitochondrial membrane integrity was tested by adding 0.01 mm cytochrome c following respiration stimulated by 0.5 μm rotenone, 10 mm succinate, and 5 mm ADP under multiple saponin concentrations. Results from these experiments demonstrated that a saponin concentration of 50 μg ml−1 was optimal for experiments, which is similar to that used in permeabilized fibres from contractile tissues (Veksler et al. 1987; Anderson & Neufer, 2006; Perry et al. 2011).
Optimizing respiration
Saturating substrate concentrations were confirmed through performing multiple overlapping titrations for pyruvate (30, 75, 250, 1000, 2000 μm; in the presence of 5 mm ADP and 5 mm malate), glutamate (100, 250, 500, 2000, 4000 and 6000 μm; in the presence of 5 mm ADP and 5 mm malate) and ADP (25, 50, 100, 175, 500, 2000 and 6000 μm; in the presence of 10 mm pyruvate and 5 mm malate). Respiration values for maximal substrate concentrations were confirmed through bolus additions and the additive effect of substrates was confirmed by the serial addition of 10 mm pyruvate, 5 mm malate, 5 mm ADP, 10 mm glutamate, and 10 mm succinate.
Changes in oxygen consumption were confirmed to originate from mitochondria through coupling substrate-specific changes in respiration with corresponding inhibitors. Therefore complex I-stimulated respiration through 10 mm glutamate, 5 mm malate, and 5 mm ADP was followed by the addition of either 0.5 μm rotenone (complex I inhibitor), 0.5 μm myxothiazol (downstream complex III inhibitor) or 2 μg ml−1 oligomycin (downstream complex V inhibitor). Complex II-specific respiration was stimulated by 10 mm succinate in the presence of 5 mm ADP and 0.5 μm rotenone (to prevent electron backflow), and was inhibited by the addition of 20 mm oxaloacetate (tricarboxylic acid substrate and potent inhibitor of succinate dehydrogenase at complex II).
Imaging
Transmission electron microscopy was performed as previously described (Holloway et al. 2010) to determine qualitatively mitochondrial morphology. This was conducted on freshly fixed untreated cortex, cortex from the same rodent after a full-length permeabilized respiration protocol, and on freshly isolated mitochondria (see below) without previous respiratory analysis.
Images of sample sizes were taken on fresh untreated mouse cortex (Infinity 2 attached to Zeiss Stemi 2000, Oberkocken, Germany).
Assessing mitochondrial function in ischaemia and reperfusion injuries
Mice were kept under anaesthetic with 1.5% isoflurane on a heating pad either for the duration of the surgical procedure (ischaemia or isch-aemia + reperfusion) or for 60 min in control mice (n = 6). We determined that length of time under anaesthetic does not affect respiration results. Ischaemic injury was induced through occluding the common carotid arteries for 15 min through bilateral ligation with a 5-0 surgical thread. After ischaemia, mice were either killed (n = 6) or the suture was removed to allow for 30 min of reperfusion (n = 6) under anaesthetic. Following removal of the brain, two samples of the cerebral cortex, striatum and hippocampus were quickly dissected out under a microscope, weighed on a microbalance and placed into the respiratory chamber for analysis of either pyruvate- or glutamate-stimulated respiration. After the addition of saponin, pyruvate-stimulated state IV respiration was initiated through the addition of 10 mm pyruvate and 5 mm malate, and state III was stimulated by 5 mm ADP. Maximal complex I-stimulated respiration was produced with the addition of 10 mm glutamate, and maximal mitochondrial respiration was stimulated through complex II by adding 10 mm succinate. Glutamate-stimulated respiration was produced similarly, however though adding glutamate and malate, ADP, pyruvate, and succinate in succession, respectively. All respiration experiments were performed in the same period (<40 min following saponin addition). For all treatments, a piece of frontal cortex was also flash-frozen for analysis of protein content and oxidation products (see below). Following respiration, the samples and respiration medium were collected, centrifuged for 1 min at 10,000 g, the supernatant was discarded and the remaining pellet was frozen for analysis of protein content by Western blotting.
Mitochondrial catalase transgenic (MCAT) mice (n = 5 per condition) were purchased from Jackson Laboratory (Bar Harbor, ME, USA) and treated as described above to investigate the role of oxidative stress in contributing to ischaemia–reperfusion injuries.
Experiments in isolated mitochondria
Non-synaptosomal mitochondria were isolated by the method described by Sims (1990) and modified by Panov et al. (2004) with the exception that the final mitochondrial pellets were resuspended in Mg2+-absent MiR05. Respiration in isolated mitochondria was performed in Mg2+-absent MiR05 at 25°C under the substrate conditions listed above. Maximal respiration for comparison with permeabilized brain was generated in the presence of 10 mm pyruvate, 5 mm malate, 5 mm ADP, 10 mm glutamate, and 10 mm succinate, where substrate-specific differences in respiration were created through altering pyruvate and glutamate as the initial substrate introduced. Succinate-stimulated (complex II-specific) respiration was generated with 10 mm succinate, 5 mm ADP, and 0.5 μm rotenone. Substrate control ratios were made relative to maximum respiration generated by either pyruvate or glutamate. Experiments in isolated mitochondria were performed on control (n = 5) and ischaemic (n = 5) cortex pooled from two mice to obtain sufficient tissue yield. The ADP/oxygen ratio was assessed during pyruvate-stimulated respiration with a 50 μm addition of ADP in the presence of 10 mm pyruvate and 5 mm malate, before the addition of 5 mm ADP. Glutamate respiration was performed by stimulating state IV respiration with 10 mm glutamate and 5 mm malate followed by state III respiration with 5 mm ADP.
The inability of isolated mitochondria to detect dysfunction reliably with ischaemia, was tested through examining respiration in the presence of 5 mm ADP and 5 mm malate before the addition of 10 mm glutamate. This was compared to repeated experiments (n = 4–5) in control and ischaemic cortex using the permeabilized brain preparation under both glutamate-stimulated conditions to validate the efficacy of the current preparation.
Western blotting
Western blotting was performed on recovered cortex, striatum and hippocampus in control, ischaemia and reperfusion conditions using methods described previously (Herbst et al. 2014) to determine changes in mitochondrial protein content. For this, subunits of each electron transport chain complex (complexes I–V) were analysed using an OXPHOS antibody cocktail (Mitosciences, Eugene, OR, USA). The efficacy of analysing retrieved samples was confirmed through consistent results in cortex homogenate from the same animals. The presence of lipid peroxidation products (4-hydroxy-2-nonenal; Alpha Diagnostics, San Antonio, TX, USA) and protein carbonylation (Oxyblot protein oxidation detection kit; Millipore, Billerica, MA, USA) were analysed as markers of oxidative stress in cortex homogenates from wild-type (WT) and MCAT mice in all conditions. All gels were loaded with 20 μg of protein.
Statistics
A one-way analysis of variance was used to detect differences between control, ischaemia and reperfusion time points for a single substrate condition within a brain region. If significance was detected, a Fisher least significant difference post-hoc test was applied. A Student's t test (two-way) was used to detect differences between WT and MCAT data for a single substrate and time point, as well as between control and ischaemic conditions in isolated mitochondria and repeated permeabilized brain experiments. Statistics for MCAT respiration were performed on respiration values before transformation into percentage control. Significance was determined at P < 0.05 with confidence intervals ≥95%.
Results
An in situ mitochondrial preparation
We aimed to establish a methodology that minimized tissue and time requirements for the assessment of mitochondrial function in the brain. Permeabilized preparations have previously been successfully employed to analyse in situ mitochondrial function in contractile tissues (Veksler et al. 1987; Kunz et al. 1993; Kuznetsov et al. 2008) using remarkably small sample sizes (often 0.8 mg wet weight) to assess the impact of disease on metabolic dysfunction. When establishing an in situ method in the brain, we optimized the dimensions of the tissue to <1 mm in depth and <2 mm in width and length (Fig. 1A), and the required amount of tissue to ∼1.5–3.0 mg wet weight (Fig. 1B), where smaller weights were unable to detect subtle changes in respiration, and larger sample weights demonstrated prominent diffusion limitations evidenced through a delayed ability to reach steady state and significantly enhanced respiration in divided samples. To create a permeabilized preparation we applied a mild cholesterol-binding agent, saponin, to the sample within the respirometers. As the content of cholesterol is abundant in plasma membranes and extremely low in mitochondria (Colbeau et al. 1971), saponin creates an in situ system through forming small pores to remove limitations on substrate and oxygen diffusion. We optimized the amount of saponin required for this preparation to 50 μg ml−1 (Fig. 2A and B), as alternative concentrations either were insufficient in achieving maximal respiration (<50 μg ml−1) or became detrimental to function (>50 μg ml−1). It is noteworthy that in situ muscle preparations are also optimized to 50 μg ml−1 (Kunz et al. 1993), suggesting an optimal saponin concentration when permeabilizing plasma membranes. We note that this preparation requires saponin treatment to be performed within the respirometers and therefore a short incubation period (∼10 min) in the respirometers before the addition of saponin is needed to stabilize the system and improve consistency of the data between samples (Fig. 1C). Owing to this, however, respiration begins to become compromised ∼40 min post-permeabilization and therefore the experimental duration should not exceed this time frame. As such, state III, maximal complex I and maximal respiration were stimulated at 10 ± 1.3 min, 18.5 ± 0.8 min and 27.5 ± 1.4 min, respectively. Finally, the independent addition of exogenous ADP following the addition of saponin increased respiration only by ∼3 pmol s–1 mg–1 wet weight, highlighting the minimal effects of residual endogenous substrates in this preparation.
Figure 1. Optimizing sample size.
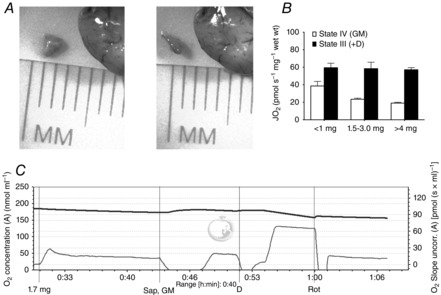
A, dimensions of an ∼2 mg sample of cortex. B, sample sizes <1 mg show an impaired ability to detect changes in respiration, where samples >4 mg show prominent diffusion limitations. C, typical GM trace for a 1.7 mg sample followed by complex I inhibition with rotenone; oxygen concentration nmol ml–1 (top thick line) and oxygen consumption (pmol (s × ml)–1 (bottom thin line). D, ADP; GM, glutamate; Rot, rotenone; Sap, saponin.
Figure 2. Creation and validation of an in situ mitochondrial preparation.
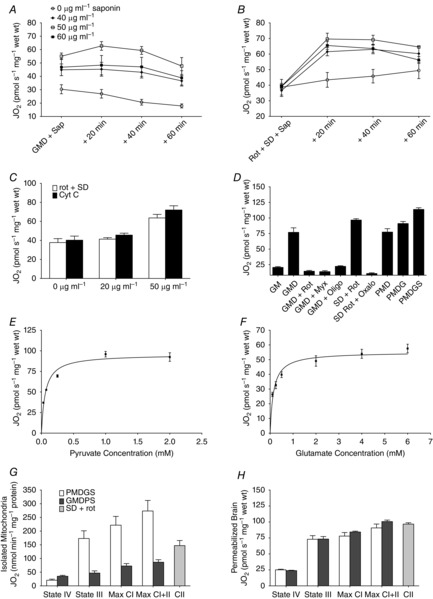
A, optimizing saponin concentrations for respiration stimulated with glutamate, malate, and ADP or (B) succinate, rotenone, and ADP. C, verification of maintained membrane integrity performed through the addition of cytochrome c following succinate-stimulated respiration at several concentrations of saponin. D, confirmation of mitochondrial-stimulated respiration through complementing glutamate, malate, and ADP with rotenone, myxothiazole, or oligomyocin as complex I-, III- and V-specific inhibitors, respectively. Succinate-stimulated complex II respiration was performed in the presence of rotenone + ADP and inhibited with oxaloacetate. Maximal mitochondrial respiration was stimulated with serial additions of pyruvate, malate, ADP, glutamate, and succinate in succession. Pyruvate (E) and glutamate (F) titrations demonstrate a system responsive to changes in substrate availability and confirm saturating substrate concentrations for respiration. In both isolated mitochondria (G) and permeabilized brain (H), substrate responses to maximum respiration initially stimulated by pyruvate (white bars), glutamate (dark bars) or succinate alone (grey bars) show substrate-specific differences between mitochondrial preparations. n = 3–5 for all experiments. D, ADP; G, glutamate; M, malate; myx, myxothiazole; oligo, oligomyocin; oxalo, oxaloacetate; P, pyruvate; rot, rotenone; S, succinate.
To evaluate the efficacy of the preparation, we confirmed that mitochondrial integrity is maintained following saponin addition through the addition of cytochrome c with several saponin concentrations (Fig. 2C). We then performed a series of classical respirometry experiments to validate the responsiveness of the system. We first determined the submaximal and maximal responses to the prominent mitochondrial substrates pyruvate (Fig. 2E) and glutamate (Fig. 2F) using multiple overlapping substrate titrations. We further confirmed maximal saturating respiration values using bolus substrate additions of either pyruvate or glutamate (Fig.2D), as maximal respiration can be affected by the manner in which substrates are made available. Where traditional experiments in isolated mitochondria display lower respiration values stimulated with glutamate than pyruvate (Lai et al. 1977), these experiments demonstrate similar fluxes with both substrates in the permeabilized preparation, which may be due to the absence of mechanical disruption and centrifugation in our in situ preparation. We next investigated the response to changes in substrate availability through stimulating complex I respiration with a single substrate, or two substrates combined to maximize electron entry through a single complex, and additionally stimulated electron entry through complex II to determine rates of maximal mitochondrial respiration (Fig. 2D) and confirm the classical additive responses to substrates. Finally, we performed mitochondrial complex-specific inhibition to interrupt multiple locations individually within the electron transport chain and confirm fluxes in respiration are of mitochondrial origin. Specifically, we complimented glutamate-stimulated complex I state III respiration with rotenone-induced complex I inhibition to impede the point of electron entry, myxothiazole-induced complex III inhibition to interrupt the midpoint of electron transport, or oligomycin-induced complex V inhibition to impede electron flow through proton build-up (Fig. 2D). These experiments demonstrate the presence of coupled electron transfer in our preparation. To target an additional site of electron entry separately, complex II-specific respiration was stimulated with succinate in the presence of rotenone to prevent backflow of electrons through complex I, and was subsequently inhibited with the addition of oxaloacetate (Fig. 2D). As build-up of oxaloacetate is a natural inhibitor of complex II activity, these results also demonstrate that residual tricarboxylic acid intermediates are depleted following the incubation period. As the inhibitors used here are specific to targeting mitochondrial complexes essential for respiration, these findings are evidence that data acquired through the permeabilized brain method are reflective of changes in mitochondrial respiratory function resulting from flux through the electron transport chain.
To compare flux rates between our preparation and the traditional method of isolated mitochondria, we compared maximal respiration stimulated initially through either pyruvate, glutamate, or by succinate alone in both preparations. In isolated mitochondria, maximal respiration varied considerably when stimulated by each substrate (Fig. 2G), whereas in permeabilized brain, maximal flux was similar among all conditions (Fig. 2H). While in contrast to permeabilized tissue protocols, work in isolated mitochondria is almost universally conducted at 25°C. Therefore, although the discrepancies observed between protocols could reflect a temperature dependency, maximal respiration was not altered in the permeabilized preparation at 25°C (data not shown), indicating temperature is not an explanation for the observed methodological differences. In addition, as the flux control ratios were similar between preparations regardless of the initial substrate (data not shown), this suggests that glutamate appears to only stimulate a fraction of its maximal potential in isolated mitochondria, identifying substrate-specific discrepancies that are not present in permeabilized brain.
Imaging
Analysis of mitochondrial respiration typically requires high-speed stirring to prevent oxygen and substrate stratification, which may endanger the reticular integrity of samples within the respirometer. We therefore used transmission electron microscopy to confirm that the morphology of the mitochondria was maintained in our preparation. As our control we imaged untreated cortex and observed clear evidence of intact tubular mitochondria (Fig. 3A), supporting conservation of the mitochondrial environment in freshly extracted brain tissue. In tissue from the same brains, we retrieved samples from the respirometers following a full-length permeabilized protocol, encompassing ∼60 min period from sample acquisition to fixation, and showed that the elongated nature of the mitochondria was fully conserved (Fig. 3B). In contrast, we compared these images with the traditional preparation of isolated mitochondria without prior respiratory analysis and show numerous individually separated and partially swollen mitochondria (Fig. 3C). Combined, these images demonstrate that the morphological nature of the mitochondria in the current permeabilized preparation better reflects the in vivo environment.
Figure 3. Imaging of mitochondrial morphology.
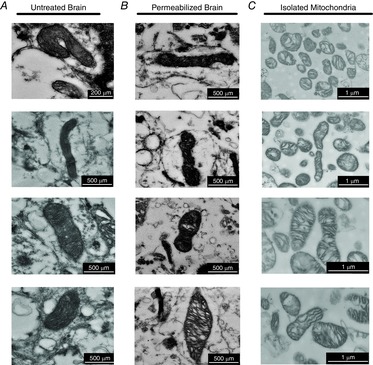
Sections of intact mitochondrial reticulum are present in untreated cortex (A) and following retrieval from a full-length permeabilized respiration protocol (B). C, isolated mitochondria are numerous, swollen and absent of mitochondrial networks. Scale bars represent 200–500 nm for in vivo (left) and in situ mitochondria (centre), 1 μm for isolated mitochondria (right).
A new approach to assessing mitochondrial dysfunction
We applied our preparation to examine changes in respiratory function that occur with acute ischaemia and reperfusion injuries. Ischaemia produces an anoxic environment that leads to changes in brain structure and mitochondrial integrity that is histologically detectible shortly after arterial occlusion (Garcia et al. 1993; Boutin et al. 1999). We induced ischaemia in the mouse through a bilateral carotid artery occlusion for 15 min and quickly dissected-out three cerebral brain regions, the cortex, striatum and hippocampus, for assessing mitochondrial respiration. Pyruvate-supported respiration is a widely analysed substrate when investigating impairments in mitochondrial function, and in this study, pyruvate respiration remained unaffected by the acute ischaemic event in both the cortex (P = 0.49) (Fig. 4A) and hippocampus (P = 0.95) (Fig. 4C). In contrast, striatal pyruvate-stimulated respiration was reduced ∼50% with ischaemia (P < 0.0001) (Fig. 4B), demonstrating that pyruvate metabolism in the striatum possesses a unique susceptibility to dysfunction with anoxia. In addition to pyruvate, glutamate is also a major substrate metabolized by brain mitochondria, although it is not as frequently assessed when investigating mitochondrial function. When we investigated glutamate-stimulated respiration following ischaemia, glutamate oxidation decreased by ∼30% in the cortex (P = 0.002) (Fig. 4D), ∼50% in the striatum (P = 0.0001) (Fig. 4E) and ∼25% in the hippocampus (P = 0.027) (Fig. 4F). This demonstrates that mitochondrial dysfunction occurs early in ischaemia across multiple brain regions through impaired glutamate metabolism, which may result in exacerbated recovery from ischaemic events due the well-established roles of glutamate in neurotransmission (Waagepetersen et al. 2003; McKenna, 2007) and the neurotoxic effects of excessive glutamate in brain disorders. Considering the difficulty of isolating mitochondria from subregions of the mouse brain, these data expand on our knowledge of the substrate- and region-specific nature by which ischaemia affects cerebral mitochondrial function by identifying glutamate-specific dysfunction early in ischaemia. These results are also the first to investigate the effect of ischaemia on brain mitochondrial function with mitochondrial morphology resembling the in vivo environment.
Figure 4. Application in acute ischaemia–reperfusion injuries.
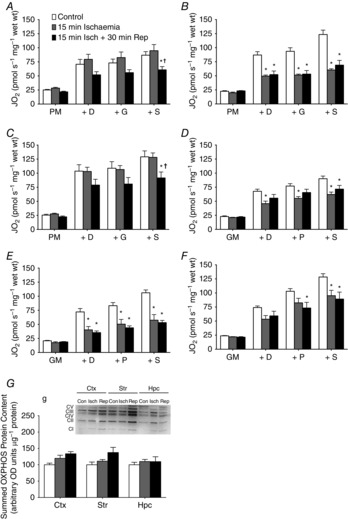
Whole brain ischaemia was induced through a bilateral carotid artery occlusion for 15 min and reperfusion was permitted for 30 min (n = 6/group). Pyruvate-stimulated respiration observed in the cortex (A), striatum (B) and hippocampus (C) in control (white bars), ischaemia (grey bars) and reperfusion (black bars). Glutamate-stimulated respiration demonstrates unique dysfunction in the cortex (D), striatum (E) and hippocampus (F). G, samples retrieved from the respirometer demonstrated that protein content of select electron transport chain subunits from complexes I to V were not affected by ischaemia–reperfusion injuries. CI-V, complex I–V; n = 6 for all groups. *P < 0.05 vs. control; †P < 0.05 vs. ischaemia. ctx, cortex; D, ADP; G, glutamate; hpc, hippocampus; isch, ischaemia; M, malate; P, pyruvate; rep, reperfusion; S, succinate; str, striatum.
Although ischaemic injury may cause initial dysfun-ction during anoxia, reperfusion is well supported to result in further damage that significantly impairs recovery (Sanderson et al. 2013). We therefore investigated the effects of a 30 min period of reperfusion following ischaemia on mitochondrial function with an intact reticulum in the same brain regions. Our results show that reperfusion caused significant impairments to both pyruvate-stimulated (cortex P = 0.036; striatum P < 0.0001; hippocampus P = 0.018) (Fig. 4A–C) and glutamate-stimulated (cortex P = 0.026; striatum P < 0.0001; hippocampus P = 0.011) (Fig. 4D–F) mitochondrial function in every brain region examined. These experiments corroborate with previous results in isolated mitochondria that show reperfusion impairs mitochondrial function across multiple brain regions (Sims, 1991) and supports that separate mechanisms appear to affect ischaemia and reperfusion injuries in a region-, substrate- and perhaps compartment-specific manner, which may require consideration when approaching protective strategies.
Finally, to validate that mitochondrial dysfunction in these injuries is not the result of proteolysis affecting mitochondrial protein content, we retrieved the permeabilized brain samples following the respirometry protocol and analysed mitochondrial protein content through Western blotting. Our results demonstrated that representative electron transport chain subunits essential for oxidative phosphorylation (complexes I–V) were not decreased by acute ischaemia–reperfusion injuries in any brain region examined (Fig. 4G), which indicates that changes in mitochondrial protein cannot explain the reduced respiratory function observed. This highlights an additional benefit to the permeabilization method, as the ability to retrieve analysed samples not only maximizes the amount of measures possible in a single mouse brain, but also permits the analysis of both function and protein content performed on the same samples for comparison in representative brain regions.
Validation with isolated mitochondria
To date, analysing respiration in isolated mitochondria represents the gold standard for assessing mitochondrial function; however, the significant volume of tissue required for isolation complicates exploring small brain regions in the mouse model. The current in situ preparation removes this complication and therefore we sought to compare results obtained from isolated mitochondria and permeabilized brain to validate our preparation. We chose to perform this comparison in control and ischaemic cortex due to sufficient tissue availability in this region and to the unique substrate-specific sensitivity found in the permeabilized cortex with ischaemia. In isolated mitochondria, pyruvate-stimulated ADP/oxygen ratios (P = 0.19) (Fig. 5A) and respiratory control ratios (control: 8.28 ± 1.2; ischaemia: 7.45 ± 0.6, P = 0.54) remained unaltered following ischaemia, demonstrating no change in the properties of mitochondrial coupling. Mitochondrial function assessed through maximal pyruvate-supported respiration was also not affected by arterial occlusion (P = 0.79) (Fig. 5B). These results in isolated mitochondria corroborate with pyruvate-stimulated respiration from our in situ preparation, thus supporting our method.
Figure 5. Comparison against isolated mitochondria.
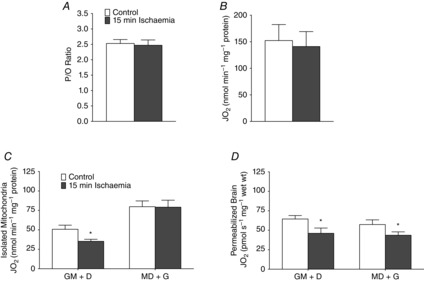
Isolated mitochondria from pooled cortex demonstrate that P/O ratios in the presence of pyruvate (A) and pyruvate respiration (B) are not affected by ischaemia. Results with glutamate-stimulated respiration in isolated mitochondria demonstrate order-dependent variability (C) where repeated experiments in the permeabilized brain preparation are consistent in demonstrating impairments with ischaemia independent of substrate addition order (D). n = 6 for all groups. *P < 0.05 vs. control. D, ADP; G, glutamate; M, malate; P/O, ADP/oxygen ratio.
When comparing glutamate-stimulated respiration in isolated mitochondria, we observed significant discrepancies during validation experiments in maximal respiration values when we altered the substrate addition order. In addition to the strong discrepancies in maximal respiration already demonstrated between glutamate- and pyruvate-stimulated respiration, we hypothesized that this could further impact the reliability and interpretation of results obtained by mitochondrial isolation and therefore assessed the impact of ischaemia under two glutamate-stimulated substrate-order conditions. We detected significant impairments in respiration in ischaemic isolated mitochondria when respiration was stimulated with glutamate before the addition of ADP (P = 0.025), corroborating with results from the permeabilized preparation; however, when ADP was provided before glutamate addition, mitochondrial function appeared unimpaired in ischaemic isolated mitochondria (P = 0.45) (Fig. 5C). We repeated these experiments using the permeabilized brain preparation and demonstrated the ability to detect impaired mitochondrial glutamate-stimulated respiration with our preparation regardless of substrate addition order (Fig.5D). It is possible that the discrepancy between methods results from the loss of intrinsic mitochondrial properties with mechanical disruption during the isolation process, and raises concerns over the reliability of using isolated mitochondria when assessing changes in glutamate oxidation in the brain. Therefore, these results demonstrate the unique reliability of the permeabilized preparation in maintaining the in vivo functional regulatory changes that affect mitochondrial bioenergetics.
Applications with genetic models
The current method increases the feasibility of exploring mitochondrial regulation with the use of genetically modified rodent models. This is particularly true in the mouse, where the process of obtaining sufficient sample for isolation becomes costly and probably deters these models from being applied to diseases affecting brain mitochondrial function. Mitochondrial hyperpolarization in ischaemia–reperfusion injuries is known to enhance production of reactive oxygen species (ROS) from the mitochondria and therefore promote further dysfun-ction through oxidative damage (Liu & Murphy, 2009; Sanderson et al. 2013). Therefore, we applied the same ischaemia–reperfusion protocol in mice transgenically expressing MCAT, which possess enhanced mitochondrial ROS-buffering capacity, to investigate the role of ROS in exacerbating mitochondrial dysfunction. Our results show that where dysfunction in pyruvate-stimulated respiration was unique to the striatum in ischaemia, the application of MCAT mice prevented this deficit (P = 0.0001), resulting in no observable impairment in pyruvate-stimulated mitochondrial function in the ischaemic brain (Fig. 6A). Further, where glutamate respiration was impaired in all examined brain regions under the same circumstances, this dysfunction was averted in the cortex (P = 0.006) and hippocampus (P = 0.019) of MCAT mice, but not in the striatum (P = 0.25) (Fig. 6B). This supports previous data where targeting mitochondrial ROS reduced ischaemic injury detected histologically (Mattiasson et al. 2003; Hains et al. 2010) and demonstrates differences in the progression of mitochondrial dysfunction in ischaemia, with glutamate being the more sensitive substrate to oxidative damage. These experiments are the first application of the MCAT mouse model in buffering ROS during ischaemic injury, and further, the permeabilized brain preparation ensures accuracy in results obtained when assessing glutamate-stimulated respiration.
Figure 6. Simplified application when approaching mechanisms with transgenic models.
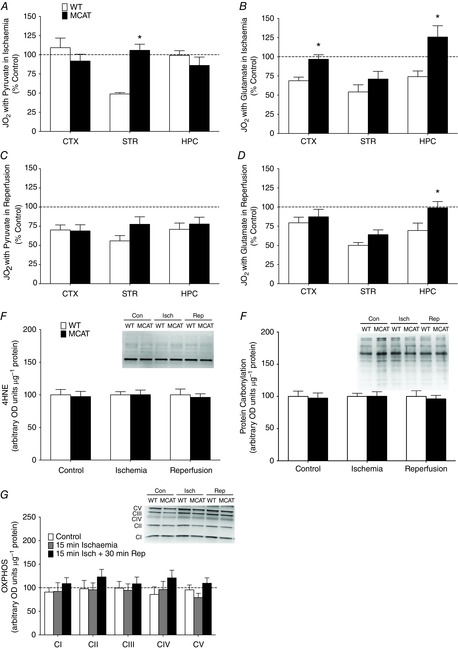
Ischaemia–reperfusion injuries were performed in mice expressing MCAT to buffer reactive oxygen species (n = 5/group). Comparing the maximal pyruvate-stimulated (A) and glutamate-stimulated (B) respiration of ischaemic cortex, striatum and hippocampus in WT and MCAT mice. Maximal pyruvate-stimulated (D) and glutamate-stimulated (E) respiration of cortex, striatum and hippocampus following ischaemia and reperfusion. Respiration values are shown as a percentage of control values within a genotype. Acute changes in function are undetectable using protein markers of oxidative damage such as lipid peroxidation (4HNE) (E) and protein carbonylation (F). G, protein content of subunits of the electron transport chain did not change in examined cortex of WT and MCAT mice in any condition. n = 5 for all groups. *P < 0.05 MCAT vs. WT percentage control under the same condition. 4HNE, 4-hydroxy-2-nonenal; con, control; ctx, cortex; hpc, hippocampus; isch, ischaemia; MCAT, mitochondrial-targeted catalyse; rep, reperfusion; str, striatum; WT, wild-type.
We then applied the MCAT model to reperfusion injuries to investigate if continued ROS buffering or previous protection during ischaemia attenuated reperfusion-induced damage to mitochondrial function. Following 30 min of blood flow return, MCAT mice displayed no protection towards pyruvate oxidation in any examined brain region (cortex, P = 0.58; striatum, P = 0.11; hippocampus, P = 0.58) (Fig. 6C) and glutamate metabolism in the cortex (P = 0.53) and striatum (P = 0.09) also remained unprotected (Fig. 6D). These results suggest either that the emission of mitochondrial ROS during reperfusion exceeds the additional buffering capacity provided in the MCAT mouse or that perhaps additional mechanisms play a role in combination with ROS that exacerbate dysfunction during reperfusion. These results are the first to identify that ROS buffering through targeting the mitochondria is insufficient in preventing mitochondrial dysfunction during reperfusion.
In these experiments we demonstrate that rapid analysis of mitochondrial function provides the ability to detect mitochondrial damage in situations where ROS damage is not detectible on an acute time scale. As perpetual ROS is known to result in oxidative damage that affects membrane integrity (Williams et al. 1998) and protein function, examination of products of lipid peroxidation (4-hydroxy-2-nonenal) and protein carbonylation in this study revealed that neither damage from ischaemia–reperfusion injuries or the protection provided by MCAT mice were detectable at the protein level (P > 0.7) (Fig. 6E and F), supporting the benefits of detecting dysfunction early through analysis of function. Finally, we confirmed that MCAT mice do not possess differences in mitochondrial content through examining representative subunits of each electron transport chain complex in both WT and MCAT mice (P > 0.1) (Fig. 6G), demonstrating that protection in MCAT mitochondria is not due to changes in protein content.
In these experiments, the permeabilized preparation refined our ability to address ROS as a mechanism of dysfunction and assess the application of targeting mitochondrial ROS to prevent mitochondrial dysfunction. We accomplished this in a mouse model using a remarkable sample size (n = 5) through assessing mitochondrial protein content and function with two substrates in three brain regions, which could not be accomplished through the traditional isolated mitochondrial preparation.
Discussion
Traditional approaches to analysing brain mitochondrial function have included either examining the maximal enzymatic activity of mitochondrial complexes or measuring respiration in isolated mitochondria. Although typically more time consuming, respiration encompasses the combined dynamics of the citric acid cycle enzymes and the electron transport chain, therefore providing a thorough representation of mitochondrial function. The preparation introduced here was designed to expand on the benefits of isolated mitochondria by enabling more rapid analysis of mitochondrial function while minimizing tissue requirements. As such, where mitochondrial isolation typically requires 1–3 h to obtain intact mitochondria with a range of 25–600 mg wet weight of brain tissue (Sims & Anderson, 2008), the permeabilized preparation requires no isolation time and produces consistent respiration with ∼2 mg, thus allowing for the analysis of small and specific brain subregions. This approach additionally improves upon isolated mitochondria, as where a set volume of isolated mitochondria may be committed only to a single analysis, samples from the permeabilized preparation may be retrieved for additional examination of protein content. Thus with this preparation, we were able to analyse mitochondrial respiration simulated by two different substrates in three unique regions within a single mouse brain and examine each sample for changes in mitochondrial electron transport chain content.
In the present study we applied our preparation to investigate the effects of acute ischaemia–reperfusion injuries on mitochondrial respiration and determined the role of mitochondrial ROS in exacerbating mitochondrial dysfunction. This approach identified ischaemic impairments in mitochondrial glutamate oxidation in all brain regions and dysfunction in pyruvate respiration in only select regions, supporting a role for region- and substrate-specific responses to mitochondrial dysfunction and disease. Further, the buffering of mitochondrial ROS prevented against ischaemic mitochondrial dysfunction only in select regions, identifying complexities in targeting prevention against ischaemic damage in the brain. It has been known for some time that ischaemia results in damage to mitochondrial function (Ozawa et al. 1967); however, the impact of mitochondrial ROS in exacerbating dysfunction has only marginally been elucidated histologically (Mattiasson et al. 2003). Determining the region- and substrate-specific benefits of buffering mitochondrial ROS through applying transgenic MCAT mice would not be a feasible approach in isolated mitochondria, due to the demanding tissue requirements of isolating small brain regions such as mouse striatum. Further, in comparing our preparation with isolated mitochondria from pooled cortex, we also revealed incongruities in the results obtained when assessing glutamate-stimulated respiration with isolated mitochondria. Therefore, the permeabilized preparation creates the ability to detect accurately the altered glutamate metabolism, which is of benefit as glutamate toxicity has been observed to impact negatively on many of these neurodegenerative diseases (Plaitakis et al. 1988; Rothstein et al. 1993). This may be due to the maintained intact mitochondrial morphology provided by the permeabilized preparation, which may be crucial when assessing mitochondrial function. Mitochondrial fission has become known as a key event leading to apoptosis (Youle & Karbowski, 2005), which results in impaired function in Parkinson's disease (Qi et al. 2013) and in pathologies expressing mutations in mitochondrial fission and fusion proteins (Chen & Chan, 2009).
Although the permeabilized technique may be applied to understand the relationship between mitochondrial function and behaviour, or investigate the effects of mitochondrial-targeted pharmacokinetics, the advantages of this approach lie most notably in detecting changes in mitochondrial function in models of neurological disease. Amyotrophic lateral sclerosis, Alzheimer's, Parkinson's and Huntington's disease are among the most commonly studied neurological diseases, which all appear to have a causal link with mitochondrial abnormalities. These disorders are unique to humans, and therefore investigating these neuropathologies, particularly in their early stages, require relying more predominantly on genetically manipulated rodent models to pair in vivo symptomology with physiological relevance. These diseases tend to manifest in specific subregions of the brain before affecting their surrounding environments and therefore the current technique may be easily applied to detect the location and onset of bioenergetic dysfunction in models of this disease. Application of this methodology is therefore expected to have an impact on understanding disease progression and novel treatment options in various neurodegenerative pathologies.
Glossary
Abbreviations
- MCAT
mitochondrial catalase transgenic
- ROS
reactive oxygen species
- WT
wild-type
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
E.A.F.H. and G.P.H. designed experiments, analysed the data and proofed the manuscript. E.A.F.H. performed experiments and wrote the manuscript. Both authors approved the final version for publication.
Funding
This research was funded by the Natural Sciences and Engineering Research Council of Canada (NSERC).
References
- Amchenkova AA, Bakeeva LE, Chentsov YS, Skulachev VP. Zorov DB. Coupling membranes as energy-transmitting cables. I. Filamentous mitochondria in fibroblasts and mitochondrial clusters in cardiomyocytes. J Cell Biol. 1988;107:481–495. doi: 10.1083/jcb.107.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EJ. Neufer PD. Type II skeletal myofibers possess unique properties that potentiate mitochondrial H2O2 generation. Am J Physiol Cell Physiol. 2006;290:C844–C851. doi: 10.1152/ajpcell.00402.2005. [DOI] [PubMed] [Google Scholar]
- Boutin H, Dauphin F, MacKenzie ET. Jauzac P. Differential time-course decreases in nonselective, mu-, delta-, and kappa-opioid receptors after focal cerebral ischemia in mice. Stroke. 1999;30:1271–1277. doi: 10.1161/01.str.30.6.1271. Discussion 1278. [DOI] [PubMed] [Google Scholar]
- Chen H. Chan DC. Mitochondrial dynamics – fusion, fission, movement, and mitophagy – in neurodegenerative diseases. Hum Mol Genet. 2009;18:R169–R176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbeau A, Nachbaur J. Vignais PM. Enzymic characterization and lipid composition of rat liver subcellular membranes. Biochim Biophys Acta. 1971;249:462–492. doi: 10.1016/0005-2736(71)90123-4. [DOI] [PubMed] [Google Scholar]
- Dubinsky JM. Heterogeneity of nervous system mitochondria: location, location, location! Exp Neurol. 2009;218:293–307. doi: 10.1016/j.expneurol.2009.05.020. [DOI] [PubMed] [Google Scholar]
- Fiskum G, Murphy AN. Beal MF. Mitochondria in neurodegeneration: acute ischemia and chronic neurodegenerative diseases. J Cereb Blood Flow Metab. 1999;19:351–369. doi: 10.1097/00004647-199904000-00001. [DOI] [PubMed] [Google Scholar]
- Garcia JH, Yoshida Y, Chen H, Li Y, Zhang ZG, Lian J, Chen S. Chopp M. Progression from ischemic injury to infarct following middle cerebral artery occlusion in the rat. Am J Pathol. 1993;142:623–635. [PMC free article] [PubMed] [Google Scholar]
- Hains DS, Sims-Lucas S, Carpenter A, Saha M, Murawski I, Kish K, Gupta I, McHugh K. Bates CM. High incidence of vesicoureteral reflux in mice with Fgfr2 deletion in kidney mesenchyma. J Urol. 2010;183:2077–2084. doi: 10.1016/j.juro.2009.12.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst EA, Paglialunga S, Gerling C, Whitfield J, Mukai K, Chabowski A, Heigenhauser GJ, Spriet LL. Holloway GP. Omega-3 supplementation alters mitochondrial membrane composition and respiration kinetics in human skeletal muscle. J Physiol. 2014;592:1341–1352. doi: 10.1113/jphysiol.2013.267336. (6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway GP, Gurd BJ, Snook LA, Lally J. Bonen A. Compensatory increases in nuclear PGC1alpha protein are primarily associated with subsarcolemmal mitochondrial adaptations in ZDF rats. Diabetes. 2010;59:819–828. doi: 10.2337/db09-1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz WS, Kuznetsov AV, Schulze W, Eichhorn K, Schild L, Striggow F, Bohnensack R, Neuhof S, Grasshoff H, Neumann HW. Gellerich FN. Functional characterization of mitochondrial oxidative phosphorylation in saponin-skinned human muscle fibers. Biochim Biophys Acta. 1993;1144:46–53. doi: 10.1016/0005-2728(93)90029-f. [DOI] [PubMed] [Google Scholar]
- Kuznetsov AV, Veksler V, Gellerich FN, Saks V, Margreiter R. Kunz WS. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc. 2008;3:965–976. doi: 10.1038/nprot.2008.61. [DOI] [PubMed] [Google Scholar]
- Lai JC, Walsh JM, Dennis SC. Clark JB. Synaptic and non-synaptic mitochondria from rat brain: isolation and characterization. J Neurochem. 1977;28:625–631. doi: 10.1111/j.1471-4159.1977.tb10434.x. [DOI] [PubMed] [Google Scholar]
- Legros F, Malka F, Frachon P, Lombes A. Rojo M. Organization and dynamics of human mitochondrial DNA. J Cell Sci. 2004;117:2653–2662. doi: 10.1242/jcs.01134. [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y. Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Liu RR. Murphy TH. Reversible cyclosporin A-sensitive mitochondrial depolarization occurs within minutes of stroke onset in mouse somatosensory cortex in vivo: a two-photon imaging study. J Biol Chem. 2009;284:36109–36117. doi: 10.1074/jbc.M109.055301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Weaver D, Shirihai O. Hajnoczky G. Mitochondrial ‘kiss-and-run’: interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 2009;28:3074–3089. doi: 10.1038/emboj.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, Atkin TA. Kittler JT. Mitochondrial trafficking and the provision of energy and calcium buffering at excitatory synapses. Eur J Neurosci. 2010;32:231–240. doi: 10.1111/j.1460-9568.2010.07345.x. [DOI] [PubMed] [Google Scholar]
- Manji H, Kato T, Di Prospero NA, Ness S, Beal MF, Krams M. Chen G. Impaired mitochondrial function in psychiatric disorders. Nat Rev Neurosci. 2012;13:293–307. doi: 10.1038/nrn3229. [DOI] [PubMed] [Google Scholar]
- Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, Warden CH, Castilho RF, Melcher T, Gonzalez-Zulueta M, Nikolich K. Wieloch T. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat Med. 2003;9:1062–1068. doi: 10.1038/nm903. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Partin J. Evidence for mitochondrial control of neuronal polarity. J Neurosci Res. 1999;56:8–20. doi: 10.1002/(SICI)1097-4547(19990401)56:1<8::AID-JNR2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- McKenna MC. The glutamate-glutamine cycle is not stoichiometric: fates of glutamate in brain. J Neurosci Res. 2007;85:3347–3358. doi: 10.1002/jnr.21444. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Seta K, Araki H. Handa H. The effect of ischemia on mitochondrial metabolism. J Biochem. 1967;61:512–514. doi: 10.1093/oxfordjournals.jbchem.a128576. [DOI] [PubMed] [Google Scholar]
- Panov AV, Andreeva L. Greenamyre JT. Quantitative evaluation of the effects of mitochondrial permeability transition pore modifiers on accumulation of calcium phosphate: comparison of rat liver and brain mitochondria. Arch Biochem Biophys. 2004;424:44–52. doi: 10.1016/j.abb.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Perry CG, Kane DA, Lin CT, Kozy R, Cathey BL, Lark DS, Kane CL, Brophy PM, Gavin TP, Anderson EJ. Neufer PD. Inhibiting myosin-ATPase reveals a dynamic range of mitochondrial respiratory control in skeletal muscle. Biochem J. 2011;437:215–222. doi: 10.1042/BJ20110366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Taivassalo T, Ritchie D, Wright KJ, Thomas MM, Romestaing C. Hepple RT. Mitochondrial structure and function are disrupted by standard isolation methods. PLoS One. 2011;6:e18317. doi: 10.1371/journal.pone.0018317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaitakis A, Constantakakis E. Smith J. The neuroexcitotoxic amino acids glutamate and aspartate are altered in the spinal cord and brain in amyotrophic lateral sclerosis. Ann Neurol. 1988;24:446–449. doi: 10.1002/ana.410240314. [DOI] [PubMed] [Google Scholar]
- Qi X, Qvit N, Su YC. Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci. 2013;126:789–802. doi: 10.1242/jcs.114439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Jin L, Dykes-Hoberg M. Kuncl RW. Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc Natl Acad Sci U S A. 1993;90:6591–6595. doi: 10.1073/pnas.90.14.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson TH, Reynolds CA, Kumar R, Przyklenk K. Huttemann M. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol. 2013;47:9–23. doi: 10.1007/s12035-012-8344-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims NR. Rapid isolation of metabolically active mitochondria from rat brain and subregions using Percoll density gradient centrifugation. J Neurochem. 1990;55:698–707. doi: 10.1111/j.1471-4159.1990.tb04189.x. [DOI] [PubMed] [Google Scholar]
- Sims NR. Selective impairment of respiration in mitochondria isolated from brain subregions following transient forebrain ischemia in the rat. J Neurochem. 1991;56:1836–1844. doi: 10.1111/j.1471-4159.1991.tb03438.x. [DOI] [PubMed] [Google Scholar]
- Sims NR. Anderson MF. Isolation of mitochondria from rat brain using Percoll density gradient centrifugation. Nat Protoc. 2008;3:1228–1239. doi: 10.1038/nprot.2008.105. [DOI] [PubMed] [Google Scholar]
- Veksler VI, Kuznetsov AV, Sharov VG, Kapelko VI. Saks VA. Mitochondrial respiratory parameters in cardiac tissue: a novel method of assessment by using saponin-skinned fibers. Biochim Biophys Acta. 1987;892:191–196. doi: 10.1016/0005-2728(87)90174-5. [DOI] [PubMed] [Google Scholar]
- Waagepetersen HS, Sonnewald U. Schousboe A. Compartmentation of glutamine, glutamate, and GABA metabolism in neurons and astrocytes: functional implications. Neuroscientist. 2003;9:398–403. doi: 10.1177/1073858403254006. [DOI] [PubMed] [Google Scholar]
- Williams MD, Van Remmen H, Conrad CC, Huang TT, Epstein CJ. Richardson A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J Biol Chem. 1998;273:28510–28515. doi: 10.1074/jbc.273.43.28510. [DOI] [PubMed] [Google Scholar]
- Youle RJ. Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol. 2005;6:657–663. doi: 10.1038/nrm1697. [DOI] [PubMed] [Google Scholar]