

Abstract
The roles of CB1 cannabinoid receptors in regulating neuronal activity have been extensively characterized. Although early studies show that CB1 receptors are present in the nervous system and CB2 cannabinoid receptors are in the immune system, recent evidence indicates that CB2 receptors are also expressed in the brain. Activation or blockade of CB2 receptors in vivo induces neuropsychiatric effects, but the cellular mechanisms of CB2 receptor function are unclear. The aim of this study is to determine how activation of CB2 receptors present in the hippocampus regulates synaptic function. Here, we show that when organotypic cultures of rodent hippocampal slices were treated with a CB2 receptor agonist (JWH133 or GP1a) for 7–10 days, quantal glutamate release became more frequent and spine density was increased via extracellular signal-regulated kinases. Chronic intraperitoneal injection of JWH133 into mice also increased excitatory synaptic transmission. These effects were blocked by a CB2 receptor antagonist (SR144528) or absent from hippocampal slices of CB2 receptor knock-out mice. This study reveals a novel cellular function of CB2 cannabinoid receptors in the hippocampus and provides insights into how cannabinoid receptor subtypes diversify the roles of cannabinoids in the brain.
Key points
The effects of cannabinoids are primarily mediated by two types of cannabinoid receptors, CB1 receptors in the nervous system and CB2 receptors in the immune system.
Recent evidence indicates that CB2 receptors are also widely expressed in the brain and involved in neuropsychiatric functions, such as schizophrenia-like behaviours, anxiety, memory, vomiting and pain.
The cellular mechanisms by which CB2 receptors regulate neuronal functions are unknown.
We show that chronic activation of CB2 receptors in the hippocampus for 7–10 days increases excitatory synaptic transmission, whereas short-term activation of CB2 receptors has little effect on synaptic activity.
This study reveals a novel role of CB2 receptors in the brain, which is clearly distinct from that of CB1 receptors, and thus, will help us to understand better the diverse effects of cannabinoids in the nervous system.
Introduction
Growing interest in medical marijuana requires an accurate understanding of the effects of marijuana on the nervous system. This task is challenging because novel effects of cannabis are still being discovered. The presence of CB2 cannabinoid receptors (CB2Rs) in the brain is one such new finding, of which the functional significance has yet to be determined (Atwood & Mackie, 2010; Onaivi et al. 2012). Early studies showed that CB1 cannabinoid receptors (CB1Rs) are present in the nervous system and CB2Rs are in the immune system (Devane et al. 1988; Matsuda et al. 1990; Munro et al. 1993). Over the last decade, however, accumulating evidence has shown that CB2Rs are also widely present in the brain, in both microglia and neurons (Svíženská et al. 2008; Atwood & Mackie, 2010; Onaivi et al. 2012). Proteins and/or mRNA of the CB2R have been found in various areas of the CNS, such as the brainstem, pons, cerebellum, cerebral cortex, hippocampus, amygdala, striatum, substantia nigra, thalamus, hypothalamus and olfactory bulb (Svíženská et al. 2008; Atwood & Mackie, 2010). In the hippocampus, CB2R immunostaining is predominant in the soma and dendrites of pyramidal cells and some interneurons (Gong et al. 2006; Onaivi et al. 2006; Brusco et al. 2008), as well as in microglia (Svíženská et al. 2008; Atwood & Mackie, 2010). In the dendrites of hippocampal neurons, CB2Rs are located near synaptic contacts (Gong et al. 2006; Onaivi et al. 2006; Brusco et al. 2008).
While CB1Rs mediate most neurophysiological effects of cannabinoids, CB2Rs are also involved in neurological functions such as anxiety (García-Gutiérrez et al. 2012), schizophrenia-related behaviours (Ortega-Alvaro et al. 2011), impulsive behaviours (Navarrete et al. 2012), vomiting (Van Sickle et al. 2005) and pain (Jhaveri et al. 2007; Anand et al. 2009; Han et al. 2013). Evidence for the presence and function of CB2Rs in the brain is growing, but the cellular mechanisms of CB2R function are largely unknown. To understand accurately the variety of roles that cannabinoids play in the brain, it would be essential to characterize how cannabinoids act via CB2Rs.
It is important to understand the function of both CB1Rs and CB2Rs because major endocannabinoids, e.g. 2-arachidonoylglycerol (2-AG) and anandamide, and constiuents of cannabis, e.g. Δ9-tetrahydrocannabinol (Δ9-THC), can activate both types of receptors (Mechoulam et al. 1995; Showalter et al. 1996; Sugiura et al. 2000). The role of CB1Rs in the nervous system has been studied in detail (Alger, 2002; Chevaleyre et al. 2006; Kano et al. 2009; Castillo et al. 2012; Katona & Freund, 2012). Presynaptic CB1Rs mediate retrograde regulation of synaptic transmission in response to endocannabinoids, which originate from postsynaptic cells. Both CB1Rs and CB2Rs are coupled with Gi/o proteins, stimulate mitogen-activated protein kinases and Akt and inhibit adenylyl cyclase (Demuth & Molleman, 2006; Fernández-Ruiz et al. 2007; Svíženská et al. 2008). The two types of receptors also have sharp contrasts. While CB1Rs are mostly expressed on presynaptic axon terminals, CB2Rs have been proposed to be postsynaptic (Gong et al. 2006; Onaivi et al. 2006; Brusco et al. 2008). The expression level of CB1Rs in the brain is much higher than that of CB2Rs (Onaivi et al. 2006). It is unknown whether such differences in receptor expression lead to functional divergence.
The proximity of neuronal CB2Rs to synapses raises the possibility that they could be involved in the regulation of synaptic function. Indeed, the spine density of hippocampal neurons in CB2R knock-out (KO) mice is lower than that of wild-type mice (García-Gutiérrez et al. 2013), implying that endocannabinoids contribute to the maintenance of spines via CB2Rs. It is still unclear whether and how the activation of CB2Rs regulates synaptic transmission. Furthermore, it needs to be determined whether CB2Rs in the brain are involved in synaptic modulation. We hypothesize here that the activation of CB2Rs located in the hippocampus increases spine density and excitatory synaptic transmission.
Methods
Ethical approval
All experiments were conducted in accordance with the animal use protocol that was approved by the Institutional Animal Care and Use Committee of Georgia Regents University.
Animals and hippocampal slices
Organotypic slice cultures were prepared from the hippocampus of rats and mice. Male Sprague–Dawley rats (Harlan Laboratories, Indianapolis, IN, USA) at 13–15 days of age were anaesthetized with isoflurane and decapitated. The hippocampi were dissected out and sliced at 350 μm thickness with a vibrating slicer (VT1200S, Leica, Wetzlar, Germany) in ice-cold slicing saline, which consisted of 108 mm NaCl, 2.5 mm KCl, 45 mm NaHCO3, 1 mm NaH2PO4, 0.5 mm CaCl2, 5 mm MgSO4, 10 mm glucose and 0.5 mm ascorbic acid (300 mosmol kg−1; bubbled with 95% O2 and 5% CO2). After being washed with 37°C culture medium, the slices were placed on culture membranes (EMD Millipore, Billerica, MA, USA) at the interface of the culture medium and air (with 5% CO2) at 37°C. The medium was exchanged every 2–3 days and was composed of 50% Basal Medium Eagle (Invitrogen, Grand Island, NY, USA), 25% Earle's balanced salt solution, 25% horse serum (HyClone, Logan, UT, USA), 2 mm l-glutamine, 10 mm Hepes and an additional 5 mm glucose. Mouse slice cultures were prepared from the hippocampus of 7- to 8-day-old C57BL/6J or CB2R KO mice (Jackson Laboratory, Bar Harbor, ME, USA) of either sex with the same method as for rat cultures, except slice thickness (400 μm) and medium composition, as follows: 50% Minimum Essential Media (Invitrogen; 11090-081), 25% Hank's balanced salt solution (Invitrogen; 24020-117), 25% horse serum (HyClone), 2 mm l-glutamine, 10 mm Hepes and an additional 5 mm glucose. Slice cultures were used for experiments at 18–23 days in vitro (DIV). Sister slice cultures, which were made from a single animal, were divided into two groups for chronic treatment with drugs (JWH133, GP1a, SR144528 and/or PD0325901) or vehicle (0.01–0.02% ethanol). The treatment started at 11–16 DIV and lasted for 7–10 days unless otherwise noted. Slice cultures used for Figs1A, B and C and 5D were not chronically treated.
Figure 1. Chronic activation of CB2 cannabinoid receptors (CB2Rs) increases the frequency of miniature excitatory postsynaptic current (mEPSC).
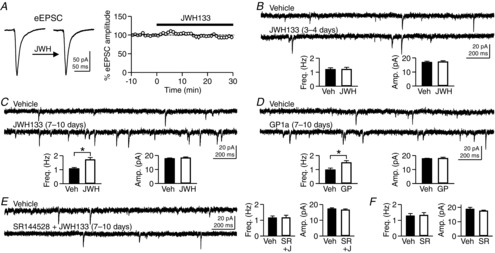
A, evoked excitatory postsynaptic currents (eEPSCs) were recorded from CA1 pyramidal neurons in rat hippocampal slice cultures, and JWH133 (200 nm), a CB2R agonist, was applied to the bath solution. B, rat hippocampal slice cultures were treated with JWH133 (200 nm) for 3–4 days. Miniature EPSCs were recorded from CA1 pyramidal neurons in the absence of JWH133 in the bath solution. C, after treatment of slice cultures with JWH133 (200 nm) for 7–10 days, mEPSCs were recorded from CA1 pyramidal neurons. *P = 0.0019, Student's unpaired t test. D, mEPSCs in cells treated with GP1a (10 nm), another CB2R agonist, for 7–10 days were more frequent than those in control cells. *P = 0.011, Student's unpaired t test. E, treatment of slice cultures with both SR144528 (100 nm), a CB2R antagonist, and JWH133 (200 nm) for 7–10 days had no effect on mEPSCs. F, treatment with SR144528 (100 nm) alone did not alter mEPSCs. Error bars indicate SEM.
Figure 5. Activation of CB2Rs does not affect presynaptic release properties or the resting chloride conductance.
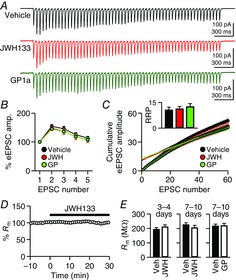
A, rat hippocampal slice cultures were treated with vehicle, JWH133 (200 nm) or GP1a (10 nm) for 7–10 days. Trains of eEPSCs (20 Hz for 3 s) were recorded from CA1 pyramidal cells. An ensemble average was made from 10–20 traces. B, short-term facilitation of the first five eEPSC amplitudes in the 20 Hz train shown in A. The eEPSC amplitudes were normalized to the first eEPSC amplitude of the train. C, from the 20 Hz trains, cumulative amplitudes of eEPSCs, after being normalized to the first eEPSC, were plotted against the eEPSC number. For each cell, the data points from the 36th–40th EPSCs were extrapolated to the y-axis. Inset, the mean sizes of the readily releasable pool (RRP), in multiples of the first eEPSC amplitude. D, membrane resistance (Rm) was measured from CA1 pyramidal cells, and JWH133 was applied to the bath solution. The data were analysed and pooled from the cells in Figs 1A and 3B. E, Rm was measured from cells treated with JWH133 or GP1a. The treatment duration is shown above each graph. The data are from the cells in Fig.1B–D. Error bars indicate SEM.
Acute hippocampal slices were made from 41- to 46-day-old C57BL/6J or CB2R KO mice of either sex (Fig.8). Slices were cut at 300 μm thickness in the ice-cold slicing saline, collected in a submerging chamber filled with artificial cerebrospinal fluid (ACSF), and incubated in ACSF for 30 min at 34°C and then for 1–8 h at 23°C. The ACSF contained 127 mm NaCl, 2.5 mm KCl, 26 mm NaHCO3, 1 mm NaH2PO4, 2 mm CaCl2, 2 mm MgSO4 and 10 mm glucose (300 mosmol kg−1) and was equilibrated with 95% O2 and 5% CO2. Mice used for acute slice preparation were injected once per day with JWH133 (5 mg kg−1, i.p.; ∼200 μl per injection) for 7–9 days starting at postnatal day 34–39. Control mice were injected with vehicle (0.9% NaCl solution with 5% DMSO and 5% Tween 80). A JWH133 stock solution for in vivo injection was made in DMSO at 30 mm and diluted immediately before injection in a NaCl–Tween 80 solution. CB2R KO mice (Figs2 and 8) were bred from a pair of homozygous CB2R KO mice (Jackson Laboratory; 005786) and genotyped using REDExtract-N-Amp Tissue PCR Kit (Sigma-Aldrich, St. Louis, MO, USA) and the following primers: GGGGATCGATCCGTCCTGTAAGTCT, GACTAGAGCTTTGTAGGTAGGCGGG and GGAGTTCAACCCCATGAAGGAGTAC.
Figure 8. Chronic activation of CB2Rs in intact mice enhances excitatory synaptic transmission.
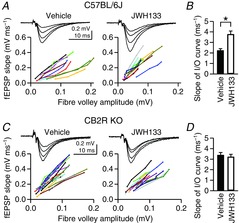
A, field excitatory postsynaptic potentials (fEPSPs) were recorded from the CA1 stratum radiatum in acute hippocampal slices of C57BL/6J mice. Input–output (I/O) curves of fEPSPs were constructed from the rising slope of fEPSP against the amplitude of the presynaptic fibre volley. Mice were injected with JWH133 (5 mg kg−1, i.p.) or vehicle once per day for 7–9 days. Acute hippocampal slices were made 24 h after the last injection. Each line represents one slice. B, the mean slope of the I/O curves was obtained from linear regression of individual I/O curves shown in A. *P = 0.0008, Student's unpaired t test. C and D, experiments were done as in A and B, but acute hippocampal slices were made from CB2R KO mice injected with JWH133 (5 mg kg−1, i.p.) or vehicle once per day for 7–9 days.
Figure 2. JWH133-induced increase in mEPSC frequency is absent from slice cultures made from CB2R knock-out (KO) mice.
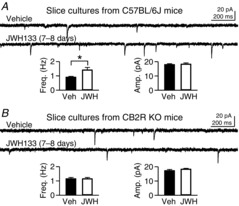
A, hippocampal slice cultures made from C57BL/6J mice were treated with JWH133 (200 nm) for 7–8 days. Miniature EPSCs were recorded from CA1 pyramidal neurons. *P = 0.009, Student's unpaired t test. B, slice cultures were made from the hippocampus of CB2R KO mice. Treatment with JWH133 (200 nm) for 7–8 days had no effect on mEPSCs in CA1 pyramidal cells. Error bars indicate SEM.
Electrophysiology
Hippocampal slices in a recording chamber were perfused with ACSF at a rate of 1.6–1.8 ml min−1 at 32 ± 0.5°C. All of the whole-cell voltage-clamp recordings were made from CA1 pyramidal neurons in hippocampal slice cultures at a holding potential of –70 mV. The ACSF also contained gabazine (3 μm), CGP37849 (5 μm) and tetrodotoxin (0.5 μm) for recording of miniature excitatory postsynaptic currents (mEPSCs), and 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX; 5 μm), CGP37849 (5 μm) and tetrodotoxin (0.5 μm) for recording of miniature inhibitory postsynaptic currents (mIPSCs). Evoked EPSCs (eEPSCs) were recorded in the presence of gabazine (3 μm), CGP37849 (5 μm) and NBQX (0.3 μm), and evoked IPSCs (eIPSCs) were recorded with NBQX (5 μm) and CGP37849 (5 μm) in the ACSF. Field excitatory postsynaptic potentials (fEPSPs) were recorded from the CA1 stratum radiatum of mouse acute slices without inhibitors of neurotransmitter receptors in the ACSF. The recording electrodes for fEPSPs were filled with ACSF, and their resistance was 0.5–1 MΩ.
For whole-cell recordings, the electrode resistance was 2.5–4.0 MΩ and the series resistance (<15 MΩ) was stable within 15%. The pipette solution for mEPSC recording contained 98 mm caesium methanesulfonate, 3 mm NaCl, 5 mm QX314-Cl, 4 mm MgSO4, 1 mm CaCl2, 10 mm EGTA, 3 mm ATP-Na, 0.3 mm GTP-Na, 30 mm Hepes and 5.4 mm biocytin (pH adjusted to 7.2 with ∼20 mm CsOH, 285–287 mosmol kg−1). From this pipette solution, biocytin was omitted and caesium methanesulfonate was increased to 100 mm for recording of eEPSC trains (Fig.5A). A high concentration of EGTA was used in these solutions to prevent Ca2+-dependent changes, if any, in EPSC amplitude. The eEPSCs and eIPSCs in Figs1A and 3B and C were recorded with the pipette solution containing 110 mm caesium methanesulfonate, 10 mm NaCl, 5 mm QX314-Cl, 4 mm MgSO4, 0.2 mm CaCl2, 2 mm EGTA, 3 mm ATP-Na, 0.3 mm GTP-Na and 30 mm Hepes (pH adjusted to 7.2 with ∼12 mm CsOH, 290–292 mosmol kg−1). The concentration of EGTA was lowered to 2 mm in this solution to allow for Ca2+-dependent processes. The pipette solution for mIPSCs contained 61 mm caesium methanesulfonate, 40 mm CsCl, 3 mm NaCl, 5 mm QX314-Cl, 4 mm MgSO4, 1 mm CaCl2, 10 mm EGTA, 3 mm ATP-Na, 0.3 mm GTP-Na and 30 mm Hepes (pH adjusted to 7.2 with ∼20 mm CsOH, 285 mosmol kg−1). The concentration of Cl– was elevated to 50 mm in this solution to increase the driving force for Cl– current.
Figure 3. The effects of JWH133 on GABAergic transmission and tests for the specificity of CB2R drugs.
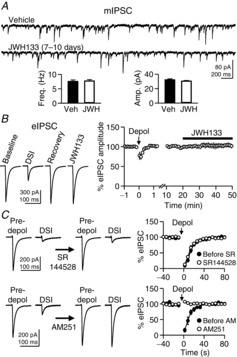
A, rat hippocampal slice cultures were treated with JWH133 (200 nm) for 7–10 days, and miniature inhibitory postsynaptic currents (mIPSCs) were recorded from CA1 pyramidal neurons. B, evoked inhibitory postsynaptic currents (eIPSCs) were recorded from CA1 pyramidal neurons in rat hippocampal slice cultures, and JWH133 (200 nm), a CB2R agonist, was applied to the bath solution. Before the JWH133 application, postsynaptic cells were depolarized to 0 mV for 5 s to induce DSI, which is CB1R-mediated suppression of IPSCs. The eIPSC trace for DSI is an average of the first two eIPSCs after depolarization. C, DSI elicited by postsynaptic depolarization to 0 mV for 5 s was measured before and during bath application of SR144528 (200 nm; 10–20 min) or AM251 (300 nm; 7–11 min). Two eIPSCs after depolarization were averaged for the DSI traces. Error bars indicate SEM.
Evoked synaptic transmission was elicited by a stimulus via a θ glass electrode (tip diameter, 10–20 μm) filled with ACSF and located in the CA1 stratum radiatum. The presynaptic stimuli were delivered every 6 s for eIPSCs, 12 s for eEPSCs (Fig.1A), 20 s for fEPSPs and 60 s for a train of eEPSCs (Fig.5A). The membrane resistance (Rm) was measured from steady-state current elicited by a 5 mV voltage step, every 2 min during mEPSC recordings and between individual traces during eEPSC/eIPSC recordings. Signals were amplified by a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA), filtered at 1 (for mEPSCs) or 2 kHz and digitized at 20 (for miniature current) or 50 kHz (for evoked current and fEPSP) by a Digidata 1440 A and the Clampex 10 program (Molecular Devices).
The mEPSCs were recorded between 6 and 25 min after membrane break-in, and 300 mEPSCs per cell were used for analysis. The mEPSCs were detected based on a template waveform in the Clampfit software (Molecular Devices). The template was 30 ms long including a 6 ms baseline. For mIPSCs, 500 events per cell were used for analysis and the template was 25 ms long including a 3 ms baseline. The peak amplitudes of eEPSCs and eIPSCs were measured over a 0.4 ms window. Stimulus artifacts were graphically removed in the figures.
In the experiments of depolarization-induced suppression of inhibition (DSI; Fig.3C), postsynaptic cells were depolarized to 0 mV for 5 s every 3 min, and two or three DSI trials were averaged for a given cell. The first IPSC after each depolarization was evoked 2 s after the termination of depolarization. A recovery of the eIPSC amplitudes from DSI was fitted with an exponential function, f(t) = Ae–t/τ + C, where t is the time of eIPSC relative to the first postdepolarization eIPSC, τ is a time constant and A and C are adjustment variables. The fitting was performed in a 90 s range that started from the first eIPSC after depolarization. The GABAergic transmission from CB1R-negative axon terminals was blocked by incubating slices with ω-agatoxin IVA (300 nm) for 15–30 min (Wilson et al. 2001; Fig.3C).
For an analysis of eEPSC trains (60 stimuli at 20 Hz; Fig.5A–C), 10–20 individual traces per cell were ensemble averaged. The eEPSC amplitudes in a train were normalized to the amplitude of the first eEPSC. To estimate the size of the readily releasable pool (RRP) of vesicles, the cumulative values of normalized eEPSC amplitudes were plotted against the EPSC number. Then, the data points from the 36th to 40th eEPSCs (i.e. 1.8–2.0 s after the first stimulus) were fitted with a straight line, and the y-intercept was obtained to estimate the RRP size. In the present study, the number of stimulated synapses varied randomly from experiment to experiment; therefore, RRP size was normalized to the number of stimulated synapses based on the following rationale. Given that I = npq (where I is the EPSC amplitude, n is the number of stimulated synapses, p is the probability of release or Pr, and q is the quantal size) and CB2R agonists changed neither p (Fig.5B) nor q (Fig.1C and D), the normalization to the first eEPSC amplitude (I) is equivalent to the normalization to the number of stimulated synapses (n) regardless of experimental group. This process allows for comparisons of the RRP sizes across experiments.
The fEPSPs were recorded with a set of four or five presynaptic stimulus intensities. The range of stimulus intensities was determined such that input–output (I/O) relationships remained linear without saturation. The fibre volley amplitude and fEPSP slope were analysed from an ensemble average of 10–15 individual traces. The slope of fEPSP was obtained in the range of 10–60% of the peak amplitude. Stimulus artifacts were graphically removed in the figures.
The NBQX, CGP37849, gabazine and tetrodotoxin were purchased from Abcam Biochemicals (Cambridge, MA, USA); JWH133 and GP1a from R&D Systems (Minneapolis, MN, USA); SR144528 and AM251 from Cayman Chemical (Ann Arbor, MI, USA); and ω-agatoxin IVA from Peptide International (Louisville, KY, USA). Other chemicals were obtained from Sigma-Aldrich.
Confocal imaging
During mEPSC recordings, the recorded cells were filled with biocytin. After recording, the electrode was retracted slowly and the slice was washed with ACSF for 5–10 min. Then, slices were fixed in PBS with 4% formaldehyde for 5–7 days at 23°C. Slices were washed five times with PBS (10 min each) and permeabilized in PBS containing Triton X-100 (0.1%) and bovine serum albumin (BSA, 2%) for 30–60 min at 23°C. Biocytin-filled cells were stained with streptavidin–Alexa 488 (1:500) in the permeabilization solution at 23°C overnight with protection from light. Slices were then washed five times with PBS (10 min each) and mounted on glass slides with mounting medium (ProLong Gold Antifade Reagent; Invitrogen). Spines were imaged with a ×63 objective lens on a laser-scanning confocal microscope (LSM700, Zeiss, Oberkochen, Germany). Dendritic segments were selected throughout the apical dendrites but only from secondary or tertiary branches. The pixel size was 0.05 μm and the optical thickness 0.345 μm. For a given image, the maximal projection of all optical sections was used for spine analysis. The NeuronStudio software (Rodriguez et al. 2008) was used for automatic detection of spines and measurement of dendritic length. Spines were classified as follows. If the ‘neck ratio’ (head diameter/neck diameter) of a spine was >1.1, the spine was classified as a thin or mushroom spine; mushroom if the head diameter was ≥0.3 μm; and thin otherwise. If the neck ratio of a spine was ≤1.1, the spine was classified as thin or stubby; thin if the ‘thin ratio’ (spine length/head diameter) is > 2.5; and stubby otherwise. The minimal size of a spine was set at 10 voxels and the maximal width of a spine at 3 μm. The height of a spine was limited to 0.2–3.0 μm.
Western blot analysis
Four or five slices per condition per experiment were scraped from the culture membrane and immediately transferred to ice-cold RIPA buffer (100–125 μl; Thermo Scientific, Waltham, MA, USA), which was supplemented with a protease inhibitor cocktail (Sigma-Aldrich) and a phosphatase inhibitor cocktail (Thermo Scientific). Then, slices were homogenized by sonication (Ultrasonic Cleaner, Branson, Danbury, CT, USA) with five 15 s pulses (one pulse every minute) at 2–5°C. The homogenates were centrifuged at 10,000g for 10 min at 4°C. The supernatants were collected and incubated with sample buffer (Thermo Scientific) at 70°C for 10 min to denature the proteins. Protein samples were run on a 10% polyacrylamide gel (Bio-Rad, Hercules, CA, USA) and then transferred onto polyvinylidene difluoride membranes (Thermo Scientific). After a brief wash with TBST (TBS + 0.1% Tween 20), the membranes were blocked for 1 h at 23°C in a TBST blocking solution, which contained 5% BSA, 50 mm NaF and 5 mm Na3VO4, and then incubated with a primary antibody overnight at 4°C. Primary antibodies (Cell Signaling Technology, Danvers, MA, USA unless otherwise noted) were diluted in the blocking solution to the following concentration: phosphorylated extracellular signal-regulated kinase (ERK) 1/2, 1:20,000; phospho-Akt, 1:50,000; pan-ERK1/2, 1:50,000; pan-Akt, 1:10,000 and β-tubulin, 1:100,000 (Thermo Scientific). The membranes were washed five times with TBST (5 min each) and incubated with HRP-conjugated anti-IgG (1:10,000 in the blocking solution; Bio-Rad) for 1 h at 23°C. Bands were visualized with a chemiluminescence detection solution (Thermo Scientific). The membranes were reused for two cycles of stripping/reprobing with different primary antibodies. Membranes were washed three times (5 min each) in stripping buffer, which contained 50 mm glycine and 0.1% SDS (pH adjusted to 2.0–2.5 with HCl), at 23°C. Western blot films were scanned on a film scanner (Epson Perfection V700) at 1200 d.p.i., and the grey scale of bands was measured with ImageJ software (NIH, Bethesda, MD, USA). The band-free background intensity was first subtracted from each band intensity, and then band intensities were normalized to the signal from sister control slices and normalized again to the β-tubulin signal from the same lane. All Western blotting experiments were duplicated with the same samples, and the duplicate values of a given sample were averaged.
Statistics
Comparisons were made only between or among sister cultures because of potential variability of data among different batches of culture. Comparisons between two groups were made using Student's t tests (either paired or unpaired) with a two-tailed confidence level of P < 0.05. Multiple comparisons were performed with one- or two-way ANOVA. Western blot data, which were normalized to control values in each experiment, were subjected to Student's paired t test vs. 100% with a two-tailed confidence level of P < 0.05.
Results
Chronic activation of CB2Rs increases mEPSC frequency
To test whether CB2Rs are involved in the regulation of excitatory synaptic transmission, we first examined an acute effect of a CB2R agonist on the amplitude of eEPSCs. The eEPSCs were recorded from CA1 pyramidal cells in slice cultures of the rat hippocampus, and JWH133 (200 nm), a specific CB2R agonist, was applied to the bath solution. The eEPSC amplitude was not significantly changed by the application of JWH133 for 25–30 min (94.1 ± 3.7% of baseline, n = 6; P = 0.20, Student's paired t test; Fig.1A), indicating a lack of an acute effect of JWH133. Next, to examine the effects of longer application of JWH133 on excitatory synaptic transmission, we treated slice cultures with JWH133 (200 nm) for 3–4 days and recorded mEPSCs. By treating hippocampal slice cultures with a CB2R agonist, only the CB2Rs within the hippocampus could be activated chronically without affecting peripheral CB2Rs. The mEPSC frequency in JWH133-treated cells (1.21 ± 0.14 Hz, n = 13) was not significantly different from that in vehicle-treated control cells (1.20 ± 0.11 Hz, n = 12; P = 0.97, Student's unpaired t test; Fig.1B). The mean amplitude of mEPSCs was not affected by JWH133 either (16.9 ± 0.8 pA in control cells and 17.2 ± 0.8 pA in JWH133-treated cells; P = 0.76, Student's unpaired t test; Fig.1B). Then, we prolonged the duration of JWH133 treatment (200 nm) to 7–10 days. After treatment for 7–10 days, the mEPSC frequency was significantly higher in JWH133-treated cells (1.71 ± 0.16 Hz, n = 14) than in vehicle-treated cells (1.08 ± 0.08 Hz, n = 14; P = 0.0019, Student's unpaired t test; Fig.1C). The mean amplitude of mEPSCs was not significantly changed by JWH133 (17.8 ± 0.5 pA in control cells and 18.4 ± 0.6 pA in JWH133-treated cells; P = 0.44, Student's unpaired t test; Fig.1C). Treatment with GP1a (10 nm), another CB2R agonist, for 7–10 days also increased mEPSC frequency (1.49 ± 0.14 Hz, n = 12) compared with the control value (0.98 ± 0.12 Hz, n = 12; P = 0.011, Student's unpaired t test; Fig.1D). The mEPSC amplitude was not affected by the GP1a treatment (17.7 ± 0.3 pA in control cells and 17.9 ± 0.8 pA in GP1a-treated cells; P = 0.84, Student's unpaired t test; Fig.1D). The increase in mEPSC frequency appeared to be mediated specifically by CB2Rs because SR144528 (100 nm), a CB2R antagonist, prevented the effect of JWH133 on mEPSC frequency [1.15 ± 0.11 Hz (n = 12) in control cells and 1.16 ± 0.16 Hz (n = 10) in cells treated with both SR144528 and JWH133 for 7–10 days; P = 0.99, Student's unpaired t test; Fig.1E]. Chronic treatment with SR144528 alone for 7–10 days had no effect on mEPSC frequency (P = 0.85, Student's unpaired t test) or amplitude (P = 0.32, Student's unpaired t test; n = 10 in each group; Fig.1F). These results suggest that chronic (7–10 days) activation of CB2Rs present in the hippocampus increases mEPSC frequency, while acute activation CB2Rs has little effect on excitatory synaptic transmission.
We further tested the specificity of JWH133 on CB2R using CB2R KO mice. First, we confirmed that JWH133 treatment could also increase mEPSC frequency in hippocampal slice cultures made from C57BL/6J mice, which are the congenic control for CB2R KO mice. In slice cultures from C57BL/6J mice, mEPSCs in cells treated with JWH133 (200 nm, 7–8 days) were more frequent (1.42 ± 0.17 Hz, n = 17) than those in vehicle-treated cells (0.91 ± 0.05 Hz, n = 16; P = 0.009, Student's unpaired t test; Fig.2A). The mEPSC amplitude (18.3 ± 0.8 pA) was not affected by the treatment compared with the control value (18.0 ± 0.5 pA). In contrast, treatment of slice cultures made from CB2R KO mice with JWH133 (200 nm, 7–8 days) did not affect mEPSCs. The mEPSC frequency in CB2R KO slices (1.15 ± 0.09 Hz, n = 14) was similar to that in vehicle-treated slices (1.16 ± 0.09 Hz, n = 14; P = 0.99, Student's unpaired t test; Fig.2B). The mEPSC amplitude was not affected by JWH133 either (17.3 ± 0.7 pA in vehicle-treated cells and 18.3 ± 0.5 pA in JWH133-treated cells; P = 0.26, Student's unpaired t test; Fig.2B). Given that a JWH133-mediated increase in mEPSC frequency was blocked by a CB2R antagonist (Fig.1E) and absent from CB2R KO tissues (Fig.2B), the effect of the CB2R agonist on mEPSC frequency is likely to be mediated specifically by CB2Rs.
Activation of CB2Rs has little effect on GABAergic synapses
To test the effects of a CB2R agonist on GABAergic transmission, we recorded mIPSCs from CA1 pyramidal cells after treating slice cultures of the rat hippocampus with JWH133 (200 nm) or vehicle for 7–10 days. The mean frequencies of mIPSCs were 7.61 ± 0.45 Hz (n = 16) in control cells and 7.78 ± 0.41 Hz (n = 16) in JWH133-treated cells (P = 0.79, Student's unpaired t test; Fig.3A). The mean amplitudes were 32.3 ± 1.2 pA in control cells and 30.6 ± 0.7 pA in JWH133-treated cells (P = 0.21, Student's unpaired t test; Fig.3A). Therefore, the effect of chronic activation of CB2Rs is specific for glutamatergic, not GABAergic, synapses. We also tested whether JWH133 has an acute effect on inhibitory synaptic transmission. Bath application of JWH133 (200 nm) for 25–30 min did not significantly change the mean amplitude of eIPSC (103 ± 6% of baseline, n = 6; P = 0.78, Student's paired t test; Fig.3B), implying a lack of an acute effect on GABAergic transmission.
Although the CB2R drugs used in this study are specific for CB2Rs, they can also bind to CB1Rs at high concentrations. We tested whether JWH133 activates or SR144528 blocks CB1Rs. Given that CB1Rs are highly expressed in GABAergic axon terminals in the hippocampus (Katona et al. 1999; Marsicano & Lutz, 1999), activation of CB1Rs should suppress eIPSC amplitudes. However, JWH133 did not reduce eIPSCs even though the eIPSCs were susceptible to CB1R-medaited reduction as assayed by the presence of DSI (Fig.3B). This result suggests that the CB2R agonist did not effectively activate CB1Rs. In addition, SR144528 (200 nm) did not block DSI, a CB1R-mediated process, when bath applied for 10–20 min (n = 5; Fig.3C), indicating that the CB2R antagonist did not inhibit CB1Rs. Neither the amplitude of the first eIPSC after depolarization nor the time course of eIPSC recovery from DSI was affected by SR144528 (P > 0.05, Student's paired t tests; Fig.3C). As a control, AM251 (300 nm; a specific CB1R antagonist) completely blocked DSI when bath applied for 7–11 min (n = 5; Fig.3C). Given that the experiment in Fig.3C was focused on DSI, DSI was maximized by incubating slices for 15–30 min with ω-agatoxin IVA (300 nm), which blocks the GABA release from CB1R-negative axon terminals (Wilson et al. 2001). These data suggest that JWH133 and SR144528 at the concentrations we used in our experiments had little off-target effect on CB1Rs.
Chronic activation of CB2Rs increases dendritic spine density
To investigate cellular mechanisms of the CB2R-induced increase in mEPSC frequency, we hypothesized that spine density might be increased by chronic activation of CB2Rs. After 7–10 days of treatment, the mean spine density of JWH133- or GP1a-treated cells (1.26 ± 0.12 μm−1, n = 12) was significantly higher than that of control cells (1.03 ± 0.12 μm−1, n = 11; P = 0.0018, Student's unpaired t test; Fig.4A). Given that the JWH133- and GP1a-treated groups (n = 6 each) had similar values of spine density (P = 0.30, Student's unpaired t test), we pooled the data from the two groups. The CB2Rs specifically mediated the increase in spine density because SR144528 blocked this effect of JWH133 [1.01 ± 0.10 μm−1 (n = 6) in control cells and 1.04 ± 0.15 μm−1 (n = 6) in cells treated with both SR144528 and JWH133; P = 0.66, Student's unpaired t test; Fig.4A]. The spine types were segregated based on morphology into stubby, thin and mushroom spines, and the CB2R-induced increase in density was limited to thin spines (Fig.4B and C).
Figure 4. Chronic activation of CB2Rs for 7–10 days increases the density of dendritic spines.
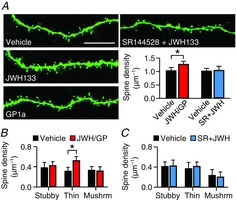
A, confocal micrographs show the dendrites of biocytin-filled CA1 pyramidal neurons in rat hippocampal slice cultures treated with vehicle, JWH133 (200 nm), GP1a (10 nm) or both SR144528 (100 nm) and JWH133. The bar graphs summarize the quantification of spine densities. Data from JWH133- and GP1a-treated cells were pooled. *P = 0.0018, Student's unpaired t test. Scale bar, 10 μm. B, spines analysed in A were divided into stubby-, thin- and mushroom-type spines based on morphology. *P = 0.000026, Student's unpaired t test. C, analysis of spine subtypes in slices treated with both SR144528 and JWH133. Error bars indicate SEM.
Activation of CB2Rs has little effect on presynaptic release properties and postsynaptic chloride conductance
We examined whether presynaptic properties, such as Pr and the size of RRP, also contributed to the increase in mEPSC frequency. From a train of eEPSCs (20 Hz for 3 s; Fig.5A), the facilitation of the first five EPSCs was used to compare Pr among vehicle-, JWH133- and GP1a-treated synapses. Short-term facilitation was not significantly different among the three groups (P = 0.29, two-way ANOVA; Fig.5B). The second eEPSC amplitudes (i.e. paired-pulse ratios) were 154 ± 6, 148 ± 8 and 145 ± 5% of the first one in vehicle-, JWH133- and GP1a-treated cells, respectively (n = 8 per group). This result suggests that Pr was not significantly altered by chronic activation of CB2Rs for 7–10 days.
The RRP size was estimated from cumulative amplitudes of eEPSCs in a 20 Hz train (Schneggenburger et al. 1999; Fig.5C). When the 36th–40th eEPSCs were used for the extrapolating method of estimation, the RRP sizes, which were normalized to the first eEPSC amplitude, were similar among the three groups, at 10.6 ± 1.7, 11.2 ± 1.4 and 12.5 ± 1.8 times the first EPSC amplitude for control, JWH133 and GP1a groups, respectively (n = 8 per group; P = 0.71, ANOVA; Fig.5C). This method for the estimation of RRP sizes is based on the kinetics of RRP depletion and/or refilling in autaptic culture synapses (Rosenmund & Stevens, 1996; Stevens & Williams, 2007). As the kinetics of RRP dynamics in organotypic culture synapses are uncertain, we also analysed eEPSCs at other time points and again found no differences. For control, JWH133 and GP1a groups, respectively, the RRP sizes estimated from the 26th–30th eEPSCs were 6.1 ± 1.3, 6.5 ± 0.7 and 7.6 ± 1.3 (P = 0.62, ANOVA); and the estimates from the 41st–50th eEPSCs were 14.0 ± 2.0, 12.7 ± 1.3 and 15.0 ± 2.1 (P = 0.69, ANOVA). Together, presynaptic properties such as Pr and RRP sizes were unaffected by chronic treatment with CB2R agonists, and thus are unlikely to be related to the changes in mEPSC frequency.
In the neocortex of 14- to 19-day-old rats, bath-applied JWH133 decreases Rm of pyramidal neurons by opening Cl– channels (den Boon et al. 2012). Reduction in the intrinsic excitability of neurons may result in an increase in mEPSC frequency via homeostatic plasticity (Burrone et al. 2002). Although our pipette solutions prevented the detection of K+-mediated changes in Rm because of Cs+ and QX314, they allowed for the measurement of any changes in Cl– conductance. In our experiment, however, bath-applied JWH133 (200 nm) did not change Rm of CA1 pyramidal neurons in slice cultures of the rat hippocampus (99.1 ± 3.9%; n = 12; P = 0.86, Student's paired t test; Fig.5D). The Rm of JWH133-treated cells (200 nm for 3–4 days; 211 ± 13 MΩ, n = 13) was not significantly different from that of vehicle-treated cells (193 ± 13 MΩ, n = 12; P = 0.33, Student's unpaired t test; Fig.5E). The Rm was not affected by 7–10 days of treatment with 200 nm JWH133 (n = 14; P = 0.38, Student's unpaired t test) or 10 nm GP1a (n = 12; P = 0.89, Student's unpaired t test) compared with vehicle-treated cells (Fig.5E). This result is not consistent with changes in Cl– conductance by a CB2R agonist applied either acutely or chronically. It remains to be determined whether CB2Rs modulate the excitability of hippocampal neurons by altering K+ conductance.
Long-term activation of CB2Rs phosphorylates ERK1/2
The CB2R is coupled with Gi/o protein to stimulate ERK and/or Akt (Demuth & Molleman, 2006; Fernández-Ruiz et al. 2007; Svíženská et al. 2008). We tested which kinase was activated, i.e. phosphorylated, by chronic treatment with a CB2R agonist. After a 7–10 day treatment with vehicle, JWH133 or GP1a, the protein levels of phosphorylated ERK1/2 and pan-ERK1/2 were analysed with the Western blotting method. The levels of phospho-ERK1/2 in JWH133- and GP1a-treated slices were 132 ± 9 (P = 0.011, Student's paired t test vs. 100%) and 156 ± 16% (P = 0.011) of the control value, respectively (n = 8 experiments; Fig.6A). However, the level of pan-ERK1/2 was affected by neither JWH133 (99 ± 5%; P = 0.86) nor GP1a (90 ± 5%; P = 0.066; Fig.6A). As a result, the ratio of phospho- to pan-ERK1/2 was significantly increased by either JWH133 (138 ± 14%; P = 0.032) or GP1a (180 ± 22%; P = 0.0078). In contrast, the levels of phospho-Akt and pan-Akt in JWH133- or GP1a-treated slices were not significantly different from those of control slices (P > 0.1; Fig.6B). The ratio of phospho- to pan-Akt was 98 ± 6% (P = 0.74, Student's paired t test vs. 100%) in JWH133-treated slices and 105 ± 6% (P = 0.41) in GP1a-treated slices (n = 8 experiments; Fig.6B). This result shows that, in our treatment conditions, CB2R agonists stimulated phosphorylation of ERK1/2 but not Akt.
Figure 6. Chronic activation of CB2Rs increases the phosphorylation of extracellular signal-regulated kinase (ERK) 1/2.
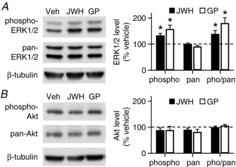
A, Western blot analysis of phosphorylated ERK1/2 and pan-ERK1/2 in rat hippocampal slice cultures treated with JWH133 (200 nm) or GP1a (10 nm) for 7–10 days. The data of phospho-ERK1/2 and pan-ERK1/2 are shown as a percentage of the control values in sister cultures. In each experiment, data were normalized to β-tubulin values. *P < 0.05, Student's paired t test vs. 100%. B, the levels of phosphorylated Akt or pan-Akt were not affected by treatment with JWH133 or GP1a. Error bars indicate SEM.
Phosphorylation of ERK contributes to the CB2R-medited increases in mEPSC frequency and spine density
To test whether phosphorylation of ERK contributed to the CB2R-induced increases in mEPSC frequency and spine density, we blocked ERK phosphorylation with PD0325901 (100 nm), an inhibitor of MEK1 and MEK2, both of which phosphorylate ERK. Treatment of slice cultures with PD0325901 alone (100 nm) for 7–10 days preserved glutamatergic synapses, because mEPSC frequency (1.01 ± 0.10 Hz, n = 11) was unaffected compared with the control value (1.16 ± 0.09 Hz, n = 11; Fig.7A). When co-treated with PD0325901, JWH133 did not increase mEPSC frequency (0.98 ± 0.10 Hz, n = 11; P = 0.38, ANOVA for the three groups; Fig.7A). The mEPSC amplitudes of the three groups were not significantly different from each other (17.0 ± 0.5 pA in control cells, 16.5 ± 0.6 pA in PD0325901-treated cells and 15.7 ± 1.1 pA in PD0325901/JWH133-treated cells; P = 0.50, ANOVA; Fig.7A). This result supports the idea that ERK phosphorylation might be involved in the JWH133-induced increase in mEPSC frequency. The effect of PD0325901 on spine density was also tested. The mean spine density in cells treated with PD0325901 alone (1.07 ± 0.18 μm−1, n = 7) or with PD0325901 and JWH133 (1.04 ± 0.12 μm−1, n = 8) was not significantly different from the spine density of vehicle-treated cells (1.06 ± 0.13 μm−1, n = 11; P = 0.93, ANOVA; Fig.7B). There was no difference among the three groups in the densities of stubby (P = 0.69, ANOVA), thin (P = 0.41) or mushroom spines (P = 0.49; data not shown).
Figure 7. Phosphorylation of ERK is involved in the CB2R-induced increases in mEPSC frequency and spine density.
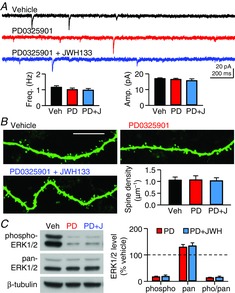
A, mEPSCs were recorded from CA1 pyramidal cells in rat hippocampal slice cultures treated with PD0325901 (100 nm) alone or with both PD0325901 and JWH133 (200 nm) for 7–10 days. B, confocal micrographs of biocytin-filled dendrites of CA1 pyramidal neurons. Rat hippocampal slice cultures were treated with the indicated drugs for 7–10 days. Scale bar represents 10 μm. C, Western blot analysis of phosphorylated ERK1/2 and pan-ERK1/2 in rat hippocampal slice cultures treated with PD0325901 or with both PD0325901 and JWH133 for 7–10 days. Error bars indicate SEM.
We examined whether PD0325901 indeed blocked the JWH133-induced increase in ERK phosphorylation. Treatment with PD0325901 alone for 7–10 days reduced the level of phosphorylated ERK1/2 compared with vehicle treatment (17 ± 2%, n = 5; P < 0.0001, Student's paired t test vs. 100%; Fig.7C), suggesting that PD0325901 blocked the basal phosphorylation of ERK1/2. The mean level of phospho-ERK1/2 in slice cultures treated with PD0325901 and JWH133 (19 ± 5%) was similar to that of PD0325901-treated slices (P = 0.75, Student's paired t test; Fig.7C), indicating that PD0325901 also blocked a JWH133-induced increase in ERK phosphorylation. Levels of pan-ERK1/2 were increased by a similar amount (∼30%) in both PD0325901-treated and PD0325901/JWH133-treated slices (P = 0.38, Student's paired t test; Fig.7C). As a result, the ratio of phospho- to pan-ERK1/2 levels in PD0325901/JWH133-treated slices was not significantly different from that of PD0325901-treated slices (P = 0.69, Student's paired t test; Fig.7C). Interestingly, in slices treated with PD0325901 alone, a reduction in the basal phosphorylation of ERK1/2 did not induce a significant decrease in mEPSC frequency or spine density (Fig.7A and B), implying that the ERK-mediated regulation of mEPSCs and spines may not be bidirectional.
Administration of a CB2R agonist in vivo increases excitatory synaptic transmission
Use of hippocampal slice cultures is essential for activation of CB2Rs only within the hippocampus. However, it is uncertain whether a CB2R agonist in vivo can also enhance excitatory synaptic transmission when administered in intact animals. We used mice for this experiment to use a CB2R KO strain. C57BL/6J mice (P34–39) were injected with JWH133 (5 mg kg−1, i.p.) once per day for 7–9 days. The efficacy of excitatory synaptic transmission was assayed by constructing I/O curves of fEPSPs recorded from the CA1 area of acute hippocampal slices made from treated mice 24 h after the last injection. The presynaptic activity of afferent input was measured as the amplitude of the presynaptic fibre volley, and the postsynaptic response was assessed by the rising slope of fEPSP. The I/O relationship was steeper in JWH133-injected C57BL/6J mice (n = 11 slices from five mice) than in vehicle-injected mice (n = 12 slices from four mice; P = 0.0009, Student's unpaired t test on the slope of the I/O curves; Fig.8A and B), implying that the synaptic response per action potential was increased. However, in CB2R KO mice, administration of JWH133 for 7–9 days had no effect on the I/O relationship (n = 16 from three mice) compared with the vehicle effect in CB2R KO mice (n = 13 from three mice; P = 0.67, Student's unpaired t test; Fig.8C and D). This result suggests that chronic activation of CB2Rs in intact mice also increased excitatory synaptic transmission. Our study, however, leaves it to be determined whether CB2R KO per se affects synaptic transmission, because CB2R KO mice could not be compared directly with C57BL/6J mice, which are only congenic controls for CB2R KO mice but not littermate controls.
Discussion
The present study shows that chronic activation of CB2Rs in the hippocampus increases mEPSC frequency and spine density via ERK-dependent signalling pathways. These effects of CB2Rs on synapses are specific for glutamatergic synapses after chronic, but not acute, activation of CB2Rs (e.g. for 7–10 days) and do not involve changes in presynaptic properties such as Pr or RRP size.
Chronic treatment of rodents in vivo with Δ9-THC reduces the expression of ionotropic glutamate receptors (e.g. GluA1, GluN2A and GluN2B) and the magnitude of long-term potentiation (LTP) of glutamatergic transmission by specifically activating CB1Rs (Hoffman et al. 2007; Fan et al. 2010). In addition to the downregulation of excitatory synapses, long-term exposure to Δ9-THC increases the I/O relationship of fEPSP and/or glutamatergic Pr (Hoffman et al. 2007; Fan et al. 2010). It has not been determined whether CB1Rs also mediate the Δ9-THC-induced increases in excitatory synaptic transmission. Given that Δ9-THC is a co-agonist for CB1Rs and CB2Rs, it is possible that some of the effects of chronic exposure to Δ9-THC may be mediated by CB2Rs. Our data suggest that in particular, upregulation of glutamatergic synapses by chronic treatment with Δ9-THC needs to be examined carefully in relationship to CB2R because chronic activation of CB2Rs can increase mEPSC frequency (Figs1 and 2), spine density (Fig.4) and the I/O relationship of fEPSP (Fig.8). When rodents are repeatedly injected with a CB1R/CB2R co-agonist, e.g. Δ9-THC or WIN55212-2, the synaptic density of CA1 pyramidal cells may be increased (Tagliaferro et al. 2006) or decreased (Chen et al. 2013), but it is unclear which type of cannabinoid receptor is involved in such effects. Once the role of CB1Rs in regulating synaptic density is determined, our results of the CB2R-mediated increase in spine density may help to reconcile these contradictory observations.
Both 2-AG and anandamide can activate CB2Rs, though 2-AG is a full agonist and anandamide is a partial agonist (Mechoulam et al. 1995; Showalter et al. 1996; Sugiura et al. 2000). 2-Arachidonoylglycerol and anandamide have 3- to 4-fold higher affinity for the CB1R than for the CB2R (Mechoulam et al. 1995; Showalter et al. 1996), and Δ9-THC binds to the CB1R and the CB2R with the same affinity (Showalter et al. 1996). Therefore, it is conceivable that both receptors in the brain may be activated if the levels of endocannabinoids are chronically elevated in pathological situations (Di Marzo & Petrosino, 2007) or with long-term intake of marijuana. Endocannabinoids are indeed implicated to induce CB2R-dependent signalling cascades. For example, CB2R KO (Racz et al. 2008; Ortega-Alvaro et al. 2011; García-Gutiérrez et al. 2013), knock-down (Onaivi et al. 2008) or overexpression (Aracil-Fernández et al. 2012; Romero-Zerbo et al. 2012) in mice induces neuropsychiatric phenotypes, and blocking endocannabinoid degradation reduces anxiety via CB2R (Busquets-Garcia et al. 2011). Further investigation is necessary to determine the physiological and pathological conditions in which endocannabinoids stimulate CB2Rs to modulate synaptic and neuronal functions.
In contrast to CB1Rs, CB2Rs exert their effects only after long-term stimulation, as shown by our data. In agreement with our observation, while acute activation of CB2Rs has little effect in intact mice, chronic administration of a CB2R agonist increases anxiety-like behaviour and reduces the expression of GABAA receptors (García-Gutiérrez et al. 2012). It is yet unclear why prolonged stimulation is required for CB2Rs to exert their effects. In principle, CB2R-induced phosphorylation of ERK is not slow, because activation of CB2Rs in HEK 293 cells and neuronal cell lines can phosphorylate ERK in minutes (Franklin & Carrasco, 2013; Zheng et al. 2013). The ERK-mediated changes in spine morphology can also occur rapidly, e.g. within hours during LTP (Segal, 2005; Bosch & Hayashi, 2012) or 24 h after treatment with brain-derived neurotrophic factor (Alonso et al. 2004). The amount of CB2R mRNA in the brain is only <1% of that of CB1R mRNA (Onaivi et al. 2006). It would be interesting to test whether such a low level of CB2R expression necessitates prolonged activation of CB2Rs for the accumulation of downstream signalling effectors. Although ERK-mediated increases in glutamatergic transmission and/or spine density are common consequences of LTP (Segal, 2005; Bosch & Hayashi, 2012), brain-derived neurotrophic factor treatment (Alonso et al. 2004) and chronic stimulation of CB2Rs, it is possible that the signalling cascade of CB2R–ERK–spine density might be distinct from the ERK signalling pathways for LTP and brain-derived neurotrophic factor treatment. For example, both homeostatic plasticity and LTP are expressed in the form of increases in AMPA receptors, but these two forms of plasticity have sharp contrasts in the time course and signalling molecules (Thiagarajan et al. 2007). The signalling cascades from CB2R to spines need to be determined in future studies.
Both ERK1 and ERK2 are involved in synaptic plasticity and memory (Thiels & Klann, 2001; Sweatt, 2004; Giovannini, 2006). ERK2 is critical for normal development, because ERK2 KO in mice is embryonically lethal (Aouadi et al. 2006). However, when ERK2 is knocked out in the CNS only, mice can survive, and the number of neurons and spine density are normal in the brain (Satoh et al. 2011). Reduced expression of ERK2 also has little effect on mouse survival and spine density in the hippocampus (Satoh et al. 2007). ERK1 KO mice display normal phenotypes in many aspects of synaptic transmission, e.g. I/O relationship of fEPSP, paired-pulse ratio of fEPSP and LTP of excitatory synapses (Selcher et al. 2001; Mazzucchelli et al. 2002). Therefore, the functions of glutamatergic synapses appear to be preserved by downregulation of ERK1 or ERK2 in the brain in spite of the importance of the kinases in synaptic plasticity. In accord with these reports, a reduction in the basal phosphorylation of ERK1/2 by treatment with PD0325901 alone (Fig.7C) did not significantly downregulate mEPSC or spine density (Fig.7A and B). If a decrease in ERK activity does not reduce spine density, as our data suggest (Fig.7), the mechanism of reduced spine density in CB2R KO mice (García-Gutiérrez et al. 2013) might not simply be the reversed signalling of the CB2R-mediated increase in spine density. Mechanistic comparisons between CB2R-mediated increase and decrease in spine density will be another topic for intensive investigation.
Long-term stimulation of CB2Rs increased the density of thin spines selectively (Fig.4B). Mushroom spines can transform to thin spines, e.g. during long-term depression of glutamatergic transmission (Nägerl et al. 2004; Zhou et al. 2004), but this transformation seemed unlikely to have occurred after chronic treatment with a CB2R agonist, because CB2R activation did not reduce the density of mushroom spines (Fig.4B). The increase in the density of total spines suggests that thin spines might be generated anew, because the formation, elimination and structural changes of thin spines are more dynamic compared with those of mushroom spines (Bourne & Harris, 2007). Filopodia are formed de novo during development or LTP, and often lack presynaptic partners (Bourne & Harris, 2007; Bosch & Hayashi, 2012). The thin spines in our data can be distinguished from filopodia because the upper limit of spine length in our analysis (3 μm) must have excluded most filopodia, which are often >3 μm (Maletic-Savatic et al. 1999). Activation of CB2Rs increased spine density by ∼20% (Fig.4A), whereas it increased mEPSC frequency by ∼50% (Fig.1C and D). It is possible that factors other than spine density might have contributed to the increase in mEPSC frequency. For instance, an increase in the total dendritic length of a neuron would also make mEPSCs more frequent.
Although chronic blockade of CB2Rs with SR144528 for 7–10 days had no effect on mEPSC frequency in slice cultures (Fig.1F), electron micrographs show that the KO of CB2Rs in mice reduces hippocampal spine density (García-Gutiérrez et al. 2013). This discrepancy could be because CB2Rs are non-functional in CB2R KO mice for a longer period of time from birth. It should be tested whether spine density is also reduced by treatment of wild-type tissue with a CB2R antagonist for a more extended period of time with an early onset. Our data show that inhibitory synaptic transmission was not affected by acute or chronic activation of CB2Rs, but in other studies acute stimulation of CB2Rs has reduced the amplitude of spontaneous IPSCs (but not mIPSCs) in the entorhinal cortex (Morgan et al. 2009) and inhibited K+-evoked GABA release from synaptosomes (Ando et al. 2012). Chronic activation of CB2Rs increases the expression of GABAA receptors (García-Gutiérrez et al. 2012). The reason for this discrepancy is unclear, but the differences in experimental conditions might be a possibility. For example, spontaneous IPSCs (Morgan et al. 2009) were recorded with excitatory and inhibitory networks intact, i.e. with only postsynaptic glutamate receptors blocked, and therefore, any changes in the excitability or Pr of a subset of neurons might alter the network activity, resulting in changes in spontaneous IPSCs. The in vivo route of drug administration (García-Gutiérrez et al. 2012), brain areas (Morgan et al. 2009; García-Gutiérrez et al. 2012) and the intactness of neurons (Ando et al. 2012) could also have contributed to the differences in the results.
In the hippocampus, CB2Rs are present in microglia (Svíženská et al. 2008; Atwood & Mackie, 2010), pyramidal neurons and interneurons (Gong et al. 2006; Onaivi et al. 2006; Brusco et al. 2008). It is critical to locate the CB2Rs that mediate the increases in mEPSC frequency and spine density to understand the mechanisms of CB2R action, e.g. cell-autonomous and network-effect mechanisms. If CB2Rs increase spine density in a cell-autonomous manner, the activation of CB2Rs in pyramidal cells may exert its effect only within the cell where those CB2Rs are present. Then, effector molecules downstream of CB2R can be intracellular messengers. A non-cell-autonomous or network-effect scenario is also possible; activation of CB2Rs in other pyramidal cells, interneurons and/or microglia might cause changes in network activity and hence increases in spine density more broadly. Intercellular signalling molecules may play a role in this case. To determine the mechanisms, it will be essential to stimulate CB2Rs only in a specific type of cell, as by knocking out or expressing CB2Rs in a certain type of cell.
The CB2R KO mice show impaired aversive memory consolidation (García-Gutiérrez et al. 2013), increased neuropathic pain (Racz et al. 2008) and schizophrenia-like behaviours (Ortega-Alvaro et al. 2011). Overexpression of CB2R in mice decreases the motor response to cocaine (Aracil-Fernández et al. 2012) and reduces food consumption (Romero-Zerbo et al. 2012). In humans, the polymorphism of CNR2, which encodes CB2R, is related to schizophrenia (Ishiguro et al. 2010; Tong et al. 2013), depression (Onaivi et al. 2008) and bipolar disorder (Minocci et al. 2011). A decrease in spine density is one of the hallmarks of individuals with schizophrenia (Jones et al. 2011), anxiety and depression (Wang et al. 2013), and CB2Rs are implicated in all of these disorders. It will be interesting to determine whether CB2Rs are involved in the spine pathology of such disorders and whether CB2R agonists can be used as a therapeutic tool for pathological spine loss.
Acknowledgments
The authors thank Drs Bo-Shiun Chen, Bradley Alger, Thomas Abrams, Scott Thompson and Jianhua Xu and members of the Kim laboratory for advice and/or technical support.
Glossary
Abbreviations
- ACSF
artificial cerebrospinal fluid
- 2-AG
2-arachidonoylglycerol
- BSA
bovine serum albumin
- CB1R
CB1 cannabinoid receptor
- CB2R
CB2 cannabinoid receptor
- Δ9-THC
Δ9-tetrahydrocannabinol
- DIV
days in vitro
- DSI
depolarization-induced suppression of inhibition
- eEPSC
evoked excitatory postsynaptic current
- eIPSC
evoked inhibitory postsynaptic current
- ERK
extracellular signal-regulated kinase
- fEPSP
field excitatory postsynaptic potential
- I/O
input–output
- KO
knock-out
- LTP
long-term potentiation
- mEPSC
miniature excitatory postsynaptic current
- mIPSC
miniature inhibitory postsynaptic current
- Pr
probability of release
- Rm
membrane resistance
- RRP
readily releasable pool
- TBST
Tris-buffered saline with Tween 20
Additional information
Competing interests
None declared.
Author contributions
J.K. and Y.L. conceived and designed the research, performed experiments, collected, analysed and interpreted data, prepared figures, wrote the manuscript and approved the final version to be published.
Funding
This work was supported by the National Institute on Aging Grant R01AG036794.
References
- Alger BE. Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol. 2002;68:247–286. doi: 10.1016/s0301-0082(02)00080-1. [DOI] [PubMed] [Google Scholar]
- Alonso M, Medina JH. Pozzo-Miller L. ERK1/2 activation is necessary for BDNF to increase dendritic spine density in hippocampal CA1 pyramidal neurons. Learn Mem. 2004;11:172–178. doi: 10.1101/lm.67804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand P, Whiteside G, Fowler CJ. Hohmann AG. Targeting CB2 receptors and the endocannabinoid system for the treatment of pain. Brain Res Rev. 2009;60:255–266. doi: 10.1016/j.brainresrev.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando RD, Biro J, Csolle C, Ledent C. Sperlagh B. The inhibitory action of exo- and endocannabinoids on [3H]GABA release are mediated by both CB1 and CB2 receptors in the mouse hippocampus. Neurochem Int. 2012;60:145–152. doi: 10.1016/j.neuint.2011.11.012. [DOI] [PubMed] [Google Scholar]
- Aouadi M, Binetruy B, Caron L, Le Marchand-Brustel Y. Bost F. Role of MAPKs in development and differentiation: lessons from knockout mice. Biochimie. 2006;88:1091–1098. doi: 10.1016/j.biochi.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Aracil-Fernández A, Trigo JM, García-Gutiérrez MS, Ortega-Álvaro A, Ternianov A, Navarro D, Robledo P, Berbel P, Maldonado R. Manzanares J. Decreased cocaine motor sensitization and self-administration in mice overexpressing cannabinoid CB2 receptors. Neuropsychopharmacology. 2012;37:1749–1763. doi: 10.1038/npp.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK. Mackie K. CB2: a cannabinoid receptor with an identity crisis. Br J Pharmacol. 2010;160:467–479. doi: 10.1111/j.1476-5381.2010.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch M. Hayashi Y. Structural plasticity of dendritic spines. Curr Opin Neurobiol. 2012;22:383–388. doi: 10.1016/j.conb.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne J. Harris KM. Do thin spines learn to be mushroom spines that remember? Curr Opin Neurobiol. 2007;17:381–386. doi: 10.1016/j.conb.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Brusco A, Tagliaferro P, Saez T. Onaivi ES. Postsynaptic localization of CB2 cannabinoid receptors in the rat hippocampus. Synapse. 2008;62:944–949. doi: 10.1002/syn.20569. [DOI] [PubMed] [Google Scholar]
- Burrone J, O'Byrne M. Murthy VN. Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature. 2002;420:414–418. doi: 10.1038/nature01242. [DOI] [PubMed] [Google Scholar]
- Busquets-Garcia A, Puighermanal E, Pastor A, de la Torre R, Maldonado R. Ozaita A. Differential role of anandamide and 2-arachidonoylglycerol in memory and anxiety-like responses. Biol Psychiatry. 2011;70:479–486. doi: 10.1016/j.biopsych.2011.04.022. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Younts TJ, Chávez AE. Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Zhang J, Fan N, Teng ZQ, Wu Y, Yang H, Tang YP, Sun H, Song Y. Chen C. Δ9-THC-caused synaptic and memory impairments are mediated through COX-2 signaling. Cell. 2013;155:1154–1165. doi: 10.1016/j.cell.2013.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA. Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Demuth DG. Molleman A. Cannabinoid signalling. Life Sci. 2006;78:549–563. doi: 10.1016/j.lfs.2005.05.055. [DOI] [PubMed] [Google Scholar]
- den Boon FS, Chameau P, Schaafsma-Zhao Q, van Aken W, Bari M, Oddi S, Kruse CG, Maccarrone M, Wadman WJ. Werkman TR. Excitability of prefrontal cortical pyramidal neurons is modulated by activation of intracellular type-2 cannabinoid receptors. Proc Natl Acad Sci USA. 2012;109:3534–3539. doi: 10.1073/pnas.1118167109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devane WA, Dysarz FA, 3rd, Johnson MR, Melvin LS. Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- Di Marzo V. Petrosino S. Endocannabinoids and the regulation of their levels in health and disease. Curr Opin Lipidol. 2007;18:129–140. doi: 10.1097/MOL.0b013e32803dbdec. [DOI] [PubMed] [Google Scholar]
- Fan N, Yang H, Zhang J. Chen C. Reduced expression of glutamate receptors and phosphorylation of CREB are responsible for in vivo Δ9-THC exposure-impaired hippocampal synaptic plasticity. J Neurochem. 2010;112:691–702. doi: 10.1111/j.1471-4159.2009.06489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Ruiz J, Romero J, Velasco G, Tolón RM, Ramos JA. Guzmán M. Cannabinoid CB2 receptor: a new target for controlling neural cell survival? Trends Pharmacol Sci. 2007;28:39–45. doi: 10.1016/j.tips.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Franklin JM. Carrasco GA. Cannabinoid receptor agonists upregulate and enhance serotonin 2A (5-HT2A) receptor activity via ERK1/2 signaling. Synapse. 2013;67:145–159. doi: 10.1002/syn.21626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Gutiérrez MS, García-Bueno B, Zoppi S, Leza JC. Manzanares J. Chronic blockade of cannabinoid CB2 receptors induces anxiolytic-like actions associated with alterations in GABAA receptors. Br J Pharmacol. 2012;165:951–964. doi: 10.1111/j.1476-5381.2011.01625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Gutiérrez MS, Ortega-Álvaro A, Busquets-García A, Pérez-Ortiz JM, Caltana L, Ricatti MJ, Brusco A, Maldonado R. Manzanares J. Synaptic plasticity alterations associated with memory impairment induced by deletion of CB2 cannabinoid receptors. Neuropharmacology. 2013;73:388–396. doi: 10.1016/j.neuropharm.2013.05.034. [DOI] [PubMed] [Google Scholar]
- Giovannini MG. The role of the extracellular signal-regulated kinase pathway in memory encoding. Rev Neurosci. 2006;17:619–634. doi: 10.1515/revneuro.2006.17.6.619. [DOI] [PubMed] [Google Scholar]
- Gong JP, Onaivi ES, Ishiguro H, Liu QR, Tagliaferro PA, Brusco A. Uhl GR. Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res. 2006;1071:10–23. doi: 10.1016/j.brainres.2005.11.035. [DOI] [PubMed] [Google Scholar]
- Han S, Thatte J, Buzard DJ. Jones RM. Therapeutic utility of cannabinoid receptor type 2 (CB2) selective agonists. J Med Chem. 2013;56:8224–8256. doi: 10.1021/jm4005626. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Oz M, Yang R, Lichtman AH. Lupica CR. Opposing actions of chronic Δ9-tetrahydrocannabinol and cannabinoid antagonists on hippocampal long-term potentiation. Learn Mem. 2007;14:63–74. doi: 10.1101/lm.439007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiguro H, Horiuchi Y, Ishikawa M, Koga M, Imai K, Suzuki Y, Morikawa M, Inada T, Watanabe Y, Takahashi M, Someya T, Ujike H, Iwata N, Ozaki N, Onaivi ES, Kunugi H, Sasaki T, Itokawa M, Arai M, Niizato K, Iritani S, Naka I, Ohashi J, Kakita A, Takahashi H, Nawa H. Arinami T. Brain cannabinoid CB2 receptor in schizophrenia. Biol Psychiatry. 2010;67:974–982. doi: 10.1016/j.biopsych.2009.09.024. [DOI] [PubMed] [Google Scholar]
- Jhaveri MD, Sagar DR, Elmes SJ, Kendall DA. Chapman V. Cannabinoid CB2 receptor-mediated anti-nociception in models of acute and chronic pain. Mol Neurobiol. 2007;36:26–35. doi: 10.1007/s12035-007-8007-7. [DOI] [PubMed] [Google Scholar]
- Jones CA, Watson DJ. Fone KC. Animal models of schizophrenia. Br J Pharmacol. 2011;164:1162–1194. doi: 10.1111/j.1476-5381.2011.01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M. Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Katona I. Freund TF. Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci. 2012;35:529–558. doi: 10.1146/annurev-neuro-062111-150420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Sperlágh B, Sík A, Käfalvi A, Vizi ES, Mackie K. Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R. Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Marsicano G. Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci. 1999;11:4213–4225. doi: 10.1046/j.1460-9568.1999.00847.x. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC. Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pagès G, Valverde O, Marowsky A, Porrazzo A, Orban PC, Maldonado R, Ehrengruber MU, Cestari V, Lipp HP, Chapman PF, Pouysségur J. Brambilla R. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron. 2002;34:807–820. doi: 10.1016/s0896-6273(02)00716-x. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, Pertwee RG, Griffin G, Bayewitch M, Barg J. Vogel Z. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- Minocci D, Massei J, Martino A, Milianti M, Piz L, Di Bello D, Sbrana A, Martinotti E, Rossi AM. Nieri P. Genetic association between bipolar disorder and 524A>C (Leu133Ile) polymorphism of CNR2 gene, encoding for CB2 cannabinoid receptor. J Affect Disord. 2011;134:427–430. doi: 10.1016/j.jad.2011.05.023. [DOI] [PubMed] [Google Scholar]
- Morgan NH, Stanford IM. Woodhall GL. Functional CB2 type cannabinoid receptors at CNS synapses. Neuropharmacology. 2009;57:356–368. doi: 10.1016/j.neuropharm.2009.07.017. [DOI] [PubMed] [Google Scholar]
- Munro S, Thomas KL. Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Nägerl UV, Eberhorn N, Cambridge SB. Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Navarrete F, Pérez-Ortiz JM. Manzanares J. Cannabinoid CB2 receptor-mediated regulation of impulsive-like behaviour in DBA/2 mice. Br J Pharmacol. 2012;165:260–273. doi: 10.1111/j.1476-5381.2011.01542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onaivi ES, Ishiguro H, Gong JP, Patel S, Meozzi PA, Myers L, Perchuk A, Mora Z, Tagliaferro PA, Gardner E, Brusco A, Akinshola BE, Hope B, Lujilde J, Inada T, Iwasaki S, Macharia D, Teasenfitz L, Arinami T. Uhl GR. Brain neuronal CB2 cannabinoid receptors in drug abuse and depression: from mice to human subjects. PLoS One. 2008;3:e1640. doi: 10.1371/journal.pone.0001640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onaivi ES, Ishiguro H, Gong JP, Patel S, Perchuk A, Meozzi PA, Myers L, Mora Z, Tagliaferro P, Gardner E, Brusco A, Akinshola BE, Liu QR, Hope B, Iwasaki S, Arinami T, Teasenfitz L. Uhl GR. Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann NY Acad Sci. 2006;1074:514–536. doi: 10.1196/annals.1369.052. [DOI] [PubMed] [Google Scholar]
- Onaivi ES, Ishiguro H, Gu S. Liu QR. CNS effects of CB2 cannabinoid receptors: beyond neuro-immuno-cannabinoid activity. J Psychopharmacol. 2012;26:92–103. doi: 10.1177/0269881111400652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Alvaro A, Aracil-Fernández A, García-Gutiérrez MS, Navarrete F. Manzanares J. Deletion of CB2 cannabinoid receptor induces schizophrenia-related behaviors in mice. Neuropsychopharmacology. 2011;36:1489–1504. doi: 10.1038/npp.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racz I, Nadal X, Alferink J, Baños JE, Rehnelt J, Martín M, Pintado B, Gutierrez-Adan A, Sanguino E, Manzanares J, Zimmer A. Maldonado R. Crucial role of CB2 cannabinoid receptor in the regulation of central immune responses during neuropathic pain. J Neurosci. 2008;28:12125–12135. doi: 10.1523/JNEUROSCI.3400-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR. Wearne SL. Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One. 2008;3:e1997. doi: 10.1371/journal.pone.0001997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Zerbo SY, Garcia-Gutierrez MS, Suárez J, Rivera P, Ruz-Maldonado I, Vida M, Rodriguez de Fonseca F, Manzanares J. Bermúdez-Silva FJ. Overexpression of cannabinoid CB2 receptor in the brain induces hyperglycaemia and a lean phenotype in adult mice. J Neuroendocrinol. 2012;24:1106–1119. doi: 10.1111/j.1365-2826.2012.02325.x. [DOI] [PubMed] [Google Scholar]
- Rosenmund C. Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Satoh Y, Endo S, Ikeda T, Yamada K, Ito M, Kuroki M, Hiramoto T, Imamura O, Kobayashi Y, Watanabe Y, Itohara S. Takishima K. Extracellular signal-regulated kinase 2 (ERK2) knockdown mice show deficits in long-term memory; ERK2 has a specific function in learning and memory. J Neurosci. 2007;27:10765–10776. doi: 10.1523/JNEUROSCI.0117-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh Y, Endo S, Nakata T, Kobayashi Y, Yamada K, Ikeda T, Takeuchi A, Hiramoto T, Watanabe Y. Kazama T. ERK2 contributes to the control of social behaviors in mice. J Neurosci. 2011;31:11953–11967. doi: 10.1523/JNEUROSCI.2349-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC. Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6:277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- Selcher JC, Nekrasova T, Paylor R, Landreth GE. Sweatt JD. Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn Mem. 2001;8:11–19. doi: 10.1101/lm.37001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Showalter VM, Compton DR, Martin BR. Abood ME. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): identification of cannabinoid receptor subtype selective ligands. J Pharmacol Exp Ther. 1996;278:989–999. [PubMed] [Google Scholar]
- Stevens CF. Williams JH. Discharge of the readily releasable pool with action potentials at hippocampal synapses. J Neurophysiol. 2007;98:3221–3229. doi: 10.1152/jn.00857.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura T, Kondo S, Kishimoto S, Miyashita T, Nakane S, Kodaka T, Suhara Y, Takayama H. Waku K. Evidence that 2-arachidonoylglycerol but not N-palmitoylethanolamine or anandamide is the physiological ligand for the cannabinoid CB2 receptor. Comparison of the agonistic activities of various cannabinoid receptor ligands in HL-60 cells. J Biol Chem. 2000;275:605–612. doi: 10.1074/jbc.275.1.605. [DOI] [PubMed] [Google Scholar]
- Svíženská I, Dubový P. Šulcová A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures — a short review. Pharmacol Biochem Behav. 2008;90:501–511. doi: 10.1016/j.pbb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Sweatt JD( Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Tagliaferro P, Javier Ramos A, Onaivi ES, Evrard SG, Lujilde J. Brusco A. Neuronal cytoskeleton and synaptic densities are altered after a chronic treatment with the cannabinoid receptor agonist WIN 55,212-2. Brain Res. 2006;1085:163–176. doi: 10.1016/j.brainres.2005.12.089. [DOI] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, Malgaroli A. Tsien RW. LTP and adaptation to inactivity: overlapping mechanisms and implications for metaplasticity. Neuropharmacology. 2007;52:156–175. doi: 10.1016/j.neuropharm.2006.07.030. [DOI] [PubMed] [Google Scholar]
- Thiels E. Klann E. Extracellular signal-regulated kinase, synaptic plasticity, and memory. Rev Neurosci. 2001;12:327–345. doi: 10.1515/revneuro.2001.12.4.327. [DOI] [PubMed] [Google Scholar]
- Tong D, He S, Wang L, Jin L, Si P. Cheng X. Association of single-nucleotide polymorphisms in the cannabinoid receptor 2 gene with schizophrenia in the Han Chinese population. J Mol Neurosci. 2013;51:454–460. doi: 10.1007/s12031-013-0062-0. [DOI] [PubMed] [Google Scholar]
- Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makriyannis A, Piomelli D, Davison JS, Marnett LJ, Di Marzo V, Pittman QJ, Patel KD. Sharkey KA. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- Wang G, Cheng Y, Gong M, Liang B, Zhang M, Chen Y, Zhang C, Yuan X. Xu J. Systematic correlation between spine plasticity and the anxiety/depression-like phenotype induced by corticosterone in mice. Neuroreport. 2013;24:682–687. doi: 10.1097/WNR.0b013e32836384db. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Kunos G. Nicoll RA. Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron. 2001;31:453–462. doi: 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]
- Zheng C, Chen L, Chen X, He X, Yang J, Shi Y. Zhou N. The second intracellular loop of the human cannabinoid CB2 receptor governs G protein coupling in coordination with the carboxyl terminal domain. PLoS One. 2013;8:e63262. doi: 10.1371/journal.pone.0063262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ. Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]