

Abstract
Vitamin D receptor (VDR) knockout (KO) mice had fewer Citrobacter rodentium in the feces than wild-type (WT) mice and the kinetics of clearance was faster in VDR KO than WT mice. VDR KO mice had more IL-22 producing innate lymphoid cells (ILC), and more anti-bacterial peptides than WT mice. The increased ILC in the VDR KO mice was a cell autonomous effect of VDR deficiency on ILC frequencies. BM transplantation from VDR KO mice into WT resulted in higher ILC and colonization resistance of the WT mice. Disruption of the gut microbiota using antibiotics in VDR KO mice reversed colonization resistance to C. rodentium infection. Confirming the role of the microbiota in the colonization resistance of VDR KO mice, transfer of the VDR KO microbiota to WT GF mice, resulted in colonization resistance. Once colonization resistance is overcome, VDR KO mice had increased susceptibility to C. rodentium. VDR expression is a regulator of ILC frequencies, IL-22, dysbiosis and C. rodentium susceptibility.
Keywords: vitamin D receptor, microbiota, innate lymphoid cells
INTRODUCTION
All cells of the immune system that have been analyzed express the vitamin D receptor (VDR) including dendritic cells (DCs), macrophages, T cells and B cells1. The VDR is a nuclear receptor that is part of the steroid hormone superfamily of receptors that regulate gene transcription. The active form of vitamin D and the high affinity VDR ligand is 1,25-dihydroxyvitamin D3 (1,25(OH)2D3)2, 3. The effects of 1,25(OH)2D3 include direct effects on IFN-γ and IL-17 production by T cells and indirect effects of 1,25(OH)2D3 on DC and macrophage that reduce the Th1 and Th17 response in vivo4. The effects of vitamin D include inhibition of the generation of Th1 and Th17 responses, induction of regulatory T cells and amelioration of experimental immune-mediated disease.
Because of the suppressive effects of 1,25(OH)2D3 on Th1 and Th17 responses, it seemed possible that vitamin D might compromise the anti-infectious response. Paradoxically, when therapeutic doses of 1,25(OH)2D3 (for immune-mediated disease) were tested, there was no effect of the 1,25(OH)2D3 treatment on the ability of the host to fight a Herpes simplex or Candida albicans infection5. In addition, vitamin D supplementation of humans during tuberculosis treatment was shown to be beneficial in some studies and not to have an effect in others6–9. In vitro and in vivo vitamin D induced the production of anti-bacterial peptides that might be beneficial in protecting the host from infection10, 11. VDR-KO mice infected with Schistosoma mansoni showed no effect of VDR expression on worm burdens, weight and fibrosis compared to wild-type (WT) mice12. VDR-KO mice had decreased Leishmania major parasite burdens compared to WT controls13. VDR-KO mice exhibited a delayed clearance of Listeria monocytogenes following infection but the VDR-KO mice were able to clear the infection14. How vitamin D could inhibit Th1 and Th17 responses in one context (immune-mediated disease) but not another (infectious disease) has not been established.
Citrobacter rodentium is a gram-negative murine pathogen that naturally colonizes and infects mice and forms attaching and effacing lesions15. In WT mice, C. rodentium colonizes the cecum and colon transiently and is cleared over three weeks 16. Th17-mediated immune responses are required for clearing the infection17. T cell, B cell and T and B cell (Rag) KO mice fail to clear a C. rodentium infection16. In the gut, innate lymphoid cells (ILC) that produce IL-22 and IL-17 (ILC3) are critical for early protection against C. rodentium infection17, ILC3 cells are the major source of IL-22 during the first 6 days following infection18. The innate immune system induces the development of protective acquired immunity required for clearance of C. rodentium.
Here we determined the role of the VDR on host resistance to C. rodentium infection. Surprisingly, VDR-KO mice were resistant to colonization with C. rodentium. The colonization resistance of the VDR-KO mice was associated with increased IL-22, increased ILCs, higher expression of RegIIIγ, Ang-4 and dysbiosis of the bacterial microbiota. Antibiotic (ABX) disruption of the gut microbiota reversed colonization resistance to C. rodentium in VDR-KO mice. Cecal transplantation of the microbiota from VDR-KO mice to WT germfree (GF) mice conferred colonization resistance compared to recipients of WT microbiota. In addition, VDR-KO bone marrow (BM) transplantation transferred colonization resistance to WT mice, which was associated with increased IL-22 and increased ILCs. Double VDR/Rag (D)KO mice had increased ILCs and IL-22 production in the gut. However, colonization resistance was not observed in the DKO mice. Both the DKO mice and the ABX treated VDR-KO mice developed more severe infections, including increased mortality, than their respective controls (Rag KO and ABX WT) to C. rodentium infection. Our data demonstrate that increased IL-22-producing ILCs contribute to dysbiosis and C. rodentium colonization resistance of VDR-KO mice. Taken together, our data suggest that the VDR regulates the gut microbiota, colonization and susceptibility to C. rodentium.
RESULTS
Reduced bacterial shedding and inflammation in VDR-KO mice
In WT mice, C. rodentium was detectable in the feces at d1 and increased through d7 when it peaked and then declined until d21 (Figure 1A). At d1 post-infection, VDR-KO mice had 3 logs fewer bacteria in the feces than WT mice (Figure 1A). The bacterial shedding in the feces was lower in VDR-KO mice compared to WT and the VDR-KO mice cleared the infection by d18 (Figure 1A). VDR-KO mice were less susceptible to C. rodentium than WT mice (Figure 1A). VDR-KO mice were resistant and WT mice were susceptible to C. rodentium infection, regardless of ancestry (breeders), sex or housing of the mice.
Figure 1. The kinetics of C. rodentium infection in WT and VDR KO mice.

(A) C. rodentium numbers in the feces. The frequencies of (B) inflammatory monocytes, (C) neutrophils and (D) T cells in the colonic LP. mRNA expression for (E) Ifn-γ, (F) Il-17A and (G) Il-6 in the colon. Data is from n=6–12 mice per group and the values represent the mean of three independent experiments ± SEM. Two-way ANOVA with Bonferroni post-hoc tests (A–G), *** P<0.001. Groups without a common letter differ at the indicated time point (B–G), P<0.05.
The recruitment of inflammatory cells into the colonic lamina propria (LP) was measured in VDR KO and WT mice. In uninfected (d0) WT and VDR-KO mice, the frequencies of either inflammatory monocytes (CD11b+Gr-1highF4/80+, Figure 1B) or neutrophils (CD11b+Gr-1highF4/80−, Figure 1C) were low (0.1%). Frequencies of inflammatory monocytes and neutrophils increased at d10 and fell significantly at d21 in WT mice (Figure 1B, C). Conversely, the frequencies of inflammatory monocytes remained low in VDR-KO mice following infection (Figure 1B). The neutrophils in VDR-KO mice were significantly lower than WT mice at d10 post-infection (Figure 1C). Infection increased the frequency of CD3+T cells in the WT colonic LP at d10 and d21 post-infection (Figure 1D). The increase in CD3 frequencies occurred after d21 of infection in VDR-KO mice (Figure 1D). VDR-KO mice had fewer CD3+T cells compared to WT mice at d10 post-infection (Figure 1D). The total numbers of LP lymphocytes isolated from WT and VDR-KO mice were not different. The expression of Ifng, Il17a and Il6 were low in uninfected colon from WT mice and increased significantly at d10 post-infection in WT mice (Figure 1E–1G). There was no increase in Ifng, Il17a and Il6 expression with infection of VDR-KO mice (Figure 1E–1G). In addition, VDR-KO mice had significantly lower expression of Ifng, Il17a and Il6 than WT mice at d10 (Figure 1E–1G). Lower colonization of VDR-KO mice with C. rodentium was associated with lower numbers of immune cell infiltrates and reduced cytokine expression.
Disruption of the microbiota in VDR KO mice reverses colonization resistance
The intestinal commensal bacteria compete with C. rodentium for colonization19. Previously we had shown that VDR-KO and WT mice had different commensal bacteria in the feces and that the difference in the microbiota occurred between VDR-KO and WT littermates suggesting an effect of VDR expression on the microbiota20. Here we used quantitative real-time PCR to determine the frequencies of bacterial phyla in the feces from mice used for these experiments (Figure 2A). Confirming the published data, VDR-KO mice had decreased quantities of bacteria from the Firmicutes phylum, and increased quantities of bacteria from the Bacteroidetes and the Proteobacteria phyla in the feces compared to WT mice (Figure 2A)20. In addition, several bacterial genus members were also different in VDR-KO mice compared to WT, including Eubacterium, Bacteroides and Salmonella (Supplementary Figure S1A–S1C). Similar phyla differences were also shown in the colon and small intestine (SI) from VDR-KO and WT mice (Supplementary Figure S1D–SII).
Figure 2. The effect of gut microbiota on infection with C. rodentium.

(A) The relative quantity of bacteria from the Firmicutes phylum, the Bacteroidetes phylum and the Proteobacteria phylum in the feces of WT and VDR KO mice. (B) The relative total amount of universal 16S rDNA in the feces of WT and VDR KO mice before and after ABX treatment. The values were normalized to the fecal weight. (C) Shedding of C. rodentium in the feces of WT, VDR KO, ABX WT and ABX VDR KO mice. (D) The survival rate of the WT, VDR KO, ABX WT and ABX VDR KO mice following infection with C. rodentium. Data shown is mean ± SEM using n=4–10 mice/group. Two-tailed student’s t tests (A), two-way ANOVA with Bonferroni post-hoc tests (B, C), and log-rank test (D), *P <0.05. Values (B and C) without a common letter are significantly different.
To determine the role of commensal bacteria in the colonization resistance of VDR-KO mice to C. rodentium infection, ABX was used to disrupt the bacteria. The one-dose ABX treatment significantly reduced the total amount of bacterial 16S rDNA in the feces of the ABX-treated WT and ABX-treated VDR-KO mice compared to the values before the ABX treatment (Figure 2B). In addition, ABX-treated WT and VDR-KO mice had decreased bacterial diversity with ABX treatment (fewer bands, Supplementary Figure S2). ABX treatment eliminated the bacterial differences in feces, colon and SI (Supplementary Figure S1). There was no effect of ABX on the WT clearance or susceptibility to C. rodentium infection, and none of the ABX-treated WT mice died following infection (Figure 2C, 2D). ABX VDR-KO mice had significantly higher bacterial shedding than VDR-KO mice starting at d2 post-infection and throughout the infection (Figure 2C). In addition, the ABX VDR KO mice took longer to clear the infection than VDR-KO, ABX WT or WT mice (Figure 2C). ABX VDR KO mice were extremely susceptible to C. rodentium infection and at peak infection 35% of them died following infection (Figure 2D). ABX treatment reduced total bacterial numbers, changed the bacterial composition and removed C. rodentium colonization resistance in VDR KO mice.
Changes in the expression of antimicrobial peptides and mucin in VDR KO mice
To determine the cause of the altered microbiota and C. rodentium colonization resistance, the expression of mRNA for several antimicrobial peptides and mucin were measured in WT and VDR KO mice. The expression of mRNA for RegIIIγ, angiogenin (Ang) 4, mucins (Muc) 1–4, RegIIIβ, cathelicidin-related antimicrobial peptide (CRAMP) and mouse β-defensin (mBD) 3 were not different at d0 in WT and VDR-KO mice (Figure 3A–3C and Supplementary Figure S3). By d10, the amount of RegIIIγ, Ang-4, RegIIIβ, mBD-3 were significantly increased in infected WT and VDR-KO mice, while the amount of Muc2, CRAMP, Muc1 were only increased in infected VDR-KO mice but not infected WT mice (Figure 3 and Supplementary Figure S3). The amount of Muc3 and Muc4 were not affected by infection or genotype (Figure 3A–3C and Supplementary Figure S3). At d10, VDR-KO mice had higher expression levels of RegIIIγ, Ang-4 and Muc 2 than WT mice at d10 post-infection (Figure 3A–3C). ABX treatment eliminated the d10 differences in the expression for RegIIIγ, Ang-4 and Muc2 between VDR-KO and WT mice (Figure 3A–3F). VDR-KO mice had increased expression of anti-microbial peptides compared to WT mice following C. rodentium infection and ABX treatment eliminated those changes.
Figure 3. Expression of antimicrobial peptides and mucins in the colon of WT and VDR KO mice.

mRNA expression for (A,D) RegIIIγ, (B,E) Ang-4 and (C,F) Muc2 in the colons of WT and VDR KO (A–C) and ABX treated WT and VDR KO (D–F) mice. Data is from n=6–8 mice per group and the values represent the mean of two independent experiments ± SEM. Two-way ANOVA with Bonferroni post-hoc tests (A–C), two-tailed Student’s t tests (D–F). Values (A–C) without a common letter are significantly different.
Increased IL-22+ ILCs in VDR-KO mice
IL-22 has been shown to induce epithelial cell production of antimicrobial peptides. RORγt+ ILCs (predominately ILC3 cells) have been identified to be the main source of IL-22 production during the early stage of C. rodentium infection21. The frequencies of ILCs (CD3−RORγt+) were measured in VDR-KO and WT mice. There were higher frequencies of ILCs in the SI of VDR-KO mice than WT mice, but the amount of ILCs were not different in the colons of these mice (Figure 4A). In addition, VDR-KO mice had more ILC3 (IL-22 producing) in the SI than WT mice (Figure 4B). Furthermore, VDR-KO mice also had more CD3−RORγt+NKp46+ (ILC1 or NK22) cells in the SI LP than WT mice (Supplementary Figure S4A). The frequencies of other ILC (LTi4, CD3−RORγt+NKp46−CD4+ and LTi0, CD3−RORγt+NKp46−CD4−) cells were increased in both SI and colonic LP in VDR-KO mice (Supplementary Figure S4). The total cell numbers isolated from the SI or colonic LP were not different between WT and VDR-KO mice, so the changes in frequencies reflect the changes in absolute numbers.
Figure 4. Cell-autonomous VDR control of ILC number and colonization resistance.

The frequencies of (A) ILCs (CD3-RORγt+) and (B) IL-22 producing ILCs in the SI and colon LP of uninfected WT and VDR KO mice. BM cells from WT, VDR KO, or a 1:1 ratio of WT and VDR KO mice were transplanted into lethally irradiated WT or VDR KO recipient mice. The frequency of VDR KO (CD45.2) and WT (CD45.1) cells in the (C) blood and colon LP and the donor derived-ILC (D) in the colon of recipient mice. WT donor BM was transplanted into WT (WT-WT) or VDR KO recipient (WT-VDR KO) mice. VDR KO BM was transplanted into WT (VDR KO-WT) or VDR KO recipient (VDR KO-VDR KO). 7–8 weeks post-BM transplantation recipient mice were infected with C. rodentium and the (E) CFU in the feces were determined. The frequencies of (F) ILCs (CD3-RORγt+), (G) IL-22+ ILCs and (H) total IL-22+ cells in the SI LP of the recipients of WT BM or VDR KO BM at d10 post-C. rodentium infection. (I) The relative quantities of bacteria in the feces of mice 7 wks post-BM transplantation and without C. rodentium infection. Values are the mean ± SEM of n=3–8 recipient mice/group. Two-tailed Student’s t tests (A–D, F–I), Two-way ANOVA (E), * P<0.05, **P<0.01.
A 1:1 ratio of WT and VDR-KO BM was injected into lethally irradiated WT recipients. 8 wks later the WT recipients had 1/2 of the blood and colonic lymphocytes of WT origin and the other 1/2 of VDR-KO origin (Figure 4C). The frequencies of ILCs in the colonic LP showed preferential reconstitution with the VDR-KO ILC compared to WT resulting in higher ILC of VDR-KO origin in the chimeric WT mice (Figure 4D). VDR deficiency results in more IL-22 producing ILCs in the GI tract and BM reconstitution of WT mice with VDR-KO BM demonstrates a cell autonomous effect of the VDR on ILC frequencies in the gut.
Colonization resistance is induced in WT mice by VDR-KO BM
WT BM was transplanted into WT (WT-WT) and VDR-KO (WT-VDR KO) recipients and VDR-KO BM was transplanted into VDR-KO (VDR KO-VDR KO), and WT (VDR KO-WT) recipients. Following confirmation of reconstitution the recipient mice were infected with C. rodentium. WT recipient mice receiving WT BM cells had 5 logs of C. rodentium at d2 post-infection and the numbers of C. rodentium shed increased to 8 logs by d7 (Figure 4E). The VDR KO-VDR KO mice had only 4 logs of C. rodentium at d2 post-infection and the numbers remained the same at d4 and 7 post-infection (Figure 4E). Like the VDR KO-VDR KO mice the VDR KO-WT mice had reduced numbers of C. rodentium in the feces at d4 and d7 compared to the WT-WT mice (Figure 4E). Conversely, the WT-VDR KO mice had higher numbers of C. rodentium shed in the feces and the shedding resembled the WT-WT mice more than the VDR KO-VDR KO (Figure 4E). The difference in the C. rodentium between recipients of VDR-KO BM and WT BM was less (Figure 4E) than that between VDR-KO and WT mice (Figure 1). This may be due to the incomplete reconstitution of the colonic LP cells in the gut (70–75%) of the BM recipients and the older age of the BM recipients (16–20wk versus 8–12wk) at the time of infection. However, partial colonization susceptibility and resistance to C. rodentium could be transferred by BM transplantation.
The frequencies of donor-derived ILCs from the mice in Figure 4E were measured at d10 post-infection. Recipients of WT BM (both WT and VDR-KO) had fewer ILCs, fewer IL-22+ ILCs and less total IL-22+ cells in the LP of the SI compared to recipients of VDR-KO BM (both WT and VDR-KO, Figure 4F–4H). The frequencies of ILCs, IL-22+ ILCs and total IL-22+ cells were significantly higher in the SI of VDR-KO BM recipients and this corresponded with colonization resistance to C. rodentium (Figure 4). Surprisingly the colonization resistance was not associated with measurable changes in the bacterial phyla in the feces of the BM reconstituted mice (Figure 4I) and this may be another reason that the colonization resistance of the VDR-KO BM recipients was less than that in the VDR-KO mice. Partial colonization resistance and susceptibility to C. rodentium was transferred via the BM. In addition, the increased numbers of IL-22 producing ILCs in the VDR-KO mice was a cell-autonomous effect of VDR deficiency.
Colonization resistance is transferred to GF WT mice by cecal transplants from VDR KO mice
In order to determine whether the changes in the VDR-KO microbiota could mediate colonization resistance, WT GF mice received cecal transplants from WT and VDR-KO mice for 48h and then were infected with C. rodentium. WT recipients of WT cecal contents had significantly higher fecal shedding of C. rodentium compared to WT recipients of VDR-KO cecal contents (Figure 5A). The reduced C. rodentium fecal shedding in the WT recipients of VDR-KO microbiota was not due to a change in ILC numbers since there was no difference in the frequencies of ILC or IL-22+ ILC compared to WT mice that received WT microbiota (Figure 5B, 5C). The WT mice with the VDR-KO microbiota did have higher total IL-22+ cells in the SI than the WT recipients of WT microbiota (Figure 5D). VDR-KO microbiota transferred C. rodentium colonization resistance to GF WT mice.
Figure 5. Cecal transplants into WT GF mice transfers colonization susceptibility to C. rodentium.
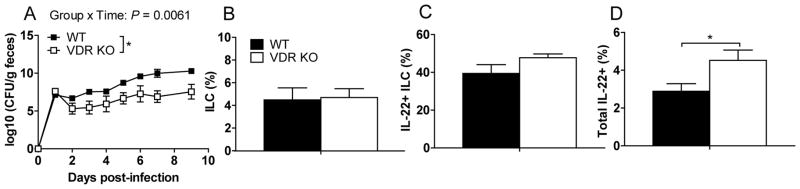
Cecal contents from WT or VDR KO mice were orally inoculated into WT GF mice. 48 hours after cecal transplantation the WT recipient mice were infected with C. rodentium. (A) C. rodentium CFU in the feces of WT GF mice receiving WT or VDR KO cecal contents. The frequencies of (B) ILCs (CD3-RORγt+), (C) IL-22+ ILCs and (D) total IL-22+ cells in the SI LP of mice in (A) at d10 post-C. rodentium infection. Values are the mean ± SEM of n=3–8 mice/group. Two-tailed Student’s t tests (B–D), one-way ANOVA (A) with Bonferroni post-hoc tests, * P<0.05.
VDR/Rag KO mice are more susceptible to C. rodentium than Rag KO mice
RORγt+ ILCs are also present in Rag KO mice. The uninfected Rag KO mice had 7.5% ILCs in the SI LP which is higher than those in WT mice (5%). ILC1, LTi4 and LTi0 cells were significantly higher in the SI (no differences in the colon) of DKO as compared to Rag KO mice (Suppl. Figure 4D–F). Despite the differences in ILC numbers between uninfected DKO and Rag KO mice, there were no differences in the Firmicutes or Bacteroidetes phyla members in the feces from DKO and Rag KO mice (Supplemental Figure S5A). There were increased numbers of the Proteobacteria phylum in the feces from DKO mice compared to Rag KO mice (Supplemental Figure S5A). DKO and Rag KO mice were infected with C. rodentium to determine whether DKO mice would be colonization resistant. Bacterial shedding in Rag KO mice increased and peaked at d7 post-infection and the Rag KO mice failed to clear the infection (Supplemental Figure S5B). Rag KO mice remained colonized with large numbers of C. rodentium that decreased survival beginning at d21 post-infection (Supplemental Figure S5B,S5C). Early post-infection DKO mice had significantly higher bacterial shedding than Rag KO mice and the DKO mice died quickly following systemic spread of the infection and weight loss (Supplemental Figure S5). DKO mice were not colonization resistant and instead were significantly more susceptible to C. rodentium infection than Rag KO mice.
DISCUSSION
VDR-KO mice were found to be significantly more resistant to C. rodentium infection than WT mice. The intestinal microbiota is in competition with C. rodentium. A previous study demonstrated that fecal transplants from resistant mice into susceptible mice conferred partial resistance to C. rodentium infection, and this protection was associated with increased IL-22 and anti-microbial peptides22. Consistent with this study, we demonstrate that transplant of the microbiota from VDR-KO mice into WT GF mice conferred protection from C. rodentium infection compared to WT microbiota. In addition, overproduction of IL-22 predominantly by ILCs in the VDR-KO gut induced antimicrobial peptides that altered the microbiota in the gut. The data further demonstrate that the VDR-KO microbiota induced more IL-22 than WT microbiota in GF WT recipients. However, the number of ILCs was not changed with VDR-KO cecal transplant and the colonization resistance was less than that seen in the VDR-KO controls. Since the microbiota don’t respond to changes in vitamin D (they don’t express a vitamin D receptor), it seems likely that the change in IL-22 and anti-microbial peptides precedes the change in microbiota. However, our results fall short of showing a direct link between higher IL-22 producing ILC and the shifts in the microbiota that result in colonization resistance. The VDR-KO mice overproduced IL-22 and ILCs that likely caused a shift in the commensal bacteria such that the VDR-KO host is resistant to infection with C. rodentium.
BM transplantation studies demonstrated that the higher number of ILCs in the VDR-KO mouse was due to a cell-autonomous effect of the VDR deficiency on ILC numbers. ILCs are critical for the control of homeostasis in the gut, but during intestinal inflammation, ILC function is dysregulated23. In particular, ILC3 cells that make IL-22 can be both protective and pathogenic23. Other nutrients and the microbiota influence ILC development and function23, 24. Our results suggest that the VDR regulates ILC3 cell numbers and possibly function. It might be possible that simple vitamin D interventions could normalize ILC function help control dysbiosis to maintain homeostasis.
Interestingly, vitamin D deficiency did not result in colonization resistance to C. rodentium (data not shown)25. There are several circumstances where deficiencies of the vitamin D ligand result in different outcomes than VDR deficiency26, 27. VDR-KO BM transferred partial colonization resistance to WT mice. Conversely, WT BM transplantation into VDR-KO mice resulted in C. rodentium colonization. The VDR is important for maintaining tight junction protein expression and barrier function in gut epithelial cells28. Furthermore, the VDR negatively regulates NF-κB expression in gut epithelial cells29. Our study now demonstrates an epithelial-independent effect of the VDR in controlling the microbiota and IL-22 production in the gut.
VDR-KO mice were extremely susceptible to C. rodentium infection after ABX treatment. The ABX treatment disrupted the microbiota and eliminated colonization resistance. A previous study demonstrated that GF mice infected with C. rodentium were unable to clear the infection, suggesting the critical role of commensal bacteria in eradicating C. rodentium in the gut19. However, none of the monoassociated GF mice died of systemic infection despite high C. rodentium numbers in the gut19. Consistent with this, ABX-treated WT mice did not succumb to systemic infection. In contrast, our data showed that ABX VDR-KO mice were unable to control the C. rodentium and 35% of them died. In addition, large numbers of C. rodentium were detected in systemic organs of ABX VDR-KO mice, including the spleen, kidney and liver (data not shown), suggesting that the VDR must be important to control infection and prevent systemic spread. T cells and B cells are important for containing C. rodentium in the gut. Mice deficient in T cells or B cells are unable to clear C. rodentium infection and develop lethal systemic infections16. The increased lethality of the ABX VDR-KO occurs within a time frame that suggests that the failure to induce a protective acquired T and/or B cell response might be preventing the VDR-KO mice from containing the infection to the gut. The data suggest that acquired immunity in the VDR-KO host is impaired such that once colonized the VDR-KO mouse is unable or too slow in mounting a protective T/B cell response required to clear C. rodentium.
The role of the VDR and vitamin D in the control of gastrointestinal infections is complicated. Salmonella infection was more severe in VDR-KO mice due in part to a change in NFkB activation in colonic epithelial cells29. It should be noted that in order for Salmonella to infect mice antibiotic pretreatment is required in both WT and VDR-KO mice29. Similarly here following antibiotic pretreatment VDR-KO mice were more susceptible to GI infection than WT mice. Recently Assa et. al demonstrated that feeding mice vitamin D deficient diet for 5 wk was associated with increased barrier dysfunction, dysbiosis of the microbiota and more intestinal inflammation following C. rodentium infection25. Paradoxically, 1,25(OH)2D3 injections increased C. rodentium numbers in the colon and spleen of mice30. The 1,25(OH)2D3-mediated increase in C. rodentium numbers was attributed to the suppression of the mucosal Th17 response30. Our data demonstrate that the VDR is a negative regulator of ILC, and infection-induced IL-22. ILC are important for the generation of both T and B cell responses in the gut23, 31. Interestingly diseases like IBD result in the reduced expression of the VDR in the gut 32. Our data predicts that with disease development the expression of the VDR will go down creating local areas of VDR deficiency, which might induce alterations in ILC and/or dysbiosis.
Our data highlights a role for the VDR in regulation of the composition of the microbial flora in the gut. Dysbiosis in the VDR-KO mouse was associated with increased ILCs and IL-22 producing cells in the gut that induced the production of anti-bacterial peptides. The increase in ILC numbers in the VDR-KO gut is a cell autonomous effect of the VDR. Understanding the role of vitamin D and the VDR in regulating ILC function and/or development would be critical for management of gastrointestinal homeostasis. In addition, expression of the VDR is a critical regulator of both the innate and acquired immune response since ABX VDR-KO and VDR/Rag DKO mice had high numbers of C. rodentium in the gut that led to the premature lethality of the mice following infection. VDR expression and regulation of VDR expression is critical for protection from infection and the maintenance of gastrointestinal homeostasis.
METHODS
Mice
Age and sex matched WT, VDR-KO, Rag KO and DKO mice on the C57BL/6 background were bred (breeders were VDR+/−) and housed in the same room at the Pennsylvania State University (University Park, PA). Experimental procedures were approved by the Office of Research Protection, Institutional Animal Care and Use Committee at the Pennsylvania State University.
Citrobacter rodentium infection
The C. rodentium strain ICC169 was a kind gift of Gad Frankel (London School of Medicine and Dentistry, London UK). C. rodentium was cultured in Luria-Bertani (LB) broth containing 20 μg/ml nalidixic acid (EMD Chemicals). Mice were infected by oral gavage with 200 μl of C. rodentium suspension, which contained 5×109 CFU. Mice were housed 1 per cage for the infection to prevent mouse to mouse transmission of C. rodentium. The C. rodentium numbers in the feces were determined by plating on LB agar plates with 50 μg/ml nalidixic acid. Some mice were treated with the ABX vancomycin (20mg/ml) the day before infection.
BM transplantation
BM cells from WT (CD45.1) and VDR KO (CD45.2) donor mice were transplanted either alone or in a 1:1 ratio into lethally irradiated WT (CD45.1/CD45.2) or VDR KO (CD45.2) recipient mice as previously described33. 7–8 wks following BM transplantation mice were evaluated for reconstitution in the blood (98–99%). The reconstitution of the gut was much less (70–75% of the total colon LP) but consistent with what has been described previously for reconstitution of the gut34.
Cecal transplant
Fresh cecal contents from 1 donor WT or 1 donor VDR KO mouse was collected and placed in PBS (0.1 g cecal content/1 ml PBS) on ice. The cecal contents from WT or VDR KO mice were homogenized and 200 μl of the homogenates were orally inoculated to WT GF mice. 48 h after cecal transplant, the recipient mice were orally infected with C. rodentium (5×109 CFU per mouse).
Cell isolation and flow cytometry
Isolation of intestinal LP lymphocytes were done as described previously 35. Pieces of the SI or colon were incubated twice in HBSS containing 5 mM EDTA, 0.15 μg/ml DTT and 5% FBS for 20 min at 37°C under 250 rpm rotation with stirring bar. The supernatant was discarded and the tissue was further incubated in RPMI-1640 containing 1 mg/ml collagenase type 1 (Worthington, Lakewood, NJ) and 10% FBS for 1.5 h at 37°C under 250 rpm rotation to obtain LP cells. The LP cells were collected from the interface of 40/80% Percoll gradients (Sigma-Aldrich). Cells were stained with FITC CD11b, FITC CD4, PE Gr-1, APC NKp46, PECy5 F4/80, PECy7 CD3 or istotype controls (eBiosciences, San Diego, CA). Sample staining histograms are shown in Supplementary Figure S6.
Cells were stimulated with PMA (0.1μg/ml, Sigma-Aldrich), ionomycin (0.5μg/ml, Sigma-Aldrich) for 5 h and for the final 3 h, Brefeldin A (10μg/ml, Sigma-Aldrich) was added to the culture medium. For the measurement of IL-22, cells were stimulated with mouse recombinant IL-23 (0.04μg/ml, R&D systems) for 5 h and for the final 3 h, Brefeldin A (10μg/ml, Sigma-Aldrich) was added to the culture medium. After surface staining, cells were fixed with 4% paraformadehyde (Sigma-Aldrich), permeabilized with 0.1% saponin (Sigma-Aldrich), and stained with FITC IFNγ, PE IL-17A, APC IL-22 or the FITC/PE/APC labeled isotype controls (eBiosciences). For RORγt staining was done using the transcription factor staining buffer kit and the manufacturer’s instructions (eBioscience). Cells were analyzed on BD Fortessa LSRII (BD Biosciences) and the data was analyzed with FlowJo 7.6.5 software (TreeStar, Ashland, OR).
PCR
Fecal DNA was isolated using QIAamp DNA stool minikit (Qiagen, Valencia, CA). Fecal DNA was amplified with universal 16S rDNA primers or specific primers for different bacterial phyla or genus using SYBR green mix (BioRad, Hercules, CA). Relative 16S rDNA quantities were calculated using ΔΔCt method and were normalized to the amount of universal bacteria. The fecal DNA was amplified with universal 16S rDNA primers and DGGE was done exactly as described 36.
Total RNA was isolated (Qiagen). cDNA was synthesized using the TaqMan reverse transcription reagents kit (Applied Biosystems, Carlsbad, CA) with SYBR green mix (BioRad) by MyiQ Single-Color Real-Time PCR machine (BioRad). Expression levels of these molecules were normalized by GAPDH and calculated with the ΔΔCt method. The primer sequences are listed in Supplementary Table 1.
Statistics
Statistical analyses were performed using GraphPad software (PRISM software, La Jolla, CA). Two-tailed Student’s t tests were used to test differences between genotype (WT vs. KO). Two-way ANOVA with Bonferroni post-hoc tests were used to test the effects of experimental groups, time and their interactions. Log-rank tests were used to test the survival rates. Some of the data was transformed (square root transformation) to eliminate unequal variances, followed by a repeated-measures (mix model) two-way ANOVA.
Supplementary Material
Acknowledgments
Supported by the National Institutes of Health/National Institute of Neurologic and Stroke Grant NS067563 and National Center for Complementary and Alternative Medicine and the Office of Dietary Supplements AT005378.
Footnotes
CONFLICTS OF INTEREST: The authors have nothing to disclose.
Supplementary Material is linked to the online version of the paper at http://www.nature.com/mi
References
- 1.Veldman CM, Cantorna MT, DeLuca HF. Expression of 1,25-dihydroxyvitamin D-3 receptor in the immune system. Arch Biochem Biophys. 2000;374:334–338. doi: 10.1006/abbi.1999.1605. [DOI] [PubMed] [Google Scholar]
- 2.Pike JW, Meyer MB. The vitamin D receptor: new paradigms for the regulation of gene expression by 1,25-dihydroxyvitamin D(3) Endocrinology and metabolism clinics of North America. 2010;39:255–269. doi: 10.1016/j.ecl.2010.02.007. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiellini G, DeLuca HF. The importance of stereochemistry on the actions of vitamin D. Current topics in medicinal chemistry. 2011;11:840–859. doi: 10.2174/156802611795165016. [DOI] [PubMed] [Google Scholar]
- 4.Cantorna MT. Mechanisms underlying the effect of vitamin D on the immune system. Proc Nutr Soc. 2010;69:286–289. doi: 10.1017/S0029665110001722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cantorna MT, Hullett DA, Redaelli C, Brandt CR, Humpal-Winter J, Sollinger HW, et al. 1,25-Dihydroxyvitamin D3 prolongs graft survival without compromising host resistance to infection or bone mineral density. Transplantation. 1998;66:828–831. doi: 10.1097/00007890-199810150-00003. [DOI] [PubMed] [Google Scholar]
- 6.Sasidharan PK, Rajeev E, Vijayakumari V. Tuberculosis and vitamin D deficiency. The Journal of the Association of Physicians of India. 2002;50:554–558. [PubMed] [Google Scholar]
- 7.Salahuddin N, Ali F, Hasan Z, Rao N, Aqeel M, Mahmood F. Vitamin D accelerates clinical recovery from tuberculosis: results of the SUCCINCT Study [Supplementary Cholecalciferol in recovery from tuberculosis]. A randomized, placebo-controlled, clinical trial of vitamin D supplementation in patients with pulmonary tuberculosis’. Bmc Infect Dis. 2013;13:22. doi: 10.1186/1471-2334-13-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martineau AR, Honecker FU, Wilkinson RJ, Griffiths CJ. Vitamin D in the treatment of pulmonary tuberculosis. J Steroid Biochem. 2007;103:793–798. doi: 10.1016/j.jsbmb.2006.12.052. [DOI] [PubMed] [Google Scholar]
- 9.Wejse C, Gomes VF, Rabna P, Gustafson P, Aaby P, Lisse IM, et al. Vitamin D as Supplementary Treatment for Tuberculosis A Double-blind, Randomized, Placebo-controlled Trial. Am J Resp Crit Care. 2009;179:843–850. doi: 10.1164/rccm.200804-567OC. [DOI] [PubMed] [Google Scholar]
- 10.Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770–1773. doi: 10.1126/science.1123933. [DOI] [PubMed] [Google Scholar]
- 11.Hewison M, Burke F, Evans KN, Lammas DA, Sansom DM, Liu P, et al. Extra-renal 25-hydroxyvitamin D3-1alpha-hydroxylase in human health and disease. J Steroid Biochem Mol Biol. 2007;103:316–321. doi: 10.1016/j.jsbmb.2006.12.078. [DOI] [PubMed] [Google Scholar]
- 12.Froicu M, Weaver V, Wynn TA, McDowell MA, Welsh JE, Cantorna MT. A crucial role for the vitamin D receptor in experimental inflammatory bowel diseases. Mol Endocrinol. 2003;17:2386–2392. doi: 10.1210/me.2003-0281. [DOI] [PubMed] [Google Scholar]
- 13.Ehrchen J, Helming L, Varga G, Pasche B, Loser K, Gunzer M, et al. Vitamin D receptor signaling contributes to susceptibility to infection with Leishmania major. Faseb J. 2007;21:3208–3218. doi: 10.1096/fj.06-7261com. [DOI] [PubMed] [Google Scholar]
- 14.Bruce D, Whitcomb JP, August A, McDowell MA, Cantorna MT. Elevated non-specific immunity and normal Listeria clearance in young and old vitamin D receptor knockout mice. Int Immunol. 2009;21:113–122. doi: 10.1093/intimm/dxn129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luperchio SA, Schauer DB. Molecular pathogenesis of Citrobacter rodentium and transmissible murine colonic hyperplasia. Microbes and infection / Institut Pasteur. 2001;3:333–340. doi: 10.1016/s1286-4579(01)01387-9. [DOI] [PubMed] [Google Scholar]
- 16.Mundy R, MacDonald TT, Dougan G, Frankel G, Wiles S. Citrobacter rodentium of mice and man. Cell Microbiol. 2005;7:1697–1706. doi: 10.1111/j.1462-5822.2005.00625.x. [DOI] [PubMed] [Google Scholar]
- 17.Rubino SJ, Geddes K, Girardin SE. Innate IL-17 and IL-22 responses to enteric bacterial pathogens. Trends in immunology. 2012;33:112–118. doi: 10.1016/j.it.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 18.Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity. 2011;34:122–134. doi: 10.1016/j.immuni.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, et al. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science. 2012;336:1325–1329. doi: 10.1126/science.1222195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ooi JH, Li Y, Rogers CJ, Cantorna MT. Vitamin D regulates the gut microbiome and protects mice from dextran sodium sulfate-induced colitis. J Nutr. 2013;143:1679–1686. doi: 10.3945/jn.113.180794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 22.Willing BP, Vacharaksa A, Croxen M, Thanachayanont T, Finlay BB. Altering host resistance to infections through microbial transplantation. PloS one. 2011;6:e26988. doi: 10.1371/journal.pone.0026988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sonnenberg GF. Regulation of intestinal health and disease by Innate lymphoid cells. Int Immunol. 2014 doi: 10.1093/intimm/dxu052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van de Pavert SA, Ferreira M, Domingues RG, Ribeiro H, Molenaar R, Moreira-Santos L, et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature. 2014;508:123–127. doi: 10.1038/nature13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Assa A, Vong L, Pinnell LJ, Avitzur N, Johnson-Henry KC, Sherman PM. Vitamin D Deficiency Promotes Epithelial Barrier Dysfunction and Intestinal Inflammation. The Journal of infectious diseases. 2014 doi: 10.1093/infdis/jiu235. [DOI] [PubMed] [Google Scholar]
- 26.Mayne CG, Spanier JA, Relland LM, Williams CB, Hayes CE. 1,25-Dihydroxyvitamin D3 acts directly on the T lymphocyte vitamin D receptor to inhibit experimental autoimmune encephalomyelitis. Eur J Immunol. 2011;41:822–832. doi: 10.1002/eji.201040632. [DOI] [PubMed] [Google Scholar]
- 27.Driver JP, Lamont DJ, Gysemans C, Mathieu C, Serreze DV. Calcium insufficiency accelerates type 1 diabetes in vitamin D receptor-deficient nonobese diabetic (NOD) mice. Endocrinology. 2011;152:4620–4629. doi: 10.1210/en.2011-1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kong J, Zhang Z, Musch MW, Ning G, Sun J, Hart J, et al. Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. American journal of physiology Gastrointestinal and liver physiology. 2008;294:G208–216. doi: 10.1152/ajpgi.00398.2007. [DOI] [PubMed] [Google Scholar]
- 29.Wu S, Liao AP, Xia Y, Li YC, Li JD, Sartor RB, et al. Vitamin D receptor negatively regulates bacterial-stimulated NF-kappaB activity in intestine. The American journal of pathology. 2010;177:686–697. doi: 10.2353/ajpath.2010.090998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryz NR, Patterson SJ, Zhang Y, Ma C, Huang T, Bhinder G, et al. Active vitamin D (1,25-dihydroxyvitamin D3) increases host susceptibility to Citrobacter rodentium by suppressing mucosal Th17 responses. Am J Physiol Gastrointest Liver Physiol. 2012;303:G1299–1311. doi: 10.1152/ajpgi.00320.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsuji M, Suzuki K, Kitamura H, Maruya M, Kinoshita K, Ivanov II, et al. Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity. 2008;29:261–271. doi: 10.1016/j.immuni.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 32.Liu W, Chen Y, Golan MA, Annunziata ML, Du J, Dougherty U, et al. Intestinal epithelial vitamin D receptor signaling inhibits experimental colitis. J Clin Invest. 2013;123:3983–3996. doi: 10.1172/JCI65842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu S, Cantorna MT. The vitamin D receptor is required for iNKT cell development. Proc Natl Acad Sci U S A. 2008;105:5207–5212. doi: 10.1073/pnas.0711558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bruce D, Cantorna MT. Intrinsic requirement for the vitamin D receptor in the development of CD8 alphaalpha-expressing T cells. J Immunol. 2011;186:2819–2825. doi: 10.4049/jimmunol.1003444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weigmann B, Tubbe I, Seidel D, Nicolaev A, Becker C, Neurath MF. Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nat Protoc. 2007;2:2307–2311. doi: 10.1038/nprot.2007.315. [DOI] [PubMed] [Google Scholar]
- 36.Muyzer G, Dewaal EC, Uitterlinden AG. Profiling of Complex Microbial-Populations by Denaturing Gradient Gel-Electrophoresis Analysis of Polymerase Chain Reaction-Amplified Genes-Coding for 16s Ribosomal-Rna. Appl Environ Microb. 1993;59:695–700. doi: 10.1128/aem.59.3.695-700.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.