

Abstract
Ion channels control ionic fluxes across biological membranes by residing in any of three functionally distinct states: deactivated (closed), activated (open) or inactivated (closed). Unlike many of their cousin K+ channels, cyclic nucleotide-gated (CNG) channels do not desensitize or inactivate. Using patch recording techniques, we show that when extracellular pH (pHo) is decreased from 7.4 to 6 or lower, wild-type CNGA1 channels inactivate in a voltage-dependent manner. pHo titration experiments show that at pHo < 7 the I–V relationships are outwardly rectifying and that inactivation is coupled to current rectification. Single-channel recordings indicate that a fast mechanism of proton blockage underlines current rectification while inactivation arises from conformational changes downstream from protonation. Furthermore, mutagenesis and ionic substitution experiments highlight the role of the selectivity filter in current decline, suggesting analogies with the C-type inactivation observed in K+ channels. Analysis with Markovian models indicates that the non-independent binding of two protons within the transmembrane electrical field explains both the voltage-dependent blockage and the inactivation. Low pH, by inhibiting the CNGA1 channels in a state-dependent manner, may represent an unrecognized endogenous signal regulating CNG physiological functions in diverse tissues.
Key points
Desensitization and inactivation provide a form of short-term memory controlling the firing patterns of excitable cells and adaptation in sensory systems.
Unlike many of their cousin K+ channels, cyclic nucleotide-gated (CNG) channels are thought not to desensitize or inactivate.
Here we report that CNG channels do inactivate and that inactivation is controlled by extracellular protons.
Titration of a glutamate residue within the selectivity filter destabilizes the pore architecture, which collapses towards a non-conductive, inactivated state in a process reminiscent of the usual C-type inactivation observed in many K+ channels.
These results indicate that inactivation in CNG channels represents a regulatory mechanism that has been neglected thus far, with possible implications in several physiological processes ranging from signal transduction to growth cone navigation.
Introduction
Cyclic nucleotide-gated (CNG) channels conduct a Ca2+-permeable non-selective cation current mediating signal transduction in photoreceptor and olfactory sensory neurons (Hille, 1992; Kaupp & Seifert, 2002; Craven & Zagotta, 2006). Although CNG channels belong to the superfamily of voltage-gated ion channels (VGICs), they open in response to binding of cyclic nucleotides (CNs) and are only barely modulated by membrane voltage (Hille, 1992; Kaupp & Seifert, 2002; Yu et al. 2005; Craven & Zagotta, 2006; Mazzolini et al. 2010). CNG channel activity is also regulated by a variety of molecules and ions, including protons. Changes in extracellular hydrogen ion concentration (pHo) occur in a variety of physiological and pathophysiological conditions, such as neuronal activity, ischaemia and inflammation (Kellum et al. 2004; Isaev et al. 2008; Magnotta et al. 2012). In the retina, pHo follows a circadian rhythm (Dmitriev & Mangel, 2001), and pH changes could play a role in adaptation of the retinal response to different light intensities. Moreover, acidosis has been associated with changes in cell excitability in vascular tissues as well as in the CNS (Tolner et al. 2011; Pavlov et al. 2013), in which CNG channels are widely expressed (Zufall et al. 1997; Kaupp & Seifert, 2002; Leung et al. 2010; Lopez-Jimenez et al. 2012). Therefore, determining the effect of pHo on CNG channel gating has great clinical and physiological significance.
It is well established that extracellular protons inhibit CNG currents, although conflicting mechanisms have been proposed (Root & MacKinnon, 1994; Rho & Park, 2013; Morrill & MacKinnon, 1999; Martínez-François et al. 2010). For instance, the outward rectification observed at low pHo has been attributed to a voltage-dependent proton blockage (Rho & Park, 2013) and more recently to an enhancement of the wild-type (WT) channel inherent mild voltage-dependent gating (Martínez-François et al. 2010). Here we show that when pHo is decreased from 7.4 to 6 or lower, WT CNGA1 channels inactivate or desensitize. The term ‘desensitization’ usually refers to a mechanism of activity attenuation following sustained exposure to a chemical messenger, such as a neurotransmitter in ligand-gated channels, whereas inactivation is commonly used for VGICs. In the present investigation, mutagenesis and ionic substitution experiments indicated that the current decline is reminiscent of the usual C-type inactivation observed in Kv channels and consequently we refer to the observed loss of conduction as inactivation. A simple kinetic model in which two protons bind within the selectivity filter provides a conceptual framework to rationalize both voltage-dependent proton block and inactivation. Inactivation thus represents a novel regulatory mechanism of CNG channels that has been neglected.
Methods
Ethical approval
All studies were approved by the SISSA's Ethics Committee according to the Italian and European guidelines for animal care (d.l. 116/92; 86/609/C.E.). Oocytes were harvested from female Xenopus laevis frogs using an aseptic technique. All X. laevis surgeries were performed under general anaesthesia, obtained by immersion in a 0.2% solution of tricaine methane sulfonate (MS-222) adjusted to pH 7.4 for 15–20 min. Depth of anaesthesia was assessed by loss of the righting reflex and loss of withdrawal reflex to a toe pinch. After surgery animals were singly housed for 48 h. Frogs were monitored daily for 1 week post-operatively to ensure the absence of any surgery-related stress. Post-operative analgesics were not routinely used. Considering the simplicity of the procedure, the lack of complications, the effectiveness of anaesthetic regimens and reductions in the number of animals likely to occur compared to the number that would be required if only one surgery were permitted, multiple surgeries on a single animal were performed. Individual donors were used up to five times, conditional upon the relative health of an individual animal. Recovery time between oocyte collection from the same animal was maximized by rotation of the frogs being used. A minimum recovery period of 1 month was ensured between ovarian lobe resection from the same animal to avoid distress. Evidence of surgery-related stress resulted in an extended rest period based on recommendations from the veterinary staff. After the fifth terminal surgery frogs were humanely killed through anaesthesia overdose via 2 h of immersion in a 5 g l−1 MS-222 solution adjusted to pH 7.4.
Molecular biology
The CNGA1 channel from bovine rods consisting of 690 amino acids was used (Kaupp et al. 1989). Selected residues were replaced as described (Becchetti et al. 1999) using the Quick Change Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA, USA). Point mutations were confirmed by sequencing, using a LI-COR sequencer (4000 l; LI-COR Biosciences, Lincoln, NE, USA). cDNAs were linearized and were transcribed to cRNA in vitro using the mMessage mMachine kit (Ambion, Austin, TX, USA).
Oocyte preparation and chemicals
Mutant channel cRNAs were injected into X. laevis oocytes (‘Xenopus express’ Ancienne Ecole de Vernassal, Le Bourg 43270, Vernassal, Haute-Loire, France). Oocytes were prepared as described (Nizzari et al. 1993). Injected eggs were maintained at 18°C in a Barth solution supplemented with 50 μg ml–1 gentamycin sulfate and containing (in mm): 88 NaCl, 1 KCl, 0.82 MgSO4, 0.33 Ca(NO3)2, 0.41 CaCl2, 2.4 NaHCO3 and 5 Tris-HCl, pH 7.4 (buffered with NaOH). During the experiments, oocytes were kept in a Ringer solution containing (in mm): 110 NaCl, 2.5 KCl, 1 CaCl2, 1.6 MgCl2 and 10 Hepes, pH 7.4 (buffered with NaOH). Usual salts and reagents were purchased from Sigma Chemicals (St Louis, MO, USA).
Recording apparatus
cGMP-gated currents from excised patches (Hamill et al. 1981) were recorded with a patch-clamp amplifier (Axopatch 200; Axon Instruments Inc., Foster City, CA, USA), 2–6 days after RNA injection, at room temperature (20–24°C). The perfusion system was as described (Sesti et al. 1995) and allowed a complete solution change in less than 0.1 s. Macroscopic and single-channel current recordings were obtained with borosilicate glass pipettes which had resistances of 2–5 MΩ in symmetrical standard solution. The standard solution on both sides of the membrane consisted of (in mm) 110 NaCl, 10 Hepes and 0.2 EDTA (pH 7.4). For solutions buffered at pH lower than 7, 10 mm MES instead of 10 mm Hepes was used and for solution at pH 9, 10 mm CHES was used. Solutions were buffered with tetramethylammonium hydroxide at the desired pH. When the cation X+ was used as the charge carrier, NaCl in the standard solution on both sides of the membrane patch was replaced by an equimolar amount of the cation X+. We used Clampex version 10.0 for data acquisition. Recordings from macroscopic currents to elicit I–V relationships (Figs2A, B, G and 4I) and to perform noise analysis (Fig.2D) were low-pass filtered at 10 kHz. Current signals were sampled with a 16-bit A/D converter (Digidata 1440A; Axon Instruments), using a sampling rate of 50 kHz. All other macroscopic current recordings were low-pass filtered at 1 kHz and sampled at 2.5 kHz if not otherwise indicated. Single-channel recordings were low-pass filtered at 2 kHz and sampled at 5 kHz.
Figure 2. Proton block and current rectification in WT CNGA1 channel.

A and B, macroscopic current recordings in the inactivated state obtained 2 min after the addition of 1 mm cGMP at 0 mV elicited by voltage steps from −200 to +200 mV (ΔV = 20 mV) at pHo 5 (A) and 7.4 (B). Leak and capacitative components were removed by subtracting from the cGMP-activated current those records obtained in response to the same voltage protocol but without cGMP. C, single-channel i–V (black circles) and macroscopic I–V relationships (red circles) obtained from noise analysis (D) and from the recording in A, respectively. The I–V relationship was scaled to the i flowing at +180 mV. D, stationary fluctuation analysis at the indicated positive (upper panels) and negative (lower panels) voltages. A linear regression fit throughout each dataset provided the indicated unitary current i. Currents were evoked by 30 μm intracellular cGMP and leak currents and variance observed in the absence of cGMP subtracted. Each point indicates the mean current (I) and the variance of the corresponding mean current (σ2) measured from one independent excised membrane patch. Recordings for each patch were of duration between 0.5 and 2 s. E, single-channel current recordings at +100 mV at the indicated pHo. Amplitude histogram from recordings at pHo 5 is shown at the right (grey area). Dashed brown and blue lines represent a two-component Gaussian fit to histograms obtained at pHo 5 and 7.4, respectively. Black dashed lines indicate the 0 current level. C, Ob and O refer to the closed, blocked and open states respectively; i indicates the unitary current. F, as in E but at −100 mV. G, macroscopic current recording in the inactivated state elicited by a voltage prepulse held at +200 mV followed by test potentials ranging from −200 to +200 mV (ΔV = 20 mV). No tail currents were observed at negative membrane potentials, suggesting a fast mechanism of proton block. Black dashed line indicates the 0 current level. H, a sketch of the energy landscape for a symmetrical two-barrier Woodhull model of block. U±i, k(0)±i and δ indicate the heights of energy barriers, rate constants at 0 mV and the electrical distance, respectively. I, dependence of the fraction of unblocked channels (see Methods) on V at different pHo (filled circles, open circles, filled triangles, open triangles, open squares and filled squares refer to pHo 9, 8, 7, 6, 5 and 4, respectively). The red lines represent simultaneous fits of all the data with the model shown in F with k(0)1, k(0)–1, k(0)2, k(0)–2 and δ equal to 7.3 × 107 m−1 s−1, 40.8 s−1, 8.9 s−1, 1.6 × 107 m−1s−1 and 0.41, respectively.
Figure 4. Properties of inactivation in pore mutant channels.
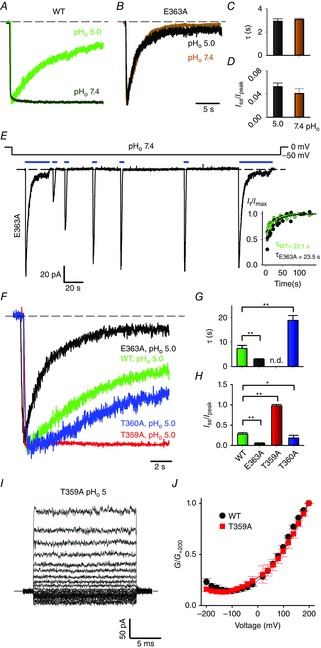
A and B, current recordings during a voltage step at −60 mV at pHo 5 and 7.4, in symmetrical 110 mm Na+ for the WT (A) and for the E363A mutant (B) channels. Currents were evoked by 1 mm cGMP and scaled for comparison. C and D, inactivation time constant τ (C) and residual fractional current Iss/Ipeak (D) for the E363A mutant channel at pHo 5 and 7.4. An unpaired two-tailed t-test was performed to compare the two conditions. No statistically significant differences were found. E, current recording during a voltage step at −50 mV at pHo 7.4 in symmetrical Na+ for the E363A mutant channel. Currents were evoked by the application of 1 mm cGMP as indicated by solid black bars. The inset shows the fractional recovery It/Imax for the E363A mutant channel at pHo 7.4 (black circles) and WT channel at pHo 5 (green circles) plotted as a function of the cumulative interpulse interval. Data were fitted to a single exponential function providing the indicated time constants τ. F, current recordings during a voltage step at −60 mV in symmetrical 110 mm Na+ for E363A (black trace), WT (green trace), T360A (blue trace) and T359A (red trace) channels, at pHo 5. Currents were evoked by 1 mm cGMP and scaled for comparison. G and H, inactivation time constant τ (G) and residual fractional current Iss/Ipeak (H) for WT, E363A, T359A and T360A channels. ANOVA and Holm–Sidak post hoc tests were performed to compare each ion against each other (*P < 0.05; **P < 0.01). I, macroscopic current recording in symmetrical 110 mm Na+, evoked by 1 mm cGMP and elicited by voltage steps from −200 to +200 mV (ΔV = 20 mV) at pHo 5 for the T359A mutant channel. J, dependence of G/G+200 on V for WT (black dots) and T359A mutant (red squares) channels at pHo 5. While abolishing desensitization (F), Thr359 replacement with an alanine does not affect voltage dependency of proton blockage (J). Dashed black lines in A, B, E and F indicate the 0 current level.
Data analysis
Inactivation time constants (τ) were obtained from fitting cGMP activated currents with one-component exponential functions. Single-channel currents (i) were estimated from patches containing only one CNGA1 channel and fitting normalized all-point histograms with two-component Gaussian functions as previously described (Bucossi et al. 1997). The dependency of the fraction of unblocked channels (Punblocked/Pmax, Fig.2I) on voltage at different pHo was determined from the ratios of the normalized conductance G/G+200 at the indicated pHo and at pHo = 9. If not otherwise indicated, data are presented as mean ± SEM, with n indicating the number of patches. Statistical significance for parametric analysis was determined using unpaired two-tailed t-test or single-variable ANOVA, as indicated. For pairwise comparisons, a Holm–Sidak test was used as post hoc test. P < 0.01 or P < 0.05 were considered significant, as indicated. Data analysis and figures were made with Clampfit version 10.1 (Molecular Devices, Sunnyvale, CA, USA) and Sigmaplot 12.0 (Systat Software, Chicago, IL, USA). Kinetic models were generated and evaluated using MatLab 7.9.0 (MathWorks, Natick, MA, USA). The voltage-dependent association constant for protons shown in Fig.6 was assumed to be of the form:
where X(0) is the association constant at 0 mV, z is the valence (+1), V is the voltage across the membrane, δ is the fraction of the membrane voltage at the binding site (the electrical distance) and F/RT = 25.5 mV at 23°C.
Figure 6. Proposed mechanism for proton action in CNGA1 channels.
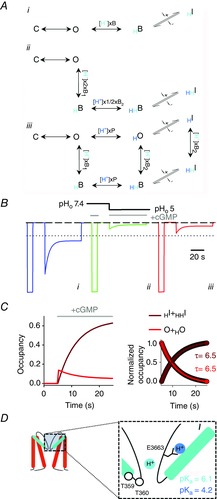
A, schemes showing the proposed mechanisms for proton binding and inactivation with one proton binding site (i), two equivalent proton binding sites (ii) and two non-equivalent and non-independent binding sites (iii). In iii columns depict protons (cyan circle and symbols) binding within permeation pathway and blocking ion conduction (D). A second proton (blue circle and symbols) binds to E363 carboxylate, possibly impairing an electrostatic interaction between E363 side chain and neighbouring residues in the P-helix (D) (Mazzolini et al. 2009; Martínez-François et al. 2010). The open conformation of the pore is now metastable and collapses to inactivated states HI and HHI. C, O, B and I indicate closed, open, blocked and inactivated states, respectively. H subscripts indicate proton transfer to the pore. P and Bi are the association constants for protons to the two non-equivalent binding sites within the pore lumen. i+ and i– are the forward and backward inactivation rate constants. B, simulated currents with schemes i (blue trace), ii (green trace) and iii (red trace) shown in A in response to a pHo step from 7.4 to 5 (black line) at −50 mV. The dotted black line indicates the mean fractional peak current observed in outside-out patches at pHo 5 (I(pHo5)/I(pHo7.4) = 0.21 ± 0.02, n = 3; Fig.1F). The dashed black line indicates the 0 current level. Rate constants are listed in the legend to Fig.5. C, time course of occupancies for the conductive states (O + HO; solid red line) and inactivated states (HI + HHI; solid brown line). The occupancies were calculated from the scheme iii shown in A at −50 mV and pHo 5 after a step in cGMP concentration from 0 to 1 mm. The inset at the right shows the decaying and rising phase of these occupancies, highlighting the same time constants (τ = 6.5 s). cGMP application in B and C is indicated by the solid grey lines. D, cartoon illustrating CNGA1 pore domain. The inset shows an enlargement of the selectivity filter region. Residues E363, T359 and T360 are shown. Cyan and blue circles represent two protons binding, respectively, within the permeation pathway and to the E363 side chain. The logarithmic acidic dissociation constants (pKa) for the two binding sites were determined from P(0) and B1(0) association constants by fitting the model in A to the data shown in Fig.5.
Results
Extracellular protons induce inactivation and outward rectification
WT CNG channels are known not to inactivate or to desensitize in the presence of a steady CN concentration. Rather unexpectedly, when CNGA1 channels were exposed to a low pHo, they inactivated (Fig.1A). We filled the patch pipette with the usual concentration of 110 mm NaCl buffered at pHo 5 and stepped the voltage command to −50 mV (Fig.1A). After a few seconds we added 1 mm cGMP to the medium bathing the cytoplasmic side of the membrane patch (indicated by solid blue bars in Fig.1A) and very quickly the expected cGMP-gated current appeared (Fig.1A). This current, however, was not stable and declined to a much lower level corresponding to approximately 25% of the current initially observed. After 30–40 s of exposure to cGMP, the CN was removed from the medium and we monitored the current amplitude by exposing the membrane patch to brief pulses of cGMP. The amplitude of the cGMP-activated current recovered to its original level, provided that cGMP was removed for at least 60–90 s from the bathing medium.
Figure 1. Properties of macroscopic currents at different pHo in WT CNGA1 channel.
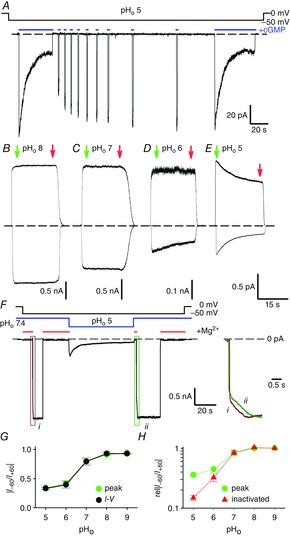
A, current recording during a voltage step at −50 mV at pHo 5 (pHi 7.4) in symmetrical 110 mm Na+ solutions. Currents were evoked by the application of 1 mm cGMP as indicated by solid blue bars. Brief cGMP pulses were used to monitor channel recovery from inactivation. A second prolonged application of cGMP after recovery produced an almost identical time-dependent current decline. B–E, current recordings during voltage steps at +60 and −60 mV evoked by 1 mm cGMP jumps at pHo 8 (B), 7 (C), 6 (D) and 5 (E). F, current recording obtained from a representative outside-out patch, during a prolonged voltage step at −50 mV, in symmetrical 110 mm Na+ solutions. The experiment was performed varying pHo from 7.4 to 5 and back to 7.4 (solid blue line). CNG channels were continuously activated by 1 mm cGMP present in the patch pipette. Reference current was obtained by adding 20 mm Mg2+ to the bath solution as indicated (solid red lines). Kinetics of current activation at pHo 7.4 prior to (i) and after inactivation at pHo 5 (ii) are shown to the right. The areas in the green and red boxes are expanded along the time scale and superimposed for comparison. G, dependence of current rectification at +60 and −60 mV (|I–60/I+60|) on pHo determined before inactivation onset (green circles) and from I–V relationships such as in Fig.2A (black circles). H, dependence of relative current rectification (rel|I–60/I+60|) on pHo before (green circles) and after (red circles) current inactivation. rel|I–60/I+60| was determined from the ratio of I–60/I+60 at the indicated pHo and I–60/I+60 at pHo 9. Green and red arrows in B–E refer to the currents prior to and after inactivation development. Dashed black lines indicate the 0 current level.
Inactivation was not observed when pHo was reduced from 7.4 to 7 (Fig.1C) or when increased to 8 (Fig.1B). When pHo was reduced to 6, an appreciable inactivation was observed only at negative voltages (Fig.1D). At pHo 5, CNG channels inactivate significantly also at positive voltages (Fig.1E). At steady state, currents declined to 72 ± 6 and 28 ± 2% (n = 4) of their initial values at +60 and −60 mV, respectively. When inactivation fully developed, after a steady exposure to cGMP for 1–2 min, the I–V relationships were clearly outward rectifying at pHo 5 (Fig.2A), in agreement with previous observations (Martínez-François et al. 2010). Thus, extracellular acidification not only induces current rectification (Rho & Park, 2013; Martínez-François et al. 2010) but is also responsible for a slow, state-dependent loss of conduction modulating CNG channel activity.
The effect of extracellular acidification is best seen in outside-out membrane patches where different proton concentrations could be assessed during the same experiment (Fig.1F). Mg2+ at 20 mm was used (indicated by solid red bars in Fig.1F) to block CNG channels – which are continuously exposed to 1 mm cGMP present in the patch pipette – and estimate the leak current (Liu & Siegelbaum, 2000). Upon pHo switch to 5, a current corresponding to approximately 20% of the current at pHo 7.4 is observed (Fig.1F). Notably, this current is not stable and exhibits a slow run-down as observed in inside-out patches at the same pHo (Fig. 1E). The current decline is fully reversible when pHo was reverted from 5 to 7.4 (Fig.1F, right panel).
As inactivation appears to be stronger at negative potentials (Fig.1E) and both inactivation and rectification depend on pHo (Fig.1B–E and G), the observed outward rectification (Fig.2A) could arise from the voltage and pH dependency of the current decline. However, several observations suggest that this is not the case. First, while rectification develops almost instantaneously (Fig.2A), inactivation occurs within several seconds (Fig.1D, E), and thus within the 20 ms voltage steps used to elicit the I–V relationship there would not be enough time for a significant inactivation to develop. Indeed, the rectification measured at +60 and −60 mV (I–60/I+60) from the I–V relationships (Fig.1G, black circles) is very similar to the rectification observed before the current decline onset (Fig.1G, green circles). Second, inactivation and rectification appear to have a different pH dependency: while a significant increase in rectification was already observed when pHo is lowered from 8 to 7 (Fig.1B, C, G), an appreciable inactivation was not detected until pHo was lowered to 6 (Fig.1B–D). Lastly, the voltage dependency of inactivation adds to the instantaneous rectification resulting in an enhancement of the steady-state current rectification (Fig.1H). Thus, the voltage- and time-dependent loss of conductance is not at the origin of the outward rectification observed in the I–V relationship, and the two processes of rectification and inactivation could have different underlying molecular mechanisms.
Outward rectification arises from voltage dependency of proton blockage
The almost instantaneous rectification at pHo 5 (Fig.2A) was substantially relieved at the usual pHo 7.4 (Fig.2B) and could reflect obstruction of the permeation pathway by protons within the transmembrane electrical field. Indeed, at hyperpolarized membrane potentials, protons are expected to be driven into the channel pore, enhancing the block and resulting in an outward rectification. To confirm this mechanism, we estimated the single-channel current i by noise analysis of macroscopic currents at different voltages (Fig.2D). Estimates of the corresponding i thus obtained are plotted as a function of voltage in Fig.2C (black dots). As with the macroscopic I–V relationship (Fig.2C, red dots), the i–V plot is outwardly rectifying and the two curves are almost overlapping. Indeed, the ratio of the macroscopic and unitary currents flowing at +180 and −180 mV (I+180/I–180 and i+180/i–180) were 4.5 and 3, respectively, suggesting that the voltage dependency of proton blockage is responsible for a large fraction of the observed rectification.
Estimates of single-channel currents based on stationary fluctuation analysis could be biased by filtering settings (Hille, 1992), and therefore we also attempted to measure single-channel events from membrane patches containing possibly only one channel. Such electrical recordings are shown in Fig.2E and F and have been obtained at +100 (Fig.2E) and −100 mV (Fig.2F) at the usual pHo of 7.4 (right traces) and when pHo was lowered to 5 (left traces). Proton elevation resulted in a reduction of the single-channel amplitude that was more prominent at negative voltages (Fig.2F). At −100 mV, amplitude histograms indicate that the i reduced from −2.3 to −0.58 pA when pHo was lowered from 7.4 to 5 (Fig.2F), and a substantial relief of block was observed at +100 mV (Fig.2E, left trace). Indeed, the ratio of the single-channel conductance at +100 and −100 mV (γ+100/γ–100) is equal to 3.6 at pHo 5. These data suggest that a fast mechanism of proton blockage is the leading cause of the current rectification, a notion that is further substantiated by the lack of tail currents when classical voltage jump experiments are considered to uncover the voltage dependency of channel deactivation (Fig.2G).
Like divalent cations, protons appear to reduce unitary currents by acting inside the conduction pathway of the channel (Dzeja et al. 1999; Seifert et al. 1999), and therefore we examined whether the Woodhull model (Woodhull, 1973) describes proton blockage (Fig.2H). We assumed that H+ binds to a binding site (BS) located at a certain electrical distance δ from the extracellular side of the membrane (Fig.2H). Figure2I illustrates the normalized G/G+200 relationship recorded at different pHo, the shape of which could be described by the Woodhull model, yielding a δ of ∼0.41. In this framework, the negative slope of the conductance observed at negative voltages (Fig.2I) reflects the punch-through of H+ from the pore. The model, however, does not satisfactorily fit the data at a pHo equal to or lower than 5 (filled and open squares, respectively, in Fig.2I), in which the predicted proton blockage is consistently larger than effectively measured. A very similar model has been described to explain extracellular divalent cation blockage and the blocking site is located at almost the same electrical distance δ of 0.45 (Seifert et al. 1999). It is thus conceivable that protons and divalent cations compete for the same BS within the selectivity filter of CNG channels.
The inactivation gate is located at the selectivity filter
In K+ channels, the intimate relationship between the so-called C-type inactivation and the selectivity filter is illustrated by the substantial effects of the permeant ions on inactivation gating (López-Barneo et al. 1993; Starkus et al. 1997). Therefore, we examined whether ionic substitution could also affect the extent and the rate of inactivation in CNGA1 channels. Current decline was observed in the presence of Na+, NH4+ and methylammonium (MA+) (Fig.3A), although inactivation was appreciably slower in the presence of NH4+ (τ = 10.5 ± 1.7 s, n = 5) compared to Na+ (τ = 7.5 ± 1.2 s, n = 5) and MA+ (τ = 7.2 ± 1.2 s, n = 3) conditions (Fig.3B). Moreover, in the presence of either NH4+ or MA+ ions, current decline was significantly less complete (Fig.3C). When Na+ ions were replaced with Rb+ and Cs+, inactivation properties did not differ among the different conditions (Fig.3A–C). Thus, the pore occupancy of specific ions appears to be one of the microscopic factors controlling inactivation. Recently we showed that the presence of large cations such as Rb+ and Cs+ in the bathing medium inhibits the Na+ inward current by interacting with Thr359 and Thr360 at the intracellular mouth of the selectivity filter. Indeed, the ring of these threonines contributes to an ion binding site at the intracellular entrance of the selectivity filter, and cations present in the intracellular medium could potentially interact with this site and affect channel gating and permeation (Marchesi et al. 2012). Therefore, we investigated whether occupancy of this binding site by certain ions compared to others could affect inactivation properties. When NH4+ ions in the bathing medium were replaced by an equimolar amount of Na+, the NH4+ current not only appeared to be inhibited by the presence of intracellular Na+ as the NH4+ peak current reduced by 32% in the presence of intracellular Na+ (I(NH4+)i/I(Na+)i = 0.68 ± 0.06; n = 4), but inactivation also proceeded faster (τ = 5.38 ± 0.98 s, n = 4) and was more complete (Fig.3D). It is therefore conceivable that Na+ occupancy of this intracellular site, signalled by the NH4+ current blockage, prompted channel collapse towards the non-conductive, inactivated state.
Figure 3. Properties of inactivation under different ionic conditions in WT CNGA1 channel.
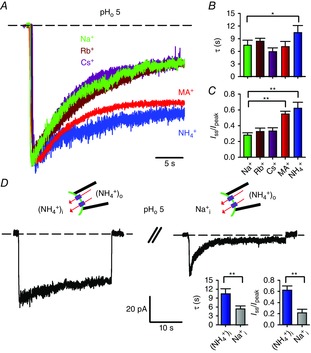
A, current recordings during a voltage step at −60 mV at pHo 5 in symmetrical 110 mm Na+ (green trace), Rb+ (brown trace), Cs+ (purple trace), MA+ (red trace) and NH4+ (blue trace) solutions. Currents were evoked by 1 mm cGMP and scaled for comparison. B and C, inactivation time constant τ (B) and residual fractional current Iss/Ipeak (C) in the presence of Na+, Rb+, Cs+, MA+ and NH4+. ANOVA and Holm–Sidak post hoc tests were performed to compare each ion against each other (*P < 0.05, **P < 0.01). D, current recordings during a prolonged voltage step at −60 mV at pHo 5 in symmetrical NH4+ (left trace) and when intracellular NH4+ was replaced by an equimolar amount of Na+ (right trace). A 2 min wash-out between the two recordings was performed to ensure complete recovery from inactivation. Currents were evoked by 1 mm cGMP. Inactivation time constant τ and residual fractional current Iss/Ipeak in the presence of symmetrical NH4+ and when intracellular NH4+ was replaced by an equimolar amount of Na+ are shown in the right insets. An unpaired two-tailed t-test was performed to compare Na+i to (NH4+)i (**P < 0.01). Dashed lines in A and D indicate the 0 current level.
Previous studies identified the Glu363 residue at the outer mouth of the selectivity filter as the major proton and divalent cation target (Root & MacKinnon, 1993, 1994; Eismann et al. 1994; Rho & Park, 2013; Morrill & MacKinnon, 1999). Therefore, we evaluated whether besides current rectification, proton binding to Glu363 also controls the time- and voltage-dependent loss of conductance discussed earlier. Similar to WT channels when pHo was reduced to 5, mutant E363A channels inactivated at the usual pHo of 7.4 (Fig.4A, B, E), as previously described (Mazzolini et al. 2009). Moreover, the current decline observed at pHo 7.4 and pHo 5 is almost identical in this mutant (Fig.4B); the inactivation time constant τ is equal to 3.1 ± 0.1 and 2.9 ± 0.2 s, respectively, and the residual steady-state current to 0.04 ± 0.01 and 0.05 ± 0.01 (Fig.4C, D; n ≥ 4). Thus, neutralization of Glu363 abolishes the pH dependency of inactivation, indicating that proton binding to Glu363 is also responsible for the slow current decline. Remarkably, compared to the WT channel, the onset rate of current decline was faster (Fig.4F, G) and inactivation was more pronounced in the E363A mutant channel (Fig.4F, H), while the time course of recovery did not significantly differ in the two channels (Fig.4E, inset). Inactivation kinetics is from two to three orders of magnitude slower than the gating transitions: indeed CNGA1 channels open in a few milliseconds in response to cGMP concentration jumps (Nache et al. 2006). Relying on this separation of timescales, the onset rate of inactivation depends on both the forward and the backward inactivation rate constants, while the recovery process depends solely on the backward rate constant. As the current decline onset in the E363A mutant is faster but recovery from this effect is not different from the WT channel, the mutation appears to affect only the forward inactivation rate constant, i.e. mutation of Glu363 destabilizes the open conformation of the pore.
To further substantiate the notion that the occupancy of an ion binding site in close proximity to the central cavity controls inactivation, we studied the current decline in Thr359 and Thr360 mutant channels (Marchesi et al. 2012). Inactivation kinetics for WT, T359A and T360A at pHo 5 are shown in Fig.4F. Current decline developed significantly more slowly in the T360A mutant channel (τ = 19.0 ± 1.9 s, n = 3, Fig.4G) compared to the WT channel (τ = 7.5 ± 1.2 s, n = 5, Fig.4G). When Thr359 at the intracellular entrance of the selectivity filter was mutated to an alanine, a significant inactivation was not observed within 20 s (Fig.4F, H). The lack of current decline observed in T359A channels is unlikely to arise from a decreased affinity to protons, as the outward rectification at pHo 5 is very similar in WT and T359A channels (Fig.4I, J).
Voltage dependency of inactivation is expected from proton binding within the electrical transmembrane field
We studied the effect of membrane potential on inactivation kinetics by eliciting currents in response to 1 mm cGMP at different voltages (Fig.5A). Two key differences among these records are the amplitude of steady-state currents and the rate of inactivation: the former increases as the membrane is made more depolarized, while the latter decreases. This finding is best illustrated in Fig.5B, in which current decays obtained at different voltages were normalized to the current measured and compared immediately after addition of cGMP. Fitting the time course of inactivation at +70 and −70 mV with a single exponential yields a τ of inactivation equal to 16.3 and 6.7 s, respectively, and a residual steady-state current (It/Imax) equal to 0.63 and 0.27. The I–V relationships measured immediately after exposure to cGMP (filled circles) and after development of current decline (open circles) are shown in Fig.5C and had a different degree of rectification. Indeed, at steady state, the outward rectification increased due to the additional contribution of the inactivation (Fig.5C). Finally, the pH dependency of inactivation at +60 mV (open circles) and −60 mV (solid circles) is shown in Fig.5D. The fraction of channels residing in the inactivated state appears to be larger at negative membrane potentials and low pH (Figs1H and 5D).
Figure 5. Voltage dependency of inactivation in WT CNGA1 channel.
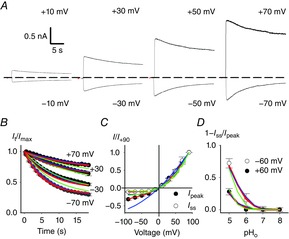
A, current recordings at pHo 5 from the same inside-out patch at the indicated membrane potentials in symmetrical Na+. Currents were evoked by 1 mm cGMP. Dashed line indicates the 0 current level. B, time course of inactivation It/Imax at pHo 5 for the indicated membrane potentials from experiments as in A where sampling points were averaged every 500 μs. Each current decay was obtained from at least three independent experiments. Imax was measured at the peak of the cGMP current. C, dependence of the cGMP-activated current on membrane voltage prior to current decline (black circles) and after completion of inactivation (white circles). Current was scaled to the current flowing at +90 mV. D, dependence of fractional inactivation (1 – Iss)/Ipeak at + 60 mV (white dots) and −60 mV (black dots) on pHo. Ipeak and Iss were measured at the beginning and termination of the cGMP pulse, respectively. Blue, green and red lines refer to simultaneous fits of all the data with schemes i, ii, and iii shown in Fig.6A, respectively. For scheme i association constant B(0) was 1.59 × 104 m−1 (pKa 4.2). The electrical distance δB was 0.37. The forward (i+) and backward (i–) inactivation rate constants were 0.41 and 0.05 s−1, respectively. For scheme ii association constants B(0)1 and B(0)2 were 2.8 × 105 m−1 (pKa 5.44) and 2.5 × 104 m−1 (pKa 4.4), respectively. The electrical distance δB was 0.22. The forward (i+) and backward (i–) inactivation rate constants were 0.51 and 0.035 s−1, respectively. For scheme iii association constants P(0), B(0)1 and B(0)2 were 1.59 × 104 m−1 (pKa 4.2), 1.25 × 106 m−1 (pKa 6.1) and 5 × 104 m−1 (pKa 4.7), respectively. The electrical distances δP and δBi were 0.37 and 0.45, respectively. The forward (i+) and backward (i–) inactivation rate constants were 0.41 and 0.05 s−1, respectively.
The slow time course of current decline described here might reflect a reduced accessibility of Glu363 carboxylate to extracellular protons. Indeed, previous structure–function studies suggested that this residue is not directly accessible to extracellular solvent (Sun et al. 1996; Mazzolini et al. 2009; Martínez-François et al. 2009). Yet, this simple interpretation is hard to reconcile with several experimental observations. First, according to this idea, the disruption of the proton binding site should have resulted in a non-inactivating phenotype at pHo 5. This is in contrast to the finding that neutralization of Glu363 results in inactivating currents at the usual pHo of 7.4 (Fig.4E). Indeed, the inactivation onset and recovery in the E363A mutant are still slow and comparable to those observed in the WT channel (Fig.4E, F). Second, recovery from inactivation is fast – less than 1 s as no obvious kinetics could be resolved – when pHo is reverted from 5 to 7.4 in outside-out patches (Fig.1F, right panel). If the slow current run-down reflected the low accessibility of Glu363 to protons, then one would have expected a similar slow recovery upon pHo switch to 7.4. Therefore, we suggest that current decline reflects proton-induced conformational changes, of which the kinetics are limited by a transition downstream from protonation. In this view, protons first bind to Glu363 within the electrical transmembrane field, quickly blocking ion conduction and destabilizing the pore, which subsequently undergoes a slow structural collapse leading to a virtually closed channel. This molecular picture is captured by the simple linear scheme shown in Fig.6Ai, involving transitions among an open, blocked and inactivated state, which could successfully describe the pH and voltage dependency of inactivation, yielding a pKa of 4.2 (Fig.5B, D blue lines). Although we could not rule out the possibility that membrane voltage might affect the conformation of charged residues and/or structures located within the electrical field as suggested for bacterial K+ channel KcsA (Cordero-Morales et al. 2006), the properties of the observed current decline are well accounted by the voltage-dependent concentration of H+ within the electrical field alone, without the necessity of taking explicitly into account a voltage-dependent conformational change. The fraction of the membrane voltage at the binding site thus obtained (δ ∼0.37) is consistent with the expected location of Glu363 within the electrical field (Contreras et al. 2010). This scheme, however, appears to be critically deficient in reproducing the instantaneous and steady-state I–V relationship (Figs5C and 6B, blue lines). The basis of this shortcoming is the different pH dependency of proton blockage and inactivation. Indeed, proton blockage has been associated with an apparent acidic dissociation constant (pKa) between 6 and 7 in several reports (Root & MacKinnon, 1994; Seifert et al. 1999). It is therefore possible that the binding of at least two protons underlines proton blockage and inactivation in CNGA1 channels.
Discussion
Desensitization and inactivation are widespread phenomena in ion channels, in which they modulate the amplitude, duration and frequency of electrical signalling within and between cells. Unlike many of their cousin K+ channels and other membrane receptors, CNG channels do not desensitize or inactivate at physiological pH when a network of chemical interactions between residues in the pore region is maintained (Mazzolini et al. 2009). We have shown that in response to changes of extracellular pH, CNG channels also inactivate, representing a possible novel regulatory mechanism that has so far been neglected.
Previous studies suggested the presence of multiple H+-binding sites in the CNG channel pore. Single-channel current analysis in olfactory CNG channels indicated that the binding of two protons is required to model the proton monovalent current inhibition (Root & MacKinnon, 1994). Moreover, Seifert et al. (1999, fig. 5B, C) showed that a Hill coefficient of ∼2 describes the pH dependency of divalent cation blockage in CNGA1 channels, suggesting the binding of at least two protons. In agreement with these earliest observations we were unable to describe the experimental data when the binding of only one proton was considered (Fig.5). Similarly, a linear scheme assuming two equivalent proton binding sites (Fig.6Aii) failed to satisfactory reproduce the experimental data as it overestimates the current inhibition observed at low pHo (compare Figs1F and 6B, green trace). Therefore, we evaluated a more general kinetic scheme in which two non-equivalent and non-independent proton binding reactions were modelled (Fig.6Aiii). This model appears to adequately describe key features of the reported current decline, such as the time course of inactivation (Fig.5B, red lines), its pH dependency (Fig.5D, red lines), the instantaneous and steady-state rectification (Fig.5C, red lines) and the fractional current blockage observed at pHo 5 (Fig.6B, red trace). As we were unable to resolve the kinetics of H+ unbinding in outside-out patches (Fig.1F) as well as the kinetics of proton blockage from voltage jump experiments (Fig.2G), the proton binding reactions are not likely to contribute to the observed slow current relaxation kinetics and the two binding reactions are therefore assumed to be diffusion limited in this model. Once protons have bound, the slow current decline reflects the very slow inactivation rate constants, which are the rate-limiting reactions in the scheme. Indeed, the time courses of the occupancies for the conductive states (O + HO, red lines) and the inactivated states (HI + HHI, brown lines) are symmetrical (Fig.6C), yielding at −50 mV the same time constant τ of 6.5 s. Thus, we propose that the non-equivalent binding of two protons within the transmembrane electrical field underlies voltage-dependent blockage and inactivation.
It has also been suggested that voltage-dependent gating contributes to the rectification of the macroscopic currents at very low pHo (pHo = 4) as a consequence of Glu363 side chain titration (Martínez-François et al. 2010). Based on noise analysis (Fig.2C, D), single-channel recordings (Fig.2E, F) and voltage-jump experiments (Fig.2G), this mechanism does not appear to significantly contribute to the outward rectification observed at higher pHo (pHo ≥ 5) and has therefore not been included (Fig.6A).
How could the proposed kinetic mechanism be interpreted in the light of the available structural and functional data? The crystal structure of a recently solved bacterial channel mimicking the CNG channel pore (Derebe et al. 2011) suggests that Ca2+ ions are chelated by backbone carbonyl oxygens and not by the Glu363 side chain, which is engaged in hydrogen bonding interactions with its neighbouring residues and buried underneath the external surface of the protein. The authors of this study suggested that the Glu363 carboxylate negative charge could perturb the electron shell distribution along the backbone of the filter residues, making certain carbonyl oxygen atoms, such as those of Gly65, more electronegative and suited for Ca2+ binding (Derebe et al. 2011). These observations are consistent with several recent structure–function studies, suggesting that interactions between the Glu363 side chain and residues in the P-helix are necessary for normal gating in CNG channels (Mazzolini et al. 2009; Martínez-François et al. 2009, 2010). In this view proton blockage could arise from the high-affinity binding to the main chain carbonyl oxygens within the selectivity filter (Fig.6D, pKa = 6.1). A second, low-affinity binding site for protons could be constituted by the Glu363 side chain (pKa = 4.2), titration of which results in the neutralization of the negatively charged carboxylate and in the weakening of key interactions anchoring the selectivity filter to the surrounding channel moiety (Fig.6D) (Mazzolini et al. 2009; Martínez-François et al. 2009, 2010). After Glu363 protonation, glycine carbonyl oxygen specificity for protons and divalent cations is lost – the pKa of the proton high-affinity binding site reduces from 6.1 to 4.7 – and thus a substantial relief from blockage is observed at low extracellular pHo (Fig.2I). It then follows a slow pore collapse toward a non-conductive, inactivated state. These observations suggest that the two binding sites are non-independent, and explain why neutralization of the same residue (Glu363) affects both proton blockage (Root & MacKinnon, 1994) and the pH dependency of inactivation (Fig.4).
One of the hallmarks of the C-type inactivation is its intimate dependence on the permeant ion. Indeed, it has long been known that the rate and extent of C-type inactivation is governed by the occupancy of an ion binding site near the external mouth of the pore via a foot-in-the-door mechanism (Yellen, 1998; Kurata & Fedida, 2006). Our results indicate that such a relationship between the permeant ion and inactivation also exists in CNG channels (Fig.3), although the underlying mechanism appears to be somewhat different. In fact, inactivation appears to be primarily modulated by the nature of the intracellular cation, being faster in the presence of intracellular Na+ (Fig.3D). Furthermore, inactivation is abolished when the intracellular ion binding site is disrupted (Fig.4F), thus suggesting that the occupancy of an ion binding site possibly equivalent to the fourth ion binding site within the selectivity filter (S4 site) of the K+ channel might control the rate and extent of inactivation in CNG channels. Therefore, it is conceivable that the pore collapse triggered by the protonation of Glu363 implies fine conformational changes also of the intracellular vestibule involving the two threonines Thr359 and Thr360. Although the molecular basis underpinning the C-type inactivation is still of debate (Devaraneni et al. 2013; Armstrong & Hoshi, 2014), according to the predominant view inactivation is associated with a conformational change close to the external mouth of the K+ channel pore (Yellen, 1998; Kurata & Fedida, 2006; Hoshi & Armstrong, 2013). Nonetheless, rearrangements near the intracellular face of the selectivity filter have been implied as well. Indeed, Posson et al. (2013) demonstrated that the binding affinity of certain intracellular blockers depends on the conformational state of the selectivity filter (open-conductive versus closed-inactivated). The authors of this study argued that the observed state-dependent affinities reflect a conformational change at the intracellular face of the selectivity filter upon inactivation. These rearrangements close to the intracellular end of the selectivity filer could be more pronounced during the CNG channel inactivation process as they may reflect the allosteric connection between the CN-binding domain and the selectivity filter gate. In this view, residues Thr359 and Thr360 might play an important role in the observed current decline. Overall, inactivation appears to be associated with a local remodelling confined to the selectivity filter and neighbouring regions in CNG as well as K+ channels (Roncaglia & Becchetti, 2001; Kurata & Fedida, 2006; Mazzolini et al. 2009).
In the present study, we demonstrate that the outward rectification observed at pHo ≥ 5 at stationary conditions (i.e. after a steady exposure to cGMP for 1–2 min) is mainly caused by asymmetries in the unitary current (Fig.2). This finding is consistent with several earlier studies suggesting that proton elevation is associated with the stabilization of low conductance states primarily at negative voltages (Root & MacKinnon, 1994; Rho & Park, 2013; Morrill & MacKinnon, 1999). We propose that a Woodhull model of block, alongside divalent cation blockage (Seifert et al. 1999), underlies proton block in CNG channels. This is directly opposite to the conclusions reached by a recent report suggesting that the outward rectification is due to an inherent channel voltage-dependent gating (Martínez-François et al. 2010). The reasons for these discrepancies are not clear. Several of the Glu363 mutants display voltage-dependent gating in addition to inactivation (Bucossi et al. 1996; Martínez-François et al. 2009). It is thus likely that voltage gating additively contributes to the observed rectification when extracellular pH is further lowered to 4 and slow activation kinetics are observed (Martínez-François et al. 2010). Determining the extent that such a mechanism contributes to the outward rectification at very low pHo will require further experimentation. Nonetheless, demonstration of common gating features (inactivation and voltage gating) as the result of pore mutation and protonation further illustrates the gating role of the outer pore in CNG channels.
Changes in pHo can arise in a variety of physiological and pathophysiological conditions, such as neuronal activity, ischaemia and inflammation (Kellum et al. 2004; Isaev et al. 2008; Magnotta et al. 2012). Low pH acts as a negative feedback mechanism that inhibits the CNGA1 channel in a state-dependent manner and may represent an unrecognized endogenous signal regulating CNG physiological functions in diverse tissues.
Glossary
Abbreviations
- BS
binding site
- CN
cyclic nucleotide
- CNG
cyclic nucleotide-gated
- VGIC
voltage-gated ion channel
Additional Information
Competing interests
The authors declare that there are no competing interests.
Author contributions
M.A. and A.M. performed experiments on oocytes. M.M. performed mutagenesis. All authors participated in designing experiments, and analysing and interpreting the data. A.M. wrote the paper, which was revised by all authors in collaboration. A.M. and V.T. supervised the project. All authors approved the final version of the manuscript.
Funding
We acknowledge the financial support of the following projects within the Seventh Framework Programme for Research of the European Commission: the SI-CODE project for Future and Emerging Technologies (FET) no. FP7-284553, the FOCUS Project no. FP7-ICT-270483 and the NEUROSCAFFOLDS Project no. 604263.
References
- Armstrong CM. Hoshi T. K+ channel gating: C-type inactivation is enhanced by calcium or lanthanum outside. J Gen Physiol. 2014;144:221–230. doi: 10.1085/jgp.201411223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becchetti A, Gamel K. Torre V. Cyclic nucleotide-gated channels. Pore topology studied through the accessibility of reporter cysteines. J Gen Physiol. 1999;114:377–392. doi: 10.1085/jgp.114.3.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucossi G, Eismann E, Sesti F, Nizzari M, Seri M, Kaupp UB. Torre V. Time-dependent current decline in cyclic GMP-gated bovine channels caused by point mutations in the pore region expressed in Xenopus oocytes. J Physiol. 1996;493:409–418. doi: 10.1113/jphysiol.1996.sp021392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucossi G, Nizzari M. Torre V. Single-channel properties of ionic channels gated by cyclic nucleotides. Biophys J. 1997;72:1165–1181. doi: 10.1016/S0006-3495(97)78765-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras JE, Chen J, Lau AY, Jogini V, Roux B. Holmgren M. Voltage profile along the permeation pathway of an open channel. Biophys J. 2010;99:2863–2869. doi: 10.1016/j.bpj.2010.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero-Morales JF, Cuello LG. Perozo E. Voltage-dependent gating at the KcsA selectivity filter. Nat Struct Mol Biol. 2006;13:319–322. doi: 10.1038/nsmb1070. [DOI] [PubMed] [Google Scholar]
- Craven KB. Zagotta WN. CNG and HCN channels: two peas, one pod. Annu Rev Physiol. 2006;68:375–401. doi: 10.1146/annurev.physiol.68.040104.134728. [DOI] [PubMed] [Google Scholar]
- Derebe MG, Zeng W, Li Y, Alam A. Jiang Y. Structural studies of ion permeation and Ca2+ blockage of a bacterial channel mimicking the cyclic nucleotide-gated channel pore. Proc Natl Acad Sci USA. 2011;108:592–597. doi: 10.1073/pnas.1013643108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraneni PK, Komarov AG, Costantino CA, Devereaux JJ, Matulef K. Valiyaveetil FI. Semisynthetic K+ channels show that the constricted conformation of the selectivity filter is not the C-type inactivated state. Proc Natl Acad Sci USA. 2013;110:15698–15703. doi: 10.1073/pnas.1308699110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmitriev AV. Mangel SC. Circadian clock regulation of pH in the rabbit retina. J Neurosci. 2001;21:2897–2902. doi: 10.1523/JNEUROSCI.21-08-02897.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzeja C, Hagen V, Kaupp UB. Frings S. Ca2+ permeation in cyclic nucleotide-gated channels. EMBO J. 1999;18:131–144. doi: 10.1093/emboj/18.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eismann E, Müller F, Heinemann SH. Kaupp UB. A single negative charge within the pore region of a cGMP-gated channel controls rectification, Ca2+ blockage, and ionic selectivity. Proc Natl Acad Sci USA. 1994;91:1109–1113. doi: 10.1073/pnas.91.3.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B. Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer Associates Inc; 1992. , 2 Sub. [Google Scholar]
- Hoshi T. Armstrong CM. C-type inactivation of voltage-gated K+ channels: pore constriction or dilation? J Gen Physiol. 2013;141:151–160. doi: 10.1085/jgp.201210888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaev NK, Stelmashook EV, Plotnikov EY, Khryapenkova TG, Lozier ER, Doludin YV, Silachev DN. Zorov DB. Role of acidosis, NMDA receptors, and acid-sensitive ion channel 1a (ASIC1a) in neuronal death induced by ischemia. Biochemistry (Mosc) 2008;73:1171–1175. doi: 10.1134/s0006297908110011. [DOI] [PubMed] [Google Scholar]
- Kaupp UB, Niidome T, Tanabe T, Terada S, Bönigk W, Stühmer W, Cook NJ, Kangawa K, Matsuo H. Hirose T. Primary structure and functional expression from complementary DNA of the rod photoreceptor cyclic GMP-gated channel. Nature. 1989;342:762–766. doi: 10.1038/342762a0. [DOI] [PubMed] [Google Scholar]
- Kaupp UB. Seifert R. Cyclic nucleotide-gated ion channels. Physiol Rev. 2002;82:769–824. doi: 10.1152/physrev.00008.2002. [DOI] [PubMed] [Google Scholar]
- Kellum JA, Song M. Li J. Science review: extracellular acidosis and the immune response: clinical and physiologic implications. Crit Care. 2004;8:331–336. doi: 10.1186/cc2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurata HT. Fedida D. A structural interpretation of voltage-gated potassium channel inactivation. Prog Biophys Mol Biol. 2006;92:185–208. doi: 10.1016/j.pbiomolbio.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Leung YK, Du J, Huang Y. Yao X. Cyclic nucleotide-gated channels contribute to thromboxane A2-induced contraction of rat small mesenteric arteries. PLoS ONE. 2010;5:e11098. doi: 10.1371/journal.pone.0011098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. Siegelbaum SA. Change of pore helix conformational state upon opening of cyclic nucleotide-gated channels. Neuron. 2000;28:899–909. doi: 10.1016/s0896-6273(00)00162-8. [DOI] [PubMed] [Google Scholar]
- López-Barneo J, Hoshi T, Heinemann SH. Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Lopez-Jimenez ME, González JC, Lizasoain I, Sánchez-Prieto J, Hernández-Guijo JM. Torres M. Functional cGMP-gated channels in cerebellar granule cells. J Cell Physiol. 2012;227:2252–2263. doi: 10.1002/jcp.22964. [DOI] [PubMed] [Google Scholar]
- Magnotta VA, Heo H-Y, Dlouhy BJ, Dahdaleh NS, Follmer RL, Thedens DR, Welsh MJ. Wemmie JA. Detecting activity-evoked pH changes in human brain. Proc Natl Acad Sci USA. 2012;109:8270–8273. doi: 10.1073/pnas.1205902109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesi A, Mazzolini M. Torre V. A ring of threonines in the inner vestibule of the pore of CNGA1 channels constitutes a binding site for permeating ions. J Physiol. 2012;590:5075–5090. doi: 10.1113/jphysiol.2012.238352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-François JR, Xu Y. Lu Z. Mutations reveal voltage gating of CNGA1 channels in saturating cGMP. J Gen Physiol. 2009;134:151–164. doi: 10.1085/jgp.200910240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-François JR, Xu Y. Lu Z. Extracellular protons titrate voltage gating of a ligand-gated ion channel. J Gen Physiol. 2010;136:179–187. doi: 10.1085/jgp.201010444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzolini M, Anselmi C. Torre V. The analysis of desensitizing CNGA1 channels reveals molecular interactions essential for normal gating. J Gen Physiol. 2009;133:375–386. doi: 10.1085/jgp.200810157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzolini M, Marchesi A, Giorgetti A. Torre V. Gating in CNGA1 channels. Pflügers Arch. 2010;459:547–555. doi: 10.1007/s00424-009-0751-2. [DOI] [PubMed] [Google Scholar]
- Morrill JA. MacKinnon R. Isolation of a single carboxyl-carboxylate proton binding site in the pore of a cyclic nucleotide-gated channel. J Gen Physiol. 1999;114:71–83. doi: 10.1085/jgp.114.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nache V, Kusch J, Hagen V. Benndorf K. Gating of cyclic nucleotide-gated (CNGA1) channels by cGMP jumps and depolarizing voltage steps. Biophys J. 2006;90:3146–3154. doi: 10.1529/biophysj.105.078667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizzari M, Sesti F, Giraudo MT, Virginio C, Cattaneo A. Torre V. Single-channel properties of cloned cGMP-activated channels from retinal rods. Proc Biol Sci. 1993;254:69–74. doi: 10.1098/rspb.1993.0128. [DOI] [PubMed] [Google Scholar]
- Pavlov I, Kaila K, Kullmann DM. Miles R. Cortical inhibition, pH and cell excitability in epilepsy: what are optimal targets for antiepileptic interventions. J Physiol. 2013;591:765–774. doi: 10.1113/jphysiol.2012.237958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posson DJ, McCoy JG. Nimigean CM. The voltage-dependent gate in MthK potassium channels is located at the selectivity filter. Nat Struct Mol Biol. 2013;20:159–166. doi: 10.1038/nsmb.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho SH. Park CS. Extracellular proton alters the divalent cation binding affinity in a cyclic nucleotide-gated channel pore. FEBS Lett. 2013;440:199–202. doi: 10.1016/s0014-5793(98)01353-2. [DOI] [PubMed] [Google Scholar]
- Roncaglia P. Becchetti A. Cyclic-nucleotide-gated channels: pore topology in desensitizing E19A mutants. Pflügers Arch. 2001;441:772–780. doi: 10.1007/s004240000480. [DOI] [PubMed] [Google Scholar]
- Root MJ. MacKinnon R. Identification of an external divalent cation-binding site in the pore of a cGMP-activated channel. Neuron. 1993;11:459–466. doi: 10.1016/0896-6273(93)90150-p. [DOI] [PubMed] [Google Scholar]
- Root MJ. MacKinnon R. Two identical noninteracting sites in an ion channel revealed by proton transfer. Science. 1994;265:1852–1856. doi: 10.1126/science.7522344. [DOI] [PubMed] [Google Scholar]
- Seifert R, Eismann E, Ludwig J, Baumann A. Kaupp UB. Molecular determinants of a Ca2+-binding site in the pore of cyclic nucleotide-gated channels: S5/S6 segments control affinity of intrapore glutamates. EMBO J. 1999;18:119–130. doi: 10.1093/emboj/18.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesti F, Eismann E, Kaupp UB, Nizzari M. Torre V. The multi-ion nature of the cGMP-gated channel from vertebrate rods. J Physiol. 1995;487:17–36. doi: 10.1113/jphysiol.1995.sp020858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkus JG, Kuschel L, Rayner MD. Heinemann SH. Ion conduction through C-type inactivated Shaker channels. J Gen Physiol. 1997;110:539–550. doi: 10.1085/jgp.110.5.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun ZP, Akabas MH, Goulding EH, Karlin A. Siegelbaum SA. Exposure of residues in the cyclic nucleotide-gated channel pore: P region structure and function in gating. Neuron. 1996;16:141–149. doi: 10.1016/s0896-6273(00)80031-8. [DOI] [PubMed] [Google Scholar]
- Tolner EA, Hochman DW, Hassinen P, Otáhal J, Gaily E, Haglund MM, Kubová H, Schuchmann S, Vanhatalo S. Kaila K. Five percent CO₂ is a potent, fast-acting inhalation anticonvulsant. Epilepsia. 2011;52:104–114. doi: 10.1111/j.1528-1167.2010.02731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhull AM. Ionic blockage of sodium channels in nerve. J Gen Physiol. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G. The moving parts of voltage-gated ion channels. Q Rev Biophys. 1998;31:239–295. doi: 10.1017/s0033583598003448. [DOI] [PubMed] [Google Scholar]
- Yu FH, Yarov-Yarovoy V, Gutman GA. Catterall WA. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol Rev. 2005;57:387–395. doi: 10.1124/pr.57.4.13. [DOI] [PubMed] [Google Scholar]
- Zufall F, Shepherd GM. Barnstable CJ. Cyclic nucleotide gated channels as regulators of CNS development and plasticity. Curr Opin Neurobiol. 1997;7:404–412. doi: 10.1016/s0959-4388(97)80070-0. [DOI] [PubMed] [Google Scholar]