

Abstract
The aim of this study was to determine the role of AKT as a therapeutic target in ovarian clear cell carcinoma (CCC), an aggressive, chemoresistant histological subtype of ovarian cancer. AKT activation was assessed by immunohistochemistry (IHC) using human tissue microarrays of primary ovarian cancers, comprised of both CCC and serous adenocarcinoma (SAC). The growth-inhibitory effect of AKT-specific targeting by, the small molecule inhibitor, perifosine was examined using ovarian CCC cell lines in vitro and in vivo. Finally, the activity of perifosine was examined using in CCC-derived tumors that had acquired resistance to anti-VEGF or chemotherapeutics like bevacizumab or cisplatin, respectively. Interestingly, AKT was frequently activated both in early-stage and advanced-stage CCCs. Treatment of CCC cells with perifosine attenuated the activity of AKT-mTORC1 signaling, inhibited proliferation, and induced apoptosis. The effect of perifosine was more profound under conditions of high AKT activity compared to low AKT activity. Increased AKT activation and enhanced sensitivity to perifosine were observed in the context of cisplatin-resistant CCC. Treatment with perifosine concurrently with cisplatin significantly enhanced the anti-tumor effect of cisplatin. Moreover, perifosine showed significant anti-tumor activity in CCC-derived tumors that had acquired resistance to bevacizumab or cisplatin. Collectively, these data reveal that AKT is frequently activated in ovarian CCCs and is a promising therapeutic target in aggressive forms of ovarian cancer.
Implications
AKT-targeted therapy has value in a front-line setting as well as a second-line treatment for recurrent disease developing after platinum-based chemotherapy or bevacizumab treatment.
Keywords: AKT, perifosine, ovarian cancer, clear cell carcinoma, chemotherapy
Introduction
Ovarian carcinoma is the fourth most common cause of cancer deaths among women in the United States, with 22,240 new cases diagnosed and approximately 14,030 deaths in 2013 (1). Cytoreductive surgery followed by platinum-based chemotherapy combined with paclitaxel has been the standard initial treatment in patients with epithelial ovarian cancer (2). Recently, it has been reported that addition of bevacizumab during and after carboplatin and paclitaxel chemotherapy prolongs progression free survival (PFS) by about 4 months in patients with advanced epithelial ovarian cancer. (3,4). However, many clinical problems still exist in the treatment of epithelial ovarian cancer.
One of the most important problems that needs to be resolved is the management of clear cell carcinoma (CCC) of the ovary, which was first recognized by the World Health Organization as a distinct histological subtype in 1973 (5). The major clinical problems in the management of CCCs are its poor sensitivity to first-line platinum-based chemotherapy and the lack of effective chemotherapy for recurrent CCCs (6). Therefore, to improve survival of patients with CCC, a better understanding of the mechanism of platinum-resistance and the identification of effective treatment strategies for both advanced and recurrent disease are needed.
The sensitivity of cancer cells to chemotherapeutic drug-induced apoptosis depends on the balance between pro-apoptotic and anti-apoptotic signals (7,8). Therefore, inhibition of anti-apoptotic signals, such as those mediated by the AKT pathway, has been proposed as a promising strategy to enhance the efficacy of conventional chemotherapeutic agents (7,8).
AKT is a serine–threonine protein kinase that has a crucial role in cellular processes including glucose metabolism, apoptosis, and cell proliferation (7,8). AKT is known to be activated by both phosphoinositide-dependent kinase 1 (PDK1) and mTORC2, and activated AKT in turn phosphorylates multiple downstream targets via its kinase activity (7,8). It has been reported that AKT is frequently activated in epithelial ovarian cancer (9,10). Moreover, AKT inhibition therapy has been shown to inhibit the proliferation of ovarian cancer cells with hyperactivation of AKT both in vitro and in vivo (10-12). However, since most tumor specimens and tumor-derived cell lines used in these prior investigations have been ovarian SACs, the role of AKT in CCC remains largely unknown.
It has been reported that activating mutations of PIK3CA occur in about 40% of ovarian CCCs, which is more frequent than in any other histological subtype of epithelial ovarian cancer (13). It has also been reported that loss of PTEN expression is common in CCC of the ovary (14). Moreover, it has also been recently reported that mTORC2 is activated in ∼70% of CCCs (15). Since these genetic and epigenetic changes results in the hyperactivation of AKT signaling, CCCs may be more strongly dependent on AKT signaling for tumor progression than are other histological subtypes of epithelial ovarian cancer, and thus AKT may be a very promising therapeutic target in CCC. Given that patients with CCC have poor prognosis, hopes are high for the development of AKT-targeting therapy in this patient population.
Perifosine is a synthetic alkylphospholipid that inhibits the activation of AKT through preventing cell membrane recruitment of the N-terminal AKT pleckstrin homology (PH) domain (16). Previous studies with perifosine demonstrated antitumor activities in multiple human trials (16). Perifosine have also shown significant anti-tumor activity either as a single agent (17) or in combination with paclitaxel (18) in preclinical studies ovarian cancer. However, the activity of perifosine in CCC remains unknown.
In the current investigation, we examined the activation status of AKT both in early stage and advanced stage CCC, and determined whether perifosine has anti-neoplastic efficacy in both in vitro and in vivo models of CCC. Moreover, we investigated the potential role of AKT-inhibition therapy in CCCs that had acquired resistance after treatment with cisplatin or bevacizumab treatments.
Materials and Methods
Reagents/antibodies
Perifoine was obtained from Aeterna Zentaris GmbH (Frankfurt, Germany). Antibodies recognizing AKT, phospho-AKT (Ser473), S6K1, phospho-S6K1 (Thr389), poly ADP ribose polymerase (PARP) and β-actin, were obtained from Cell Signaling Technology (Beverly, MA). Anti-rabbit secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The Cell Titer 96-well proliferation assay kits were obtained from Promega (Madison, WI). Cisplatin was purchased from Sigma (St. Louis, MO). Bevacizumab was kindly provided by Chugai Pharmaceutical Co., Ltd. (Kanagawa, Japan).
Cell lines and cultures
Human ovarian CCC cell lines RMG1, RMG2, KOC7C, and HAC2 were kindly provided by Dr. H. Itamochi (Tottori University, Tottori, Japan). Human ovarian CCC cell line OVISE, human SAC cell lines SKOV-3 and A2780 were purchased from the American Type Culture Collection. Human ovarian adenocarcinoma cell lines OVCAR4 and OVCAR5 were kindly provided by Cell Culture Facility at Fox Chase Cancer Center (Fox Chase Cancer center, PA, USA). We tested these cells lines in our laboratory for their authentication by morphologic observation. No further cell line authentication was conducted by the authors. Each cell line was never continuously passaged in culture for more than 3 months, and after that, a new vial of frozen cells was thawed. Cells were cultured in DMEM/Ham's F-12, (Gibco, Carlsbad, CA) with 10% fetal bovine serum, as reported previously (15).
Establishment of cisplatin-resistant cell lines
Cisplatin-resistant sublines from RMG1 and RMG2 were developed by continuous exposure to cisplatin as described previously (19). Briefly, cells of both lines were exposed to stepwise increases in cisplatin concentrations. Initial cisplatin exposure was at a concentration of 1 μM. After the cells had regained their exponential growth rate, the cisplatin concentration was doubled and then the procedure was repeated until selection at 10 μM was attained. The resulting cisplatin-resistant sublines, designated as RMG1-CR and RMG2-CR, were cultured in DMEM/Ham's F-12 containing 10 μM cisplatin to maintain a high level of cisplatin-resistance.
Cell proliferation assay
MTS assay was used to analyze the effect of perifosine and/or cisplatin on cell viability as described (20). Cells were cultured overnight in 96-well plates (1×104 cells/well). Cell viability was assessed 48 h after addition of perifosine or/and cisplatin at the indicated concentrations. The number of surviving cells was assessed by determination of the A490 nm of the dissolved formazan product after addition of MTS for 1 h as described by the manufacturer (Promega, Madison, WI). Cell viability is expressed as follows: Aexp group/Acontrol × 100.
Clonogenic Survival Assay
The cells were plated into 6 cm dishes and then perifosine was added for 24 h. All of the cells were trypsinized and counted. The cells (5 × 102/well) were plated in 6-well tissue culture plates in medium containing 10% fetal bovine serum. The plates were incubated for 10-14 days. Cells were fixed and stained using Diff-Quik (Dade Behring, Newark, DE), and the number of colonies, consisting of ≥50 cells, in triplicate wells were counted as surviving colonies.
Cell cycle analysis
Cells were incubated with perifosine at the indicated concentrations for 24 h. Cells were then fixed with 75% ethanol overnight at 4°C, and stained with propidium iodide (50 μg/ml) in the presence of RNase A (100 μg/ml; Roth, Karlsruhe, Germany) for 20 min at 4°C. Cell cycle distribution was determined by analyzing 10,000 cells using a FACScan flow cytometer and Cell Quest software (Becton Dickinson, San Jose, CA) as reported previously (15).
Western blotting
Cells treated as indicated were lysed for 10 min at 4°C. Equal amounts of proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes. Blocking was done in 5% nonfat milk in 1×Tris-buffered saline. Western blot analyses were performed using specific primary antibodies. Immunoblots were visualized with horseradish peroxidase-coupled immunoglobulin by using an enhanced chemiluminescence Western blotting system (Perkin Elmer, Boston, MA).
In vitro VEGF protein quantitation
RMG2 cells (2 × 106) were incubated in DMEM Ham's F-12 medium containing 10% FBS for 24 h under normoxia or hypoxia (1% O2). Then, the culture supernatants were collected and levels of VEGF were determined using the Quantikine Human Vascular Endothelial Growth Factor Immunoassay (R&D Systems) according to the manufacturer's protocol. The remaining monolayers were trypsinized, and the cells were counted to normalize VEGF protein values. VEGF values were derived from a standard curve of known concentrations of recombinant human VEGF. Each sample was analyzed in duplicate and averaged.
Tube formation assay
Tube formation assay was done as described previously (21). The surfaces of 96-well plates were coated with 30 μL of growth factor–reduced Matrigel matrix (BD Biosciences). Then, 1 × 104 serum-starved human umbilical vein endothelial cells (HUVECs) in 100 μL of M199 medium containing 0.5% bovine serum albumin were plated. Perifosine was added at the time of plating. After 8-hour incubation, tube formation was visualized under an inverted microscope (×40) and the images were analyzed.
HUVEC proliferation assay
Serum-starved HUVECs were plated at 1 × 104 cells per 96 well with perifosine at the indicated concentrations. The cell were incubated at 37°C and 5% CO2 for 24 h in HuMedia-EG2 supplemented with 2% fetal bovine serum (Kurabo Industries, Osaka, Japan) and proliferation was assessed by MTS assay (Promega, Madison, WI).
Detection of apoptosis
CCC cells were treated with 30 μM of perifosine for 24 h. Then, cells were harvested, and stained with propidium iodide (PI) and annexin V using the annexin V-FITC apoptosis detection kit (BioVision, San Francisco, CA), according to the manufacturer's instructions. Fluorescence data were collected using flow cytometry. The sum total of early apoptopic cells, annexin V(+) PI(-), and late apoptopic cells, annexin V(+) PI (+), was defined as the total number of apoptotic cells.
Clone Selection
The plasmid encoding constitutively active AKT2 (HA-Myr-AKT2) or the control vector (pcDNA3) used in this study have been described previously (22). The plasmids encoding the kinase-dead AKT (HA-AktK179M) and control vector (pCMV6) have been described previously (11, 23). RMG1 and RMG2 cells were transfected in 6-well tissue culture plates with 1 μg of the pcDNA3, HA-Myr-AKT2, pCMV6, or HA-AktK179M using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen, Carlsbad, CA). Clonal selection was performed by adding G418 (Famingdale, Enzo Life Sciences). The resulting stable transfectants expressing control plasmid or HA-Myr-AKT2 were designated as RMG1-control, RMG1-HA-Myr-AKT2, RMG1-HA-AKT-KM, RMG2-control, RMG2-HA-Myr-AKT2 and RMG2-HA-AKT-KM.
Clinical samples
All surgical specimens were collected and archived according to protocols approved by the institutional review boards of the parent institutions. Appropriate informed consent was obtained from each patient. The tumors included 52 CCCs and 46 SACs. Based on criteria of the international Federation of Gynecology and Obstetrics (FIGO), 27 CCCs were stage I-II tumors and 25 were stage III-IV tumors. Among SACs, 22 were stage I-II tumors and 24 were stage III-IV tumors. Tumor samples were fixed in 10% neutral buffered formalin overnight and then embedded in paraffin. Ovarian cancer tissue microarrays consisting of two cores from each tumor sample were prepared, as described previously (15,19).
Immunohistochemistry
Tissue sections were cut at 4 μm, mounted on slides, and processed for immunohistochemical staining. Sections were incubated with the primary antibody, followed by the appropriate peroxidase-conjugated secondary antibody. The slides were scored semiquantitatively by two pathologists who were blinded to the clinical outcome. Surrounding non-neoplastic stroma served as an internal negative control for each slide. A score of 0 indicated no staining, +0.5 was weak focal staining (<10% of the cells stained), +1 was indicative of focal positive staining (10-50% of the cells stained), +2 indicated clearly positive staining (>50% of the cells stained), and a score of +3 was intensely positive. Tumors with staining of +1, +2 or +3 comprised the positive-staining groups. When the two cores from the same tumor sample showed different positively results, the lower score was considered valid.
In vivo tumor studies
All procedures involving animals and their care were approved by the Institutional Animal Care and Usage Committee of Osaka University, in accordance with institutional and NIH guidelines.
Initial experiments were conducted to examine the anti-tumor activity of perifosine monotherapy in CCC. Then 5 to 7-week-old nude mice (n=10) were inoculated s.c into the right flank either with 5×106 RMG2 cells in 200 μl of PBS. When tumors reached about 50 mm3, the mice were assigned into two treatment groups: placebo (n=5) or perifosine (n=5).
The second set of experiments was conducted to examine the anti-tumor effect of perifosine in CCC that had acquired resistance to bevacizumab. To confirm the anti-tumor effect of bevacizumab on the growth of CCCs, 5 to 7-week-old nude mice (n=10) were inoculated s.c into the right flank either with 5×106 RMG2 cells in 200 μl of PBS. When tumors reached about 50 mm3, mice were assigned into two treatment groups; bevacizumab (n=5) or placebo (n=5). To confirm the anti-tumor effect of perifosine on CCCs that had acquired resistance to bevacizumab treatment, 5 to 7-week-old nude mice (n=10) were inoculated s.c into the right flank either with 5×106 RMG2 cells in 200 μl of PBS. When tumors reached about 50 mm3, all mice were treated with bevacizumab. After 3 weeks of treatment, the mice were assigned into two treatment groups: bevacizumab continuation (n=5) or perifosine treatment (n=5). Perifosine was administered intragastrically using an animal-feeding needle. Perifosine was given in a loading dose of 75 mg/kg (2 × 37.5 mg/kg separated by 12 h) followed by daily maintenance dose of 25 mg/kg for 14 days. Bevacizumab was administered intra-peritoneally twice-weekly at a dose of 5 mg/kg. Caliper measurements of the longest peripendicular tumor diameters were done every week to estimate tumor volume using the following formula: V = L × W × D × π/6, where V is the volume, L is the length, W is the width, and D is the depth.
Statistical analysis
The effect of AKT inhibition on cell proliferation and apoptosis was analyzed by Student's t test. Tumor volume was analyzed by Student's t test and Wilcoxon exact test. Immunoreactivity was analyzed using Fisher's exact test.
Results
AKT is frequently activated in CCCs
To determine the activation of AKT, tissue microarrays consisting of 52 ovarian CCCs and 46 ovarian SACs were examined immunohistochemically for phospho-AKT (Ser473). As shown in Fig. 1A, phospho-AKT (Ser473) expression was observed in 68% of advanced stage CCCs and in 70% of early stage CCCs, compared to 66% and 49% in advanced stage and early stage SACs, respectively. In earlier stage (stage I-II), although the frequency of phospho-AKT expression is slightly higher in CCCs and in SACs, the difference was not statistically significant.
Figure 1. AKT is frequently activated and can be a therapeutic target in ovarian CCCs.
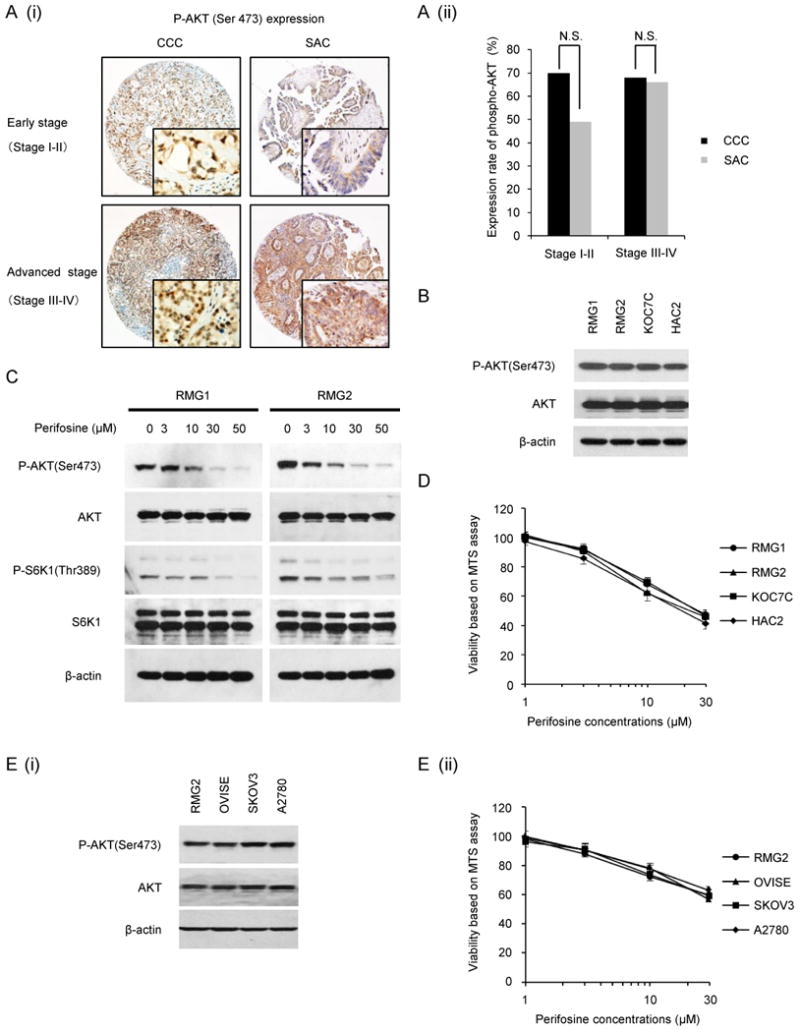
A, (i) Ovarian cancer tissue microarrays were stained with phospho-AKT (Ser473) antibody, and representative photographs of ovarian tissue microarray cores are shown. Magnifications: ×100, and ×400 (insets). (ii) Histogram indicating the frequency of phospho-AKT staining by clinical stage and histological type. N.S., no statistically significant difference. B, AKT activation status in four ovarian CCC cell lines. CCC cells were incubated in the presence of 10% FBS, after which the AKT activity was determined by western blotting. Actin expression was used as a loading control. C, The effect of perifosine on AKT-mTOR activation in vitro. RMG1 and RMG2 cells were treated with 0, 3, 10, 30 or 50 μM perifosine for 6 h in the presence of 10% FBS. Cells were harvested, and equivalent amounts (30 μg) of protein were subjected to SDS-PAGE and blotted with anti-phospho-AKT (Ser473), anti-AKT, anti-phospho-S6K1 (Thr389), anti-S6K1, or anti-β-actin antibodies. D, Sensitivity of CCC cells to perifosine. CCC cells were treated with the indicated concentrations of perifosine in the presence of 10% FBS for 72 h. Cell viability was assessed by MTS assay. The experiment was performed six times. Data are shown as the mean of six experiments. E, (i), Sensitivity of SAC cells and CCC cells to perifosine. SAC cells (OVISE and RMG2) and CCC cells (SKOV-3 and A2780) were treated with the indicated concentrations of perifosine in the presence of 10% FBS for 48 h. Cell viability was assessed by MTS assay. The experiment was performed three times. Data are shown as the mean of three experiments. (ii) AKT activation status in four cell lines. CCC cells were incubated in the presence of 10% FBS, after which the AKT activity was determined by western blotting. Actin expression was used as a loading control.
In vitro growth-inhibitory effect of perifosine on CCC cell lines
Given the frequent AKT activation found in human CCC tumor specimens (Fig. 1A), we evaluated the activation of AKT in four human CCC cell lines. As shown in Fig. 1B, AKT activation are observed in all CCC cell lines tested, which is consistent with immunohistochemical results observed with tumor samples.
Using these cell lines, we examined the anti-tumor effect of AKT-targeting therapy in vitro. For this purpose, we employed the AKT inhibitor perifosine, which has been shown to inhibit the activity of AKT by preventing cell membrane recruitment of the AKT. We first confirmed the effect perifosine on the activation of AKT signaling in CCC cells. As shown, treatment of RMG1 and RMG2 cells with perifosine significantly attenuated the phosphorylation of AKT and S6K1 in a dose dependent manner, indicating that treatment with perifosine effectively inhibited the AKT-mTORC1 signaling pathway (Fig. 1C). Moreover, perifosine treatment significantly inhibited the proliferation of all CCC cells tested to the same extent (Fig. 1D). To investigate whether the growth-inhibitory effect of perifosine is specific for CCC, we next examined the anti-proliferative effect of perifosine using 2 CCC cell lines and 2 SAC cell lines that express similar levels of phospho-AKT (Fig. 1E). As shown, these 4 cell lines showed similar sensitivity to perifosine, indicating that the perifosine have anti-tumor effect for ovarian cancers exhibiting activation of AKT irrespective of tumor-histology.
To investigate the mechanism by which perifosine inhibits the proliferation of CCC cells, we first examined the effect of perifosine on cell cycle progression by flow cytometry. As shown, treatment of both RMG1 and RMG2 cells with 30 μM perifosine resulted in an increase in the percentage of cells in G1 phase (Fig. 2A, Table1). Moreover, the percentage of apoptotic cells in sub-G1 was also significantly increased after treatment with perifosine in both cell lines, which is consistent with a western blotting (Fig. 2B (i)) or a flow cytometry (Fig. 2B (ii), Table 1). Collectively, these results suggest that perifosine inhibits proliferation of CCC cells by inducing both cell cycle arrest and apoptosis.
Figure 2. Perifosine induces both cell cycle arrest and apoptosis.
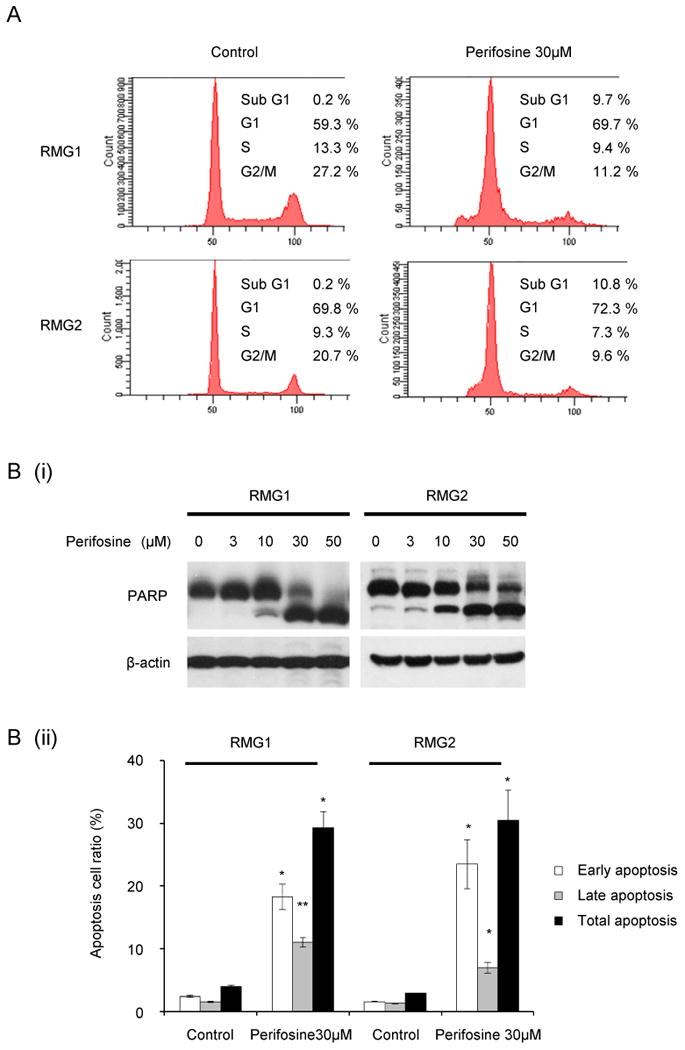
A, Perifosine induces both cell cycle arrest in G1 phase. RMG1 and RMG2 cells were treated with or without 30 μM perifosine for 24 h. Cell cycle analyses were performed with a fluorescence activated cell sorter (FACS) can as described in the “Materials and Methods.” B, Perifosine induces apoptosis. RMG1 and RMG2 cells were treated with or without 30 μM perifosine for 24 h. Cells were harvested, and the effect of perifosine was evaluated by western blotting (B, (i)) or flow cytometry (B, (ii)) as described in “Materials and Methods”.
Table 1.
Effects of perifosine on cell cycle progression
| Cell lines | Treatments | Sub G1 % (SD) |
G1 % (SD) |
S % (SD) |
G2/M % (SD) |
|---|---|---|---|---|---|
| RMG1 | Control | 0.2 (±0.06) | 59.0 (±0.30) | 13.8 (±0.46) | 27.0 (±0.32) |
| Perifosine 30 μM | 9.2** (±0.76) | 71.3** (±2.18) | 9.3 (±1.00) | 10.2 (±1.50) | |
| RMG2 | Control | 0.2 (±0.06) | 70.6 (±0.66) | 9.1 (±0.18) | 20.1 (±0.46) |
| Perifosine 30 μM | 10.6** (±1.56) | 74.1* (±1.70) | 6.8 (±0.50) | 8.5 (±2.18) |
p<0.05,
p < 0.01, as compared with control
AKT activation as a predictor of perifosine sensitivity
It has been previously reported that the basal levels of phosphorylation of AKT correlate with sensitivity of cancer cells to PI3K-AKT inhibitors (7-12). Thus, we investigated whether AKT activity is associated with the sensitivity of CCC cells to perifosine. As shown in Fig. 3A, a clear differential sensitivity was demonstrated depending on the phospho-AKT status. OVCAR4 and OVCAR5 that have low AKT activity were insensitive to perifosine, versus the RMG1 cells with AKT activity showed high sensitivity to perifosine. To further investigate the potential of phospho-AKT expression as a biomarker to predict the sensitivity to perifosine, we next established CCC cell lines stably transfected constitutively active AKT2, which resulted in constitutive phosphorylation of AKT under serum-starvation conditions (Fig. 3B). As predicted, CCC cell lines stably transfected with HA-Myr-AKT2 showed relatively lower sensitivity to cisplatin compared to those transfected with empty vector (pcDNA3) (Fig. 3C (i)). However, as shown, overexpression of HA-Myr-AKT2 resulted and higher sensitivity to perifosine (Fig. 3C (ii)). Moreover, inhibition of AKT by the overexpression of kinase-dead AKT decreased the AKT phosphorylation levels, and resulted in decreased sensitivity to perifosine (Fig. 3D). These results suggest that AKT activity may be a biomarker to predict sensitivity of CCC cells to perifosine. These results suggest that AKT activity may be a biomarker to predict sensitivity of CCC cells to perifosine.
Figure 3. AKT activation as a predictor of perifosine sensitivity.
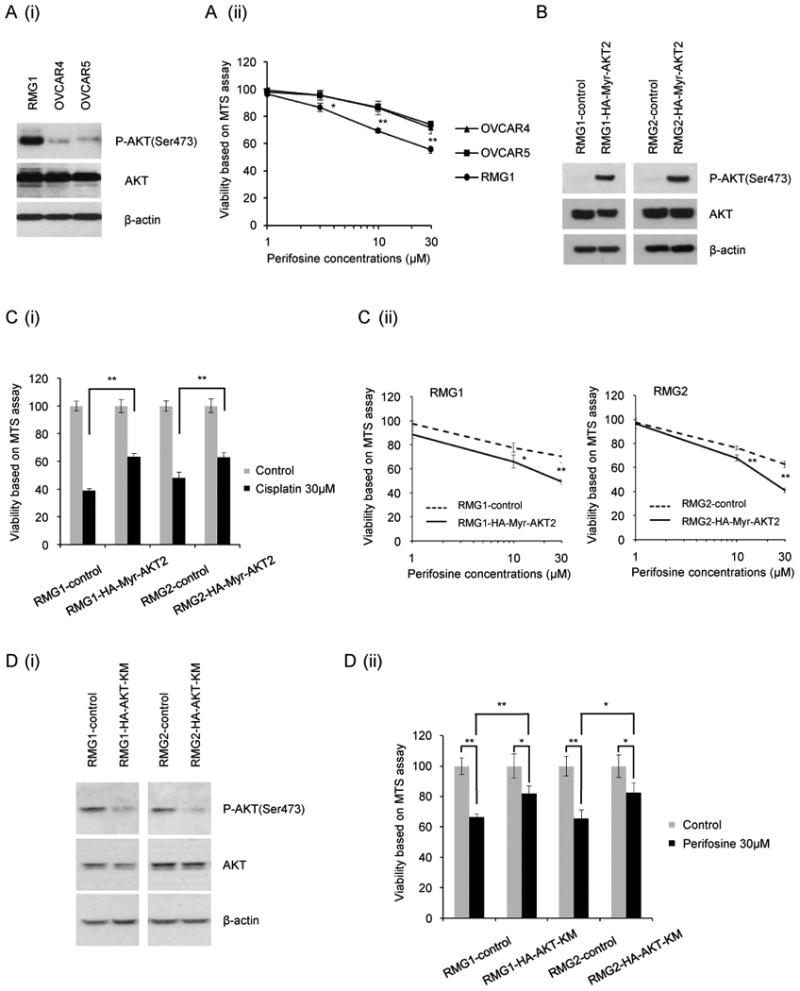
A, Sensitivity to perifosine according to AKT activation status. Ovarian cancer cells with differential AKT activation were treated with the indicated concentrations of perifosine and incubated in medium containing 10% FBS for 48 h. Cell viability was assessed by an MTS assay as described in the “Materials and Methods”. (i), AKT activation status. (ii), Cell viability was assessed by an MTS assay. The experiment was performed six times. Data are shown as the mean of six experiments. B, Activation status in CCC cells after transfection with control vector (pcDNA3) or constitutively active AKT2 (HA-Myr-AKT2). Note that in panel B, the .cells were harvested after serum-starvation overnight. Western blotting was carried out with anti-phospho-AKT (Ser473), anti-AKT, or anti-β-actin antibody. C, Sensitivity to cisplatin (i) or perifosine (ii) according to AKT activation status. CCC cells stably transfected with control vector (pcDNA3) or constitutively active AKT2 (HA-Myr-AKT2) were treated with the indicated concentrations of perifosine and incubated in medium containing 10% FBS for 48 h. Cell viability was assessed by an MTS assay. D, (i), Activation status in CCC cells after transfection with control vector (pCMV6) or kinase-dead AKT (HA-AktK179M). (ii), Sensitivity to perifosine according to AKT activation status. CCC cells stably transfected with control vector (pCMV6) or kinase-dead AKT (HA-AktK179M) were treated with the indicated concentrations of perifosine and incubated in medium containing 10% FBS for 48 h. Cell viability was assessed by an MTS assay. *, P< 0.05.
Perifosine inhibits tumor growth in a subcutaneous xenograft model
We next examined the growth-inhibitory effect of perifosine on ovarian CCC cells in vivo. As shown in Fig. 4A, treatment with perifosine decreased tumor burden compared to placebo, indicating that perifosine has a marked anti-tumor activity, even when used as a monotherapy. Importantly, drug treatment was well tolerated, with no apparent toxicity throughout the study. To investigate the mechanism by which perifosine inhibits the growth of tumors derived from subcutaneously-inoculated human ovarian CCC cell, tumors harvested from the placebo- or perifosine-treated mice were evaluated immunohistochemically. As shown in Fig. 4B, strong immunoreactivity for phosphorylated AKT was detected in tumors from mice treated with placebo but was reduced in tumors from mice treated with perifosine. These data suggest that the growth inhibitory effect of perifosine in vivo is associated with inhibition of AKT-signaling, confirming appropriate drug targeting in vivo. Given that the AKT-mTOR pathway is known to stimulate tumor-related angiogenesis, we next examined the anti-angiogenic activity of perifosine both in vitro and in vivo. As shown, treatment of hypoxic RMG2 cells with perifosine attenuated the expression of VEGF (Fig. 4C). Moreover, treatment with perifosine significantly inhibited the proliferation and the tube formation activity of the HUVEC in vitro (Fig. 4D). Consistent with these findings, as shown in Fig. 4E, large CD31-immunopositive vessels were observed in tumors from placebo-treated mice, whereas the CD31-immunopositive vessels were smaller and fewer in tumors from perifosine-treated mice. Collectively, these results indicate that the anti-tumor effect of perifosine is associated, at least in part, with inhibition of tumor angiogenesis.
Figure 4. Effect of perifosine on the growth of CCC-derived tumor cells in vivo.
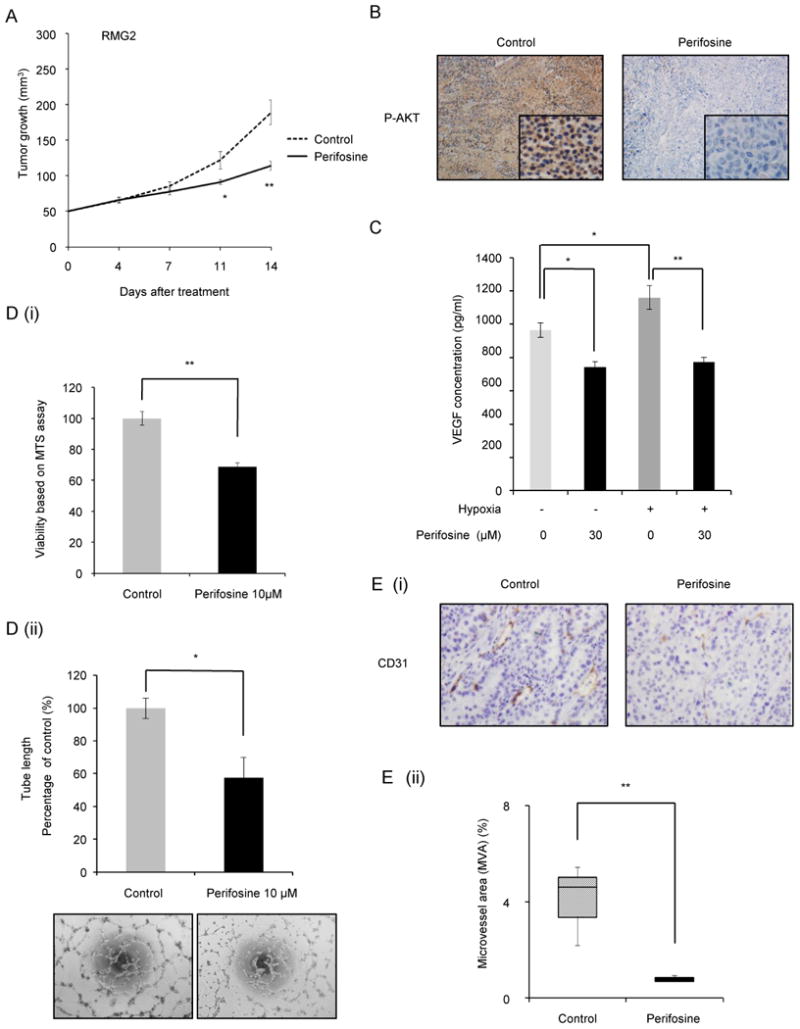
A, Graph depicting weekly tumor volumes. Athymic nude mice were inoculated s.c. with RMG2 cells. When the tumors reached an average size of about 50 mm3, mice were treated with placebo or perifosine for 2 wks. Points, mean; bars, SD. *, P< 0.05, **, P<0.01 significantly different from placebo-treated mice. B, Effect of perifosine on the expression of phospho-AKT in vivo. Representative subcutaneous tumors from placebo- or perifosine-treated mice were excised, and stained with anti-phospho-AKT antibody. C, RMG2 cells were treated with indicated concentrations of perifosine and incubated for 24 h under nonhypoxic (20% O2) or hypoxic (1% O2) conditions. The conditioned medium was collected, and the VEGF concentrations were determined by ELISA. D, (i), Anti-proliferative activity of perifosine on HUVEC in vitro. HUVEC were cultured with indicated concentration of perifosine in the presence of 2% FBS for 24 h. Cell viability was assessed by MTS assay. (ii), Anti-angiogenic activity of perifosine determined using the tube formation assay. Three random fields per sample were recorded, and the tube length of every field was measured. Each experiment was performed at least three times, and data from one representative experiment are shown. E, Effect of perifosine on tumor-angiogenesis. (i) Serial sections of subcutaneous tumors were stained with anti-CD31 antibody, and representative photographs are shown. Note the decreased microvessel staining (dark brown) in the representative perifosine-treated tumor compared with representative placebo-treated tumor. (ii) A box blot indicating CD-31 positive microvesel area (MVA) of placebo- or perifosine-treated tumors. Columns, mean; bars, SD. **, P<0.01.
Anti-tumor effect of perifosine on cisplatin-resistant cell lines
Cisplatin-resistance is regarded as a major clinical problem in the management of CCC of the ovary, both in front-line and recurrent settings (6). Relative platinum-resistance of CCC cells compared to SAC cells has been demonstrated in preclinical studies (24, 25). It has been previously reported that hyperactivation of AKT is involved in the resistance of ovarian SAC cells to cisplatin (26), however, whether AKT is involved in resistance to cisplatin in CCC remains unknown. We then established cisplatin-resistant sublines from RMG1 and RMG2 cells (Fig. 5A), and investigated the activity of AKT in both cisplatin-resistant sublines and parental cisplatin-sensitive cells by western blotting. As shown in Fig. 5B, higher levels of phospho-AKT were observed in both cisplatin-resistant cell lines compared with their respective parental cell lines.
Figure 5. Effect of perifosine on cisplatin-resistant CCC.
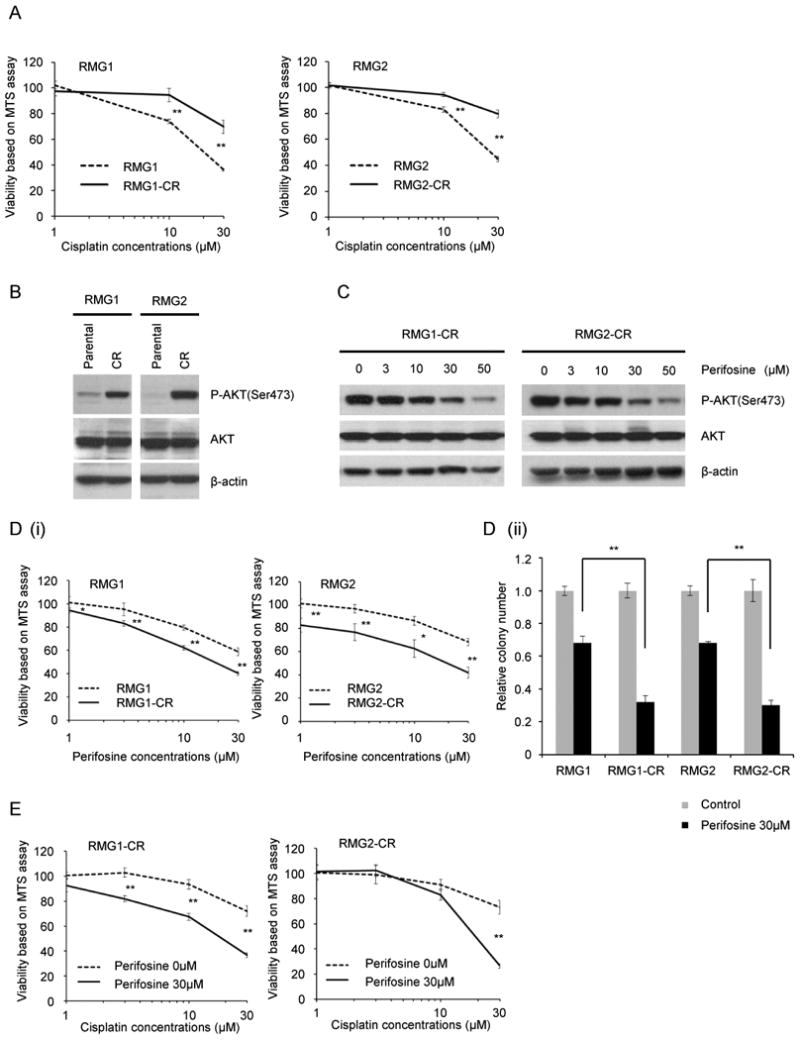
A, Establishment of cisplatin-resistant variant cell lines. Cisplatin-resistant sublines were established as described in “Materials and Methods.” Cisplatin-sensitive parental (RMG1 and RMG2) and cisplatin-resistant variant (RMG1-CR and RMG2-CR) cells were treated with the indicated concentrations of cisplatin in the presence of 10% FBS for 48 h. Cell viability was assessed by MTS assay. Points, mean; bars, SD (**, P<0.01). B, Activation of AKT in cisplatin-sensitive parental and cisplatin-resistant variant cells in vitro. RMG1, RMG1-CR, RMG2 and RMG2-CR cells were serum starved overnight. Cells were harvested, and equivalent amounts (30 μg) of protein were subjected to SDS-PAGE and blotted with anti-phospho-AKT (Ser473), anti-AKT, or anti-β-actin antibodies. C, Perifosine attenuates the phosphorylation of AKT in cisplatin-resistant CCC cells. RMG1-CR and RMG2-CR cells were treated with the indicated concentrations of perifosine for 6 h in the presence of 10% FBS. Cells were harvested, and equivalent amounts (30 μg) of protein were subjected to SDS-PAGE and blotted with anti-phospho-AKT (Ser473), anti-AKT, or anti-β-actin antibodies. D, Enhanced sensitivity to perifosine in cisplatin-resistant CCC cells in vitro. Cisplatin-sensitive parental (RMG1 and RMG2) and cisplatin-resistant variant (RMG1-CR and RMG2-CR) cells were treated with the indicated concentrations of perifosine in the presence of 10% FBS for 48 h. Cell viability was assessed by MTS (i) or by clonogenic survival assays (ii). Points, mean; bars, SD (*, p<0.05, **, P<0.01). E, Perifosine enhances the therapeutic efficacy of cisplatin in CCC cells. RMG1-CR and RMG2-CR cells were treated with various concentrations of cisplatin with or without 30 μM perifosine in the presence of 10% FBS. Cell viability was assessed by MTS assay. Points, mean; bars, SD (*, P<0.05, **, P<0.01).
Since increased AKT activity resulted in enhanced sensitivity of CCC cells to perifosine (Fig. 3C), we considered cisplatin-resistant sublines to be good candidates for treatment with perifosine. Thus, we next examined the inhibitory effect of perifosine on cisplatin-resistant and parental cisplatin-sensitive CCC cell lines. We first confirmed that treatment with perifosine effectively inhibited phosphorylation of AKT in vitro (Fig. 5C). We next examined the inhibitory effect of perifosine on CCC cell viability by MTS assay, which revealed a clear differential effect dependent on sensitivity to cisplatin (Fig. 5D). Cisplatin-resistant RMG1-CR and RMG2-CR cells were found to be significantly more sensitive to perifosine than their respective parental cell lines RMG1 and RMG2. Since RMG1-CR and RMG2-CR are insensitive to cisplatin treatment (Fig. 5A), perifosine may hold promise for the treatment for recurrent CCCs developing after cisplatin treatment.
We also used RMG1-CR and RMG2-CR cells to determine whether treatment with perifosine can sensitize RMG1-CR and RMG2-CR cells to cisplatin. As shown in Fig. 5E, in the presence of 30 μM of perifosine, the ability of cisplatin to inhibit cell proliferation and survival was significantly enhanced in both cell lines.
Activity of perifosine on the growth of bevacizumab-resistant CCCs
Based on recent phase III clinical trials (3,4), bevacizumab combined with carboplatin plus paclitaxel is becoming one of the standard front-line treatments for patients with advanced epithelial ovarian cancer. However, in these studies, improvement in PFS by the addition of bevacizumab was only about 4 months (3,4). Thus, development of improved treatments for recurrent cancers following bevacizumab treatment are urgently needed. Thus, we next decided to evaluate the anti-tumor efficacy of perifosine on CCC xenografts following bevacizumab treatment. As shown in Fig. 6A, treatment with bevacizumab significantly decreased CCC tumor burden. However, when we continued the bevacizumab treatment, after roughly 3 weeks of exposure, the subcutaneous tumors started to grow rapidly, presumably as a result of acquired resistance to bevacizumab (Fig. 6A). Thus, in a subsequent experiment, after 3 weeks of bevacizumab treatment, mice were assigned into two treatment groups receiving either bevacizumab or perifosine alone. We found that when treatment was switched from bevacizumab to perifosine after the initial treatement with bevacizumab for 3 weeks, growth of the subcutaneous tumors was significantly inhibited (Fig. 6B). These results indicate that perifosine may have therapeutic efficacy for recurrent CCCs after bevacizumab treatment.
Figure 6. In vivo growth-inhibitory effect of perifosine on bevacizumab-resistant CCC.
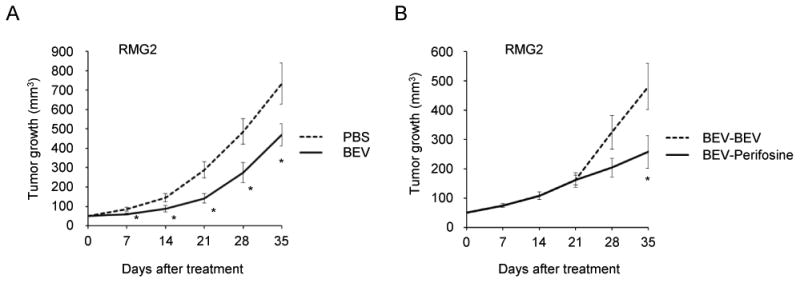
A, The effect of bevacizumab (BEV) on CCC growth in vivo. Athymic nude mice inoculated s.c. with RMG2 cells were assigned into two treatment groups; bevacizumab (n=5) or placebo (n=5). B, The effect of perifosine on CCC that had acquired resistance to bevacizumab. Athymic nude mice that were inoculated s.c. with RMG2 cells and received bevacizumab treatment for 3 wks. Then, the mice were assigned into two treatment groups; bevaizumab (n=5) or perifosine (n=5). Tumor volumes were monitored weekly, and growth curves of each treatmen groups are shown. Points, mean; bars, SD (*, P<0.05).
Discussion
Despite advances in platinum-based combination chemotherapy, patients with CCC of the ovary, especially in advanced stage or recurrent disease, have a worse PFS and overall survival when compared with patients with a serous histology (6). Thus, to improve survival of ovarian CCC patients, novel treatments need to be developed.
One possible strategy to inhibit tumor progression and to enhance the efficacy of platinum-based chemotherapy in CCC is to target anti-apoptotic signals, such as those mediated by the AKT pathway. AKT is known to regulate various cellular pathways that promote cell survival, cell proliferation, angiogenesis, and invasion (7,8). We and others have previously reported that AKT is frequently activated in epithelial ovarian cancer and that inhibition of AKT activity by PI3K inhibitors significantly inhibits cell proliferation (12) and enhances the activity of conventional anticancer agents, including cisplatin (12,26) and paclitaxel (11) in preclinical models of ovarian cancer. A more recent investigation suggested that phospho-AKT expression correlated with the overall survival and the platinum-response in ovarian cancer patients (27). However, since most tumor specimens and tumor-derived cell lines used in these investigations have been ovarian SACs, the role of AKT as a therapeutic target in CCC has remained unknown.
The Cancer Genome Atlas (TCGA) Research Network has recently reported activation of the PI3K/AKT pathway in high-grade serous ovarian cancer in approximately 40 percent of these tumors, mostly through somatic copy number alterations and not through point mutations (28). In contrast, in ovarian CCC, a previous study revealed activating mutations of the PIK3CA gene in approximately 40% of cases (13), which is among the most frequent in solid malignancies of various origins. Moreover, previous investigations indicated that mTORC1 (a downstream effector of AKT) and mTORC2 (an upstream stimulator of AKT) are frequently activated in CCC of the ovary, and in vitro and in vivo studies have suggested that both mTORC1 and mTORC2 are promising therapeutic targets in CCC (19, 21, 25, 29). Collectively, these results strongly indicate that inhibition of the AKT pathway is a promising strategy for the clinical management of ovarian carcinomas of the CCC type.
Perifosine is an orally bioavailable AKT inhibitor that is currently being evaluated in phase I/II clinical studies in a variety of solid tumors (16), and this drug has shown encouraging clinical activity when used in combination with capecitabine in patients with metastatic colorectal cancer (30). In ovarian cancer, a phase II trial designed to evaluate the efficacy of perifosine in combination with docetaxel was conducted in the recurrent setting (31). Although satisfactory efficacy of perifosine could not be demonstrated in this study, treatment with perifosine plus docetaxel appeared to be more effective in those cases in which the PI3K/AKT pathway was mutationally activated (31), suggesting that clinical activity of perifosine should be tested in patients whose tumors display activated AKT signaling in future clinical trials.
CCC patients have a high frequency of PIK3CA mutations (13), mTORC2 activation (15), and loss of PTEN expression (14). Consistent with these previous findings, the frequent AKT activation was observed in CCCs in the current study: the expression of phospho-AKT was observed in both stage III-IV CCCs (68%) and stage I-II CCCs (70%). Consistent with previous findings using other PI3K-AKT inhibitors (Fig. 2), perifosine treatment had more robust anti-tumor activity in CCC cells with high AKT activity than in cells with low AKT activity. Moreover, treatment of CCC cells with perifosine significantly enhanced the anti-tumor effect of cisplatin (Fig. 4E). Taken together, these findings indicate that AKT is a rational target for the treatment of CCC of the ovary. Moreover, our results indicate that an AKT inhibitor, such as perifosine, could have significant anti-tumor effects as a single agent or in combination with cisplatin for both previously untreated CCCs and recurrent CCCs developing after cisplatin treatment.
Perifosine has shown significant anti-tumor activity in a genetically engineered mouse model of ovarian endometrioid adenocarcinoma displaying hyperactivation of AKT (32). Perifosine also showed significant antiproliferative activity and enhanced the activity of cisplatin in cisplatin-resistant SAC cells with elevated AKT activity in vitro (33). Moreover, in the current study, CCC cells and SAC cells exhibiting similar levels of AKT activation showed similar sensitivity to perifosine (Fig. 1E). These results suggest that ovarian cancers exhibiting hyperactivation of AKT would represent good candidates for AKT inhibition therapy irrespective of histological subtypes.
An additional important finding of our study is the potent anti-tumor activity of perifosine in recurrent CCC following bevacizumab treatment (Fig. 5C). Given that ovarian cancer patients treated with bevacizumab in combination with standard platinum-based chemotherapy experienced disease progression with a median progression free interval of 4 months in a phase III study (3,4), it would be very important to explore the salvage treatment for recurrent tumors that develop after bevacizumab treatment. Since the experimental in vivo model used in this study mimics resistance development in bevacizumab-treated patients, our results suggest that AKT inhibitors might be efficacious for the clinical management of recurrent CCCs following treatment with bevacizumab.
The limitations of our study need to be addressed. Although our in vitro investigations suggested that AKT is involved in the cisplatin-resistance in CCC of the ovary (Fig. 5), due to the limited clinical data, we could not investigate the association between immunoreactivity for phospho-AKT and the tumor response to platinum-based chemotherapies. Another potential weakness is our experimental design, subcutaneous xenograft model. Peritoneal dissemination is the main process of progression in human ovarian cancer, thus an orthotopic xenograft model of clear cell ovarian cancer may accurately model advanced disease. Although we tried to examine the anti-tumor effect of perifoine in an orthotopic model, the CCC cells lines used in the current study did not developed ovarian tumors in nude mice (data not shown). To validate the results from the current study and make definitive conclusions, further investigations may be needed.
In summary, the results presented here indicate that AKT is frequently activated in ovarian CCC and is a promising therapeutic target for this disease both as a front-line therapy and as a salvage treatment for recurrence after cisplatin or bevacizumab treatment. This work provides scientific rationale for future clinical trials of AKT-targeting therapies in patients with ovarian CCC, a chemoresistant histological subtype characterized by frequent activation of the AKT pathway.
Acknowledgments
This work was supported by grant-in-aid for General Scientific Research, from the Ministry of Education, Culture, Sports, Science and Technology of Japan (no. 26462523). J.R.T. received support from NCI Grant R01 CA77429.
Abbreviations
- CCC
clear cell carcinoma
- SAC
serous adenocarcinoma
- mTOR
mammalian target of rapamycin
- mTORC
mTOR complexes
- S6K1
S6 kinase 1
- IHC
immunohistochemistry
- PBS
phosphate buffered saline
- PFS
progression free survival
Footnotes
Conflicts of interest statement: The authors declare that they have no conflicts of interest to disclose.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 63(1):11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Ozols RF, Bundy BN, Greer BE, Fowler JM, Clarke-Pearson D, Burger RA, et al. Gynecologic Oncology Group. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2003;21:3194–200. doi: 10.1200/JCO.2003.02.153. [DOI] [PubMed] [Google Scholar]
- 3.Burger RA, Brady MF, Bookman MA, Fleming GF, Monk BJ, Huang H, et al. Gynecologic Oncology Group. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365:2473–83. doi: 10.1056/NEJMoa1104390. [DOI] [PubMed] [Google Scholar]
- 4.Perren TJ, Swart AM, Pfisterer J, Ledermann JA, Pujade-Lauraine E, Kristensen G, et al. ICON7 Investigators. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365:2484–96. doi: 10.1056/NEJMoa1103799. [DOI] [PubMed] [Google Scholar]
- 5.Serov SF, Scully RE, Sobin LH. Histologic Typing of Ovarian Tumors. Vol. 9. Geneva: World Health Organization; 1973. International histologic classification of tumors. [Google Scholar]
- 6.Del Carmen MG, Birrer M, Schorge JO. Clear cell carcinoma of the ovary: A review of the literature. Gynecol Oncol. 2012;126:481–90. doi: 10.1016/j.ygyno.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 7.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 8.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–62. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 9.Cheng JQ, Godwin AK, Bellacosa A, Taguchi T, Franke TF, Hamilton TC, et al. AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomas. Proc Natl Acad Sci U S A. 1992;89:9267–71. doi: 10.1073/pnas.89.19.9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altomare DA, Wang HQ, Skele KL, De Rienzo A, Klein-Szanto AJ, Godwin AK, et al. AKT and mTOR phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene. 2004;23:5853–7. doi: 10.1038/sj.onc.1207721. [DOI] [PubMed] [Google Scholar]
- 11.Mabuchi S, Ohmichi M, Kimura A, Hisamoto K, Hayakawa J, Nishio Y, et al. Inhibition of phosphorylation of BAD and Raf-1 by Akt sensitizes human ovarian cancer cells to paclitaxel. J Biol Chem. 2002;277:33490–500. doi: 10.1074/jbc.M204042200. [DOI] [PubMed] [Google Scholar]
- 12.Ohta T, Ohmichi M, Hayasaka T, Mabuchi S, Saitoh M, Kawagoe J, et al. Inhibition of phosphatidylinositol 3-kinase increases efficacy of cisplatin in in vivo ovarian cancer models. Endocrinology. 2006;147:1761–9. doi: 10.1210/en.2005-1450. [DOI] [PubMed] [Google Scholar]
- 13.Kuo KT, Mao TL, Jones S, Veras E, Ayhan A, Wang TL, et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol. 2009;174:1597–601. doi: 10.2353/ajpath.2009.081000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hashiguchi Y, Tsuda H, Inoue T, Berkowitz RS, Mok SC. PTEN expression in clear cell adenocarcinoma of the ovary. Gynecol Oncol. 2006;101:71–75. doi: 10.1016/j.ygyno.2005.09.047. [DOI] [PubMed] [Google Scholar]
- 15.Hisamatsu T, Mabuchi S, Matsumoto Y, Kawano M, Sasano T, Takahashi R, et al. Potential Role of mTORC2 as a Therapeutic Target in Clear Cell Carcinoma of the Ovary. Mol Cancer Ther. 2013;12:1367–77. doi: 10.1158/1535-7163.MCT-12-1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gills JJ, Dennis PA. Perifosine: update on a novel Akt inhibitor. Curr Oncol Rep. 2009;11:102–10. doi: 10.1007/s11912-009-0016-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Al Sawah E, Chen X, Marchion DC, Xiong Y, Ramirez IJ, Abbasi F, et al. Perifosine, an AKT inhibitor, modulates ovarian cancer cell line sensitivity to cisplatin-induced growth arrest. Gynecol Oncol. 2013;131:207–12. doi: 10.1016/j.ygyno.2013.07.088. [DOI] [PubMed] [Google Scholar]
- 18.Sun H, Yu T, Li J. Co-administration of perifosine with paclitaxel synergistically induces apoptosis in ovarian cancer cells: more than just AKT inhibition. Cancer Lett. 2011;310:118–28. doi: 10.1016/j.canlet.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 19.Mabuchi S, Kawase C, Altomare DA, Morishige K, Sawada K, Hayashi M, et al. mTOR is a promising therapeutic target both in cisplatin-sensitive and cisplatin-resistant clear cell carcinoma of the ovary. Clin Cancer Res. 2009;15:5404–13. doi: 10.1158/1078-0432.CCR-09-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mabuchi S, Ohmichi M, Kimura A, Ikebuchi Y, Hisamoto K, Arimoto-Ishida E, et al. Tamoxifen inhibits cell proliferation via mitogen-activated protein kinase cascades in human ovarian cancer cell lines in a manner not dependent on the expression of estrogen receptor or the sensitivity to cisplatin. Endocrinology. 2004;145:1302–13. doi: 10.1210/en.2003-0709. [DOI] [PubMed] [Google Scholar]
- 21.Mabuchi S, Kawase C, Altomare DA, Morishige K, Hayashi M, Sawada K, et al. Vascular endothelial growth factor is a promising therapeutic target for the treatment of clear cell carcinoma of the ovary. Mol Cancer Ther. 2010;9:2411–22. doi: 10.1158/1535-7163.MCT-10-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanno S, Tanno S, Mitsuuchi Y, Altomare DA, Xiao GH, Testa JR. AKT activation up-regulates insulin-like growth factor I receptor expression and promotes invasiveness of human pancreatic cancer cells. Cancer Res. 2001;61:589–93. [PubMed] [Google Scholar]
- 23.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 24.Itamochi H, Kigawa J, Sultana H, Iba T, Akeshima R, Kamazawa S, et al. Sensitivity to anticancer agents and resistance mechanisms in clear cell carcinoma of the ovary. Jpn J Cancer Res. 2002;93:723–8. doi: 10.1111/j.1349-7006.2002.tb01312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mabuchi S, Hisamatsu T, Kawase C, Hayashi M, Sawada K, Mimura K, et al. The activity of trabectedin as a single agent or in combination with everolimus for clear cell carcinoma of the ovary. Clin Cancer Res. 2011;17:4462–73. doi: 10.1158/1078-0432.CCR-10-2987. [DOI] [PubMed] [Google Scholar]
- 26.Mabuchi S, Ohmichi M, Nishio Y, Hayasaka T, Kimura A, Ohta T, et al. Inhibition of NFkappaB increases the efficacy of cisplatin in in vitro and in vivo ovarian cancer models. J Biol Chem. 2004;279:23477–85. doi: 10.1074/jbc.M313709200. [DOI] [PubMed] [Google Scholar]
- 27.Al Sawah E, Chen X, Marchion DC, Xiong Y, Ramirez IJ, Abbasi F, et al. Perifosine, an AKT inhibitor, modulates ovarian cancer cell line sensitivity to cisplatin-induced growth arrest. Gynecol Oncol. 2013;131:207–12. doi: 10.1016/j.ygyno.2013.07.088. [DOI] [PubMed] [Google Scholar]
- 28.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mabuchi S, Hisamatsu T, Kimura T. Targeting mTOR signaling pathway in ovarian cancer. Curr Med Chem. 2011;18:2960–8. doi: 10.2174/092986711796150450. [DOI] [PubMed] [Google Scholar]
- 30.Bendell JC, Nemunaitis J, Vukelja SJ, Hagenstad C, Campos LT, Hermann RC, et al. Randomized placebo-controlled phase II trial of perifosine plus capecitabine as second- or third-line therapy in patients with metastatic colorectal cancer. J Clin Oncol. 2011;29:4394–400. doi: 10.1200/JCO.2011.36.1980. [DOI] [PubMed] [Google Scholar]
- 31.Fu S, Hennessy BT, Ng CS, Ju Z, Coombes KR, Wolf JK, et al. Perifosine plus docetaxel in patients with platinum and taxane resistant or refractory high-grade epithelial ovarian cancer. Gynecol Oncol. 2012;126:47–53. doi: 10.1016/j.ygyno.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu R, Hu TC, Rehemtulla A, Fearon ER, Cho KR. Preclinical testing of PI3K/AKT/mTOR signaling inhibitors in a mouse model of ovarian endometrioid adenocarcinoma. Clin Cancer Res. 2011;17:7359–72. doi: 10.1158/1078-0432.CCR-11-1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Engel JB, Schönhals T, Häusler S, Krockenberger M, Schmidt M, Horn E, et al. Induction of programmed cell death by inhibition of AKT with the alkylphosphocholine perifosine in in vitro models of platinum sensitive and resistant ovarian cancers. Arch Gynecol Obstet. 2011;283:603–10. doi: 10.1007/s00404-010-1457-6. [DOI] [PubMed] [Google Scholar]