

Abstract
The axonal voltage-gated Ca2+ channels (VGCCs) that catalyse dopamine (DA) transmission are incompletely defined. Yet, they are critical to DA function and might prime subpopulations of DA neurons for parkinsonian degeneration. Previous studies of VGCCs will have encompassed those on striatal cholinergic interneurons, which strongly influence DA transmission. We identify which VGCCs on DA axons govern DA transmission, we determine their dynamic properties and reveal an underlying basis for differences between the caudate putamen (CPu) and nucleus accumbens (NAc). We detected DA release evoked electrically during nicotinic receptor blockade or optogenetically by light activation of channel rhodopsin-expressing DA axons in mouse striatal slices. Subtype-specific VGCC blockers indicated that N-, Q-, T- and L-VGCCs govern DA release in CPu, but in NAc, T and L-channels are relatively silent. The roles of the most dominant channels were inversely frequency-dependent, due to low-pass filtering of DA release by Ca2+-dependent relationships between initial release probability and short-term plasticity. Ca2+ concentration–response curves revealed that differences between CPu and NAc were due to greater underlying Ca2+ sensitivity of DA transmission from CPu axons. Functions for ‘silent’ L- and T-channels in NAc could be unmasked by elevating extracellular [Ca2+]. Furthermore, we identified a greater coupling between BAPTA-sensitive, fast Ca2+ transients and DA transmission in CPu axons, and evidence for endogenous fast buffering of Ca2+ in NAc. These data reveal that a range of VGCCs operate dynamically on DA axons, depending on local driving forces. Furthermore, they reveal dramatic differences in Ca2+ handling between axonal subpopulations that show different vulnerability to parkinsonian degeneration.
Key points
The voltage-gated Ca2+ channels (VGCCs) that catalyse striatal dopamine transmission are critical to dopamine function and might prime subpopulations of neurons for parkinsonian degeneration.
However, the VGCCs that operate on mesostriatal axons are incompletely defined; previous studies encompassed channels on striatal cholinergic interneurons that strongly influence dopamine transmission.
We define that multiple types of axonal VGCCs operate that extend beyond classic presynaptic N/P/Q channels to include T- and L-types.
We reveal differences in VGCC function between mouse axon types that in humans are vulnerable versus resistant to Parkinson's disease. We show for the first time that this is underpinned by different sensitivity of dopamine transmission to extracellular Ca2+ and by different spatiotemporal intracellular Ca2+ microdomains.
These data define key principles of how Ca2+ and VGCCs govern dopamine transmission in the healthy brain and reveal differences between neuron types that might contribute to vulnerability in disease.
Introduction
Release of dopamine (DA) from mesostriatal DA neurons is critical to the selection and learning of our actions and motivations. Release of transmitters is catalysed by presynaptic VGCCs that provide a transient Ca2+ microdomain but the VGCCs that govern DA transmission have not previously been resolved. Typically, N-type (Cav2.2) and P/Q-type (Cav2.1) VGCCs dominate in neurotransmission at CNS synapses (Rusakov, 2006), but it is increasingly evident that other VGCCs, including T-types (Cav3) and L-types (Cav1.2–4), may regulate neurotransmitter release from some neuron types (Pan et al. 2001; Tippens et al. 2008; Holmgaard et al. 2009; Zanazzi & Matthews, 2009; Tang et al. 2011).
Mesostriatal DA neurons possess a uniquely extensive arbour of DA release sites: axonal fields form the vast majority (∼99%) of the total membrane area of DA neurons (Matsuda et al. 2009; Henny et al. 2012) with individual rat DA neurons comprising approximately 0.5 m of axons (Matsuda et al. 2009) and half a million release sites (Arbuthnott & Wickens, 2007). These striking figures will be orders of magnitude higher in the human brain (Pissadaki & Bolam, 2013). The VGCCs that support axonal DA release will be critical to DA function, and might contribute to the Ca2+-dependent vulnerability of DA neurons to degeneration in Parkinson's disease (Mosharov et al. 2009; Collier et al. 2011). Subpopulations of DA neurons of the substantia nigra pars compacta (SNc) that innervate caudate putamen (CPu) are more vulnerable than those of the adjacent ventral tegmental area (VTA) innervating nucleus accumbens (NAc), and to date a somatodendritic L-type conductance (Cav1.3) in SNc but not VTA has been identified as a contributing factor (Chan et al. 2007; Guzman et al. 2009). The additional handling of Ca2+ by axons might generate a substantial ionic load and metabolic or proteostatic burden (Surmeier et al. 2010; Harris et al. 2012; Pissadaki & Bolam, 2013).
Previous studies in CPu identified that N-, P/Q-, R- and T-channels but not L-channels govern DA transmission (Phillips & Stamford, 2000; Chen et al. 2006; Sgobio et al. 2014) and suggested that VGCC subtypes might differ between striatal regions. It was also suggested that VGCC roles might differ during low tonic and high phasic firing frequencies to encode DA functions attributed to specific firing patterns (Phillips & Stamford, 2000). However, new insights suggest other factors could account for those findings. There are underlying short-term dynamics in DA release probability that govern frequency dependence that depend on Ca2+ and vary between striatal regions (Cragg, 2003; Montague et al. 2004). Furthermore, an input from cholinergic interneurons (ChIs) to nicotinic acetylcholine receptors (nAChRs) on DA axons governs DA release probability, plasticity and frequency dependence (Zhou et al. 2001; Rice & Cragg, 2004; Zhang & Sulzer, 2004; Cragg, 2006; Exley & Cragg, 2008; Threlfell et al. 2012) and these effects, alongside VGCCs on ChIs, will have confounded interpretation.
Here, we identify which VGCCs on mouse DA axons govern DA transmission in CPu versus NAc and the principles that underlie their dynamic participation. Furthermore, we reveal significant differences in the dynamic coupling of Ca2+ to DA transmission.
Methods
Slice preparation
Male adult mice were C57Bl6/J wild-type (Charles River) or DA transporter (DAT)-Cre heterozygote mice used previously (Threlfell et al. 2012) of mean age 8 weeks, which did not differ for experiments between different regions. Mice were killed by cervical dislocation, the brains removed, and 300 μm coronal slices containing CPu and NAc prepared as described previously (Exley et al. 2012; Threlfell et al. 2012) in ice-cold Hepes-based buffer saturated with 95% O2/5% CO2, containing (in mm): 120 NaCl, 20 NaHCO3, 6.7 Hepes acid, 5 KCl, 3.3 Hepes salt, 2 CaCl2, 2 MgSO4, 1.2 KH2PO4 and 10 glucose. Slices were incubated at room temperature for ≥1 h in Hepes-based buffer before transferral to the recording chamber. All procedures were carried out according to institutional guidelines and conformed to the UK Animals (Scientific Procedures) Act 1986.
Fast-scan cyclic voltammetry
DA release was monitored in acute slices using fast-scan cyclic voltammetry (FCV) as we have described previously (Exley et al. 2012; Threlfell et al. 2012). Slices were superfused in a recording chamber with bicarbonate-buffered artificial cerebrospinal fluid (aCSF) saturated with 95%O2/5%CO2 at 31–33°C, containing (in mm): 124 NaCl, 26 NaHCO3, 3.8 KCl, 2.4 CaCl2, 1.3 MgSO4, 1.3 KH2PO4 and 10 glucose. Evoked extracellular DA concentration ([DA]o) was monitored using FCV at 7–10 μm diameter carbon fibre microelectrodes fabricated inhouse (tip length 50–100 μm) and a Millar voltammeter (Julian Millar, Barts and the London School of Medicine and Dentistry). In brief, a triangular voltage waveform (range −700 mV to +1300 mV vs. Ag/AgCl) was applied at 800 V s–1 at a scan frequency of 8 Hz. Electrodes were switched out of circuit between scans. Electrodes were calibrated using 1–2 μm DA in each experimental medium. Calibration solutions were prepared immediately before calibration from a 2.5 mm stock solution in 0.1 m HClO4 stored at 4°C. Signals were attributable to DA by the potentials for peak oxidation and reduction currents (oxidation peak: +500–600 mV, reduction peak: ∼−200 mV).
Electrical stimulation
Recordings were obtained from both dorsolateral CPu and NAc core. DA release was evoked by a local bipolar concentric Pt/Ir electrode (25 μm diameter; FHC Inc., Bowdoin, ME, USA) placed approximately 100 μm from the carbon fibre microelectrodes as previously (Threlfell et al. 2012; Exley et al. 2013). Stimulus pulses (200 μs duration) were given at 0.6 mA (perimaximal in control conditions). DA neurons in vivo exhibit a range of firing frequencies from ∼1–40 Hz or higher. We applied either single pulses (1p) or five pulses (5p) at 5, 25, 40 and 100 Hz to span a full range of firing frequencies. Mean peak [DA]o evoked by 1p was equivalent to that of a 1 Hz train; 1p is used in frequency comparison to indicate maximum 1 Hz data. A frequency of 100 Hz may be supraphysiological but is useful as a tool for exposing changes in short-term plasticity (STP) that arise through changes in initial release probability (Rice & Cragg, 2004). Electrical stimulations were repeated at 2.5 min intervals, which allow stable release to be sustained over several hours. Each stimulus type was repeated in triplicate in a random order.
All data were obtained in the presence of the nAChR antagonist, dihydro-β-erythroidine (DHβE, 1 μm) added to the recording aCSF, to inhibit nAChRs on DA axons and remove the confounding effects of VGCCs on cholinergic interneurons that regulate ACh release and ACh effects on DA (Rice & Cragg, 2004; Exley & Cragg, 2008). Experiments were conducted in the presence of 2.4 mm extracellular Ca2+ unless otherwise stated. Muscarinic acetylcholine receptors do not regulate DA transmission during the stimulation protocols used here (Threlfell et al. 2010) and therefore it was not necessary to include a muscarinic acetylcholine receptor antagonist. Data were acquired without the use of DAT inhibitors, which are used in some studies to boost DA signals, as the DAT may modify the Ca2+ dependence of DA release (Kile et al. 2010). Release was tetrodotoxin-sensitive as previously (Threlfell et al. 2010, 2012), and was not modulated by glutamate or GABA antagonists as shown previously (Threlfell et al. 2010), and confirmed here (not illustrated).
Optogenetic stimulation
The use of optogenetic stimuli is this study was limited judiciously to corroborating data obtained with electrical stimuli, and was not a first choice for a stimulus since the Ca2+ conductance associated with channel rhodopsin may interfere with endogenous Ca2+-dependent processes. We incorporated the light-activated channelrhodopsin2 (ChR2) into DAT-positive neurons with a Cre-LoxP approach we have used previously (Threlfell et al. 2012). In brief, we injected a Cre-inducible recombinant AAV vector containing ChR2-eYFP (pAAV5-double floxed-hChR2(H134R)-EYFP-WPRE-pA) into the SNc (anteroposterior −3.5 mm, mediolateral ±1.2 mm, dorsoventral −4.0 mm) of mice expressing Cre-recombinase under the control of the DAT (DAT-IRES-Cre, B6.SJL-Slc6a3tm1.1(cre)Bkmn/J; Jackson Laboratory, Bar Habor, ME, USA). Approximately 2 weeks post-surgery, striatal slices were prepared for FCV recordings, as described above. ChR2-expressing DA fibres were activated using either a 473 nm diode laser (DL-473; Rapp optoElectric, Wedel, Germany), coupled to the microscope with a fibre optic cable (200 μm multimode; NA 0.22), which illuminated a 60 μm diameter spot through the immersion objective (10×), or a 470 nm LED (OptoLED; Cairn Research, Kent, UK), which illuminated the full field of view (2.2 mm, 10× water-immersion objective). TTL-driven light pulses (2 ms duration, 4.5–8 mW, optical power meter; Thor labs, Ely, UK) were applied singly or in trains of five pulses at 5–25 Hz.
We confirmed that the specificity of ChR2-eYFP expression in DAT-Cre animals was restricted to tyrosine hydroxylase-immunoreactive neurons, as shown previously (Threlfell et al. 2012). Midbrain slices were fixed in 4% paraformaldehyde before re-sectioning to 40 μm. Free-floating sections were washed in phosphate-buffered saline (PBS) and incubated in 0.5% Triton X-100 with 10% normal goat serum and 10% fetal bovine serum. Slices were incubated overnight at 4°C in primary antibody (1:2000 rabbit anti-tyrosine hydroxylase; Sigma-Aldrich, Dorset, UK) diluted in PBS with 0.5% Triton X-100, 1% normal goat serum and 1% fetal bovine serum. Slices were washed in PBS then incubated in secondary antibody (1:1000 DyLight 594 goat antirabbit; Jackson ImmunoResearch, Suffolk, UK) diluted in PBS with 0.5% Triton X-100 and 1% normal goat serum and 1% fetal bovine serum for 2 h at room temperature. Slices were then washed in PBS then mounted on to gelled slides and cover-slipped with hard mount Vectashield (Vector Labs, Peterborough, UK) and imaged with an Olympus BX41 microscope with Olympus UC30 camera and filters for appropriate excitation and emission wavelengths (Olympus Medical, Southend-on-Sea, UK).
Incubation of EGTA-AM or BAPTA-AM
Coronal sections (300 μm) containing both CPu and NAc were bisected. Each hemisphere was incubated for 30 min in aCSF at room temperature (either 2.4 or 3.6 mm [Ca2+]) containing 2-hydroypropyl-β-cyctodextrin (70 μm), probenecid (175 μm), pluronic acid (0.1%) and for EGTA-AM or BAPTA-AM conditions also contain EGTA-AM or BAPTA-AM (100 μm) (Ouanounou et al. 1999; Kukley et al. 2007). Following incubation, hemispheres were placed in the recording chamber with the recording electrode for 30 min before recordings were taken. Equivalent anatomical recording sites were chosen in each of the hemispheres, alternating between the control slice and EGTA-AM/BAPTA-AM-incubated slice: whether the control or EGTA/BAPTA slice was started with was alternated between animals. DA release data in EGTA-AM or BAPTA-AM were expressed as the percentage of paired control recording site.
Drugs and solutions
DHβE, ω-Agatoxin IVA, ω-Conotoxin GVIA, NNC 55-0396, isradipine and BAPTA-AM were purchased from Abcam (Cambridge, UK) or Tocris (Bristol, UK); Pluronic acid from Life Technologies (Paisley, UK); EGTA-AM from Millipore (Hertfordshire, UK). All other reagents were purchased from Sigma-Aldrich. Stock solutions were made to 1000–2000× final concentrations in H2O (DHβE, ω-Conotoxin GVIA, ω-Agatoxin IVA and NNC 55-0396) or dimethyl sulphoxide (isradipine) and stored at −20°C. Drugs were diluted to their required concentrations in aCSF immediately before use. Drug concentrations were chosen in accordance with previous studies (Phillips & Stamford, 2000; Chen et al. 2006; Guzman et al. 2009; Tai et al. 2011) and with pilot concentration–response curves, which corroborated maximal drug effects.
For T-type inhibition, NNC 55-0396 was chosen in preference to mibefradil or Ni2+ because of confounding effects: mibefradil obscures electrode sensitivity to DA (Chen et al. 2006) and Ni2+ inhibits DA uptake (Brimblecombe & Cragg, 2014). NNC 55-0396 has fewer off-target effects than mibefradil (Li et al. 2005), and we show that its effects vary predictably with Ca2+ concentration (see Fig. 7) corroborating its VGCC site of action. We also trialled the R-type blocker SNX 482 (100 nm), which was found to be electroactive, to reduce electrode sensitivity to DA and to lead to a paradoxical increase in evoked [DA]o in CPu and NAc (all stimulation frequencies, data not shown). As there are no alternative R-selective compounds commercially available to corroborate these paradoxical effects, it is difficult to interpret these data and they were excluded from further consideration here.
Figure 7. ‘Silent’ L- and T-voltage-gated Ca2+ channels in NAc can be unmasked.
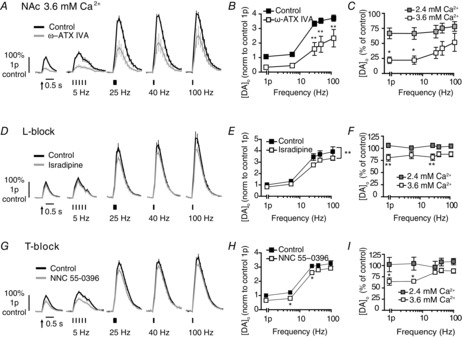
A, D and G, mean profiles of [DA]o ± SEM vs. time evoked by one or five electrical pulses (5–100 Hz) in NAc in 3.6 mm extracellular Ca2+ (control, black) or the presence (grey) of P/Q block (A), L-block (D) or T-block (G). Data are normalized to 1p peak [DA]o. B, E and H, mean peak [DA]o ± SEM vs. frequency (5p) in NAc in 3.6 mm extracellular Ca2+, in control conditions (filled) or block (unfilled). Bonferroni post-tests vs. control: *P < 0.05 **P < 0.01. C, F and I, mean peak [DA]o ± SEM in NAc after P/Q block (C), L-block (F) or T-block (I) expressed as percentage of control at each frequency compared for 2.4 mm [Ca2+]o (filled) and 3.6 mm [Ca2+]o (unfilled) shows a significantly greater effect of each voltage-gated Ca2+ channel blocker at higher [Ca2+]o. Bonferroni post-tests, *P < 0.05 **P < 0.01, n = 5–6 animals. Data for 2.4 mm [Ca2+]o in (C), (F) and (I) is taken from Figs 2, 4 and 5. DA, dopamine; NAc, nucleus accumbens.
Data and statistical analysis
Data were acquired and analysed using Axoscope 10.2 (Molecular Devices, Berkshire, UK), Whole Cell Program (University of Strathclyde, Glasgow) and Excel macros written locally. Data are expressed as means ± SEM, and n = number of animals (for data in Figs 7) or number of sites per region (for data in Fig. 8). Data from each animal were obtained by averaging at least three recordings for each stimulus type and normalizing to mean control 1p conditions for each animal. Data presented are normalized for greater clarity and for more ready comparisons between regions. Note that the outcomes reported here in each region were the same using raw data.
Figure 8. Intracellular Ca2+ chelators reveal different Ca2+ dynamics in CPu and NAc.
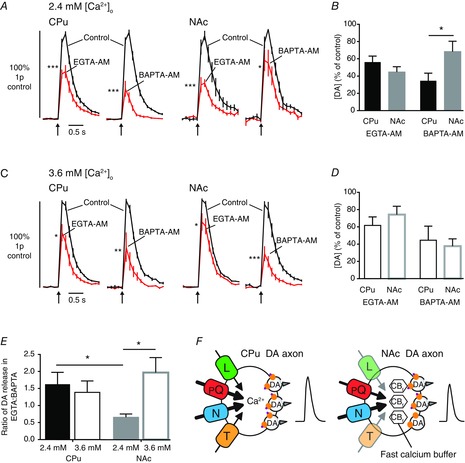
A and D, mean profiles of [DA]O ± SEM vs. time evoked by 1p for 2.4 mm [Ca2+]o (A) or 3.6 mm [Ca2+]o (C) in control conditions (black lines) and after incubation with EGTA-AM (100 μm) or BAPTA-AM (100 μm) (red lines). B and D, mean peak [DA]o ± SEM following EGTA-AM or BAPTA-AM incubation in 2.4 mm [Ca2+]o (B) or 3.6 mm [Ca2+]o (D). E, ratio of DA release in EGTA-AM:BAPTA-AM (expressed as a percentage of control ± SEM) in CPu (black) and NAc (grey) at 2.4 mm (filled) and 3.6 mm [Ca2+]o (unfilled). Bonferroni post-tests vs. control: *P < 0.05, **P < 0.01, ***P < 0.001, n = 7–10 sites per region, n = 3–4 animals. F, schematic summary depicting the relative regulation of DA transmission by different voltage-gated Ca2+ channels and Ca2+ in CPu versus NAc. Arrow weight and channel opacity indicate relative role of voltage-gated Ca2+ channels, CBf indicates an apparent additional fast Ca2+ buffer. CPu, caudate putamen; DA, dopamine; NAc, nucleus accumbens.
Population means were compared using one- or two-way ANOVA with post-hoc Bonferroni's t test where appropriate using GraphPad Prism. Curve fits and EC50 analysis were done using GraphPad Prism without constraints; EC50 values were compared using an F test. Raw control data passed Shapiro–Wilk normality test.
Results
N-type voltage-gated Ca2+ channels have a major role in dopamine transmission that is frequency-dependent
We first explored the VGCC dependence of striatal DA release, evoked electrically by single pulses (1p) and short trains of five pulses (5p) at frequencies ranging from 5 to 100 Hz, in the presence of 2.4 mm extracellular Ca2+. The nAChR antagonist DHβE (1 μm) was present throughout to eliminate the confounding effects of the VGCCs on striatal ACh. We chose electrical stimulation (with nAChR block) in preference to optogenetic activation, as the Ca2+ conductance associated with ChR2 might interfere with endogenous Ca2+-dependent processes. Peak-evoked [DA]o varied with stimulus frequency in CPu (Fig. 1A and B; one-way ANOVA, F4,26 = 62.7, P < 0.001) and NAc (Fig. 1C and D; one-way ANOVA, F4,14 = 36.3, P < 0.001) as shown previously (Rice & Cragg, 2004; Exley & Cragg, 2008; Exley et al. 2012). Single pulses evoked a mean peak [DA]o of 0.97 ± 0.10 μm in CPu and 0.67 ± 0.10 μm in NAc, in line with previous studies (Threlfell et al. 2010; Hartung et al. 2011).
Figure 1. N-type voltage-gated Ca2+ channels modulate DA release in CPu and NAc.
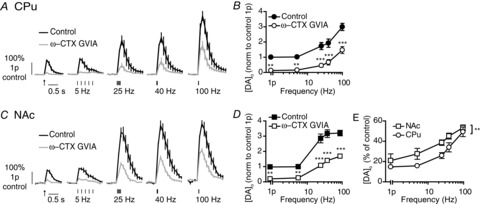
A and C, mean profiles of [DA]o ± SEM vs. time evoked by one or five pulses (5–100 Hz) in CPu (A) or NAc (C) in control or the presence of ω-CTX GVIA (100 nm). Data are normalized to 1p peak [DA]o in control conditions. Control conditions (black lines), N-block (grey lines). B and D, mean peak [DA]o ± SEM vs. frequency (5p) in CPu (B) and NAc (D) in control conditions (filled) or N-block (unfilled). Bonferroni post-test vs. control: **P < 0.01, ***P < 0.001. E, mean peak [DA]o ± SEM after voltage-gated Ca2+ channel block expressed as a percentage of control at each frequency in CPu (circles) and NAc (squares) shows variable effect of block with frequency and a greater effect in CPu than NAc. Two-way ANOVA, CPu vs. NAc: **P < 0.01, n = 3 animals. CPu, caudate putamen; ω-CTX ω-Conotoxin GVIA; DA, dopamine; NAc, nucleus accumbens.
Application of the N-type blocker ω-Conotoxin GVIA (100 nm) dramatically reduced peak [DA]o to ∼15–50% of control [DA]o in CPu (Fig. 1A and B; two-way ANOVA, effect of drug F1,20 = 121.5, P < 0.001) and NAc (Fig. 1C and D; two-way ANOVA, effect of drug F1,20 = 210.1, P < 0.001). The effect size varied inversely and significantly with stimulus frequency in CPu (Fig. 1A and B; two-way ANOVA, drug × frequency interaction F4,44 = 5.3, P < 0.01) and NAc (Fig. 1C and D; two-way ANOVA, drug × frequency interaction F4,20 = 6.6, P < 0.001). The reduction of evoked [DA]o by N-type channel blockade was greater at lower frequency stimulations. Furthermore, N-channel inhibition had a significantly greater effect in CPu than NAc (Fig. 1E; two-way ANOVA, region: F1,20 = 13.7, P < 0.001).
P/Q-type voltage-gated Ca2+ channels participate in dopamine transmission, with frequency dependence in caudate putamen
To block P-type channels we applied a low concentration of ω-Agatoxin IVA (15 nm), which had no effect on evoked [DA]o in either CPu or NAc (data not illustrated). A higher concentration of ω-Agatoxin IVA (200 nm) was subsequently used to block P + Q-type channels and significantly reduced peak evoked [DA]o in the CPu (Fig. 2A and B; two-way ANOVA, effect of drug F1,40 = 93.7, P < 0.001) and NAc (Fig. 2C and D; two-way ANOVA, effect of drug F1,40 = 24.1, P < 0.001). Evoked [DA]o was reduced by P+Q-block in CPu to ∼30–50% of control, depending inversely on stimulation frequency (Fig. 2A and B; two-way ANOVA, drug × frequency interaction F4,40 = 3.8, P < 0.05) but in NAc was only reduced to ∼70–80% of control and did not vary significantly with stimulation frequency (Fig. 2C and D; two-way ANOVA, drug × frequency interaction F4,40 = 0.9, P > 0.05). The effect of P + Q channel blockade on [DA]o was significantly greater in CPu than NAc (Fig. 2E, two-way ANOVA, region F1,40 = 29.3, P < 0.001).
Figure 2. P/Q-type voltage-gated Ca2+ channels modulate DA release in CPu and NAc.

A and C, mean profiles of [DA]o ± SEM vs. time evoked by 1 or 5 pulses (5–100 Hz) in CPu (A) or NAc (C) in control or the presence of ω-ATX IVA (200 nm). Data are normalized to 1p peak [DA]o in control conditions. Control conditions (black lines), P/Q block (grey lines). B and D, mean peak [DA]o ± SEM vs. frequency (5p) in CPu (B) and NAc (D) in control conditions (filled) or P/Q block (unfilled). Bonferroni post-tests vs. control: *P < 0.05, **P < 0.01, ***P < 0.001. E, mean peak [DA]o ± SEM after voltage-gated Ca2+ channel block expressed as percentage of control at each frequency in CPu (circles) and NAc (squares) shows variable effect of block with frequency in CPu and a greater effect in CPu than NAc. Two-way ANOVA, CPu vs. NAc: ***P < 0.001, n = 5 animals. ω-ATX IVA, ω-Agatoxin IVA; CPu, caudate putamen; DA, dopamine; NAc, nucleus accumbens.
T-type voltage-gated Ca2+ channels control dopamine release in the caudate putamen
Application of the T-type VGCC blocker NNC 55-0396 (1 μm) (Tai et al. 2011) significantly reduced peak-evoked [DA]o in CPu (Fig. 3A and B; two-way ANOVA, drug F1,20 = 28.7, P < 0.001), but not in NAc under these conditions (Fig. 3C and D; two-way ANOVA, drug F1,20 = 0.03, P > 0.05) (but see also Fig. 7). In the CPu, NNC 55-0396 reduced evoked [DA]o to 57–65% of control levels, which did not vary with frequency (Fig. 3A and B; two-way ANOVA, drug × frequency interaction F4,20 = 2.1, P > 0.05). These effects in the CPu were significantly different from those in the NAc (Fig. 3E; two-way ANOVA, region F1,20 = 37.9, P < 0.001). Higher drug concentrations (10 μm, NNC 55-0396) had no further effect in either region (not illustrated). The effect of T-block was unaffected by the presence of GABA and glutamate blockers (data not illustrated) confirming that VGCC effects were not via actions of GABA/Glu. Ni2+ or mibefradil are used commonly to block T-type channels, but their use is precluded here: we have recently identified that Ni2+ inhibits DA uptake (Brimblecombe & Cragg, 2014) while mibefradil renders electrodes insensitive to DA (Chen et al. 2006).
Figure 3. T-type voltage-gated Ca2+ channels modulate DA release in CPu but apparently not in NAc.
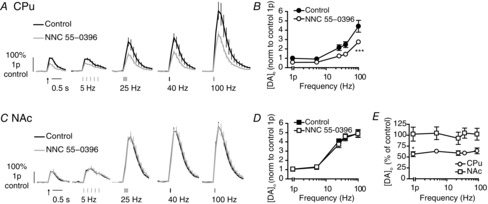
A and C, mean profiles of [DA]o ± SEM vs. time evoked by one or five pulses (5–100 Hz) in CPu (A) or NAc (C) in control or the presence of NNC 55-0396 (1 μm). Data are normalized to 1p peak [DA]o evoked in control conditions. Control conditions (black lines), NNC 55-0396 (grey lines). B and D, mean peak [DA]o ± SEM vs. frequency (5p) in CPu (B) and NAc (D) in control conditions (filled) or T-block (unfilled). Bonferroni post-test vs. control: ***P < 0.001. E, mean peak [DA]o ± SEM after voltage-gated Ca2+ channel block expressed as percentage of control at each frequency in CPu (circles) and NAc (squares) shows a greater effect in CPu than NAc. Bonferroni post-test: *P < 0.05, n = 3 animals. CPu, caudate putamen; DA, dopamine; NAc, nucleus accumbens.
L-type voltage-gated Ca2+ channels regulate dopamine release in the caudate putamen
In the CPu, blockade of L-type Ca2+ channels with isradipine 5 μm (or nifedipine 10 μm, data not shown) modestly but significantly reduced electrically evoked [DA]o to ∼75% of controls in the CPu for all stimulation frequencies (Fig. 4A and B; two-way ANOVA, F1,30 = 20.38, P < 0.001). These effects were reversible on drug washout (Fig. 4C). By contrast, in the NAc, isradipine did not modify evoked [DA]o under these conditions (Fig. 4D and E; two-way ANOVA, F1,20 = 1.18) (but see Fig. 7). These different outcomes of L-block in the CPu and NAc were statistically different (Fig. 4F; two-way ANOVA, region F1,20 = 206.7, P < 0.001). Higher isradipine concentrations (10 μm) did not unmask an effect in the NAc (not illustrated). Non-specific effects of isradipine were minimal in these experiments: The lack of effect of isradipine in the NAc suggest that any non-specific effects on other channels are negligible, and to confirm specificity, lower concentrations of isradipine (500 nm) in the CPu also significantly reduced 1p DA release, however with a protracted wash-on time (n = 3, t4 = 3.9, P < 0.05, not illustrated). The effect of the L-block was unaffected by the presence of GABA and glutamate blockers (data not illustrated) confirming that VGCC effects were not via actions of GABA/Glu.
Figure 4. L-type voltage-gated Ca2+ channel blocker modulates DA release in CPu but apparently not NAc.
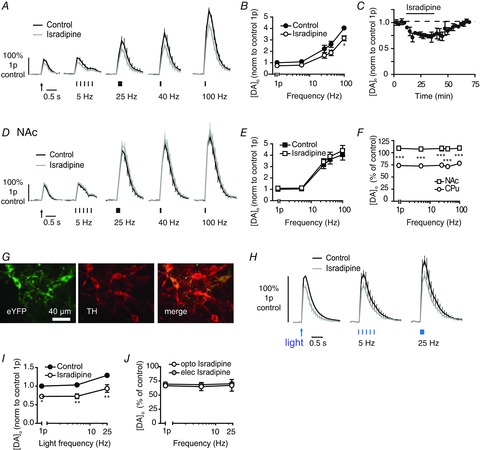
A and D, mean profiles of [DA]o ± SEM vs. time evoked by one or five electrical pulses (5–100 Hz) in CPu (A) or NAc (D) in controls or the presence of isradipine (5 μm). Data are normalized to 1p peak [DA]o evoked in control conditions. Control conditions (black lines), isradipine (grey lines). B and E, mean peak [DA]o ± SEM vs. frequency (5p) in CPu (B) and NAc (E) in control conditions (filled) or L-block (unfilled). Bonferroni post-test vs. control: *P < 0.05. C, mean peak [DA]o ± SEM evoked by a single pulse during wash-on (black bar) and wash-off of isradipine (5 μm). F, mean peak [DA]o ± SEM after voltage-gated Ca2+ channel block expressed as percentage of control at each frequency in CPu (circles) and NAc (squares) shows a significantly greater effect in CPu than NAc. Bonferroni post-test, CPu vs. NAc: ***P < 0.001, n = 3–4 animals. G, fluorescence images in substantia nigra pars compacta of ChR2-eYFP (left), anti-TH (middle) and merged images (right), showing expression of ChR2-eYFP restricted to TH-positive cells. Scale bar, 40 μm. H, mean profiles of [DA]o ± SEM vs. time evoked by one or five blue light pulses (5–25 Hz) in CPu of ChR2-expressing DAT-Cre mice in control and presence of isradipine (5 μm). Data are normalized to 1p peak [DA]o evoked in control conditions. Control conditions (black lines), isradipine (grey lines). I, mean peak [DA]o ± SEM vs. frequency (5p) of blue light pulses in CPu of ChR2-expressing DAT-Cre mice, in control conditions (filled) or L-block (unfilled). Bonferroni post-test vs. control: *P < 0.05, **P < 0.01, n = 3 animals. J, mean peak [DA]o ± SEM after L-block expressed as percentage of control at each frequency in CPu after electrical (filled) or optogenetic stimuli (unfilled). CPu, caudate putamen; DA, dopamine; NAc, nucleus accumbens.
To confirm that the L-type channels that regulate DA release in the CPu are located to DA axons rather than to other neuromodulatory inputs driven by the electrical field stimulus, we used an optogenetic approach to drive DA axons selectively. ChR2-eYFP was expressed by DA axons after injection of a Cre-inducible floxed construct into the SNc of DAT-Cre mice (Fig. 4G), as previously described (Threlfell et al. 2012). In drug-free control conditions, pulses of blue light (2 ms, one or five pulses at 5 or 25 Hz) evoked [DA]o of ∼0.75–1.5 μm, which varied slightly with frequency (Fig. 4H and I, effect of frequency, P < 0.01) as shown previously (Threlfell et al. 2012). Light-evoked [DA]o was not modulated by tonic ACh as the nAChR antagonist DHβE (1 μm) had no effect (data not illustrated). Isradipine (5 μm) significantly reduced light-evoked [DA]o to ∼75% of control, invariantly with stimulus frequency (Fig. 4H and I; two-way ANOVA effect of drug, F1,6 = 23.6, P < 0.01) as seen for electrical stimuli (Fig. 4I). These data corroborate a role in DA release for L-type VGCCs located on DA axons.
Do voltage-gated Ca2+ channel subtypes operate preferentially at specific frequencies?
Previous work suggested that the role of particular types of VGCCs in DA transmission might by differently recruited with frequency (Phillips & Stamford, 2000) and differently tune DA transmission during phasic or tonic modes of DA neuron activity. That study, however, did not account for the effects of the VGCC blockers on ACh release, which acts at nAChRs to modify DA release in a frequency-dependent manner (Zhou et al. 2001; Rice & Cragg, 2004; Zhang & Sulzer, 2004; Threlfell et al. 2012). Our study excluded the effects of ACh by including nAChR antagonists, and none the less, detected variation in the effects of some VGCC blockers with stimulus frequency (e.g. N- and P/Q-blockers; Figs 1 and 2): The dynamic role of P/Q channels was significant in the CPu but not NAc, indicating it was not simply an intrinsic property of these channels, e.g. time constants for activation/inactivation. This dynamic property was detectable only for blockers that caused a large (>50%) suppression of DA release evoked by single pulses. At many synapse types, reductions in initial release probability is expected to relieve short-term depression of release and, at sufficiently short interpulse intervals, enhance short-term facilitation (STF) (Katz & Miledi, 1968; Schneggenburger & Neher, 2005). We hypothesized that the apparent frequency-specific functions of some VGCCs might then arise from the inverse relationship between initial DA release probability (PR) and its dynamic STP during trains, that depends on Ca2+ availability and interpulse intervals at DA synapses (Cragg, 2003; Rice & Cragg, 2004) as at non-DA synapses.
To explore whether frequency-specific effects of VGCC blockers were due to classical Ca2+-dependent changes to initial PR and STP, we tested the following three predictions that: (i) activity-specific outcomes of specific VGCC blockers should be mimicked by a sufficient reduction in extracellular Ca2+; (ii) there should be an overall inverse relationship across the data sets as a whole between STP and initial PR; and therefore (iii) a channel that does not ordinarily show activity-specific modulation of DA release should do so if PR is already sufficiently low to have modified STP. We explored each prediction in turn.
First, we compared the effect of N-channel blockade to that of a decrease in extracellular Ca2+. We lowered extracellular Ca2+ from 2.4 mm to 1.0 mm to reduce peak [DA]o to levels comparable with that seen previously with the N-block (∼15–30% of control). Lowered [Ca2+]o reduced peak [DA]o for all stimuli and, like the effect of N-block, did so in a manner that varied inversely with frequency in the CPu (Fig. 5A and B; two-way ANOVA, effect of treatment F1,20 = 1.16, P > 0.05; effect of frequency F4,20 = 10.2, P < 0.001) or NAc (Fig. 5C and D; two-way ANOVA, effect of treatment F1,20 = 3.75, P > 0.05; effect of frequency F4,20 = 7.12, P < 0.01).
Figure 5. The activity dependence of voltage-gated Ca2+ channels is due to Ca2+-dependent changes to release probability and plasticity.
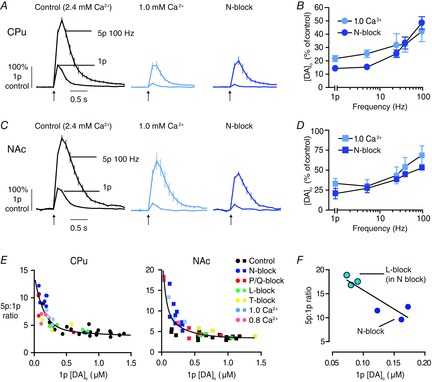
A and C, mean profiles of [DA]o ± SEM vs. time evoked by one or five pulses at 100 Hz in CPu (A) or NAc (C) in control conditions (2.4 mm Ca2+, left), 1.0 mm Ca2+ (middle), compared to N-block (right, data from Fig. 1). Data are normalized to 1p peak [DA]o in control conditions. B and D, mean peak [DA]o ± SEM in low Ca2+ (pale) or N-block (dark) expressed as percentage of control at each frequency in CPu (B) and NAc (D), show effects of low Ca2+ varies with frequency like N-block: CPu, two-way ANOVA effect of treatment: F1,20 = 1.16, P > 0.05; effect of frequency: F4,20 = 10.2, P < 0.001; NAc, two-way ANOVA effect of treatment: F1,20 = 3.75, P > 0.05; effect of frequency: F4,20 = 7.12, P < 0.01. E, plots of 5p:1p ratio vs. 1p peak [DA]o for CPu (left) and NAc (right), for individual means for each experiment with each voltage-gated Ca2+ channel blocker and for 1.0 [Ca2+]o and 0.8 mm [Ca2+]o. Control is 2.4 mm [Ca2+]o. Curve fits, two phase exponential decays, CPu R2 = 0.73, NAc R2 = 0.76. F, plot of 5p:1p ratio vs. 1p peak [DA]o for CPu, for individual means for experiments with L-type blocker (green) applied in the presence of N-block (blue), showing changes to activity dependence after L-block not seen for L-block alone (see panel E) and indicating that changes to activity dependence of DA release are dependent upon previous changes to DA PR (and are not absolute effects of any one channel). Approximation to linear regression over this range, R2 = 0.79, P < 0.05. n = 3 animals. CPu, caudate putamen; DA, dopamine; NAc, nucleus accumbens.
Secondly, we explored whether for the data set as a whole, there was a relationship between release evoked by 1p and its STP. We determined the ratio of [DA]o evoked by a burst (5p at 100 Hz) compared to a single pulse (the 5p:1p ratio) as a measure of STP, and plotted this ratio against [DA]o evoked by 1p, for all data sets combined that used either VGCC blockers or a range of [Ca2+]o (2.4 mm, 1.0 mm and 0.8 mm) (Fig. 5E). These plots show that where 1p [DA]o was high, for example, in 2.4 mm Ca2+ controls, or was only weakly reduced (L- or T-block in CPu, or P/Q block in NAc), the 5p:1p ratio remained at ≤5, i.e. showed some short-term depression because release sublinearly reflected the number of pulses. As 1p [DA]o became more strongly reduced (by low [Ca2+]o, N-block or P/Q block in CPu), the 5p:1p ratios became correspondingly greater, such that when 1p [DA]o release was reduced to the lowest quartile of release levels, there was STF, i.e. 5p:1p ratio >5. Furthermore, these data across conditions could be described by a continuous inverse relationship between 5p:1p ratio and 1p [DA]o in the CPu (Fig. 5E, left: two-phase exponential curves, CPu, R2 = 0.74) and in NAc (Fig. 5E, right: two-phase exponential curve, NAc, R2 = 0.76). These findings support the hypothesis that the apparent frequency specificity of some VGCCs results from the relationships between Ca2+ entry, DA PR and Ca2+-limited STF.
Thirdly, we tested whether the L-type channel blocker isradipine, that modified DA release in the CPu in a similar manner across frequencies (no change in 5p:1p ratio; see Figs 4 and 5E), could be made to become activity-specific. We explored whether the previous reduction of DA PR with the N-blocker, to a point where any further reductions in PR should dramatically increase 5p:1p ratio, would result in isradipine increasing the 5p:1p ratio. Indeed, in the CPu, in the presence of the N-type blocker (ω-Conotoxin GIVA 100 nm), when 1p [DA]o was low and the 5p:1p ratio was >10 (Fig. 5E and F), the subsequent application of the L-blocker isradipine (5 μm) further decreased [DA]o evoked by 1p (by ∼50%) but did not reduce [DA]o evoked by 5p/100 Hz, resulting in a significant increase in 5p:1p ratio to ∼17 (Fig. 5F; linear regression R2 = 0.79, P < 0.05). Therefore, we show here that the dynamic roles for subtypes of VGCCs in gating DA release are due to classical relationships between release probability and its STP.
Different voltage-gated Ca2+ channel roles in the caudate putamen and nucleus accumbens are due to different Ca2+ dependence
Across experiments, we noted that VGCC blockers caused more pronounced reductions in DA release in the CPu than the NAc. To date, there is no evidence that expression levels of VGCC subtypes differ correspondingly between DA neurons. To understand why the control of DA release was different, we explored whether DA release in the NAc was less sensitive than in the CPu to entry of extracellular Ca2+. Ca2+ entry was required for DA release in both regions: brief application of zero extracellular Ca2+ solution (with 100 μm EGTA) abolished DA release in the NAc or CPu to below detection levels, for trains and single pulses (Fig. 6A). However, by varying [Ca2+]o we identified that 1p [DA]o varied with a steeper concentration–response in the CPu, with a lower EC50 (CPu: 1.9 mm; NAc 2.9 mm) and larger Hill coefficient (CPu: 3.7 ± 0.7; NAc: 2.0 ± 0.9) than the NAc (Fig. 6B; R2: CPu = 0.93, NAc = 0.87; comparison of fits: F2,76 = 3.2, P < 0.05). These responses suggest there could be either greater co-operativity or overlap of Ca2+ domains (Schneggenburger & Neher, 2005; Schneggenburger et al. 2012), or enhanced Ca2+ entry or a reduced Ca2+ buffering capacity in the CPu. They confirm also that DA release is more sensitive to reductions in [Ca2+]o below 2.4 mm in the CPu than NAc (see Fig. 6B), consistent with the greater effects on DA release in CPu of limiting Ca2+ entry with a wider range of VGCC blockers (Figs 4). We followed up two key implications of these observations.
Figure 6. CPu and NAc have different Ca2+ sensitivity.
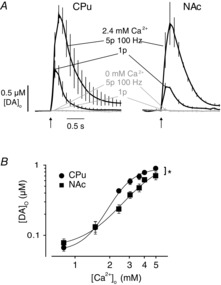
A, mean profiles of [DA]o ± SEM vs. time evoked by 1p in CPu (left) and NAc (right) in control (black) and in zero [Ca2+]o (with EGTA 100 μm) (grey), which abolishes DA release showing DA transmission is fully dependent on extracellular Ca2+. B, mean peak [DA]o ± SEM evoked by 1p, vs. [Ca2+]o. Evoked [DA]o varies with [Ca2+]o in both regions, but differently in CPu (circles) and NAc (squares). Variable slope sigmoidal curves, CPu: EC50 1.9 mm, 95% confidence limits 1.7–2.1, Hill coefficient 3.7 ± 0.7; NAc: EC50 2.9 mm, 95% confidence limits 1.6–5.1, Hill coefficient 2.0 ± 0.9. Comparison of fits test: F2,76 = 4.8, *P < 0.05, n = 6 animals. CPu, caudate putamen; DA, dopamine; NAc, nucleus accumbens.
‘Silent’ voltage-gated Ca2+ channels can be unmasked with enhanced Ca2+ availability
The steepest point on the Ca2+ response curves will be the point when changes to the number of open Ca2+ channels should be most effective in modulating transmitter release. In NAc, the steepest point is at a [Ca2+]o greater than in CPu and greater than the [Ca2+]o we used in experiments with VGCC blockers (2.4 mm Ca2+). Therefore, the lack of evident roles for T- and L-VGCCs in NAc might be due to the lesser responsiveness of DA transmission to open Ca2+ channels than in the CPu, rather than channel absence. We therefore investigated whether roles for otherwise ‘silent’ L- and T-VGCCs in the NAc could be unmasked at higher [Ca2+]o, when evoked [DA]o should be more sensitive to any changes in Ca2+ availability brought about by channel block.
We obtained proof of principle that higher [Ca2+]o could enhance the effects of channel block, by testing the effect on P/Q channel inhibition, in the NAc. At 3.6 mm [Ca2+]o, ω-Agatoxin IVA (200 nm) significantly decreased evoked [DA]o (Fig. 7A and B) and to a greater extent than at 2.4 mm [Ca2+]o (Fig. 7C; two-way ANOVA, effect of [Ca2+]o F1,40 = 19.9, P < 0.001). We then investigated the effect of the L- and T-blockers in 3.6 mm [Ca2+]o. Isradipine slightly but significantly decreased evoked [DA]o in a frequency-independent manner (Fig. 7D–F). This effect was significantly greater than in 2.4 mm [Ca2+]o (Fig. 7F; two-way ANOVA, effect of [Ca2+]o F1,50 = 41.4, P < 0.001). Similarly, an effect of T-channels was revealed at 3.6 mm [Ca2+]o: NNC 55-0396 slightly but significantly decreased evoked [DA]o (Fig. 7G–I). The effect of NNC 55-0396 on DA release was significantly greater than in 2.4 mm [Ca2+]o (Fig. 7I; two-way ANOVA, effect of [Ca2+]o F1,40 = 1.44, P < 0.05). Therefore, the greater roles for T- and L-VGCCs in the CPu versus NAc is not due to an absence of these VGCCs at DA release sites in NAc terminals. These channels are present and can operate in the CPu and the NAc, if/when appropriate local Ca2+ conditions are met. These data also corroborate the hypothesis that local handling of Ca2+ varies between the CPu and NAc.
Endogenous buffering of how Ca2+ couples to dopamine release differs in the caudate putamen and nucleus accumbens
The greater Ca2+ sensitivity of DA transmission in the CPu compared to NAc, and greater roles for more VGCC types in the CPu, might arise from underlying mechanisms that could include enhanced co-operativity or ‘domain overlap’ of the intracellular Ca2+ micro-/nanodomains of VGCCs, enhanced sensitivity of the exocytotic machinery and/or reduced intracellular buffering, which might govern spatial and temporal Ca2+ summation (Blatow et al. 2003; Zhao et al. 2011; Eggermann et al. 2012; Hoppa et al. 2012; Pan & Ryan, 2012; Schneggenburger et al. 2012). The SNc and VTA DA neurons differ in expression of endogenous Ca2+ buffers. Whereas the SNc DA neurons express higher levels of the buffer parvalbumin (Greene et al. 2005), a slow buffer (Schwaller et al. 2002), VTA DA neurons express higher levels of the buffer calbindin-D28k (Haber et al. 1995; Greene et al. 2005; Chung et al. 2005), a fast, high-affinity buffer (Nägerl et al. 2000; Schwaller et al. 2002; Eggermann et al. 2012). To explore if differences in Ca2+ dependence of DA transmission in the CPu versus NAc were due to detectable differences in Ca2+ handling we used the cell-permeable exogenous Ca2+ chelators EGTA-AM (100 μm) and BAPTA-AM (100 μm), to identify the roles of slow and fast Ca2+ transients respectively. These exogenous buffers have comparable affinity values but differ by a factor of ∼40 in their buffering rates (reviewed by Eggermann et al. 2012). In 2.4 mm [Ca2+]o, following brief incubation with EGTA-AM, evoked [DA]o was decreased to a similar extent in CPu and NAc (Fig. 8A and B) indicating a role for slow/remote coupling between Ca2+ source and sensor in both axon types. However, following brief incubation with BAPTA-AM, evoked [DA]o was decreased to a greater extent in the CPu than NAc, indicating greater coupling between fast/local Ca2+ transients and DA release in the CPu (Fig. 8B; Bonferroni post-test BAPTA P < 0.05).
To explore whether the lesser effect of BAPTA-AM in the NAc results from buffering by fast endogenous buffers in the NAc, we increased [Ca2+]o from 2.4 mm to 3.6 mm to saturate and overwhelm endogenous buffers, which should be evident through a preferential increase in effect of BAPTA-AM compared to EGTA-AM. First in the CPu, at 3.6 mm [Ca2+]o, EGTA-AM and BAPTA-AM decreased [DA]o (Fig. 8C and D) but the ratio of [DA]o remaining in the presence of EGTA-AM versus BAPTA-AM was unchanged compared to that seen at 2.4 mm [Ca2+]o (Fig. 8E; CPu post-test P > 0.05). By contrast, in the NAc at 3.6 mm [Ca2+]o, there was a markedly enhanced effect of BAPTA-AM (Fig. 8C and D), and a resulting significant increase in the ratio of [DA]o remaining in the presence of EGTA-AM vs. BAPTA-AM indicating an increase in the relative role for fast BAPTA-buffered transients, and suggesting an underlying role for endogenous fast buffers (Fig. 8F). This ratio at 3.6 mm [Ca2+]o did not then differ between the NAc and CPu (Fig. 8E; post-test NAc, 2.4 vs. 3.6 mm, P < 0.05; post-test 3.6 mm NAc vs. CPu, P > 0.05). The different dominance of VGCC subtypes and apparent greater role for fast endogenous buffer in the NAc versus CPu is illustrated in the schematic in Fig. 8F.
Discussion
Here, we reveal the VGCCs on mesostriatal DA axons that regulate DA transmission and the principles that govern their dynamic roles, revealed when we exclude the obscuring effects of VGCCs on ChIs. We identify major underlying differences in the intracellular Ca2+ dynamics that couple to DA transmission in the CPu versus NAc.
Multiple voltage-gated Ca2+ channel subtypes regulate dopamine transmission in the striatum
It is generally assumed that presynaptic Ca2+ influx required for synaptic neurotransmission occurs typically via the N-subtypes (Cav2.2) and P/Q-subtypes (Cav2.1) of the VGCCs, which, through synprint motifs, associate closely with SNARE proteins (Kim & Catterall, 1997; Vance et al. 1999; Zamponi, 2003). Properties defined at classical synapses cannot be assumed to apply universally across all synapse types, particularly for DA release sites, which can differ from classic models of point-to-point synapses in structural, junctional and functional aspects (Cragg & Rice, 2004; Arbuthnott & Wickens, 2007; Rice et al. 2011). None the less, we found major roles for N/P/Q-channels in the control of striatal DA release. The efficacy of high but not low concentrations of ω-Agatoxin IVA suggests that the P/Q component arises from Q-channels. We show also, however, that T- and L-type VGCCs participate in DA release, more readily in the CPu than NAc.
T- and L-type VGCCs are expressed by DA neurons (Takada et al. 2001; Wolfart & Roeper, 2002; Dryanovski et al. 2013) and our findings are consistent with accumulating evidence in other neuron types that L- and T-channels can regulate exocytosis. L-type VGCCs are the primary VGCC involved in neurotransmitter release from ribbon-type active zones of primary sensory neurons (Brandt et al. 2003; Zanazzi & Matthews, 2009) and they operate alongside N/P/Q-VGCCs within central areas that include the hippocampus (Murakami et al. 2002; Tippens et al. 2008) and supraoptic nucleus (Bhaukaurally et al. 2005). They regulate presynaptic plasticity in the amygdala and primary cortical neurons (Fourcaudot et al. 2009; Subramanian & Morozov, 2011). Their roles in action potential-dependent transmission are probably underestimated, however: neuronal L-type channels can be as fast and as efficient as the N-type channels at supporting calcium entry during action potential-like stimuli but they are inhibited only slowly by the dihydropyridine blockers usually applied to probe their functions (Helton et al. 2005). Previous studies of DA transmission or intracellular Ca2+ that did not resolve the L-channel activity on DA axons (Phillips & Stamford, 2000; Chen et al. 2006; Sgobio et al. 2014) will have been confounded by the overriding effects of co-stimulating the ChI input to DA axons, and by the limited ability to resolve small changes in Ca2+ that none the less translate to detectable changes in neurotransmission through a steep power relationship.
T-type VGCCs are typically thought to activate in response to subthreshold membrane depolarizations and regulate membrane excitability, oscillatory activity and rebound firing. Mounting evidence suggests T-channels also participate in vesicular exocytosis from various neurons (Pan et al. 2001; Carbone et al. 2006; Tang et al. 2011; Weiss et al. 2012) and that like the classic ‘high-voltage-activated’ N/P/Q-channels, T-type channels also directly interact with synaptic proteins (syntaxin-1A and SNAP-25) (Weiss et al. 2012).
Dynamic changes in PR and short-term plasticity underlie frequency-specific roles of voltage-gated Ca2+ channels
We showed that the VGCCs with greatest contributions to DA release had roles that depended inversely on frequency, suggesting that the VGCCs have a greater role in gating DA release by single or lower frequency activity than during phasic-like higher frequencies. We showed that this frequency-dependent role of a given VGCC could be explained, and predicted, by a classical underlying inverse relationship between initial DA PR and subsequent STF that depends on initial Ca2+ entry. The greater the inhibition of release for a single pulse by a VGCC blocker, the greater the relief from short-term depression (STD) that limits re-release (Katz & Miledi, 1968; Cragg, 2003) and the greater the STF (5p:1p ratio >5). The effect of N-block on the DA varied more strongly with frequency than other VGCCs because the N-block caused the greatest suppression of initial DA PR and led to STF at sufficiently high frequencies. Blockers for channels that only slightly reduced DA PR (L/T-type channels) did not relieve the STD sufficiently to modify frequency dependence, but could do so if applied in combination, when the cumulative reduction in PR was sufficient to promote the STF. These data therefore indicate that VGCCs contribute to low-pass filtering of DA release and that their role is dynamic. Rather than having a fixed contribution to transmission, their roles are probably therefore to vary depending on the local driving forces that modulate PR, including recent history of synapse use.
Different voltage-gated Ca2+ channel function in the caudate putamen and nucleus accumbens is due to different Ca2+ dependence and handling
Differences in VGCC function between the NAc and CPu were observed that could not readily be attributed to variable VGCC expression or levels. Expression levels of the 10 potential α1 subunit genes that define VGCC subtype show little relevant difference between DA neurons (Grimm et al. 2004; Greene et al. 2005; Chung et al. 2005; Greene, 2006; Dryanovski et al. 2013). While one study reported that Cav3.1 (T-type) expression in SNc neurons is 1.2-fold that in VTA neurons (Greene et al. 2005), another indicated Cav3.3 (T-type) expression in VTA is twice that in the SNc (Grimm et al. 2004).
We showed that the different VGCC dependence of DA transmission in the CPu and NAc could be attributed to different underlying Ca2+ sensitivity. The Ca2+ concentration–response curve in CPu is leftward-shifted and steeper (higher co-operativity/Hill slope). Different Ca2+ dependence of DA transmission in CPu and NAc prompted us to explore whether otherwise ‘silent’ L- and T-VGCCs in NAc could be unmasked at higher [Ca2+]o when the greater sensitivity to [Ca2+]o makes small changes in the number of open Ca2+ channels more effective yet in modulating transmitter release. These data reveal that L- and T-type VGCCs are not restricted to DA axons in the CPu but can be functional in NAc axons, under appropriate conditions. These conditions might be met physiologically in vivo, and dynamically. Such conditions might arise in vivo after previous synaptic activity or neuromodulatory inputs that promote Ca2+ entry and shift DA release upwards on its Ca2+ response curve, or through signalling pathways that modify phosphorylation states and conductance of the channels or their coupling to auxiliary subunits.
We explored whether differences between CPu and NAc were due to differences in how Ca2+ dynamics couple to DA release, by exploring the effects of cell-permeable exogenous Ca2+ chelators to chelate slow or fast Ca2+ transients, using EGTA-AM or BAPTA-AM respectively. Our findings reveal critical differences in endogenous Ca2+ buffering or signalling mechanisms. At 2.4 mm [Ca2+]o, DA transmission in CPu and NAc was dependent on slow EGTA-sensitive Ca2+ dynamics, suggesting that Ca2+ source and Ca2+ sensor can be remotely coupled within both axon types (Eggerman et al. 2012). Furthermore, DA transmission was coupled to an additional fast/local BAPTA-sensitive Ca2+ source, more tightly in the CPu than in the NAc. However, when we provided more Ca2+, we enhanced the dependence of DA release on faster sources compared to slower Ca2+ ones in the NAc but not the CPu. These data suggest that different Ca2+-dependent mechanisms operate in the NAc versus CPu, and that a fast mechanism normally limits the coupling of Ca2+ entry to DA exocytosis in the NAc that can be exposed by overwhelming it with higher Ca2+. One explanation is that an additional fast endogenous buffer normally operates in NAc, and becomes overwhelmed by higher Ca2+. Calbindin-D28k, a fast, high-affinity and mobile buffer expressed by DA axons in the NAc but not CPu, is a possible candidate additional fast buffer. Calbindin is expressed in VTA DA neurons at ∼2–3-fold higher levels than in the SNc (Haber et al. 1995; Greene et al. 2005; Chung et al. 2005). It can regulate Ca2+-dependent exocytosis from dissociated DA neurons (Pan & Ryan, 2012), and participate in spatial buffering in hippocampal neurons (Blatow et al. 2003; Müller et al. 2005), and because of its high Ca2+ affinity, the Ca2+-binding sites of calbindin-D28k are expected to become saturated after a sufficiently large Ca2+ influx (Neher, 1998) in keeping with our experimental observations here. In summary, these data provide the first evidence that the spatiotemporal handling of Ca2+ sources that couple to DA transmission differs significantly in DA axons in the CPu than in the NAc, and in particular, that buffering of fast Ca2+ sources may be more limited in the CPu.
Ca2+ dependence is not the only feature in which DA transmission from these two axonal populations differs. DA transmission in the CPu and NAc differs in release probability, STP and regulation by presynaptic neuromodulatory receptors (e.g. nAChRs, opioid receptors), synucleins and DA uptake (Cragg et al. 2000; Cragg, 2003; Britt & McGehee, 2008; Exley & Cragg, 2008; Anwar et al. 2011; Exley et al. 2012). We now show that this regional control of DA transmission extends to Ca2+ coupling, which may even account for some of these properties.
Summary and further implications
In summary, we reveal key principles that underscore the regulation of striatal DA transmission by Ca2+ and VGCCs. We show that multiple VGCC types in DA axons regulate DA release: N- and (P)Q-types dominate but L- and T-channels also participate. However, whether a given VGCC contributes to DA release is not fixed, but varies dynamically depending on neuron activity and factors that govern local Ca2+ and DA PR. VGCC roles appear to vary with frequency because they contribute to low-pass filtering of DA release through a classic relationship between DA PR and its STP. A wider repertoire of VGCC types more readily regulate DA transmission in the CPu than in NAc due to an underlying greater Ca2+ sensitivity in the CPu. Furthermore, we reveal the striking finding that these different axon types have different a spatiotemporal coupling of Ca2+ dynamics to DA exocytosis, with endogenous fast Ca2+ buffers apparently limiting DA transmission in the NAc more than in the CPu. These findings reveal that different Ca2+-handling mechanisms control DA function in limbic- versus motor-associated striatum.
As the axonal fields of DA neurons form the vast majority (∼99%) of the total membrane area of a DA neuron (Matsuda et al. 2009; Henny et al. 2012), their Ca2+ handling might contribute to the differential susceptibility of different populations of DA neurons to degeneration in Parkinson's disease. L-channel antagonists are currently in clinical trial as neuroprotective therapies for Parkinson's disease (Simuni et al. 2010; Parkinson Study Group, 2013), and their actions on axonal L-channels might therefore contribute to neuroprotection but also modulate DA transmission.
Glossary
Abbreviations
- ChI
cholinergic interneuron
- ChR2
channelrhodopsin 2
- CPu
caudate putamen
- Cre
Cre recombinase
- DA
dopamine
- DAT
dopamine transporter
- DHβE
dihydro-β-erythroidine hydrobromide
- FCV
fast-scan cyclic voltammetry
- NAc
nucleus accumbens
- nAChR
nicotinic acetylcholine receptor
- PR
release probability
- SNc
substantia nigra pars compacta
- STD
short-term depression
- STF
short-term facilitation
- STP
short-term plasticity
- VGCC
voltage-gated Ca2+ channel
- VTA
ventral tegmental area
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
K.B. performed experiments and their design, data analysis and co-wrote the paper; K.G. and N.P. performed experiments and data analysis; and S.C. designed experiments and co-wrote the paper. All authors approved the final version for publication.
Funding
Parkinson's UK (H-1003), and the Medical Research Council (MR/K013866/1).
References
- Anwar S, Peters O, Millership S, Ninkina N, Doig N, Connor-Robson N, Threlfell S, Kooner G, Deacon RM, Bannerman DM, Bolam JP, Chandra SS, Cragg SJ, Wade-Martins R. Buchman VL. Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J Neurosci. 2011;31:7264–7274. doi: 10.1523/JNEUROSCI.6194-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbuthnott GW. Wickens J. Space, time and dopamine. Trends Neurosci. 2007;30:62–69. doi: 10.1016/j.tins.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Bhaukaurally K, Panatier A, Poulain DA. Oliet SHR. Voltage-gated Ca2+ channel subtypes mediating GABAergic transmission in the rat supraoptic nucleus. Eur J Neurosci. 2005;21:2459–2466. doi: 10.1111/j.1460-9568.2005.04097.x. [DOI] [PubMed] [Google Scholar]
- Blatow M, Caputi A, Burnashev N, Monyer H. Rozov A. Ca2+ buffer saturation underlies paired pulse facilitation in calbindin-D28k-containing terminals. Neuron. 2003;38:79–88. doi: 10.1016/s0896-6273(03)00196-x. [DOI] [PubMed] [Google Scholar]
- Brandt A, Striessnig J. Moser T. CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J Neurosci. 2003;23:10832–10840. doi: 10.1523/JNEUROSCI.23-34-10832.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimblecombe KR. Cragg SJ. Ni2+ affects dopamine uptake which limits suitability as inhibitor of T-type voltage-gated Ca2+ channels. ACS Chem Neurosci. 2014 doi: 10.1021/cn500274g. DOI: 10.1021/cn500274g. [DOI] [PubMed] [Google Scholar]
- Britt JP. McGehee DS. Presynaptic opioid and nicotinic receptor modulation of dopamine overflow in the nucleus accumbens. J Neurosci. 2008;28:1672–1681. doi: 10.1523/JNEUROSCI.4275-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone E, Giancippoli A, Marcantoni A, Guido D. Carabelli V. A new role for T-type channels in fast ‘low-threshold’ exocytosis. Cell Calcium. 2006;40:147–154. doi: 10.1016/j.ceca.2006.04.019. [DOI] [PubMed] [Google Scholar]
- Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE. Surmeier DJ. ‘Rejuvenation’ protects neurons in mouse models of Parkinson's disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- Chen BT, Moran KA, Avshalumov MV. Rice ME. Limited regulation of somatodendritic dopamine release by voltage-sensitive Ca2+ channels contrasted with strong regulation of axonal dopamine release. J Neurochem. 2006;96:645–655. doi: 10.1111/j.1471-4159.2005.03519.x. [DOI] [PubMed] [Google Scholar]
- Chung CY, Seo H, Sonntag KC, Brooks A, Lin L. Isacson O. Cell type-specific gene expression of midbrain dopaminergic neurons reveals molecules involved in their vulnerability and protection. Hum Mol Genet. 2005;14:1709–1725. doi: 10.1093/hmg/ddi178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier TJ, Kanaan NM. Kordower JH. Ageing as a primary risk factor for Parkinson's disease: evidence from studies of non-human primates. Nat Rev Neurosci. 2011;12:359–366. doi: 10.1038/nrn3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg SJ. Variable dopamine release probability and short-term plasticity between functional domains of the primate striatum. J Neurosci. 2003;23:4378–4385. doi: 10.1523/JNEUROSCI.23-10-04378.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg SJ. Meaningful silences: how dopamine listens to the ACh pause. Trends Neurosci. 2006;29:125–131. doi: 10.1016/j.tins.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Cragg SJ, Hille CJ. Greenfield SA. Dopamine release and uptake dynamics within nonhuman primate striatum in vitro. J Neurosci. 2000;20:8209–8217. doi: 10.1523/JNEUROSCI.20-21-08209.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg SJ. Rice ME. DAncing past the DAT at a DA synapse. Trends Neurosci. 2004;27:270–277. doi: 10.1016/j.tins.2004.03.011. [DOI] [PubMed] [Google Scholar]
- Dryanovski DI, Guzman JN, Xie Z, Galteri DJ, Volpicelli-Daley LA, Lee VM-Y, Miller RJ, Schumacker PT. Surmeier DJ. Calcium entry and -synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J Neurosci. 2013;33:10154–10164. doi: 10.1523/JNEUROSCI.5311-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Bucurenciu I, Goswami SP. Jonas P. Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nat Rev Neurosci. 2012;13:7–21. doi: 10.1038/nrn3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exley R, Clements MA, Hartung H, McIntosh JM, Franklin M, Bermudez I. Cragg SJ. Striatal dopamine transmission is reduced after chronic nicotine with a decrease in α6-nicotinic receptor control in nucleus accumbens. Eur J Neurosci. 2013;38:3036–3043. doi: 10.1111/ejn.12298. [DOI] [PubMed] [Google Scholar]
- Exley R. Cragg SJ. Presynaptic nicotinic receptors: a dynamic and diverse cholinergic filter of striatal dopamine neurotransmission. Br J Pharmacol. 2008;153(Suppl):S283–S297. doi: 10.1038/sj.bjp.0707510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exley R, McIntosh JM, Marks MJ, Maskos U. Cragg SJ. Striatal α5 nicotinic receptor subunit regulates dopamine transmission in dorsal striatum. J Neurosci. 2012;32:2352–2356. doi: 10.1523/JNEUROSCI.4985-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourcaudot E, Gambino F, Casassus G, Poulain B, Humeau Y. Lüthi A. L-type voltage-dependent Ca2+ channels mediate expression of presynaptic LTP in amygdala. Nat Neurosci. 2009;12:1093–1095. doi: 10.1038/nn.2378. [DOI] [PubMed] [Google Scholar]
- Greene JG. Gene expression profiles of brain dopamine neurons and relevance to neuropsychiatric disease. J Physiol. 2006;575:411–416. doi: 10.1113/jphysiol.2006.112599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JG, Dingledine R. Greenamyre JT. Gene expression profiling of rat midbrain dopamine neurons: implications for selective vulnerability in parkinsonism. Neurobiol Dis. 2005;18:19–31. doi: 10.1016/j.nbd.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Grimm J, Mueller A, Hefti F. Rosenthal A. Molecular basis for catecholaminergic neuron diversity. Proc Natl Acad Sci U S A. 2004;101:13891–13896. doi: 10.1073/pnas.0405340101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sánchez-Padilla J, Chan CS. Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci. 2009;29:11011–11019. doi: 10.1523/JNEUROSCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN, Ryoo H, Cox C. Lu W. Subsets of midbrain dopaminergic neurons in monkeys are distinguished by different levels of mRNA for the dopamine transporter: comparison with the mRNA for the D2 receptor, tyrosine hydroxylase and calbindin immunoreactivity. J Comp Neurol. 1995;362:400–410. doi: 10.1002/cne.903620308. [DOI] [PubMed] [Google Scholar]
- Harris JJ, Jolivet R. Attwell D. Synaptic energy use and supply. Neuron. 2012;75:762–777. doi: 10.1016/j.neuron.2012.08.019. [DOI] [PubMed] [Google Scholar]
- Hartung H, Threlfell S. Cragg SJ. Nitric oxide donors enhance the frequency dependence of dopamine release in nucleus accumbens. Neuropsychopharmacology. 2011;36:1811–1822. doi: 10.1038/npp.2011.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helton TD, Xu W. Lipscombe D. Neuronal L-type calcium channels open quickly and are inhibited slowly. J Neurosci. 2005;25:10247–10251. doi: 10.1523/JNEUROSCI.1089-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henny P, Brown MTC, Northrop A, Faunes M, Ungless MA, Magill PJ. Bolam JP. Structural correlates of heterogeneous in vivo activity of midbrain dopaminergic neurons. Nat Neurosci. 2012;15:613–619. doi: 10.1038/nn.3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgaard K, Jensen K. Lambert JDC. Imaging of Ca2+ responses mediated by presynaptic L-type channels on GABAergic boutons of cultured hippocampal neurons. Brain Res. 2009;1249:79–90. doi: 10.1016/j.brainres.2008.10.033. [DOI] [PubMed] [Google Scholar]
- Hoppa MB, Lana B, Margas W, Dolphin AC. Ryan TA. α2δ expression sets presynaptic calcium channel abundance and release probability. Nature. 2012;486:122–125. doi: 10.1038/nature11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B. Miledi R. The role of calcium in neuromuscular facilitation. J Physiol. 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile BM, Guillot TS, Venton BJ, Wetsel WC, Augustine GJ. Wightman RM. Synapsins differentially control dopamine and serotonin release. J Neurosci. 2010;30:9762–9770. doi: 10.1523/JNEUROSCI.2071-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DK. Catterall WA. Ca2+-dependent and -independent interactions of the isoforms of the α1A subunit of brain Ca2+ channels with presynaptic SNARE proteins. Proc Natl Acad Sci U S A. 1997;94:14782–14786. doi: 10.1073/pnas.94.26.14782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukley M, Capetillo-Zarate E. Dietrich D. Vesicular glutamate release from axons in white matter. Nat Neurosci. 2007;10:311–320. doi: 10.1038/nn1850. [DOI] [PubMed] [Google Scholar]
- Li M, Hansen JB, Huang L, Keyser BM. Taylor JT. Towards selective antagonists of T-type calcium channels: design, characterization and potential applications of NNC 55–0396. Cardiovasc Drug Rev. 2005;23:173–196. doi: 10.1111/j.1527-3466.2005.tb00164.x. [DOI] [PubMed] [Google Scholar]
- Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R. Kaneko T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci. 2009;29:444–453. doi: 10.1523/JNEUROSCI.4029-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montague PR, McClure SM, Baldwin PR, Phillips PEM, Budygin EA, Stuber GD, Kilpatrick MR. Wightman RM. Dynamic gain control of dopamine delivery in freely moving animals. J Neurosci. 2004;24:1754–1759. doi: 10.1523/JNEUROSCI.4279-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosharov EV, Larsen KE, Kanter E, Phillips KA, Wilson K, Schmitz Y, Krantz DE, Kobayashi K, Edwards RH. Sulzer D. Interplay between cytosolic dopamine, calcium, and α-synuclein causes selective death of substantia nigra neurons. Neuron. 2009;62:218–229. doi: 10.1016/j.neuron.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller A, Kukley M, Stausberg P, Beck H, Müller W. Dietrich D. Endogenous Ca2+ buffer concentration and Ca2+ microdomains in hippocampal neurons. J Neurosci. 2005;25:558–565. doi: 10.1523/JNEUROSCI.3799-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami N, Ishibashi H, Katsurabayashi S. Akaike N. Calcium channel subtypes on single GABAergic presynaptic terminal projecting to rat hippocampal neurons. Brain Res. 2002;951:121–129. doi: 10.1016/s0006-8993(02)03148-7. [DOI] [PubMed] [Google Scholar]
- Nägerl UV, Novo D, Mody I. Vergara JL. Binding kinetics of calbindin-D(28k) determined by flash photolysis of caged Ca(2+) Biophys J. 2000;79:3009–3018. doi: 10.1016/S0006-3495(00)76537-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. Usefulness and limitations of linear approximations to the understanding of Ca++ signals. Cell Calcium. 1998;24:345–357. doi: 10.1016/s0143-4160(98)90058-6. [DOI] [PubMed] [Google Scholar]
- Ouanounou A, Zhang L, Charlton MP. Carlen PL. Differential modulation of synaptic transmission by calcium chelators in young and aged hippocampal CA1 neurons: evidence for altered calcium homeostasis in aging. J Neurosci. 1999;19:906–915. doi: 10.1523/JNEUROSCI.19-03-00906.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan ZH, Hu HJ, Perring P. Andrade R. T-type Ca2+ channels mediate neurotransmitter release in retinal bipolar cells. Neuron. 2001;32:89–98. doi: 10.1016/s0896-6273(01)00454-8. [DOI] [PubMed] [Google Scholar]
- Pan P-Y. Ryan TA. Calbindin controls release probability in ventral tegmental area dopamine neurons. Nat Neurosci. 2012;15:813–815. doi: 10.1038/nn.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson Study Group. Phase II safety, tolerability, and dose selection study of isradipine as a potential disease-modifying intervention in early Parkinson's disease (STEADY-PD) Mov Disord. 2013;28:1823–1831. doi: 10.1002/mds.25639. [DOI] [PubMed] [Google Scholar]
- Phillips PE. Stamford JA. Differential recruitment of N-, P- and Q-type voltage-operated calcium channels in striatal dopamine release evoked by ‘regular’ and ‘burst’ firing. Brain Res. 2000;884:139–146. doi: 10.1016/s0006-8993(00)02958-9. [DOI] [PubMed] [Google Scholar]
- Pissadaki EK. Bolam JP. The energy cost of action potential propagation in dopamine neurons: clues to susceptibility in Parkinson's disease. Front Comput Neurosci. 2013;7:13. doi: 10.3389/fncom.2013.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice ME. Cragg SJ. Nicotine amplifies reward-related dopamine signals in striatum. Nat Neurosci. 2004;7:583–584. doi: 10.1038/nn1244. [DOI] [PubMed] [Google Scholar]
- Rice ME, Patel JC. Cragg SJ. Dopamine release in the basal ganglia. Neuroscience. 2011;198:112–137. doi: 10.1016/j.neuroscience.2011.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov DA. Ca2+-dependent mechanisms of presynaptic control at central synapses. Neuroscientist. 2006;12:317–326. doi: 10.1177/1073858405284672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Han Y. Kochubey O. Ca2+ channels and transmitter release at the active zone. Cell Calcium. 2012;52:199–207. doi: 10.1016/j.ceca.2012.04.011. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R. Neher E. Presynaptic calcium and control of vesicle fusion. Curr Opin Neurobiol. 2005;15:266–274. doi: 10.1016/j.conb.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Schwaller B, Meyer M. Schiffmann S. ‘New’ functions for ‘old’ proteins: the role of the calcium-binding proteins calbindin D-28k, calretinin and parvalbumin, in cerebellar physiology. Studies with knockout mice. Cerebellum. 2002;1:241–258. doi: 10.1080/147342202320883551. [DOI] [PubMed] [Google Scholar]
- Sgobio C, Kupferschmidt DA, Cui G, Sun L, Li Z, Cai H. Lovinger DM. Optogenetic measurement of presynaptic calcium transients using conditional genetically encoded calcium indicator expression in dopaminergic neurons. PLoS One. 2014;9:e111749. doi: 10.1371/journal.pone.0111749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simuni T, Borushko E, Avram MJ, Miskevics S, Martel A, Zadikoff C, Videnovic A, Weaver FM, Williams K. Surmeier DJ. Tolerability of isradipine in early Parkinson's disease: a pilot dose escalation study. Mov Disord. 2010;25:2863–2866. doi: 10.1002/mds.23308. [DOI] [PubMed] [Google Scholar]
- Subramanian J. Morozov A. Erk1/2 inhibit synaptic vesicle exocytosis through L-type calcium channels. J Neurosci. 2011;31:4755–4764. doi: 10.1523/JNEUROSCI.6594-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Guzman JN. Sanchez-Padilla J. Calcium, cellular aging, and selective neuronal vulnerability in Parkinson's disease. Cell Calcium. 2010;47:175–182. doi: 10.1016/j.ceca.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai C, Yang YC, Pan MK, Huang CS. Kuo CC. Modulation of subthalamic T-type Ca2+ channels remedies locomotor deficits in a rat model of Parkinson disease. J Clin Invest. 2011;121:3289–3305. doi: 10.1172/JCI46482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada M, Kang Y. Imanishi M. Immunohistochemical localization of voltage-gated calcium channels in substantia nigra dopamine neurons. Eur J Neurosci. 2001;13:757–762. doi: 10.1046/j.1460-9568.2001.01435.x. [DOI] [PubMed] [Google Scholar]
- Tang A-H, Karson MA, Nagode DA, McIntosh JM, Uebele VN, Renger JJ, Klugmann M, Milner TA. Alger BE. Nerve terminal nicotinic acetylcholine receptors initiate quantal GABA release from perisomatic interneurons by activating axonal T-type (Cav3) Ca2+ channels and Ca2+ release from stores. J Neurosci. 2011;31:13546–13561. doi: 10.1523/JNEUROSCI.2781-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Clements MA, Khodai T, Pienaar IS, Exley R, Wess J. Cragg SJ. Striatal muscarinic receptors promote activity dependence of dopamine transmission via distinct receptor subtypes on cholinergic interneurons in ventral versus dorsal striatum. J Neurosci. 2010;30:3398–3408. doi: 10.1523/JNEUROSCI.5620-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K. Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64. doi: 10.1016/j.neuron.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Tippens AL, Pare J-F, Langwieser N, Moosmang S, Milner TA, Smith Y. Lee A. Ultrastructural evidence for pre- and postsynaptic localization of Cav1.2 L-type Ca2+ channels in the rat hippocampus. J Comp Neurol. 2008;506:569–583. doi: 10.1002/cne.21567. [DOI] [PubMed] [Google Scholar]
- Vance C, Begg C, Lee W-L, Dubel S, Copeland T, Sönnichsen F. McEnery M. N-type calcium channel/syntaxin/snap-25 complex probed by antibodies to II–III intracellular loop of the α1B subunit. Neuroscience. 1999;90:665–676. doi: 10.1016/s0306-4522(98)00420-5. [DOI] [PubMed] [Google Scholar]
- Weiss N, Hameed S, Fernandez-Fernandez JM, Fablet K, Karmazinova M, Poillot C, Proft J, Chen L, Bidaud I, Monteil A, Huc-Brandt S, Lacinova L, Lory P, Zamponi GW. De Waard M. A Cav3.2/syntaxin-1A signaling complex controls T-type channel activity and low-threshold exocytosis. J Biol Chem. 2012;287:2810–2818. doi: 10.1074/jbc.M111.290882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfart J. Roeper J. Selective coupling of T-type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons. J Neurosci. 2002;22:3404–3413. doi: 10.1523/JNEUROSCI.22-09-03404.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW. Regulation of presynaptic calcium channels by synaptic proteins. J Pharmacol Sci. 2003;92:79–83. doi: 10.1254/jphs.92.79. [DOI] [PubMed] [Google Scholar]
- Zanazzi G. Matthews G. The molecular architecture of ribbon presynaptic terminals. Mol Neurobiol. 2009;39:130–148. doi: 10.1007/s12035-009-8058-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. Sulzer D. Frequency-dependent modulation of dopamine release by nicotine. Nat Neurosci. 2004;7:581–582. doi: 10.1038/nn1243. [DOI] [PubMed] [Google Scholar]
- Zhao C, Dreosti E. Lagnado L. Homeostatic synaptic plasticity through changes in presynaptic calcium influx. J Neurosci. 2011;31:7492–7496. doi: 10.1523/JNEUROSCI.6636-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FM, Liang Y. Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4:1224–1229. doi: 10.1038/nn769. [DOI] [PubMed] [Google Scholar]