

Abstract
How does an agonist activate a receptor? In this article I consider the activation process in muscle nicotinic acetylcholine receptors (AChRs), a prototype for understanding the energetics of binding and gating in other ligand-gated ion channels. Just as movements that generate gating currents activate voltage-gated ion channels, movements at binding sites that generate an increase in affinity for the agonist activate ligand-gated ion channels. The main topics are: i) the schemes and intermediate states of AChR activation, ii) the energy changes of each of the steps, iii) the sources of the energies, iv) the three kinds of AChR agonist binding site and v) the correlations between binding and gating energies. The binding process is summarized as sketches of different conformations of an agonist site. The results suggest that agonists lower the free energy of the active conformation of the protein in stages, by establishing favorable, local interactions at each binding site independently.
1.1 Introduction
An agonist diffuses to a resting receptor and lands on a small target site. The protein changes shape and generates a cellular response. Here I discuss evidence and ideas regarding agonist activation of muscle nicotinic acetylcholine receptors (AChRs), with a focus on energy changes occur in the activation process. (For a review of structural changes see Cecchini and Changeux, this volume.) Because AChRs are ion channels I will call the resting state C (for closed-channel) and the active state O (for open-channel). These symbols distinguish only structure and dynamics rather than function; an AChR can have the O shape even if something blocks the pore.
The basic view of receptor activation by an agonist (A) derives from the Henri-Michaelis-Menten kinetic scheme for enzyme function and was encoded into a simple chemical equation for AChRs in 1957 (2):
Although all nicotinic AChRs have at least two agonist sites, here we imagine a receptor with just one. In Scheme 1 the first step is called ‘binding’, which is the formation of a ligand-protein complex and the second step is called ‘gating’, which is the resting↔active isomerization of the system. Just as substrate molecule S makes a stable ES complex before catalysis, agonist molecule A makes a stable AC complex separately from the global conformational change. The intermediate ES/AC state is short-lived and was not detected directly until ~30 years after its proposal, in an enzyme in 1943 (4) and in AChRs in 1985 (5). Below I will add states to Scheme 1, even if some have not been detected directly.
Scheme 1.
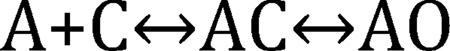
In addition to C and O, AChRs adopt stable desensitized (D) states in which the agonist is bound with high affinity but the ion channel is shut. In single-channel recordings of muscle AChRs, sojourns in D states are distinguished from those in C by virtue of their longer lifetimes (Fig. 1A) (6–9). In muscle AChRs desensitization is complex and proceeds mainly from O states, but recovery can also be directly to C. The schemes and models considered here pertain only to binding and gating, with desensitization omitted.
Figure 1. Currents and states.

A. Single-channel currents from muscle AChRs. Clusters of openings arise from binding and gating events (Scheme 1), and silent periods between clusters are sojourns in desensitized states. Arrow, 2 AChRs open at the same time. B. Cyclic model for activation of a receptor having 1 agonist site. Scheme 1 is bold. ΔG, free energy difference between states; subscripts are LA (low affinity), HA (high affinity), n (number of bound agonists). The free energy difference between C and AO is the same regardless of the connecting route: ΔGLA+ΔG1=ΔG0+ΔGHA. The free energy from the affinity change is ΔGB1=ΔGHA−ΔGLA, so ΔG1=ΔG0+ΔGB1. Agonists increase the probability that a receptor is active because the O state has the higher affinity.
1.2 Energy from the agonist
Scheme 1 (extended to two sites, for muscle AChRs) describes most of what happens in physiological conditions. However, on rare occasions wild-type (WT) receptors in muscle cells undergo spontaneous C↔O conversions in the absence of agonists (10) and agonists dissociate from the AO conformation (11). Although these events are infrequent, they demand a cyclic activation scheme that has, in addition to Scheme 1, the activation pathway C↔O↔AO (Fig. 1B). This thermodynamic cycle is sometimes called MWC, after those who first applied it to an allosteric protein (12, 13). In WT muscle AChRs the anti-clockwise activation path can be ignored when considering a concentration-response curve or a synaptic current. However, measuring the equilibrium and rate constants for this route is essential for understanding receptor activation mechanisms.
A ligand binds to the C state with a relatively low affinity (LA): ~150 µM for ACh and adult-type mouse AChRs (14). Importantly, for ligands that are agonists, the binding site affinity increases when the protein switches from C to O. A diffusing ligand delivers little more force than from a bump of a water molecule, so it cannot ‘kick’ the receptor into action. Rather, an agonist molecule floats onto its binding site as a small, side-chain-sized, reversible structural perturbation that increases the probability of a global C-to-O isomerization that occurs by thermal energy alone.
The key point is that an agonist at a binding site increases the receptor’s probability of being active (PO) simply because the O conformation of the binding site has a higher affinity for the ligand compared to the C conformation. When AC changes spontaneously to AO, favorable (negative) energy is generated from new, local interactions between the protein, the agonist molecule and water. These serve to increase the relative stability of the active form of the receptor (a ground state effect). The affinity change of a ligand-gated ion channel (LGIC) is analogous to the gating current of a voltage-gated ion channel (VGIC). Just as the movement of an S4 following depolarization stabilizes the Open ground state of a VGIC because of a more favorable disposition of charged groups, the movement of loops at an agonist binding site following the arrival of a ligand stabilizes the Open ground state of a LGIC by virtue of the higher agonist affinity.
The AC↔AO isomerization involves many different rearrangements throughout the protein that almost certainly do not occur at the exact same instant. The AChR binding sites appear to switch from low-to-high affinity early in the global transition, before the rearrangement of the conductance-regulating gate region (15, 16). Accordingly, an extra state (bold) can be inserted into the gating step of Scheme 1, to represent an AChR that has undergone the affinity switch but has not yet opened its pore:
The ″ superscript indicates a high-affinity (HA) binding site, so AC″ represents a receptor with a HA site but an overall C shape. This state (along with others) has been detected indirectly as part of the gating transition state (15, 17). In addition, brief closures in frog AChR and glycine receptor single-channel currents have been interpreted as reflecting an intermediate gating state in which the binding site has undergone a rearrangement and the gate region is shut (18, 19). The short-lived, AC″ state in Scheme 2 may have been detected directly.
Scheme 2.
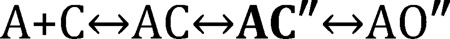
It is possible to estimate the free energy arising from the LA↔HA affinity change for the neurotransmitter, ACh. The starting point is to estimate the energy change with ACh at the binding sites, which is relatively easy. The free energy change in a chemical reaction is proportional to the log of the equilibrium constant. In the units kcal/mol, ΔGnACh=−0.59lnEnACh (at 23 °C), where EnACh is the full, C↔O gating equilibrium constant with n bound ACh molecules. In adult-type mouse muscle AChRs expressed in HEK cells and at −100 mV, E2ACh≈25, so ΔG2ACh≈−1.9 kcal/mol (hereafter just kcal). (The methods we use for estimating rate and equilibrium constants from single channel electrophysiology are described elsewhere (20)).
ΔG2ACh is the result of adding favorable free energy from the affinity change at 2 agonist sites to the unfavorable free energy of the intrinsic, C↔O isomerization (when only water occupies the binding pockets). Just as you need to know your ending and beginning bank balance to learn the deposit, you must know the O vs C energy difference in both in the presence and absence of agonists to know how much of the total free energy came from the affinity change.
In adult-type AChRs (at −100 mV) the unliganded gating equilibrium constant is E0≈7.4×10−7, or ΔG0WT≈+8.3 kcal (21). From the cycle, ΔG2=ΔG0+ΔGB2ACh (Eq. 1; Fig. 1), so we simply subtract ΔG0WT from ΔG2ACh to learn that in this receptor, the affinity change for 2 neurotransmitter molecules contributes ΔGB2ACh=−10.2 kcal towards increasing PO. The large, uphill energy gap between unliganded C and O becomes downhill because of the energy deposit arising from new, favorable interactions at two agonist sites with ACh molecules. Similar measurements with fetal-type AChRs, in which a γ subunit replaces ε, show that ΔGB2ACh=−12.2 kcal (22).
The next step is to determine how this total energy is divided between the two agonist sites. The muscle AChR’s agonist sites lie at different subunit interfaces, αδ and either αε in adult or αγ in fetal (Fig. 2). The net binding energy from the affinity change at each agonist site is ΔGB1=ΔGHA−ΔGLA (Fig. 1). This energy can be measured independently for each type of site by studying AChRs in which one site has been knocked out by a mutation (23, 24). The result is that in adult AChRs the αδ and αε sites are approximately equivalent energetically for ACh, each providing ΔGB1ACh ≈−5.1 kcal. In fetal AChRs, the αδ site still provides ~−5.1 kcal, but the αγ site provides −7.2 kcal (25). Notice that the αδ site provides the same free energy regardless of whether there is an ε or a γ subunit present, even though these differ by ~250 amino acids.
Figure 2. The AChR agonist binding sites.

Left. Torpedo AChR (pdb accession number 2bg9; (1)). Box, agonist binding site at the αγ subunit interface; horizontal lines, membrane. Right. Side view of the ligand binding regions of an acetylcholine binding protein (AChBP; pdb accession number 2uv6; (3)). Agonist-site loops A–C are labeled; CCh, carbamylcholine; residue numbering is for mouse AChRs.
The average ΔGB1 has been estimated for a number of different agonists in adulttype AChRs (Fig. 3) (20, 26, 27). These energies range from −5.1 kcal for ACh to −0.9 kcal for betaine, with the other ‘physiological’ ligands nicotine and choline falling in between. ΔGB1 is a quantitative index of how ‘partial’ an agonist is. The ~−2 kcal extra free energy from αγ allows fetal AChRs to produce a greater cell responds to low [ACh] and also to choline, an ACh precursor, breakdown product and stable component of serum. It may be that this differential sensitivity to choline is a fundamental reason for the γ to ε subunit swap that is necessary for the proper maturation and operation of the neuromuscular synapse. The structural bases for different ΔGB1 values are not known, but it could be that a small displacement of the ligand’s nitrogen atom within the binding pocket is all that is needed (26).
Figure 3. Partial agonists of the AChR.

Ligand structures and single-site ΔGB1 values in adult-type AChRs (numbers are kcal). A more negative ΔGB1 value indicates a higher-efficacy agonist. The grey bars are the average free energy, (αδ+αε)/2. Arrow, choline at the fetal, αγ site choline is ~−2 kcal more favorable than at αε/αδ.
We can also investigate the specific agonist-protein interactions that generate ΔGB1. It is well known that a group of 5 aromatic residues at the agonist site is important for activation of AChRs, some by cation-π forces (28–30). Structures of acetylcholine binding proteins show that these side chains contact the ligand (3) (Fig. 2). By measuring both liganded and unliganded gating equilibrium constants in AChRs having F or A mutations of these amino acids, it is possible to determine the relative contribute of each functional group to ΔGB1ACh and pinpoint the sources of this free energy for each type of agonist site (αδ, αε and αγ) (31).
There are 3 tyrosines on the α-subunit side of the binding pocket. F-to-A mutations suggest that the benzene rings of 190 and 198 (both in loop C) each provide about the same energy at all three kinds of site (~−2 kcal). That of 93 (in loop A) contributes little at αε and αδ, but also ~−2 kcal at αγ. Y-to-F mutations indicate that only the –OH of 190 contributes significantly to ΔGB1ACh, to about the same degree at all three kinds of site (~−2 kcal).
There are 2 agonist-site tryptophans, one in the α subunit and one in the non-α subunit. At the adult sites the indole of αW149 (in loop B) contributes about the same energy as the benzene of 198, but a bit more at the fetal αγ site (~−3 kcal). The non-α tryptophan, W55 (it is at position 57 in the δ subunit) shows the largest difference between sites. (32, 33). At αε and αδ mutation of this residue to alanine makes a modest or no dent in ΔGB1ACh, but at αγ this substitution makes ΔGB1ACh less favorable by a whopping +4.6 kcal which is nearly 40% of the total from both agonist sites combined (25). In nicotinic receptors this tryptophan is an important and variable energy source for increasing PO from the affinity change.
Mutation of a conserved glycine in loop B (αG147) to alanine or serine makes ΔGB1ACh less favorable by ~2.5 kcal at αδ/αε (34). This is only amino acid other than the aromatics discussed above that has been found so far where an alanine substitution changes ΔGB1ACh by ≥2 kcal. The agonist site, which can be defined as the set of residues that influence the energy from the affinity change, is small and discrete in muscle AChRs.
The results show that the 3 types of AChR agonist binding site are different, with αε and αδ being more similar to each other and αγ providing more energy from the agonist affinity change. Only 3 aromatic groups contribute to ΔGB1ACh at the adult agonist sites (αW149, αY190 and αY198) whereas 5 contribute at the fetal, αγ site (also αY93 and γW55). To summarize, the loop C tyrosines and the loop B tryptophan behave similarly at all of the sites; the loop A tyrosine and γW55 are larger energy sources at αγ; αY190 provides most of the free energy from the affinity change in adult AChRs, but shares the top spot with γW55 at the fetal αγ site.
Importantly, in both adult and fetal AChRs the two agonist sites appear to act nearly independently in so far as agonist energy is concerned, because the energy sum from 1-site measurements is approximately equal to the combined energy in 2-site AChRs (23, 24).
1.3 Catch and hold
The low-affinity association rate constant for ACh to a resting adult-type mouse muscle AChR (konACh) is ~1.7×108 M−1s−1 (14). Given this high value, agonist association is often thought of as being limited by diffusion. However, several observations indicate that the formation of the LA complex (A+C↔AC, in Schemes 1 and 2) requires, in addition to diffusion, crossing a chemical barrier (35).
First, agonists of similar size and charge have widely-varying association rate constants, all slower than for ACh. kon for choline is ~200 times slower, and for tetramethylammonium ~15 times slower, than for ACh. For a process that is strictly by diffusion, smaller molecules without rotatable bonds would be expected to bind faster, not slower. Second, many mutations of the binding site aromatics change the resting equilibrium dissociation constant (Kd). Regardless of the nature of the substitution, the change in affinity is caused by an almost-equivalent change in kon, with little effect on the dissociation rate constant (koff). It is hard to imagine that all of these mutations mainly effect just agonist diffusion. Third, kon has a high temperature dependence (an activation enthalpy of 34 kcal) in the construct αG153S+choline (36). Diffusional processes have a low temperature-dependence (~4–5 kcal). These three observations are not consistent with an agonist association process that is limited by diffusion. Rather, they suggest that a conformational change in the protein is required to form the LA complex.
Accordingly, an additional state, AC, can be inserted into the ‘binding’ step of Schemes 1 and 2, to separate diffusion from this conformational change.
C represents a water-filled agonist site and AC represents an ‘encounter complex’ with the ligand that is formed by diffusion only. The superscript of the next state, AC′, indicates that the conformational change that forms the LA complex (‘catch’) has taken place. As in Scheme 2, the AC″ superscript indicates the conformational change that forms the HA complex (‘hold’) has also taken place.
Both the catch and hold conformational changes refer only to local rearrangements of the agonist site that are within the global C (or O) structural ensemble. Both of these conformational changes generate short-lived intermediate states that in most experiments are undetected and, hence, remain buried within the transition states (the arrows) for binding and gating in Scheme 1.
Starting from the left in Scheme 3, the first arrow is diffusion, the second is catch, the third is hold and the last is everything else within the global isomerization, including the conductance-changing rearrangements at the gate. In Scheme 1, ‘binding’ has become A+C↔AC↔AC′ (diffusion and catch combine to produce Kd) and ‘gating’ has become AC′↔AC″↔AO″ (hold and isomerize combine to produce E1). Notice that state AC′ is both the end of the binding process and the beginning of the gating process. Below, I present cartoons of the states in Scheme 3.
Scheme 3.
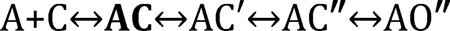
Scheme 3 only addresses activation by the primary ‘physiological’ pathway. What about the anti-clockwise pathway, that involves agonist binding to O? The HA equilibrium dissociation constant of an AChR that has the overall O shape (Jd) can be estimated in several ways, all of which give a similar answer.
Using the cyclic model (Fig. 1B) and assuming detailed balance, the total energy change from C to AO is the same by either the clockwise or anti-clockwise activation route: ΔGLA+ΔG1=ΔG0+ΔGHA. From left to right, these free energies are for low-affinity binding (ΔGLA=+0.59lnKd), gating with one bound agonist, unliganded gating and high-affinity binding (ΔGHA=+0.59lnJd). The first three energies (equilibrium constants) have been estimated experimentally for ACh so the HA binding energy for ACh can be calculated (23, 37). The result is ΔGHA≈−10.2 kcal in adult-type AChRs. This energy corresponds to JdACh≈30 nM, which is ~5000 times smaller than KdACh≈150 µM.
In the second approach, the HA agonist dissociation rate constant from O (joff) can be measured using AChRs that have a distant background mutation that increases unliganded gating (makes intrinsic gating less uphill) but has no effect on binding (11). In these constructs ACh-activated single-channel open intervals terminate either by dissociation of the agonist from AO″ or by desensitization. After separating these two processes it was estimated that joffACh≈12 s−1, which is vastly slower than koffACh≈25,000 s−1 (14). If we assume that ACh association to O is the same as to C we arrive at JdACh≈70 nM, and if we assume that it is even faster (diffusion-limited, or ~5×109 M−1s−1) we estimate JdACh≈2.5 nM. These estimates bracket the value obtained from using the cycle calculation.
In the third approach, jon and joff for ACh were estimated directly from cross-concentration fitting of single-channel shut current interval durations, much as is done for measuring kon and koff (22, 38). Here, background mutations that increase intrinsic gating and low [ACh] were used to make activation by the anti-clockwise pathway more probable than by the clockwise route. The results for ACh at either the αγ or αδ binding site show that jon≥kon. All three methods indicate that ACh association to form the HA complex is as fast, or faster, than to form the LA complex.
This result is inconsistent with the commonly-held view that ‘capping’ of loop C over the agonist site creates a steric barrier that establishes the HA complex (reviewed in (39)). If the LA↔HA switch were indeed caused by such a lid-closure, agonist dissociation and association would both be slower to O than to C. Rather, the experimental evidence suggests that whatever the local rearrangements are in the affinity change, they do not impede ACh access to the pocket. Although residues in loop C (αY190 and αY198) are indeed important for establishing both the LA and HA complexes, the approximate equivalence of the C and O association rate constants suggests that ‘capping’ does not establish a high affinity of the agonist site by shutting off a diffusional, in-out pathway.
Another interesting observation regard loop C is that the formation of a cysteine cross-link between its tip and the non-α side of the agonist site increases spontaneous openings. This led to the suggestion that ‘capping’ triggers the global C-to-O conformational change (40). However, deletion of loop C in AChRs and in the bacterial homolog GLIC does not interfere with spontaneous gating (38, 41). Loop C is crucial for stabilizing the agonist at the AChR binding site, but apparently not for initiating the overall isomerization. It is possible that stapling loop C across subunits is just a large perturbation that makes ΔG0 more favorable by jostling other nearby structural elements that indeed do initiate the isomerization.
Another idea based on the cross-linking result is a cuckoo-clock mechanism for AChR activation: loop C capping transfers energy mechanically towards the gate via perturbation of a conserved salt bridge (40, 42). This mechanism, however, is not compatible with results showing that AChRs undergo the C↔O transition normally without loop C or the salt bridge (43). The important question of how the binding sites and the gate communicate remains open to investigation, but the evidence so far suggests that loop C is not the trigger.
1.4 Correlation in binding energies
All of the rate and equilibrium constants for the entire cycle have been estimated for both fetal and adult mouse AChRs (23). These values have been obtained for AChRs expressed in HEK cells, by piecemeal analyses of single-channel currents from cell-attached patches. It is worth noting that simulations using these microscopic parameters regenerate cellular responses – concentration-response and synaptic-current profiles – that are in good agreement with those obtained using sharp electrodes and muscle cells. There do not appear to be significant artifacts arising from the patch pipette or from using heterologously-expressed proteins in tissue-cultured cells. This gives confidence that the single-channel energy measurements for different agonists, mutations, temperatures and voltages pertain to those that would prevail at synapses.
The low and high affinity binding energies, ΔGLA and ΔGHA, have been estimated for a series of agonists and mutations of several binding site residues (35). Curiously, these two energies are correlated linearly (Fig. 4A). For 8 different agonists and ~30 different mutations of binding site residues that influence ΔGB1 (Fig. 2B), the slope of the correlation (kappa) is ~0.5. For all of these structural perturbations, the change in HA binding energy was about twice that in LA binding energy (ΔΔGHA≈2ΔΔGLA; Eq. 2). In terms of equilibrium constants, the fold-change (relative to WT, ACh) in Jd was about equal to the square of that in Kd. In pharmacology terms, the effect of a perturbation on resting affinity is correlated with its effect on efficacy. Apparently, in AChRs binding and gating are not independent processes (recall that they share the AC’ state, in Scheme 3).
Figure 4. Catch-and-hold.

A. Low- and high-affinity binding are correlated linearly for different agonists and binding site mutants (values normalized to ACh, WT). A slope (kappa) of 0.5 indicates for all of the perturbations, ΔΔGHA≈2ΔΔGLA. B. Catch-and-hold energy landscape. Plots of free energy vs. reaction progress for water three different agonists. The agonist ‘tilts’ the overall chemical potential profile, to reduce the barrier heights and increase the forward catch and hold rate constants. For ACh, formation of the LA complex is almost diffusion-limited. LA and HA energies are coupled obligatorily (dotted lines).
ΔGB1 is the difference between HA and LA binding energies (Eq. 1; Fig. 1B). Combining this with Eq. 2 we find that ΔΔGLA≈ΔΔGB1, or that ΔGB1≈+.59lnKd (Eq. 3). This approximate relationship is remarkable. In muscle AChRs, the equilibrium constants that set the extremes of the concentration-response curve (zero agonist and complete saturation) allow an estimation of ΔGB1, hence Kd, hence the entire profile. For example, from the adult-type ΔGB1 values shown in Fig. 3 we use Eq. 3 to calculate Kd=180 µM (ACh), 960 µM (nicotine) and 3.7 mM (choline), all in good agreement with experimental values. Further, combining Eqs. 1–3 and the relationship EC50≈Kd/√E2 (for a strong agonist and a receptor with 2 equivalent agonist sites) we arrive at EC50≈√E0/E2. Substituting the AChR equilibrium constants for adult AChRs (7.4×10−7 and 25) we estimate EC50≈34 µM, which again agrees with the experimental value. If an agonist’s kappa-value is known, all you need to know to draw the entire dose-response curve are E0 and E2.
Kappa is an index of the relative effect of a perturbation on LA vs. HA binding energy, on a scale from 1 to 0. Not all binding site mutations have a kappa≈0.5. The kappa value for αG153 is ~0.9 (mostly affects LA binding) and that for εP121 is ~0.3 (most affects HA binding). In acetylcholine binding protein structures neither of these amino acids appears to make direct contact with the ligand. In AChRs the kappa values for loop B amino acids are slightly higher than for loop C residues, so it is possible that loop B plays a greater role in LA catch, and loop C in LA↔HA hold.
Although the agonists tested so far all have kappa~0.5, this cannot be true for all ligands. A pure antagonist, for example, binds but does not allow the LA-to-HA conversion. It is likely that agonists will be discovered that have kappa values different from 0.5.
One way to rationalize the linear correlation between catch and hold energies is by imagining a landscape for these linked processes (Fig. 4B). Accordingly, an agonist at a binding site ‘tilts’ the overall chemical potential profile to influence the rate and equilibrium constants of catch-and-hold, much as the application of voltage tilts the electrical potential profile across the membrane to influence channel block. In a WT AChR the intrinsic tilt, with water but without agonists, is steeply uphill, so that E0 is small. An agonist is a molecule that adds a downhill tilt (a negative ΔGB1) relative to water to make E1>E0, although the final tilt may still remain uphill (E1<1, as with choline in Fig. 4B). An inverse agonist makes the tilt even more uphill than in water (a positive ΔGB1), and an antagonist leaves the intrinsic tilt unchanged (ΔGB1=0). In the catch-and-hold energy landscape, the central well represents the LA complex, which in AChRs and some ligands has an energy that is a constant fraction (~50%) of the overall tilt.
This energy landscape provides a framework for understanding how an agonist increases the rate constant for channel-opening (the forward isomerization), which is determined by the height of the hold/isomerize barrier (the arrow between AC’ and AC”). In many chemical reactions, the change in a barrier height following a perturbation is a constant fraction of the change in the depth of the product energy well (15, 44). If this fraction is close to 1, then the greater the tilt of the binding chemical potential, the lower the barrier and the faster the transition. Hence, agonists that have a larger ΔGB1 will also produce a faster opening rate constant.
The catch structural change is a local rearrangement of the binding site that is possible within the C and O structural ensembles. However, it is indeed a conformational change and, as such, has the potential of moving energy over distance. This raises the possibility that some allosteric communication between the binding sites and the transmembrane domain occurs during the low-affinity catch phase of AChR activation, which likely involves backbone deformations of binding site loops. The tilted, catch-and-hold landscape provides a thermodynamic basis for the obligatory linkage between LA binding and the gating conformational change.
1.5 Discussion
Fig. 5 is an attempt to synthesize the above observations regarding the agonist binding process in the form of cartoons of a single agonist site. Catch (the LA binding rearrangement) is drawn as a rotation of the left wall of the pocket, and hold (the LA↔HA transition) as a rotation of the right wall. The status of the pore is shown as a horizontal line at the bottom. Each wall rotation provides favorable agonist binding energy. These energies are correlated in catch-and-hold, perhaps only by virtue of shared interactions with the agonist molecule. The filled circle is the agonist; water fills the binding pocket when the agonist does not. Only three main binding site sub-conformations are shown because I assume that the hold-without-catch configuration is unstable and, therefore, rare. In this scheme, agonist binding to O″ (jon) does not require passage through the intermediate catch state and thus is faster than to C.
Figure 5. Activation scheme and cartoons (one agonist binding site).

Inner square: The basic binding-gating cyclic activation model (Fig. 1); Scheme 1 is bold. Middle octagon: Extended model that distinguishes diffusion, catch, hold and the rest of a global isomerization that includes the conductance change of the pore; Scheme 3 is bold. The superscripts ′ and ″ represent sub-conformations of the agonist binding pocket (catch and catch+hold). Outer sketches: speculative conformations that correlate with the states. Arcs are binding site loops, solid circle is an agonist, hollow circles are binding site side chains; the horizontal line at the bottom reflects the shut vs. open configuration of the pore.
The sketches on the left side of Fig. 5 show an AChR binding site undergoing catch and hold rearrangements in the absence of an agonist. Several workers have noted the presence of multiple O states in unliganded gating (10, 42, 45). However, many different binding site mutations, including some that have almost no effect on ΔGB1 (for instance, αY198F), reduce or eliminate this complexity and give rise to constitutive gating that appears as a simple, two-state process (38, 46). It is therefore unlikely that the complexity of O states in WT AChRs without agonists relates directly to the 3 unliganded-O intermediates predicted by catch-and-hold (O, O’ and O” in Fig. 5). One possible explanation for the apparent simplicity of unliganded gating in mutant AChRs is that background substitutions added to increase constitutive activity, which are necessary to allow the measurements, ‘tilt’ the catch-and-hold landscape (much like an agonist molecule) to make the O and O′ states unstable and, hence, invisible. Further investigations of unliganded gating may resolve this issue.
Many of the states in Fig. 5 have been detected in single-channel currents. Others are only inferred and so far have not been observed directly in current recordings, perhaps because they are extremely short-lived. Time will tell if including these ‘virtual’ states is useful for understanding AChR activation. Someday, structures may reveal them at atomic resolution.
Agonists increase the probability of nicotinic receptor (AChR) activation because they bind with a higher affinity to O(pen-) vs. C(losed-channel) conformations.
The free energy from the affinity change can be estimated from single-channel currents.
This energy has been measured in muscle AChRs for different ligands, in fetal vs. adult receptors and after mutation of amino acids at the agonist binding sites.
Both low-affinity binding of ligands to resting receptors and the low↔high affinity switch require a conformational change at the agonist site.
The energy changes of these local rearrangements are correlated and so, too are affinity and efficacy.
In muscle AChRs a concentration-response curve can be estimated from just the extremes of zero-agonist and complete-saturation.
Acknowledgements
I thank all of the past and current members of my lab for generating the ideas, gathering the data, analyzing the results and sharing the passion. Supported by NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. Journal of molecular biology. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 2.Del Castillo J, Katz B. The identity of intrinsic and extrinsic acetylcholine receptors in the motor end-plate. Proceedings of the Royal Society of London. Series B, Containing papers of a Biological character. Royal Society (Great Britain) 1957;146:357–361. doi: 10.1098/rspb.1957.0016. [DOI] [PubMed] [Google Scholar]
- 3.Celie PH, van Rossum-Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron. 2004;41:907–914. doi: 10.1016/s0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 4.Chance B. THE KINETICS OF THE ENZYME-SUBSTRATE COMPOUND OF PEROXIDASE. Journal of Biological Chemistry. 1943;151:553–577. [Google Scholar]
- 5.Colquhoun D, Sakmann B. Fast events in single-channel currents activated by acetylcholine and its analogues at the frog muscle end-plate. J Physiol. 1985;369:501–557. doi: 10.1113/jphysiol.1985.sp015912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakmann B, Patlak J, Neher E. Single acetylcholine-activated channels show burst-kinetics in presence of desensitizing concentrations of agonist. Nature. 1980;286:71–73. doi: 10.1038/286071a0. [DOI] [PubMed] [Google Scholar]
- 7.Elenes S, Auerbach A. Desensitization of diliganded mouse muscle nicotinic acetylcholine receptor channels. J Physiol. 2002;541:367–383. doi: 10.1113/jphysiol.2001.016022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Auerbach A, Akk G. Desensitization of mouse nicotinic acetylcholine receptor channels. A two-gate mechanism. J Gen Physiol. 1998;112:181–197. doi: 10.1085/jgp.112.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Katz B, Thesleff S. A study of the desensitization produced by acetylcholine at the motor end-plate. J Physiol. 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson MB. Kinetics of unliganded acetylcholine receptor channel gating. Biophys J. 1986;49:663–672. doi: 10.1016/S0006-3495(86)83693-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grosman C, Auerbach A. The dissociation of acetylcholine from open nicotinic receptor channels. Proceedings of the National Academy of Sciences. 2001;98:14102–14107. doi: 10.1073/pnas.251402498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monod J, Wyman J, Changeux JP. On the Nature of Allosteric Transitions: A Plausible Model. Journal of molecular biology. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 13.Karlin A. On the application of "a plausible model" of allosteric proteins to the receptor for acetylcholine. Journal of theoretical biology. 1967;16:306–320. doi: 10.1016/0022-5193(67)90011-2. [DOI] [PubMed] [Google Scholar]
- 14.Chakrapani S, Bailey TD, Auerbach A. Gating dynamics of the acetylcholine receptor extracellular domain. J Gen Physiol. 2004;123:341–356. doi: 10.1085/jgp.200309004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grosman C, Zhou M, Auerbach A. Mapping the conformational wave of acetylcholine receptor channel gating. Nature. 2000;403:773. doi: 10.1038/35001586. [DOI] [PubMed] [Google Scholar]
- 16.Purohit P, Gupta S, Jadey S, Auerbach A. Functional anatomy of an allosteric protein. Nature communications. 2013;4:2984. doi: 10.1038/ncomms3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Auerbach A. Gating of acetylcholine receptor channels: brownian motion across a broad transition state. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1408–1412. doi: 10.1073/pnas.0406787102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lape R, Colquhoun D, Sivilotti LG. On the nature of partial agonism in the nicotinic receptor superfamily. Nature. 2008;454:722–727. doi: 10.1038/nature07139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Auerbach A. A statistical analysis of acetylcholine receptor activation in Xenopus myocytes: stepwise versus concerted models of gating. J Physiol. 1993;461:339–378. doi: 10.1113/jphysiol.1993.sp019517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jadey SV, Purohit P, Bruhova I, Gregg TM, Auerbach A. Design and control of acetylcholine receptor conformational change. Proceedings of the National Academy of Sciences. 2011;108:4328–4333. doi: 10.1073/pnas.1016617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nayak TK, Purohit PG, Auerbach A. The intrinsic energy of the gating isomerization of a neuromuscular acetylcholine receptor channel. The Journal of General Physiology. 2012;139:349–358. doi: 10.1085/jgp.201110752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nayak TK, Auerbach A. Asymmetric transmitter binding sites of fetal muscle acetylcholine receptors shape their synaptic response. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:13654–13659. doi: 10.1073/pnas.1308247110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jha A, Auerbach A. Acetylcholine receptor channels activated by a single agonist molecule. Biophys J. 2010;98:1840–1846. doi: 10.1016/j.bpj.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta S, Purohit P, Auerbach A. Function of interfacial prolines at the transmitter-binding sites of the neuromuscular acetylcholine receptor. The Journal of biological chemistry. 2013;288:12667–12679. doi: 10.1074/jbc.M112.443911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nayak TK, Gupta S, Auerbach A. Functional Asymmetry of Agonist Binding in Fetal and Adult Muscle Acetylcholine Receptors. Biophysical Journal. 106:339a. [Google Scholar]
- 26.Bruhova I, Gregg T, Auerbach A. Energy for wild-type acetylcholine receptor channel gating from different choline derivatives. Biophys J. 2013;104:565–574. doi: 10.1016/j.bpj.2012.11.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jadey S, Purohit P, Auerbach A. Action of nicotine and analogs on acetylcholine receptors having mutations of transmitter-binding site residue alphaG153. J Gen Physiol. 2013;141:95–104. doi: 10.1085/jgp.201210896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sine SM, Quiram P, Papanikolaou F, Kreienkamp HJ, Taylor P. Conserved tyrosines in the alpha subunit of the nicotinic acetylcholine receptor stabilize quaternary ammonium groups of agonists and curariform antagonists. Journal of Biological Chemistry. 1994;269:8808–8816. [PubMed] [Google Scholar]
- 29.Tomaselli GF, McLaughlin JT, Jurman ME, Hawrot E, Yellen G. Mutations affecting agonist sensitivity of the nicotinic acetylcholine receptor. Biophysical Journal. 1991;60:721–727. doi: 10.1016/S0006-3495(91)82102-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong W, Gallivan JP, Zhang Y, Li L, Lester HA, Dougherty DA. From ab initio quantum mechanics to molecular neurobiology: a cation-π binding site in the nicotinic receptor. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:12088–12093. doi: 10.1073/pnas.95.21.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Purohit P, Bruhova I, Auerbach A. Sources of energy for gating by neurotransmitters in acetylcholine receptor channels. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:9384–9389. doi: 10.1073/pnas.1203633109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiara DC, Cohen JB. Identification of amino acids contributing to high and low affinity d-tubocurarine sites in the Torpedo nicotinic acetylcholine receptor. The Journal of biological chemistry. 1997;272:32940–32950. doi: 10.1074/jbc.272.52.32940. [DOI] [PubMed] [Google Scholar]
- 33.Bafna PA, Jha A, Auerbach A. Aromatic Residues εTrp-55 and δTrp-57 and the Activation of Acetylcholine Receptor Channels. The Journal of biological chemistry. 2009;284:8582–8588. doi: 10.1074/jbc.M807152200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Purohit P, Auerbach A. Glycine Hinges with Opposing Actions at the Acetylcholine Receptor-Channel Transmitter Binding Site. Molecular pharmacology. 2011;79:351–359. doi: 10.1124/mol.110.068767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jadey S, Auerbach A. An integrated catch-and-hold mechanism activates nicotinic acetylcholine receptors. J Gen Physiol. 2012;140:17–28. doi: 10.1085/jgp.201210801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gupta S, Auerbach A. Temperature Dependence of Acetylcholine Receptor Channels Activated by Different Agonists. Biophysical Journal. 2011;100:895–903. doi: 10.1016/j.bpj.2010.12.3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Purohit P, Auerbach A. Unliganded gating of acetylcholine receptor channels. Proceedings of the National Academy of Sciences. 2009;106:115–120. doi: 10.1073/pnas.0809272106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Purohit P, Auerbach A. Loop C and the mechanism of acetylcholine receptor-channel gating. J Gen Physiol. 2013;141:467–478. doi: 10.1085/jgp.201210946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nys M, Kesters D, Ulens C. Structural insights into Cys-loop receptor function and ligand recognition. Biochemical pharmacology. 2013;86:1042–1053. doi: 10.1016/j.bcp.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 40.Lee WY, Sine SM. Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature. 2005;438:243–247. doi: 10.1038/nature04156. [DOI] [PubMed] [Google Scholar]
- 41.Gonzalez-Gutierrez G, Cuello LG, Nair SK, Grosman C. Gating of the proton-gated ion channel from Gloeobacter violaceus at pH 4 as revealed by X-ray crystallography. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:18716–18721. doi: 10.1073/pnas.1313156110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mukhtasimova N, Lee WY, Wang HL, Sine SM. Detection and trapping of intermediate states priming nicotinic receptor channel opening. Nature. 2009;459:451–454. doi: 10.1038/nature07923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Purohit P, Auerbach A. Acetylcholine receptor gating at extracellular transmembrane domain interface: the "pre-M1" linker. J Gen Physiol. 2007;130:559–568. doi: 10.1085/jgp.200709857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grunwald E. Structure-energy relations, reaction mechanism, and disparity of progress of concerted reaction events. Journal of the American Chemical Society. 1985;107:125–133. [Google Scholar]
- 45.Grosman C, Auerbach A. Kinetic, mechanistic, and structural aspects of unliganded gating of acetylcholine receptor channels: a single-channel study of second transmembrane segment 12' mutants. J Gen Physiol. 2000;115:621–635. doi: 10.1085/jgp.115.5.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Purohit P, Auerbach A. Energetics of gating at the apo-acetylcholine receptor transmitter binding site. J Gen Physiol. 2010;135:321–331. doi: 10.1085/jgp.200910384. [DOI] [PMC free article] [PubMed] [Google Scholar]