

Abstract
CLC transporters catalyze the exchange of Cl- for H+ across cellular membranes. To do so, they must couple Cl- and H+ binding and unbinding to protein conformational change. However, the sole conformational changes distinguished crystallographically are small movements of a glutamate side chain that locally gates the ion-transport pathways. Therefore, our understanding of whether and how global protein dynamics contribute to the exchange mechanism has been severely limited. To overcome the limitations of crystallography, we used solution-state 13C-methyl NMR with labels on methionine, lysine, and engineered cysteine residues to investigate substrate (H+) dependent conformational change outside the restraints of crystallization. We show that methyl labels in several regions report H+-dependent spectral changes. We identify one of these regions as Helix R, a helix that extends from the center of the protein, where it forms the part of the inner gate to the Cl--permeation pathway, to the extracellular solution. The H+-dependent spectral change does not occur when a label is positioned just beyond Helix R, on the unstructured C-terminus of the protein. Together, the results suggest that H+ binding is mechanistically coupled to closing of the intracellular access-pathway for Cl-.
Keywords: Nuclear magnetic resonance, Conformational change, Reductive methylation Lysine, Methionine, Chemical modification
Introduction
Members of the CLC (“Chloride-Channel”) family play central roles in cardiovascular, neuronal, bone, and epithelial function (Denton et al. 2013; Mindell 2012; Stauber et al. 2012; Tang and Chen 2011). Despite its name, the family consists of both passive Cl- channels and active Cl-/H+ exchangers (antiporters) (Accardi and Miller 2004; Chen and Hwang 2008; Graves et al. 2008; Picollo and Pusch 2005; Scheel et al. 2005). Channels form passive pores, allowing ions to move down their electrochemical gradients. Conversely, antiporters can harness energy from movement of one ion down its electrochemical gradient to drive the other ion up its electrochemical gradient. Despite this dichotomy of energetics, it is now clear that CLC channels and transporters are similar in many aspects of their molecular mechanisms (Accardi and Picollo 2010; Lisal and Maduke 2008; Lisal and Maduke 2009; Miller 2006). Thus, studies of CLCs hold relevance for both major classes of membrane-transport proteins.
CLC channels have been studied electrophysiologically for over 30 years (Maduke et al. 2000; Miller 2014; Zifarelli and Pusch 2007), but crystallographic structures are available only for antiporters. CLC antiporters catalyze the stoichiometric exchange of two Cl- for a single H+. X-ray crystallographic structures, together with experimentation and computation, have provided invaluable information on details of Cl- coordination, H+-transport pathways, and the existence of “gates” that occlude access of bound Cl- from the intracellular and extracellular solutions (Figure 1a) (Accardi et al. 2006; Accardi and Miller 2004; Accardi et al. 2005; Basilio et al. 2014; Cheng and Coalson 2012; Dutzler 2006; Dutzler et al. 2002; Dutzler et al. 2003; Faraldo-Gomez and Roux 2004; Feng et al. 2010; Han et al. 2014; Jayaram et al. 2008; Jayaram et al. 2011; Ko and Jo 2010; Kuang et al. 2007; Lim and Miller 2009; Lim and Miller 2012; Walden et al. 2007; Zhang and Voth 2011). According to the alternating access mechanism for active transport, opening and closing of these gates must be tightly coupled to ion binding and unbinding events (Jardetzky 1966; Law et al. 2008; Tanford 1983). Several models describing proposed details of this CLC transport cycle have been published (Basilio et al. 2014; Feng et al. 2010; Lim et al. 2013; Miller and Nguitragool 2009), but our knowledge remains incomplete, in part because structural details of the conformational states are incomplete. CLC inner-gate opening has been inferred by crosslinking studies (Basilio et al. 2014), but not yet detected crystallographically. Outer-gate opening has been detected both functionally (Accardi and Miller 2004) and crystallographically (Dutzler et al. 2003). Surprisingly, the outer-gate opening observed crystallographically involves only the highly localized movement of a glutamate side chain, with no concomitant global conformational change (Figure 1b). This is in contrast to the large conformational changes detected in other active transporters as they interconvert between outward-facing, occluded, and inward-facing conformational states (Forrest et al. 2011; Forrest and Rudnick 2009; Shi 2013; Slotboom 2014) (Figure 1c). Despite many crystallization attempts, no global CLC transporter conformational change has been observed (Accardi et al. 2006; Dutzler et al. 2002; Dutzler et al. 2003; Feng et al. 2010; Jayaram et al. 2011; Lim et al. 2012; Lim et al. 2013; Robertson et al. 2010). Therefore, other biophysical approaches for studying CLC protein conformations are in order.
Fig. 1. CLC structure and conformational change compared to conformational change detected in other membrane transporters.
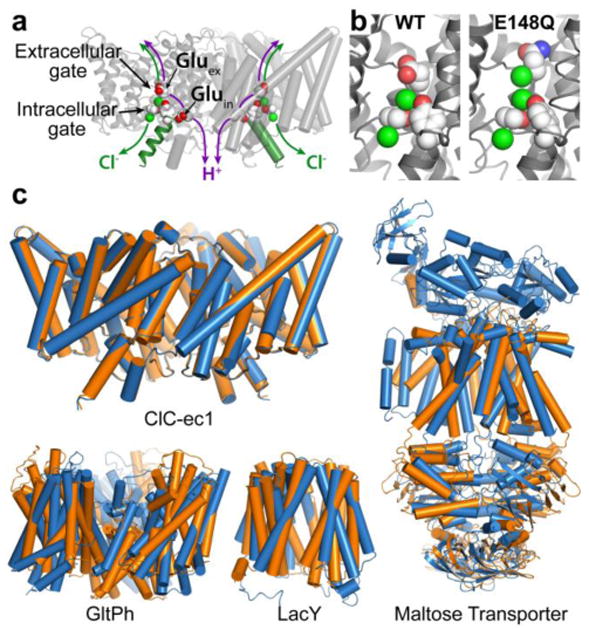
a X-ray crystallographic structure of ClC-ec1 (pdb ID: 1OTS). Each identical subunit catalyzes the exchange of 2 Cl- for 1 H+. The H+ pathway bifurcates from the Cl- pathway, passing along Gluex and Gluin. Gluex is also a “gate” that occludes Cl- from the extracellular solution. An intracellular Cl- gate is formed by a Ser/Tyr pair (shown in spacefill), the latter residing on Helix R (dark green), a subject of study here.
b Structural comparison of WT ClC-ec1 with the Gluex → Gln mutant (E148Q). Left: Close-up of the Cl--binding region in WT and E148Q. In E148Q, the glutamine side chain is flipped up out of the anion-binding site and replaced by a Cl- ion (pdb ID: 1OTU).
c Structural comparison of active transporters crystallized in different conformational states. Top left: Backbone overlay of ClC-ec1 WT (orange) and E148Q (blue) structures, 0.6 Å RMSD, illustrates the lack of global conformational change in the putative outward-facing conformational state. Bottom left: GltPh, a glutamate transporter homolog (Focke et al. 2013) in outward-facing (orange) and inward-facing (blue) states (pdb 4OYE and 4P19) (Verdon et al. 2014). Bottom middle: LacY, a member of the Major Facilitator Superfamily of transporters (Yan 2013) in occluded (orange) and inward-facing (blue) conformational states (pdb 4OAA and 2V8N) (Guan et al. 2007; Kumar et al. 2014). Right: Maltose transporter, a member of the ATP-binding cassette (ABC) transporter superfamily (Chen 2013) in inward-facing (orange) and occluded (blue) states (pdb 3FH6 and 2R6G) (Khare et al. 2009; Oldham et al. 2007).
Previously, we used 19F solution-state NMR to detect conformational change in ClC-ec1 (Elvington et al. 2009), a well-characterized prokaryotic CLC homolog of known structure (Dutzler 2006). We reasoned that the fluorine nucleus' exceptional sensitivity to chemical environment makes it well suited for detecting conformational change outside the restraints of crystallization and that its small size (nearly isosteric to hydrogen) would allow access to core regions of the protein to which larger labels such as fluorophores would be inaccessible or perturbing. Further, the high sensitivity of the 19F NMR signal and lack of endogenous fluorine in proteins would make the measurement feasible even in such a large membrane protein (50 kD per subunit). The general advantages and varied applications of 19F NMR are described in several recent reviews (Chen et al. 2013; Kitevski-LeBlanc and Prosser 2012; Yu et al. 2013).
In our previous work we labeled ClC-ec1's nine Tyr residues using fluoro-tyrosine incorporated biosynthetically in the growth media (Danielson and Falke 1996; Elvington et al. 2009). The resulting one-dimensional spectra showed significant chemical shift dispersion, reflecting 19F-tyrosine residues experiencing different chemical environments within the folded protein. Our strategy to access different conformational states involved varying [Cl-] and [H+]. Such manipulations work to enrich different conformational states in solution because active transporters tightly couple conformational changes to ion binding and unbinding events (Tanford 1983). Therefore, alterations in bulk concentrations of transported ions will alter the equilibria of transporter conformational states (Claxton et al. 2010; Ma et al. 2003; Merianos et al. 2000; Zhao et al. 2011). Manipulation of [H+] and [Cl-] resulted in spectral shifts, reflecting changes in chemical environment of the 19F reporters. In order to simplify regions of spectral overlap in the samples, we next replaced the four solvent-exposed Tyr residues with Phe. This manipulation improved resolution and enabled us to focus on the five buried Tyr residues. Using site-directed mutagenesis, we identified the two buried residues that report substrate-dependent spectral changes (Figure 2a). One of these, Y445, is located at the Cl--permeation pathway, and thus its sensitivity to substrate was expected; the other, Y419, however, is located over 20 Å away, thus suggesting that global and not merely local conformational change is occurring in response to substrate manipulations. These results indicate that solution-state NMR can detect CLC conformational change not observed in the crystal structures and strongly motivate further characterization of these changes.
Fig. 2. Summary of positions isotope-labeled for NMR studies on CLC-ec1.
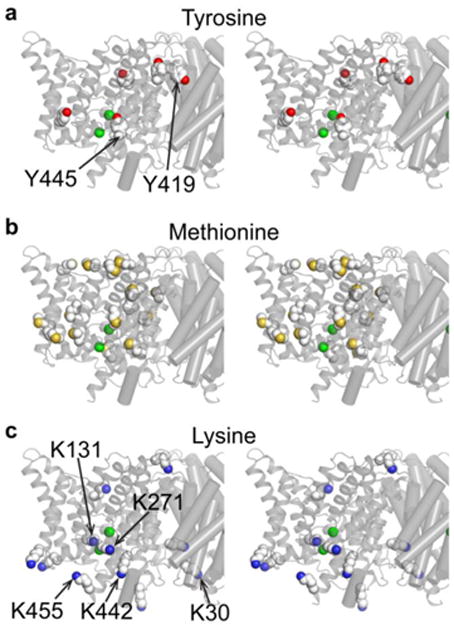
Structures are shown in stereo view, focused in on one subunit of the homodimer.
a In a previous study, the five buried tyrosines were labeled with 3-fluorotyrosine (four solvent-exposed Tyr residues were mutated to Phe). Y445 and Y419 were identified as participating in H+-dependent conformational change (Elvington et al. 2009).
b & c In this study, Met residues (b) were 13C labeled on their methyl groups and Lys residues (c) were post-translationally 13C-dimethylated. The unresolved N- and C-termini of ClC-ec1 contain one Met and 2 Lys residues that are not shown in these diagrams. Residues K30, K131, K271, K442, and K455, examined in mutagenesis studies here, are indicated.
Towards this goal, in the work reported here we applied 13C labeling to our study of ClC-ec1. Since ClC-ec1 is an alpha helical dimer of 50 kD per subunit, we aimed not to attempt the heroic feat of full NMR solution-state structure determination (Montaville and Jamin 2010), but rather to employ sparse labeling strategies that would minimize signal overlap, facilitate peak assignment and allow us to identify additional regions of conformational change. We targeted 13C-methyl groups because the intrinsic sensitivity of three protons coupled to one carbon together with favorable relaxation properties make them well suited to the solution-state study of high-molecular-weight systems (Beatty et al. 1996; Butterfoss et al. 2010; DellaVecchia et al. 2007; Mainz et al. 2013; Religa et al. 2011), including membrane proteins, where size is exacerbated by the surrounding detergent micelle or lipid bicelle (Kang and Li 2011; Kim et al. 2009; Sanders and Sonnichsen 2006). The ability to acquire spectra at low protein concentration is critical in the case of ClC-ec1, which is stable in detergent at a maximal concentration of ∼ 150 μM.
In this study, we specifically targeted 13C-methyl labeling of methionine and lysine residues. The low natural occurrence of Met residues (Brooks et al. 2002) make them attractive as our first 13C-methyl labeling target (ClC-ec1 has 18 native Met residues, compared to 67 Leu, 37 Ile, 30 Val, and 58 Ala). Further, the dispersed location of these 18 Met residues provides reporters in regions not probed in our previous 19F-Tyr labeling studies (Figure 2b). Using a monomeric ClC-ec1 variant (which is 50kD compared to the 100-kD WT ClC-ec1), we were able to obtain dispersed 13C-Met signal and to observe substrate-dependent spectral changes. However, our strategy to assign resonances by mutagenesis was foiled by the reversion to dimeric stoichiometry upon introduction of additional mutations. As an alternative labeling strategy, we added 13C methyl groups to Lys residues on WT ClC-ec1, using post-translational reductive methylation (Larda et al. 2012; Rayment 1997). The introduction of dimethyl groups onto Lys residues preserves the charge and is generally non-perturbing (Gerken et al. 1982; Kurinov et al. 2000; Larda et al. 2012; Rayment 1997; Rypniewski et al. 1993; Walter et al. 2006). The NMR spectra of reductively methylated proteins show good signal-to-noise ratio, with a 13C methyl signal from the Lys being approximately ten times as intense as the signal from an aliphatic methyl group at similar protein concentrations (Abraham et al. 2008). This is possibly due to the unobstructed rotations exhibited by 13C methyl groups attached to the relatively long side chain of Lys residues as well as the usually degenerate chemical shifts of the two 13C methyl groups on each Lys. The fast rotations may overcome the line broadening caused by the slow tumbling rate of large proteins like ClC-ec1. Furthermore, like Met, Lys occurs at relatively low levels in membrane proteins (Tourasse and Li 2000), with only 13 found in ClC-ec1 (out of 473 residues) (Figure 2c). Using 13C-Lys dimethylation, we were able to obtain dispersed spectra and to detect H+-dependent spectral changes in the ClC-ec1 dimer. The residue experiencing the most significant change in environment was identified using a Lys-to-Arg mutagenesis strategy. The location of this residue on Helix R, which forms part of the inner gate (Figure 1a), suggests that H+ binding is mechanistically coupled to either opening or closing of this gate. To further investigate the involvement of Helix R in H+-dependent conformational change, we performed a targeted labeling study of two additional nearby sites, using post-translational reaction of 13C enriched methyl methanethiosulfonate (MMTS) with thiol groups of engineered cysteines (Jaffe and Markham 1988; Religa et al. 2011). While 13C Met and Lys labeling has the advantage of adding NMR probes throughout the protein and offering the opportunity to examine different regions of the protein at the same time, MMTS labeling of engineered cysteines overcomes the limitation of multiple peak overlaps and offers a highly focused analysis of a specific region of interest. The modification of Cys with MMTS results in a S-methylthiocysteine (MTC) moiety, which is similar to the Met residue in side chain volume, and thus is generally non-perturbing. Together, our 13C-MTC and -Lys studies support the hypothesis that conformational change of Helix R is coupled to H+ binding and occurs as part of the CLC transport cycle.
Methods
Protein expression and 13C-Met labeling
Non-isotopically labeled ClC-ec1 constructs (for Lys-methylation and Cys-MTC labeling) were expressed as previously described (Accardi et al. 2004). Expression of specific 13C-Met labeled ClC-ec1 constructs was performed similar to the prior 19F NMR labeling, as follows. E. coli BL21 (DE3) competent cells (∼ 100 μL) were transformed with 6-His tagged ClC-ec1 in pASK-IBA2 (Accardi et al. 2004) (∼ 1 μg plasmid DNA) by heat shock at 42 °C for 45 sec. The cells were then added to 300 μL LB media and incubated at 37 °C for 30-60 min. Approximately 150 μL of these transformed cells were plated onto LB-agar plates containing 100 μg/mL ampicillin and then incubated at 37 °C overnight. On the next day, all colonies from the agar plates were scraped into 1 L of the nutrient-rich media, Terrific Broth (TB) (Sambrook et al. 1989) in baffled 2.8 L Fernbach flasks. One plate of cells was used for each liter of culture to be grown; for most NMR sample preparations, 6 L culture was grown. The culture was allowed to grow at 37 °C with vigorous aeration (∼220 rpm) until it reached an OD550 of 1.0 ± 0.1, and then it was pelleted by centrifugation (1720 g for 20 min at 4 °C). For every 2 L of culture in TB, the pellet was resuspended in 1 L of minimal media with M9 salts (Sambrook et al. 1989), 0.4% glycerol, 1 mM MgSO4, 2.7 mM CaCl2, 0.5 mg/L thiamine, 100 mg/L ampicillin, and 50 mg/L of each of each L-amino acid (except Met) (Sigma Aldrich). Isotopic labeling of Met residues was achieved by adding 50 mg/L 13Cε-L-Met (Cambridge Isotope Laboratories). A mix of D-lysine, D-threonine (Sigma Aldrich), D-phenylalanine, and D/L-isoleucine (MP Biomedicals) at 100 mg/L, as well as D-valine (Arcos Organics) and D-Met (MP Biomedicals) at 50 mg/L was also added to the minimal media to minimize metabolic scrambling (Tong et al. 2008). Cells were then grown with aeration (∼ 220 rpm) at room temperature for 30 min before a 17 h protein expression was induced by addition of 0.2 mg/L of anhydrotetracycline (added from a 0.2 mg/mL stock in dimethylformamide).
For partial deuteration, E. coli BL21 (DE3) competent cells were first adapted to grow in deuterated media through repetitive rounds of plating and colony selection (Paliy et al. 2003). The D2O-adapted competent cells were transformed as stated above, but then instead of using a starter culture in nutrient-rich media, all colonies from the agar plates were inoculated into 250-mL starter culture in M9 minimal media in 100% D2O with 0.4% glycerol, 1 mM MgSO4, 2.7 mM CaCl2, 0.5 mg/L thiamine, 100 mg/L ampicillin, 0.01 mM iron (II) sulfate and then shaken (∼ 220 rpm) at 37 °C until the cells reached an OD550 of 1.0 ± 0.2 (doubling time ∼2.3 h, compared to ∼0.5 h in 100% H2O and TB media). The cells were then harvested by centrifugation (1720 g for 20 min) and resuspended in the same M9 media in D2O (as above). This step provides an additional selection round to ensure the bacteria have adapted to grow in M9/D2O and also minimizes proton contamination from the overnight growth on LB (H2O) plates. The resuspended cells were inoculated into the main culture (3 L M9 media in D2O, as described above) and shaken (∼ 220 rpm) at 37 °C until the cells reached an OD550 of 1.8 ± 0.2. The culture was then transferred to room temperature (∼ 22 °C) with shaking (∼ 220 rpm) for ∼ 1 h. Protein expression with isotopic labeling of Met residues was achieved by addition of 50 mg/L of 13C-L-Met, 0.2 mg/L of anhydrotetracycline, and 100 mg/L ampicillin. At 18 h after induction, additional doses of anhydrotetracycline (0.2 mg/L), 13C-L-Met (50 mg/L) and ampicillin (100 mg/L) were added. The cells were harvested after an additional 15 h induction.
Protein purification
Both unlabeled and labeled ClC-ec1 preparations were purified as follows. Cell were harvested by centrifugation at 1720 g for 20 min, then resuspended in ∼ 50 mL of 50 mM Tris-Cl, pH 7.5 and 100 mM NaCl and broken by sonication in the presence of ∼ 2 μg/mL leupeptin (Enzo Life Sciences) and pepstatin (Santa Cruz Biotech) and ∼ 5 μg/mL phenylmethanesulfonyl fluoride (PMSF) (Sigma Aldrich) (added from aqueous stocks of 0.2 mg/mL leupeptin and pepstatin, and an ethanol stock of 0.5 mg/mL PMSF). Membrane proteins were extracted from the cell lysate with 50 mM n-decyl-β-d-maltopyranoside (DM) (Anatrace, solgrade), with gentle shaking for 2 h at room temperature. The extract was clarified by centrifugation at 20,100 g for 45 min. ClC-ec1 was purified using a Co2+-affinity column (Clonetech). The resin was equilibrated in wash buffer (WB) (20 mM Tris-Cl, pH 7.5, 100 mM NaCl, and 10 mM solgrade DM). After loading the soluble fraction of the extract, the ClC-ec1-bound resin was washed with WB, then with WB with 30 mM imidazole. ClC-ec1 was eluted using WB with 400 mM imidazole. The C-terminal polyHis-tag was removed using Endoproteinase Lys-C (Roche) (0.16 – 0.25 U per L culture, 1 h at room temperature). Lys-C protease was removed by size exclusion chromatography (Superdex 200 column, GE Healthcare Biosciences) during the final step of purification into gel filtration buffer (GFB, 150 mM NaCl, 10 mM Na-Hepes, 5 mM DM (Anatrace, anagrade), pH 7.5).
Reductive Methylation
Unlabeled ClC-ec1 protein in GFB at ∼ 1 mg/mL (∼20 μM) was reductively methylated using 13C formaldehyde (13CH2O, Cambridge Isotope Laboratories) and sodium cyanoborohydride (NaBCN, Sigma Aldrich, 95% reagent grade) (Abraham et al. 2008; Gluck and Sweeney 1990). After addition of reagents (1.8 mM of 13CH2O added from a 6.45 M aqueous stock solution and 3.6 mM of NaBCN added from a 100 mM aqueous stock solution), the reaction was incubated at 4°C for 4 h without shaking or stirring. After 4 h, an additional 1.8 mM 13CH2O and 3.6 mM NaBCN were added. After an additional 4 h (total of 8 h), the sample (1.4 - 2 mL) was dialyzed against 100 mL GFB at 4 °C. Dialysis occurred over the course of 36 h with solution changes every 8-15 h (2 changes total).
13C-MMTS labeling
Single Cys mutations in helix R (at residue positions I448 and S464) were introduced in cys-less ClC-ec1 (Nguitragool and Miller 2007) using QuikChange site-directed mutagenesis (Agilent Technologies), and resulting sequences were confirmed by DNA sequencing. For these Cys-containing mutants, purification was as described above with the following changes. 20 mM beta-mercaptoethanol (ßME) was added to the cell-extraction buffer to keep the thiols reduced. Removal of ßME occurred during the washing of the Co2+-affinity column with WB. 13C-MMTS labeling was carried out immediately after elution from the Co2+ column, by reacting ClC-ec1 (∼1 mg/ml) with a 50-fold molar excess of 13C-MMTS (Toronto Research Chemicals, Canada; 100 mM stock solution in DMSO) in the presence of layered argon gas for 2 h at room temperature with slow mixing at ∼10 rpm. After His-tag cleavage by Lys-C, the excess MMTS and cleaved His-tag were removed by size exclusion chromatorgraphy (Superdex 200), which is the final step of the standard ClC-ec1 purification procedure (described above). Methyl-group incorporation was quantified indirectly by determining the availablility of free thiols in the protein sample before and after labeling, using the thiol quantitation kit (T6060, Molecular Probes).
Liposomal Reconstitution and Flux Assays
ClC-ec1 was reconstituted into liposomes by incubation with lipid-detergent mixed-micelles followed by dialysis to removed detergent (Accardi et al. 2004; Maduke et al. 1999) E. coli polar lipids (Avanti, 25 mg/ml) were dried with a stream of argon then washed twice with pentane (with argon drying). Dried lipids were resuspended at 20 mg/ml in reconstitution buffer (RB) (300 mM KCl, 40 mM Na-citrate, pH 4.5) and mixed by rotation for 20 min. The detergent CHAPS (Amresco, Solon, OH) was then added to 35 mM (by weight), and the mixture was sonicated and then mixed by rotation at room temperature for 2 h, then additionally sonicated if necessary to obtain a clear solution. Purified ClC-ec1 (WT or mutants) was then added to the lipid-detergent solution at a concentration 0.2 - 1 μg protein per mg lipid. Detergent was removed and liposomes formed by dialysis against RB for 36 h at 16°C (∼500 μl of sample was dialyzed against 3-4 buffer changes of 1–1.3 L each). The resulting proteoliposomes were flash frozen in ethanol/dry ice and stored at -80°C.
Functional assays quantified efflux of Cl- from liposomes (Elvington et al. 2009; Walden et al. 2007). Reconstituted liposomes were thawed to room temperature. To obtain uniform sizes, these liposomes were subjected to four cycles of freezing (in dry ice-ethanol bath) and thawing (RT), followed by 15 passes through a 0.4 μm polycarbonate membrane (Avanti Polar Lipids) mounted on a mini-extruder (Avanti Polar Lipids). Spin columns were prepared by packing 1.5 mL Sephadex G-50 Fine resin (GE Healthcare Bio-Sciences, Pittsburgh, PA) equilibrated with low-chloride buffer (LCB) (300 mM potassium isethionate, 50 μM KCl, and 25 or 40 mM citrate-NaOH, pH 4.5) into empty 2 mL columns (Thermo Scientific, Rockford, IL). Immediately before each assay, a spin column was pre-spun for ∼30 sec at 7000 rpm in a clinical centrifuge (Model CL; International Equipment Co., Needham Heights, MA); then, 60 μL of extruded liposomes was loaded onto the column and buffer-exchange was achieved by centrifugation at 5000 rpm for 1 min. The buffer-exchanged liposome solution was immediately added to 600 μL LCB in a 12 mm (diameter) × 31 mm shell vial (Kimble Chase, Vineland, NJ). Bulk Cl- transport was initiated by addition of valinomycin (Vln, 5 μg/ml) and carbonyl cyanide m-chlorophenyl hydrazone (CCCP, 2 μg/ml) (from stock solutions at 2.5 mg/ml Vln dissolved in ethanol and 1 mg/ml CCCP in water). Extra-liposomal [Cl-] was monitored by a Ag/AgCl electrode connected through a Beckman Φ32 pH meter to a Axon MiniDigi 1A digitizer. Cl-transport was terminated by dissolving the vesicles with 10 μL of 10% Triton X-100. Cl-concentration data were acquired using pClamp9 software and analyzed in Igor Pro 6.0 (WaveMatrix, Portland, OR).
NMR
Protein samples for 13C-Met NMR were exchanged into deuterated water (D2O) in GFB during the last step of purification and concentrated to ∼10 mg/mL (∼100 μM) for the dimeric (WT) ClC-ec1 or ∼5-9 mg/mL (100-180 μM) for the monomeric variant. In some samples (data shown in Figure 3b and 3c) a low-salt GFB containing 100 mM instead of 150 mM NaCl was used. No significant differences were observed between spectra taken at 100 vs 150 mM NaCl. Samples for 13C-methylated Lys and 13C-MMTS NMR were concentrated to ∼10 mg/mL then spiked with 10% D2O. Lipids (E. coli polar lipid extract, Avanti Polar Lipids) were added at a 1:80 lipid:detergent ratio to stabilize ClC-ec1, except in the case of monomeric variants, which are more stable without added lipid.
Fig. 3. 1H-13C HSQC spectra of 13C-Met labeled ClC-ec1.
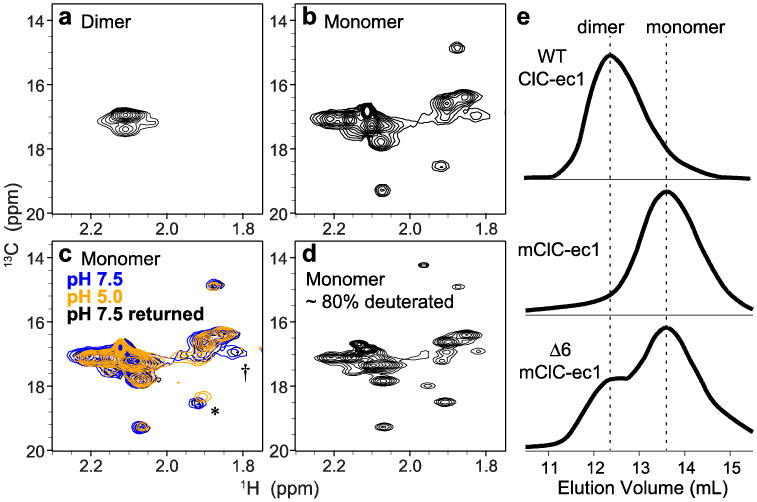
a Wildtype ClC-ec1 (homodimer, 100 kDa), methionine methyl region.
b Monomeric ClC-ec1 (I201W/I422W, 50 kDa).
c [H+]-dependence of 1H-13C HSQC spectra: blue, pH 7.5; orange, pH 5.0; black, reversal to pH 7.5. The asterisk (*) and dagger (†) indicate the most clearly resolved crosspeaks that exhibit H+-dependency.
d Partially deuterated (∼80%) monomeric ClCe-c1 with prolonged data collection. e Size exclusion chromatograms of ClC-ec1. Top WT ClC-ec1; Middle monomeric ClC-ec1 (mClC-ec1) (I201W/I422W); Bottom Δ6 mClC-ec1 (mutating six Met residues as follows: M38L, M65I, M228V, M276T, M330V, and M332L) yields a mix of dimer and monomer populations.
Sample volumes of ∼200 μL were loaded into a 3 mm NMR tube. 1H-13C heteronuclear single quantum coherence (HSQC) experiments were acquired at 25° C on 600 MHz Varian Unity INOVA and 800 MHz Varian Unity INOVA/Agilent VNMRS spectrometers at the Stanford Magnetic Resonance Laboratory, and a 900 MHz Bruker Avance spectrometer at the University of California, Berkeley. The 600 MHz spectrometer was equipped with a 5 mm, H{CN}, z-axis gradient conventional probe while the 800 and 900 MHz spectrometers were equipped with 5mm, H{CN}, z-axis gradient cryogenic probes. The 13C-Met and 13C-MTC experiments were run on the 800 and 900 MHz spectrometers. The 13C-methylated Lys experiments were run at 600 MHz. Suppression of the residual water signal was performed with the WATERGATE sequence. Spectral widths were typically 10-13.3 ppm in the 1H dimension and 30 ppm in the 13C dimension for 13C-Met and 13C-MTC experiments and 22 ppm for 13C-methylated Lys experiments. The 13C spectral window was centered at 20 ppm for 13C-Met and 13C-MTC experiments and 40 ppm for 13C-methylated Lys experiments. Experiments were acquired with 512-1024 data points in the 1H dimension and 64 increments in the 13C dimension typically for 8-32 transients per increment with 1.5s pre-scan delays. Experiments were preceded by 16 and 128-256 steady-state scans for temperature and coil equilibration for the conventional and cryogenic probes, respectively. Data collection was typically 1-2 h for each spectrum.
An extended HSQC experiment was carried out on a partially deuterated (∼80%) 13C-Met labeled sample. Experimental parameters were identical to those described above, with the following exceptions. The number of increments was doubled (128) in the 13C dimension for improved spectral resolution and the number of transients was increased to 128 to improve signal-to-noise ratio. Data collection for the 2D spectrum took approximately 14 h. In additional to water suppression, additional selective Wet pulse suppression was applied to the aliphatic region of the 1H dimension (∼ 1 ppm) to minimize streaking of detergent peaks into the Met cross-peaks in the 13C dimension (∼ 17 ppm).
Sample pH was measured using an IQ150 pH meter and ISFET NMR tube micro probe (IQ Scientific Instruments, Carlsbad, CA). Sample pH adjustments were made using 0.5 M Na-citrate, pH 4 to lower the pH from 7.5 to 4.0 and 1 M Tris-acetate, pH 9 to return the pH to 7.5.
Data were processed using VnmrJ (Varian/Agilent Technologies) and TopSpin (Bruker BioSpin). Forward linear prediction (1×) was applied to the 13C dimension. Shifted squared-sine bell functions were applied to both 1H and 13C dimensions. Spectra were analyzed using Sparky.
Results
13C-Methionine labeling and NMR
Methionine residues are distributed throughout ClC-ec1 (Figure 2b), including in regions not probed in our previous 19F-Tyr experiments (Figure 2a). The 1H-13C HSQC spectrum of 13C-Met labeled WT sample revealed signal at the expected region for 13C-methyl of Mets, δH ∼ 2.1 ppm and δC ∼ 17 ppm, but exhibited only one prominent group of peaks (Figure 3a). In contrast, a monomeric variant of ClC-ec1 (double mutant I201W/I422W) yielded greatly improved resolution and signal dispersion (Figure 3b). Using the monomeric ClC-ec1 (mClC-ec1) is reasonable as it has transport kinetics within 2-fold of the WT dimer, with the overall mechanism (2 Cl-:1 H+ stoichiometric coupling) and structure (all-atom RMSD ∼0.5 Å) perfectly preserved (Robertson et al. 2010).
In our previous work using 19F-NMR, we varied [Cl-] and [H+] to enrich different ClC-ec1 conformational states in solution (Elvington et al. 2009). Here, we focus on [H+] dependence, as the concentration of this substrate ion can be increased and reversed readily. The HSQC spectrum of 13C-Met labeled mClC-ec1 shows clear [H+] dependence (Figure 3c). Most notably, a distinct peak at δH = 1.83 ppm, δC = 17.0 ppm (marked by “†”) was observed at pH 7.5 but not pH 5.0. Another peak (δH = 1.92 ppm, δC = 18.5 ppm at pH 7.5, marked by “*”) also noticeably shifted at pH 5.0. Both spectral changes were fully reversible. Thus, 1H-13C NMR of 13C-methyl groups specifically labeled at Met residues can be used to monitor substrate-dependent structural changes in ClC-ec1.
We attempted to improve the 1H-13C HSQC spectrum by using a partially-deuterated protein, increasing the resolution of the 13C-dimension, increasing the number of transients collected, as well as applying solvent suppression to a nearby detergent peak. Although we were able to modestly enhance the quality of the spectrum (Figure 3d), spectral overlap remained a significant obstacle. Thus, we also attempted to simplify the spectrum further by removing surface-exposed Met residues, as we do not expect those residues to be interesting reporters of protein conformational change. Out of the 18 native Met residues, we identified six that are surface-exposed and made conservative substitutions by site-directed mutagenesis on the mClC-ec1 background. In Δ6 mClC-ec1, the methionines were mutated to the following residues, with substitutions chosen based on conservation within other CLC homologs: M38L, M65I, M228V, M276T, M330V, and M332L. While the yield of this construct (Δ6 mClC-ec1) was unperturbed, we found that the protein was no longer uniformly monomeric. Chromatograms from size-exclusion chromatography columns show that the Δ6 preparation contains a substantial dimer population (Figure 3e). We adopted two approaches in an attempt to correct this problem. First, we conducted a small detergent screen to search for conditions that might favor the monomeric state. However, none of three detergents tested stabilized the monomer. In decyl maltoside neopentyl glycol (Anatrace), the protein was a mix of monomer and dimer; in Cymal-6 and Cymal-7 (Anatrace), the initially monomeric protein precipitated out from solution within 24 h at 4°C (data not shown). In our second approach, we aimed to shift the monomer-dimer equilibrium using mutagenesis. Monomeric ClC-ec1 was originally achieved by substituting two Ile residues at the dimer interface with Trp, a maneuver designed to destabilize protein contacts and stabilize lipid contacts (Robertson et al, 2010). Because other Trp substitutions at the dimer interface are known to affect the monomer-dimer equilibrium (Robertson et al. 2010), we attempted to drive the equilibrium towards monomer with additional engineered Trp residues. The tested constructs (Δ6 mClC-ec1 + L406W and Δ6 mClC-ec1 + L25W + L26W), however, retained significant dimer populations. In separate studies, we discovered that even a single point mutation on the mClC-ec1 background (A404V) is sufficient to cause a shift towards dimer (∼ 30% dimer, data not shown). So unfortunately, though the 13C-Met labeling showed initial promise for probing potentially interesting regions of mClC-ec1, we have been stymied by the inability thus far to reduce NMR spectral complexity by mutagenesis, and await further development of stable monomeric constructs.
13C-Lys labeling and NMR identifies Helix R as undergoing H+-dependent conformational change
Motivated by the H+-dependent spectral changes seen in 13C-Met labeled mClC-ec1, and to overcome the limitations of spectral overlap and line-broadening, we next employed selective post-translational labeling using reductive methylation. The addition of 13C-enriched formaldehyde to protein under reducing conditions results in the introduction of two 13C-methyl groups to the ε-amino group of each Lys residue as well as the α-amino group of the N-terminus. The addition of the second methyl group is a more favorable reaction than the addition of the first methyl group, and the reaction favors production of fully dimethylated amines under standard conditions with excess reagents (Larda et al. 2012). The NMR spectra of reductively methylated proteins show good signal-to-noise ratio even for large proteins at low protein concentrations (Bokoch et al. 2010). With ClC-ec1, even the WT (dimeric) ClC-ec1 exhibits signal dispersion sufficient to reveal a specific Lys cross-peak that changes in response to shifts in [H+] (Figure 4a). At pH 7.5, one of the 13C methyl signals is noticeably distinct (δH = 2.70 ppm and δC = 44.9 ppm, highlighted by a red box) from the rest of the dimethyl-Lys signals, which cluster together. This peak exhibits a clear [H+] sensitivity, shifting towards the main peak cluster as the pH is lowered (Figure 4b). At intermediate pH values (pH 4.5 – 5.5), the peak splits into two, possibly due to a loss of degeneracy of the two 13C methyl groups. When the pH is reversed back to 7.5, the peak returns to its original chemical shift (Figure 4a).
Fig. 4. 13C-Lys ClC-ec1.
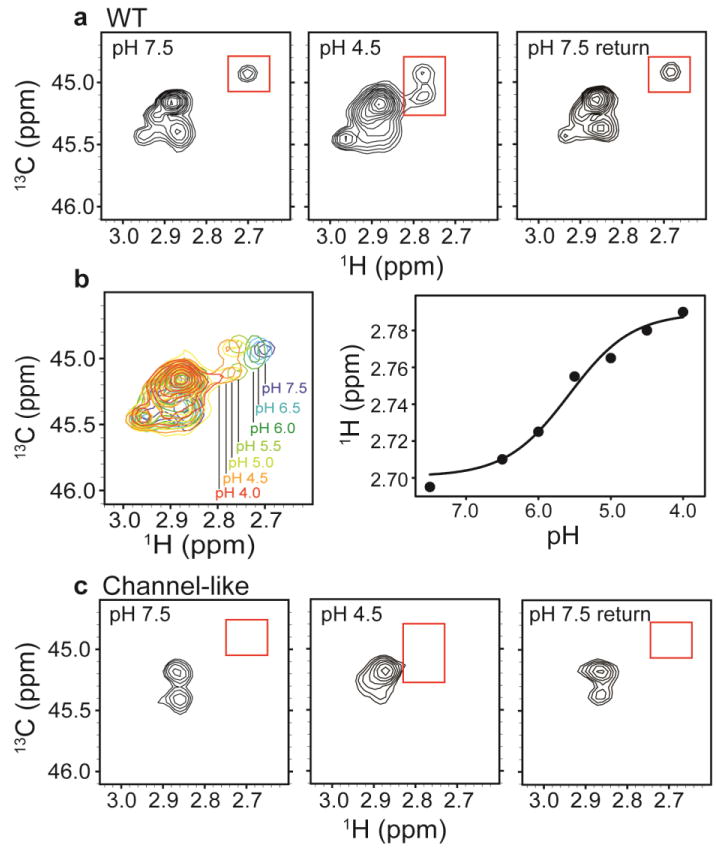
a1H-13C HSQC spectra of methylated ClC-ec1 at pH 7.5 (left), pH 4.5 (middle) and reversed to pH 7.5 (right). The spectral window is zoomed in on the 13C methyl signals arising from the Lys side chains (with the 13C signal from the methylated N-terminus not shown). Changes in [H+] induce spectral shifts, the most notable being the peak splitting and shift of the peak at δH = 2.7 ppm (highlighted by the red boxes) at low pH.
b pH titration of methylated ClC-ec1. Left: Overlaid 1H-13C HSQC spectra at pH 7.5, 6.5, 6.0, 5.5, 5.0, 4.5, and 4.0, as indicated in the figure. Right: 1H chemical shifts for the peak of interest as a function of pH. The solid curve is a fit to a single-site H+-binding isotherm, with an apparent pKa of 5.6.
c1H-13C HSQC spectra of methylated channel-like mutant at pH 7.5 (left), pH 4.5 (middle) and reversed to pH 7.5 (right). The spectrum of the channel-like mutant exhibits little [H+] sensitivity, and the peak at δH = 2.70 ppm and δC = 44.9 ppm is notably missing at both high and low pH.
The pH dependence of the NMR spectral change fits well to a single-site titration curve with an apparent pKa of 5.6 (Figure 4b). This value is comparable to the apparent pKa estimated for the extracellular gate residue Gluex (∼6.2) (Picollo et al. 2012), suggesting that the conformational change detected by NMR is coupled to opening of the gate. We therefore hypothesized that the change would be absent in the “channel-like” variant of ClC-ec1. In this variant, the Cl--permeation pathway is constitutively accessible from both the intracellular and extracellular solutions due to mutation of residues comprising the extracellular and intracellular “gates” for Cl- (Gluex and Y445 respectively, Figure 1a). The resulting double mutant (E148A/Y445S) is no longer constrained to systematically move through individual states of the transport pathway to facilitate stoichiometric antiport; instead it behaves like an electrodiffusive chloride channel that facilitates rapid transit of Cl- down (and only down) its electrochemical gradient, independent of the proton gradient (Jayaram et al. 2008). In reductively methylated channel-like ClC-ec1, the peak at δH = 2.70 ppm and δC = 44.9 ppm is notably missing at both pH 7.5 and pH 4.5, and the 1H-13C HSQC spectrum exhibits little [H+] sensitivity (Figure 4c). In addition, the main peak cluster is greatly reduced in intensity and dispersion compared to WT. This difference is likely due to increased motional averaging of the methyl probes and thus probably reflects greater conformational flexibility of the channel-like protein; a similar phenomenon is observed in the spectra of 19F-Tyr labeled channel-like ClC-ec1 (Elvington et al. 2009). The dearth of peaks in channel-like ClC-ec1 precludes conclusion that the pH-dependent conformational change is absent. Nevertheless, the notable absence of the large spectral shift observed with WT motivates identification of the residue reporting this shift.
To identify this H+-sensitive lysine residue, we made a series of single Lys-to-Arg point mutants: K30R (intracellular, Helix A-B linker); K271R (transmembrane Helix J); K442R (intracellular, Helix Q-R linker); and K455R (intracellular, Helix R). We 13C-methylated each of these mutants and compared NMR spectra at pH 7.5, 4.5, and on reversal to 7.5 with those of the WT methylated protein. The peak of interest (locations marked by red boxes) was observed in K30R, K271R, and K442R, but not K455R (Figure 5a). These results point to K455 as giving rise to the peak that displayed the largest H+-dependent shift in WT ClC-ec1.
Fig. 5. Identification of the H+-sensitive lysine residue.
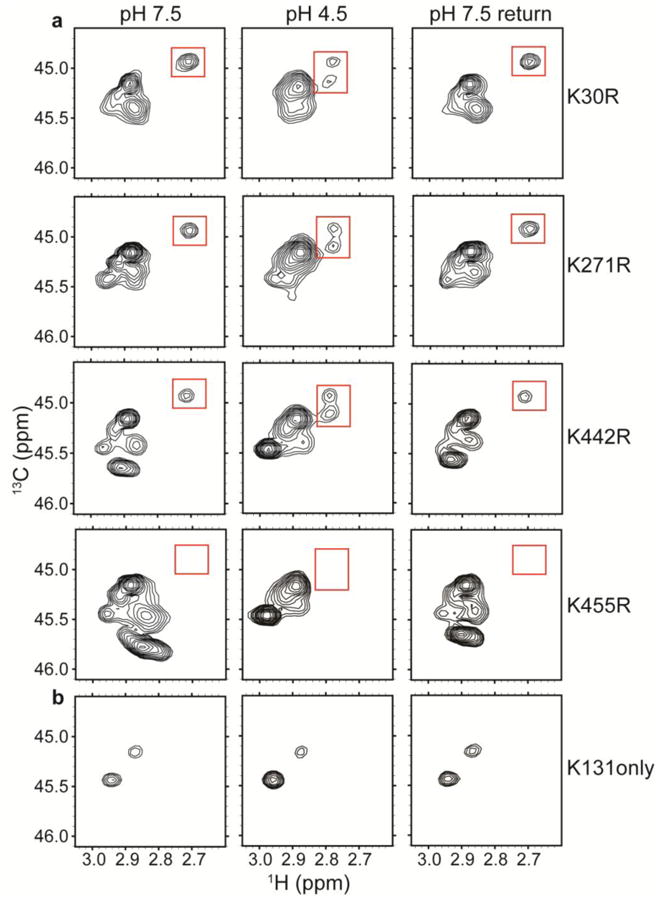
a1H-13C HSQC spectra of methylated ClC-ec1 Lys-to-Arg mutants: K30R, K271R, K442R, and K455R. Unlike the channel-like mutant, these mutants maintain the spectral dispersion observed in WT ClC-ec1. Because Arg residues do not become 13C-methylated, these spectra allow assignment of the peak of interest (highlighted by the red boxes) to K455.
b1H-13C HSQC spectra of “K131 only”, a mutant in which all other Lys residues have been mutated to Arg. The minimal H+-dependence indicates that this region of the protein (Figure 2c) is not undergoing large H+-dependent conformational change.
Many Lys residues are solvent exposed and motion-averaging in their chemical environments, so we would expect that many methylated-Lys signals would cluster together (as seen in Figures 4a, 5a between δH 2.80-3.00 ppm and δC 45.0-46.0 ppm). However, not all Lys residues are exposed, and we were especially interested in K131, the only lysine residue that is completely conserved amongst CLCs (Feng et al. 2010; Maduke et al. 1999). This buried residue influences the electrostatic profile of the Cl- binding sites (Faraldo-Gomez and Roux 2004), which are within 10 Å (Figures 1a, 2c). To investigate this residue specifically by NMR, we generated a ClC-ec1 mutant containing only one Lys residue, at K131 (with all other Lys mutated to Arg). This mutant (“K131 only”) exhibited two robust 1H-13C HSQC signals within the region of the main methylated-Lys peak cluster (Figure 5b). The observation of two peaks for this single residue could reflect that the two methyl groups are in distinct chemical environments that exchange only slowly on the NMR timescale, or that the Lys residue is slowly sampling two distinct chemical environments. These warrant further examination (e.g. by magnetization transfer experiments) but the minimal H+-dependence of these signals indicates that K131 does not participate in a large H+-dependent conformational change.
Our interpretation that the 13C-Lys spectra reveal H+-dependent conformational change at K455 (Helix R), and not at K131 (Helix E), relies on the assumption that Lys dimethylation does not perturb ClC-ec1 function. Because Lys dimethylation preserves charge and introduces only a small steric addition, it has generally been found to be non-perturbing (Larda et al. 2012). Nevertheless, the impact of reductive methylation on protein function must be evaluated on a case-by-case basis, with functional comparison of methylated and unmethylated protein. To achieve this comparison, we performed quantitative flux assays on all methylated samples. An overview of the assay (Walden et al. 2007) is shown schematically in Figure 6a. Purified ClC-ec1 is reconstituted into liposomes at high [Cl-], and then Cl- efflux is initiated by exchanging extravesicular Cl- for the impermeant isethionate and adding valinomycin (a K+ ionophore) and CCCP (a H+ ionophore) to dissipate the electrical potential. ClC-ec1 turnover rates are calculated by quantifying the initial rates of Cl- efflux. Representative raw data traces are shown in Figure 6b, with data summarized in Figure 6c. In all cases, reductive methylation has no substantial effect on ClC-ec1 function.
Fig. 6. Functional characterization of reductively methylated ClC-ec1.
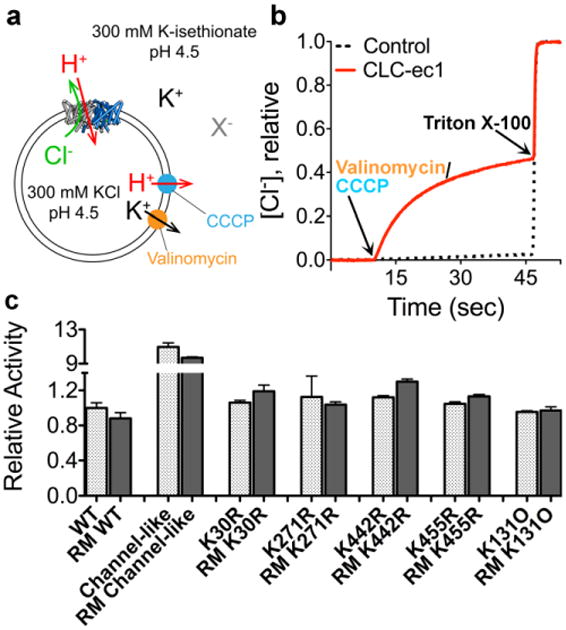
a Schematic outline of the Cl- flux assay. ClC-ec1 is reconstituted into liposomes at high [KCl], and then the extravesicular solution is exchanged for a low-[KCl] solution (replacing Cl- with the impermeant isethionate, “X-”). Bulk Cl- efflux through ClC-ec1 is initiated by valinomycin (a K+ ionophore) and CCCP (a H+ ionophore) to dissipate the electrical potential. Extravesicular [Cl-] is monitored using a Ag/AgCl electrode.
b Representative raw data traces for WT ClC-ec1 (red) and control liposomes (containing no protein) (black dashes). At the end of each experiment, the detergent Triton X-100 was added to lyse the liposomes and release all of the Cl-.
c Summary data for reductively methylated (RM) proteins studied here. The relative turnover rates (normalized to WT unlabeled ClC-ec1) were calculated from the initial slope (Δ[Cl-]/Δt) after addition of Vln/CCCP, and averages ± SEM (n=3 to 6) are reported.
13C-MMTS labeling of single cysteine mutants confirms the H+-sensitivity of Helix R
The 13C-Lys results described above identified K455 on Helix R (Figures 1a, 2c) as undergoing H+-dependent spectral change and suggest that the entire helix may participate in H+-dependent conformational change. To investigate this hypothesis, we employed a second post-translational 13C-labeling strategy involving site-specific labeling at engineered cysteine residues. Under non-reducing conditions, 13C-MMTS cross-links with Cys residues via a disulfide bond, effectively adding a highly sensitive 13C methyl probe to an otherwise unlabeled protein (Kenyon and Bruice 1977; Religa et al. 2011; Verardi et al. 2012). The resulting product, a methyl thiocysteine (MTC), closely resembles a Met residue both in volume and possible conformers, and thus is generally considered a non-perturbing substitution (Smith et al. 1975; Wynn and Richards 1993). The fact that the 13C label on the Cys is the only extraneously added NMR probe on the otherwise unlabeled protein allows specific labeling on Helix R (and hence instant peak assignment) without interference from overlapping peaks.
To further investigate the H+-dependence of helix R, we used 13C-MMTS to label a Cys residue introduced by point mutagenesis at I448C, which lies on the same face of the Helix R as K455, two turns closer towards the inner-gate residue Y445 (Figure 7a). As a control, we also labeled S464C, which follows Helix R in the unstructured C-terminus of the protein. Labeling at I448C and S464C yielded fully functional proteins as assessed by quantitative flux assays (Figure 7b) with labeling efficiencies of 93 ± 5% and 89 ± 3% respectively (estimated indirectly by thiol estimation). 1H-13C HSQC spectra acquired during the course of a pH titration revealed that the 13C methyl probe at 448-MTC experiences a reversible [H+]-dependent change in its electro-chemical environment. There is a sharp transition at pH 4.7 where a new peak emerges (δH =2.36 ppm and δC =24.7 ppm) in addition to the single peak present at pH 7.5 (δH =2.70 ppm and δC =26.1 ppm) (Figure 7c). Then, by pH 4.5, the original peak has disappeared leaving only the new peak, and this change is fully reversible (Figure 7c and left panel of 7d). In contrast, the 13C-methyl probe at 464-MTC displays no such change – the cross peak remained at δH =2.41 ppm and δC =24.5 ppm irrespective of pH (Figure 7d, compare left to right panel). Together, these results reinforce the interpretation of the Lys reductive methylation results, which indicate that H+ binding is coupled to conformational change at Helix R.
Fig. 7. 13C-methyl thiocystine (MTC) ClC-ec1.
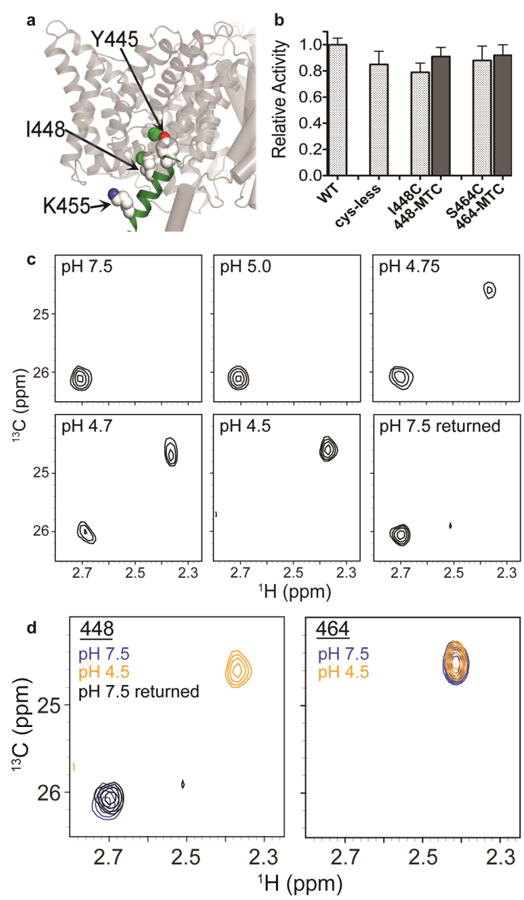
a Position of helix R and I448. The position of helix R in one subunit of the ClC-ec1 homodimer is shown in green. The intracellular Cl- gate residue Y445 (shown in spacefill) is the first residue of Helix R. 13C-MTC probes were introduced at positions I448 (shown in spacefill) and S464 (not shown; part of the unresolved C-terminus of ClC-ec1). 13C-K455 (spacefilled) was investigated by 13C-Lys methylation (Figure 4).
b Activity assays on 13C-MTC ClC-ec1 variants, with Cl- flux assays were performed as in Figure 6. Activity is normalized to WT ClC-ec1. Cys mutants were introduced into a cys-less template ClC-ec1 (Nguitragool and Miller 2007). Labeling at I448C and at S464C (with labeling efficiencies 93±5% and 89±3% respectively) has no effect on activity. Data represent averages ± SEM for n=8-11 replicates.
c1H-13C HSQC spectra of 13C-I448MTC ClC-ec1 at pH 7.5, 5.0, 4.75, 4.7, 4.5, and reversed to pH 7.5. At pH 7.5 and pH 5.0, the 13C-MTC probe exhibits a single peak (δH = 2.70 ppm). At pH 4.75 and pH 4.7, a second peak at δH = 2.36 ppm is additionally observed, and at pH 4.5 only this second peak remains. On reversal to pH 7.5, the spectrum reverts to the single peak at δH = 2.70 ppm.
d1H-13C HSQC spectra of 13C-I448MTC ClC-ec1 (left) and 13C-S464MTC (right) at pH 7.5 (blue), pH 4.5 (orange) and (for I448-MTC) reversed to pH 7.5 (black). S464 resides in the unresolved C-terminus of the protein. Its insensitivity to pH changes suggests that the peak shift in observed with I448 is likely due to structural changes in the protein rather than a general effect of pH.
Discussion
In this work, we take advantage of the favorable relaxation properties of 13C methyl groups to address a straightforward question: do the substrate-dependent conformational changes underlying the CLC transport mechanism extend beyond the Glu sidechain fluctuations observed crystallographically? Our results, which reveal H+-dependent changes with Met, Lys, and MTC labeling, support that they do.
13C-Lys and 13C-MTC NMR identify Helix R as participating in H+-dependent change
Reductive methylation of lysines is an inexpensive, straightforward method of post-translationally modifying lysines to introduce 13C-methyl NMR probes. This method has been successfully applied in a variety of systems (Chavan et al. 2013; Hattori et al. 2013; Larda et al. 2012; Mobius et al. 2013; Roberson and Macnaughtan 2014; Xie et al. 2014), including the membrane protein β-2 adrenergic receptor, where a ligand-dependent extracellular salt bridge was identified based on NMR spectral changes (Bokoch et al. 2010). Our spectra of reductively methylated ClC-ec1 identify a prominent H+ (ligand)-dependent spectral change of an isolated crosspeak (Figure 4a). We assigned this crosspeak to K455, on Helix R, based on its absence in the K455R mutant in comparison to three other K-to-R mutants (Figure 5a). This result indicates that Helix R undergoes H+-dependent conformational change.
To further investigate this region of the protein, we used engineered cysteines and MMTS to site-specifically label an additional Helix-R residue (448) as well as a control residue (464) in the unstructured C-terminal region following Helix R. This labeling method is suited to rapidly probe a region of interest where peak assignments are unknown, and is particularly attractive for studying large proteins (Religa and Kay 2010; Religa et al. 2011; Religa et al. 2010). Using this approach, we showed that position 448 on Helix R shows H+-dependent spectral changes (Figure 7c). The existence of two cross peaks at pH 4.7 suggests that the label at 448C is experiencing two different chemical environments in slow exchange on the NMR timescale. Unfortunately, attempts to assess conformational exchange by magnetization transfer thus far have been unsuccessful due to the abbreviated lifetime of the 448-MTC sample at low pH (samples became visibly cloudy within a 4-12 hour period). Future studies on the conformational dynamics of methyl labels along with other positions in Helix R will likely provide insight into the role it plays in the CLC coupling mechanism (Henzler-Wildman and Kern 2007; Rosenzweig and Kay 2014).
In all crystal structures of CLC antiporters, Helix R forms a steric barrier between bound Cl- and the intracellular solution. This barrier extends from the center of the CLC structure, where Y445 coordinates a central Cl- ion, to the intracellular solution. Together, 13C-Lys and 13C-MTC NMR identified two positions in Helix R (448 and 455) as undergoing reversible, H+- dependent spectral shifts. These results are in harmony with our previous solution 19F NMR data indicating H+-dependent changes at the inner-gate residue Y445, at the top of Helix R (Elvington et al. 2009) and with studies on ClC-ec1 fluorescently labeled at 3 positions along Helix R (446, 448, and 449) (Bell et al. 2006). The latter study showed that environmentally sensitive fluorophores attached to Helix R undergo reversible H+-dependent changes in fluorescence. Surprisingly, a fluorophore at S464C (in the unstructured C-terminus) displayed the largest H+-dependent fluorescence change. The presence of a long (5-methylene) linker attaching this fluorophore to Helix R likely provides flexibility for the fluorophore probe to interact with different regions of the protein. In contrast, the small methyl probe used on S464C in this study reported no H+-dependent change (Figure 7d). This negative control bolsters the conclusion that the H+ sensitive-change in environment is occurring directly at Helix R.
What is the nature of the conformational change underlying this change in environment? The data do not directly answer this question, but we reason that it is likely to be a closing of the intracellular vestibule (Cl- entry pathway). The increases in [H+] used here will favor protonation of Gluex and its movement out to the extracellular solution (Figure 1b). To maintain the tight coupling (2 Cl- for 1 H+) that is observed at pH 4 - 4.5, Cl- access from the intracellular solution must be restricted under these conditions. We therefore speculate that the H+ may induce a closing of the intracellular vestibule as the extracellular vestibule is opened. Such a closing could occur through movements of any region lining this pathway, including Helices R, J, C, D, or E, or the C-D loop (Engh and Maduke 2005). In support of motions occurring directly at Helix R, computational studies using functional mode analysis have found that local mobility around Gluex is coupled to collective motions at Helices Q and R (Krivobokova et al. 2012).
For methylated K455, the apparent pKa of the chemical-shift transition (∼5.6, Figure 4b) is similar to the apparent pKa for the extracellular-gate residue Gluex, as measured in the Cl-/Cl-exchange reaction (Picollo et al. 2012). This correlation is consistent with the idea that Helix R movement is coordinated with opening of the extracellular gate. For 448-MTS, the sharp transition of the chemical shift between pH 5.0 and 4.5 (Figure 7d) is in contrast to the gradual transition observed with K455 between pH 7.5 and 4.0 (Figure 4b) and suggests that the two parts of Helix R are reporting different events. Notably, the sharp transition of the 448-MTC peak coincides with the sharp increase in the turnover rate for Cl-/H+ antiport between pH 5.0 and 4.5 (Lim and Miller 2009). While we do not have sufficient evidence to identify the nature of the event 448-MTC is sensing, one possibility is that 448-MTC movement is coordinated with a cooperative opening of the (narrow) extracellular vestibule, following the titration and outward movement of Gluex. Experiments with spectroscopic probes introduced into the extracellular vestibule are needed to test this possibility.
Lysine residue K131 is unusual in being a charged residue completely buried within the transmembrane core of the protein (Dutzler et al. 2002). The side chain, positioned 7-9 Å from the Cl- binding sites (Figure 2c), electrostatically stabilizes Cl- binding at these sites and also appears to play a structural role in stabilizing the inner-gate serine residue (Faraldo-Gomez and Roux 2004). K131 is highly conserved amongst all CLCs, both transporters and channels (Feng et al. 2010; Maduke et al. 1999). In the channels, mutations at this site cause significant effects on both gating and permeation, appearing to weaken Cl- binding and Cl- movement between sites on either side of the inner-gate Ser/Tyr residues (Engh et al. 2007; Zhang et al. 2006). Given the clear importance of K131 in Cl--dependent gating, we hypothesized that K131 might also be involved in H+-dependent conformational changes associated with gate-opening in ClC-ec1. Our ability to dimethylate at only K131 in ClC-ec1 allowed us to detect its signal (Figure 5b), which is otherwise not resolved from the bulk dimethylated Lys residues in ClC-ec1 (Figure 4a). The minimal [H+]-dependence of the K131 signal suggests that this residue is not involved in the conformational change detected at Helix R. However, there may be a small shift in population between the two states observed at pH 4.5 compared to pH 7.5 (Figure 5b), and further investigation may reveal subtle but functionally important dynamics at this position.
The 13C-Lys HSQC spectrum of the K455R mutant provides an additional hint that ClC-ec1 undergoes global conformational change. At pH 7.5 (Figure 5a), this spectrum shows resonances at δC ∼ 45.8 ppm that are not seen in the WT spectra (Figure 4a). This suggests that the mutant induces a shift in conformational equilibria of global conformational states, resulting in shifts in the chemical environments of the other 13C-Lys reporters beyond Helix R. Such shifts in conformational equilibria induced by mutation are quite frequently observed in active transporters (Abramson et al. 2003; Boxenbaum et al. 1998; Kaback 2005; Krishnamurthy and Gouaux 2012; Mager et al. 2013). Further investigation of these conformational equilibria in the CLC transporters is necessary.
13C Met NMR suggests other CLC regions are involved in H+-dependent conformational change
With 13C-Met labeling, the use of monomeric ClC-ec1 allowed us to obtain vastly improved spectral resolution compared to WT (dimeric) ClC-ec1 (Figure 3a, b). [H+]-dependent spectral shifts are evident at two distinct cross peaks, and more broadly in the overlapping regions (Figure 3c). Since there are no Met residues in Helix R, these changes must arise from other regions of the protein. There are five obvious candidates. The first is M91 on Helix C (mentioned above). A second is M360, whose methyl group is ∼5.5 Å from the Helix-R residue Y445. The location of M360 on Helix N, whose backbone amides I356 and F357 coordinate the centrally bound Cl- ion, make it poised to detect changes at this site. A third is M415, near the C-terminus of Helix P and four residues from Y419 (P-Q loop) which was identified as participating in conformational change in our 19F NMR study (Elvington et al. 2009). A fourth candidate is M394, at the extracellular end of Helix O. The importance of Helix-O movement to the CLC transport cycle was recently demonstrated by inhibition of transport upon crosslinking the intracellular side (A399C) to A432C on Helix Q (Basilio et al. 2014). At the extracellular side of Helix O, Zn2+ binding to the N-O loop in the mammalian ClC-4 homolog inhibits antiport (Osteen and Mindell 2008). Finally, a fifth candidate is suggested by normal mode analysis, which predicts that opening of the extracellular vestibule is coordinated with large-scale motion at M65 (Helix B) (Miloshevsky et al. 2010). These conjectures, while compelling, remain speculative until peak assignments can be made. This would likely require a stable monomer ClC-ec1 construct amenable to mutation of surface methionines, and/or improvements to spectral quality allowing better resolution of crowded peak regions. In a preliminary attempt to improve spectral quality, we used a partially deuterated (∼80%) protein sample to reduce signal broadening due to transverse relaxation and obtained a 1H-13C HSQC spectrum with modestly narrower linewidths and improved resolution (compare Figures 3b and 3d). We also attempted to utilize the methyl-TROSY effect (Kalverda et al. 2014; Ollerenshaw et al. 2003; Xu and Matthews 2013) by using the HMQC scheme. Preliminary HMQC tests run on a partially deuterated sample showed marginal improvements in linewidths for some peaks, but other peaks became less evident (Supplementary Figure S1). Further improvements will likely require a more highly deuterated protein in combination with deuterated detergent.
Summary
In summary, using 13C methyl probes, we have detected H+-dependent spectral changes that indicate the existence of CLC conformational states not detected in the crystal structures. Some of the changes were assigned to Helix R, which lines the intracellular Cl- vestibule and forms part of the inner gate. Since increases in [H+] are known to open the outer gate, we speculate that the change detected at Helix R is a concomitant contraction of the inner-gate and vestibule. These studies set the stage for investigating the structural details and dynamics of this change.
Supplementary Material
Acknowledgments
We thank Shelley Elvington for performing the initial HSQC analysis of 13C-Met dimeric ClC-ec1, Christian Evans for assistance with some of the functional assays, Dr. Jeffrey Pelton for assistance with acquiring the 900 MHz NMR data at the University of California at Berkeley, and Martin Prieto and Tanmay Chavan for comments on the manuscript. This work was supported by the National Science Foundation (Award Number 1021472), the National Institutes of Health (U54-GM087519), and by the Mathers Foundation. The Stanford Magnetic Resonance Laboratory 800 MHz NMR spectrometer was supported in part with NIH Shared Instrumentation Grant 1 S10 RR025612-01A1. TAC was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number T32GM007365. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Abraham SJ, Hoheisel S, Gaponenko V. Detection of protein-ligand interactions by NMR using reductive methylation of lysine residues. J Biomol NMR. 2008;42:143–148. doi: 10.1007/s10858-008-9274-y. [DOI] [PubMed] [Google Scholar]
- Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science. 2003;301:610–615. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- Accardi A, Kolmakova-Partensky L, Williams C, Miller C. Ionic currents mediated by a prokaryotic homologue of CLC Cl- channels. J Gen Physiol. 2004;123:109–119. doi: 10.1085/jgp.200308935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accardi A, Lobet S, Williams C, Miller C, Dutzler R. Synergism between halide binding and proton transport in a CLC-type exchanger. J Mol Biol. 2006;362:691–699. doi: 10.1016/j.jmb.2006.07.081. [DOI] [PubMed] [Google Scholar]
- Accardi A, Miller C. Secondary active transport mediated by a prokaryotic homologue of ClC Cl- channels. Nature. 2004;427:803–807. doi: 10.1038/nature02314. [DOI] [PubMed] [Google Scholar]
- Accardi A, Picollo A. CLC channels and transporters: proteins with borderline personalities. Biochim Biophys Acta. 2010;1798:1457–1464. doi: 10.1016/j.bbamem.2010.02.022. doi:10.1016/j.bbamem.2010.02.022S0005-2736(10)00081-7 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accardi A, Walden M, Nguitragool W, Jayaram H, Williams C, Miller C. Separate ion pathways in a Cl-/H+ exchanger. J Gen Physiol. 2005;126:563–570. doi: 10.1085/jgp.200509417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basilio D, Noack K, Picollo A, Accardi A. Conformational changes required for H(+)/Cl(-) exchange mediated by a CLC transporter. Nat Struct Mol Biol. 2014;21:456–463. doi: 10.1038/nsmb.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty EJ, et al. Interlobe communication in 13C-methionine-labeled human transferrin. Biochemistry (Mosc) 1996;35:7635–7642. doi: 10.1021/bi960684g. [DOI] [PubMed] [Google Scholar]
- Bell SP, Curran PK, Choi S, Mindell JA. Site-directed fluorescence studies of a prokaryotic ClC antiporter. Biochemistry (Mosc) 2006;45:6773–6782. doi: 10.1021/bi0523815. [DOI] [PubMed] [Google Scholar]
- Bokoch MP, et al. Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature. 2010;463:108–112. doi: 10.1038/nature08650. doi:nature08650 [pii]10.1038/nature08650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxenbaum N, Daly SE, Javaid ZZ, Lane LK, Blostein R. Changes in steady-state conformational equilibrium resulting from cytoplasmic mutations of the Na,K-ATPase alpha-subunit. J Biol Chem. 1998;273:23086–23092. doi: 10.1074/jbc.273.36.23086. [DOI] [PubMed] [Google Scholar]
- Brooks DJ, Fresco JR, Lesk AM, Singh M. Evolution of amino acid frequencies in proteins over deep time: inferred order of introduction of amino acids into the genetic code. Mol Biol Evol. 2002;19:1645–1655. doi: 10.1093/oxfordjournals.molbev.a003988. [DOI] [PubMed] [Google Scholar]
- Butterfoss GL, et al. Conformational dependence of 13C shielding and coupling constants for methionine methyl groups. J Biomol NMR. 2010;48:31–47. doi: 10.1007/s10858-010-9436-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavan TS, Abraham S, Gaponenko V. Application of reductive (1)(3)C-methylation of lysines to enhance the sensitivity of conventional NMR methods. Molecules. 2013;18:7103–7119. doi: 10.3390/molecules18067103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Viel S, Ziarelli F, Peng L. 19F NMR: a valuable tool for studying biological events. Chem Soc Rev. 2013;42:7971–7982. doi: 10.1039/c3cs60129c. [DOI] [PubMed] [Google Scholar]
- Chen J. Molecular mechanism of the Escherichia coli maltose transporter. Curr Opin Struct Biol. 2013;23:492–498. doi: 10.1016/j.sbi.2013.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TY, Hwang TC. CLC-0 and CFTR: chloride channels evolved from transporters. Physiol Rev. 2008;88:351–387. doi: 10.1152/physrev.00058.2006. doi:10.1152/physrev.00058.2006 88/2/351 [pii] [DOI] [PubMed] [Google Scholar]
- Cheng MH, Coalson RD. Molecular dynamics investigation of Cl- and water transport through a eukaryotic CLC transporter. Biophys J. 2012;102:1363–1371. doi: 10.1016/j.bpj.2012.01.056. doi:S0006-3495(12)00203-2 [pii] 10.1016/j.bpj.2012.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claxton DP, Quick M, Shi L, de Carvalho FD, Weinstein H, Javitch JA, McHaourab HS. Ion/substrate-dependent conformational dynamics of a bacterial homolog of neurotransmitter:sodium symporters. Nat Struct Mol Biol. 2010;17:822–829. doi: 10.1038/nsmb.1854. doi:nsmb.1854 [pii] 10.1038/nsmb.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson MA, Falke JJ. Use of 19F NMR to probe protein structure and conformational changes. Annu Rev Biophys Biomol Struct. 1996;25:163–195. doi: 10.1146/annurev.bb.25.060196.001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DellaVecchia MJ, et al. NMR analysis of [methyl-13C]methionine UvrB from Bacillus caldotenax reveals UvrB-domain 4 heterodimer formation in solution. J Mol Biol. 2007;373:282–295. doi: 10.1016/j.jmb.2007.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton JS, Pao AC, Maduke M. Novel diuretic targets. Am J Physiol Renal Physiol. 2013;305:F931–942. doi: 10.1152/ajprenal.00230.2013. doi:10.1152/ajprenal.00230.2013 ajprenal.00230.2013 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler R. The ClC family of chloride channels and transporters. Curr Opin Struct Biol. 2006;16:439–446. doi: 10.1016/j.sbi.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- Dutzler R, Campbell EB, MacKinnon R. Gating the selectivity filter in ClC chloride channels. Science. 2003;300:108–112. doi: 10.1126/science.1082708. [DOI] [PubMed] [Google Scholar]
- Elvington SM, Liu CW, Maduke MC. Substrate-driven conformational changes in ClC-ec1 observed by fluorine. NMR EMBO J. 2009;28:3090–3102. doi: 10.1038/emboj.2009.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engh AM, Faraldo-Gomez JD, Maduke M. The role of a conserved lysine in chloride- and voltage-dependent ClC-0 fast gating. J Gen Physiol. 2007;130:351–363. doi: 10.1085/jgp.200709760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engh AM, Maduke M. Cysteine accessibility in ClC-0 supports conservation of the ClC intracellular vestibule. J Gen Physiol. 2005;125:601–617. doi: 10.1085/jgp.200509258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraldo-Gomez JD, Roux B. Electrostatics of ion stabilization in a ClC chloride channel homologue from Escherichia coli. J Mol Biol. 2004;339:981–1000. doi: 10.1016/j.jmb.2004.04.023. doi:10.1016/j.jmb.2004.04.023 S0022283604004310 [pii] [DOI] [PubMed] [Google Scholar]
- Feng L, Campbell EB, Hsiung Y, MacKinnon R. Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science. 2010;330:635–641. doi: 10.1126/science.1195230. doi:science.1195230 [pii] 10.1126/science.1195230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focke PJ, Wang X, Larsson HP. Neurotransmitter transporters: structure meets function. Structure. 2013;21:694–705. doi: 10.1016/j.str.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest LR, Kramer R, Ziegler C. The structural basis of secondary active transport mechanisms. Biochim Biophys Acta. 2011;1807:167–188. doi: 10.1016/j.bbabio.2010.10.014. doi:S0005-2728(10)00722-X [pii] 10.1016/j.bbabio.2010.10.014. [DOI] [PubMed] [Google Scholar]
- Forrest LR, Rudnick G. The rocking bundle: a mechanism for ion-coupled solute flux by symmetrical transporters. Physiology (Bethesda) 2009;24:377–386. doi: 10.1152/physiol.00030.2009. doi:24/6/377 [pii] 10.1152/physiol.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerken TA, Jentoft JE, Jentoft N, Dearborn DG. Intramolecular interactions of amino groups in 13C reductively methylated hen egg-white lysozyme. J Biol Chem. 1982;257:2894–2900. [PubMed] [Google Scholar]
- Gluck M, Sweeney WV. 13C-NMR of Clostridium pasteurianum ferredoxin after reductive methylation of the amines using [13C]formaldehyde. Biochim Biophys Acta. 1990;1038:146–151. doi: 10.1016/0167-4838(90)90197-n. [DOI] [PubMed] [Google Scholar]
- Graves AR, Curran PK, Smith CL, Mindell JA. The Cl-/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature. 2008;453:788–792. doi: 10.1038/nature06907. [DOI] [PubMed] [Google Scholar]
- Guan L, Mirza O, Verner G, Iwata S, Kaback HR. Structural determination of wild-type lactose permease. Proc Natl Acad Sci U S A. 2007;104:15294–15298. doi: 10.1073/pnas.0707688104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Cheng RC, Maduke MC, Tajkhorshid E. Water access points and hydration pathways in CLC H+/Cl- transporters. Proc Natl Acad Sci U S A. 2014;111:1819–1824. doi: 10.1073/pnas.1317890111. doi:10.1073/pnas.1317890111 1317890111 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori Y, et al. Utilization of lysine (1)(3)C-methylation NMR for protein-protein interaction studies. J Biomol NMR. 2013;55:19–31. doi: 10.1007/s10858-012-9675-9. [DOI] [PubMed] [Google Scholar]
- Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- Jaffe EK, Markham GD. 13C NMR studies of methylene and methine carbons of substrate bound to a 280,000-dalton protein, porphobilinogen synthase. Biochemistry (Mosc) 1988;27:4475–4481. doi: 10.1021/bi00412a039. [DOI] [PubMed] [Google Scholar]
- Jardetzky O. Simple allosteric model for membrane pumps. Nature. 1966;211:969–970. doi: 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- Jayaram H, Accardi A, Wu F, Williams C, Miller C. Ion permeation through a Cl--selective channel designed from a CLC Cl-/H+ exchanger. Proc Natl Acad Sci U S A. 2008;105:11194–11199. doi: 10.1073/pnas.0804503105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram H, Robertson JL, Wu F, Williams C, Miller C. Structure of a slow CLC Cl/H+ antiporter from a cyanobacterium. Biochemistry (Mosc) 2011;50:788–794. doi: 10.1021/bi1019258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaback HR. Structure and mechanism of the lactose permease. C R Biol. 2005;328:557–567. doi: 10.1016/j.crvi.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Kalverda AP, Gowdy J, Thompson GS, Homans SW, Henderson PJ, Patching SG. TROSY NMR with a 52 kDa sugar transport protein and the binding of a small-molecule inhibitor. Mol Membr Biol. 2014;31:131–140. doi: 10.3109/09687688.2014.911980. [DOI] [PubMed] [Google Scholar]
- Kang C, Li Q. Solution NMR study of integral membrane proteins. Curr Opin Chem Biol. 2011;15:560–569. doi: 10.1016/j.cbpa.2011.05.025. doi:S1367-5931(11)00094-9 [pii] 10.1016/j.cbpa.2011.05.025. [DOI] [PubMed] [Google Scholar]
- Kenyon GL, Bruice TW. Novel sulfhydryl reagents. Methods Enzymol. 1977;47:407–430. doi: 10.1016/0076-6879(77)47042-3. [DOI] [PubMed] [Google Scholar]
- Khare D, Oldham ML, Orelle C, Davidson AL, Chen J. Alternating access in maltose transporter mediated by rigid-body rotations. Mol Cell. 2009;33:528–536. doi: 10.1016/j.molcel.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Howell SC, Van Horn WD, Jeon YH, Sanders CR. Recent Advances in the Application of Solution NMR Spectroscopy to Multi-Span Integral Membrane Proteins. Prog Nucl Magn Reson Spectrosc. 2009;55:335–360. doi: 10.1016/j.pnmrs.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitevski-LeBlanc JL, Prosser RS. Current applications of 19F NMR to studies of protein structure and dynamics. Prog Nucl Magn Reson Spectrosc. 2012;62:1–33. doi: 10.1016/j.pnmrs.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Ko YJ, Jo WH. Secondary water pore formation for proton transport in a ClC exchanger revealed by an atomistic molecular-dynamics simulation. Biophys J. 2010;98:2163–2169. doi: 10.1016/j.bpj.2010.01.043. doi:S0006-3495(10)00208-0 [pii] 10.1016/j.bpj.2010.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy H, Gouaux E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature. 2012;481:469–474. doi: 10.1038/nature10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivobokova T, Briones R, Hub JS, Munk A, de Groot BL. Partial Least-Squares Functional Mode Analysis: Application to the Membrane Proteins AQP1, Aqy1, and CLC-ec1. Biophys J. 2012;103:786–796. doi: 10.1016/j.bpj.2012.07.022. doi:S0006-3495(12)00797-7 [pii] 10.1016/j.bpj.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang Z, Mahankali U, Beck TL. Proton pathways and H+/Cl- stoichiometry in bacterial chloride transporters. Proteins. 2007;68:26–33. doi: 10.1002/prot.21441. [DOI] [PubMed] [Google Scholar]
- Kumar H, Kasho V, Smirnova I, Finer-Moore JS, Kaback HR, Stroud RM. Structure of sugar-bound LacY. Proc Natl Acad Sci U S A. 2014;111:1784–1788. doi: 10.1073/pnas.1324141111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurinov IV, Mao C, Irvin JD, Uckun FM. X-ray crystallographic analysis of pokeweed antiviral protein-II after reductive methylation of lysine residues. Biochem Biophys Res Commun. 2000;275:549–552. doi: 10.1006/bbrc.2000.3329. [DOI] [PubMed] [Google Scholar]
- Larda ST, Bokoch MP, Evanics F, Prosser RS. Lysine methylation strategies for characterizing protein conformations by NMR. J Biomol NMR. 2012;54:199–209. doi: 10.1007/s10858-012-9664-z. [DOI] [PubMed] [Google Scholar]
- Law CJ, Maloney PC, Wang DN. Ins and outs of major facilitator superfamily antiporters. Annu Rev Microbiol. 2008;62:289–305. doi: 10.1146/annurev.micro.61.080706.093329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HH, Miller C. Intracellular proton-transfer mutants in a CLC Cl-/H+ exchanger. J Gen Physiol. 2009;133:131–138. doi: 10.1085/jgp.200810112. doi:jgp.200810112 [pii] 10.1085/jgp.2008.10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HH, Miller C. Intracellular proton acces in a Cl-/H+ antiporter. PLoS Biol. 2012 doi: 10.1371/journal.pbio.1001441. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HH, Shane T, Miller C. Intracellular Proton Access Mechanism of the CLC-ec1 Cl-/H+ Exchanger. Biophys J. 2012;102:214a. [Google Scholar]
- Lim HH, Stockbridge RB, Miller C. Fluoride-dependent interruption of the transport cycle of a CLC Cl-/H+ antiporter. Nat Chem Biol. 2013;9:721–725. doi: 10.1038/nchembio.1336. doi:10.1038/nchembio.1336 nchembio.1336 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisal J, Maduke M. The ClC-0 chloride channel is a ‘broken” Cl-/H+ antiporter. Nat Struct Mol Biol. 2008;15:805–810. doi: 10.1038/nsmb.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisal J, Maduke M. Review. Proton-coupled gating in chloride channels. Philos Trans R Soc Lond B Biol Sci. 2009;364:181–187. doi: 10.1098/rstb.2008.0123. doi:NX2761747066G001 [pii] 10.1098/rstb.2008.0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Inesi G, Toyoshima C. Substrate-induced conformational fit and headpiece closure in the Ca2+ATPase (SERCA) J Biol Chem. 2003;278:28938–28943. doi: 10.1074/jbc.M304120200. doi:10.1074/jbc.M304120200 M304120200 [pii] [DOI] [PubMed] [Google Scholar]
- Maduke M, Miller C, Mindell JA. A decade of CLC chloride channels: structure, mechanism, and many unsettled questions. Annu Rev Biophys Biomol Struct. 2000;29:411–438. doi: 10.1146/annurev.biophys.29.1.411. [DOI] [PubMed] [Google Scholar]
- Maduke M, Pheasant DJ, Miller C. High-level expression, functional reconstitution, and quaternary structure of a prokaryotic ClC-type chloride channel. J Gen Physiol. 1999;114:713–722. doi: 10.1085/jgp.114.5.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager T, et al. Differential effects of mutations on the transport properties of the Na+/H+ antiporter NhaA from Escherichia coli. J Biol Chem. 2013;288:24666–24675. doi: 10.1074/jbc.M113.484071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainz A, Religa TL, Sprangers R, Linser R, Kay LE, Reif B. NMR spectroscopy of soluble protein complexes at one mega-dalton and beyond. Angew Chem Int Ed Engl. 2013;52:8746–8751. doi: 10.1002/anie.201301215. [DOI] [PubMed] [Google Scholar]
- Merianos HJ, Cadieux N, Lin CH, Kadner RJ, Cafiso DS. Substrate-induced exposure of an energy-coupling motif of a membrane transporter. Nat Struct Biol. 2000;7:205–209. doi: 10.1038/73309. [DOI] [PubMed] [Google Scholar]
- Miller C. ClC chloride channels viewed through a transporter lens. Nature. 2006;440:484–489. doi: 10.1038/nature04713. [DOI] [PubMed] [Google Scholar]
- Miller C. In the beginning: A personal reminiscence on the origin and legacy of ClC-0, the “Torpedo Cl- channel”. J Physiol. 2014 doi: 10.1113/jphysiol.2014.286260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C, Nguitragool W. A provisional transport mechanism for a chloride channel-type Cl-/H+ exchanger. Philos Trans R Soc Lond B Biol Sci. 2009;364:175–180. doi: 10.1098/rstb.2008.0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miloshevsky GV, Hassanein A, Jordan PC. Antiport mechanism for Cl(-)/H(+) in ClC-ec1 from normal-mode analysis. Biophys J. 2010;98:999–1008. doi: 10.1016/j.bpj.2009.11.035. doi:S0006-3495(09)01802-5 [pii] 10.1016/j.bpj.2009.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol. 2012;74:69–86. doi: 10.1146/annurev-physiol-012110-142317. [DOI] [PubMed] [Google Scholar]
- Mobius K, et al. Investigation of lysine side chain interactions of interleukin-8 with heparin and other glycosaminoglycans studied by a methylation-NMR approach. Glycobiology. 2013;23:1260–1269. doi: 10.1093/glycob/cwt062. [DOI] [PubMed] [Google Scholar]
- Montaville P, Jamin N. Determination of Membrane Protein Structures Using Solution and Solid-State NMR. In: Lacapere JJ, editor. Membrane Protein Structure Determination: Methods and Protocols. Vol. 654. Springer; New York: 2010. pp. 261–282. [DOI] [PubMed] [Google Scholar]
- Nguitragool W, Miller C. Inaugural Article: CLC Cl/H+ transporters constrained by covalent cross-linking. Proc Natl Acad Sci U S A. 2007;104:20659–20665. doi: 10.1073/pnas.0708639104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham ML, Khare D, Quiocho FA, Davidson AL, Chen J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 2007;450:515–521. doi: 10.1038/nature06264. [DOI] [PubMed] [Google Scholar]
- Ollerenshaw JE, Tugarinov V, Kay LE. Methyl TROSY: explanation and experimental verification. Magnetic Resonance in Chemistry. 2003;41:843–852. [Google Scholar]
- Osteen JD, Mindell JA. Insights into the ClC-4 transport mechanism from studies of Zn2+ inhibition. Biophys J. 2008;95:4668–4675. doi: 10.1529/biophysj.108.137158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliy O, Bloor D, Brockwell D, Gilbert P, Barber J. Improved methods of cultivation and production of deuteriated proteins from E. coli strains grown on fully deuteriated minimal medium. J Appl Microbiol. 2003;94:580–586. doi: 10.1046/j.1365-2672.2003.01866.x. doi:1866 [pii] [DOI] [PubMed] [Google Scholar]
- Picollo A, Pusch M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature. 2005;436:420–423. doi: 10.1038/nature03720. [DOI] [PubMed] [Google Scholar]
- Picollo A, Xu Y, Johner N, Berneche S, Accardi A. Synergistic substrate binding determines the stoichiometry of transport of a prokaryotic H(+)/Cl(-) exchanger. Nat Struct Mol Biol. 2012;19:525–531. doi: 10.1038/nsmb.2277. doi:10.1038/nsmb.2277 nsmb.2277 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayment I. Reductive alkylation of lysine residues to alter crystallization properties of proteins. Methods Enzymol. 1997;276:171–179. [PubMed] [Google Scholar]
- Religa TL, Kay LE. Optimal methyl labeling for studies of supra-molecular systems. J Biomol NMR. 2010;47:163–169. doi: 10.1007/s10858-010-9419-7. [DOI] [PubMed] [Google Scholar]
- Religa TL, Ruschak AM, Rosenzweig R, Kay LE. Site-directed methyl group labeling as an NMR probe of structure and dynamics in supramolecular protein systems: applications to the proteasome and to the ClpP protease. J Am Chem Soc. 2011;133:9063–9068. doi: 10.1021/ja202259a. [DOI] [PubMed] [Google Scholar]
- Religa TL, Sprangers R, Kay LE. Dynamic regulation of archaeal proteasome gate opening as studied by. TROSY NMR Science. 2010;328:98–102. doi: 10.1126/science.1184991. doi:328/5974/98 [pii] 10.1126/science.1184991. [DOI] [PubMed] [Google Scholar]
- Roberson KJ, Macnaughtan MA. Review of methods to assign the nuclear magnetic resonance peaks of reductively methylated proteins. Anal Biochem. 2014;466C:76–82. doi: 10.1016/j.ab.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson JL, Kolmakova-Partensky L, Miller C. Design, function and structure of a monomeric ClC transporter. Nature. 2010;468:844–847. doi: 10.1038/nature09556. doi:nature09556 [pii] 10.1038/nature09556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig R, Kay LE. Bringing dynamic molecular machines into focus by methyl-TROSY NMR. Annu Rev Biochem. 2014;83:291–315. doi: 10.1146/annurev-biochem-060713-035829. [DOI] [PubMed] [Google Scholar]
- Rypniewski WR, Holden HM, Rayment I. Structural consequences of reductive methylation of lysine residues in hen egg white lysozyme: an X-ray analysis at 1.8-A resolution. Biochemistry (Mosc) 1993;32:9851–9858. doi: 10.1021/bi00088a041. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning, A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- Sanders CR, Sonnichsen F. Solution NMR of membrane proteins: practice and challenges. Magnetic resonance in chemistry : MRC. 2006;44(Spec No):S24–40. doi: 10.1002/mrc.1816. [DOI] [PubMed] [Google Scholar]
- Scheel O, Zdebik AA, Lourdel S, Jentsch TJ. Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature. 2005;436:424–427. doi: 10.1038/nature03860. [DOI] [PubMed] [Google Scholar]
- Shi Y. Common folds and transport mechanisms of secondary active transporters. Annual review of biophysics. 2013;42:51–72. doi: 10.1146/annurev-biophys-083012-130429. [DOI] [PubMed] [Google Scholar]
- Slotboom DJ. Structural and mechanistic insights into prokaryotic energy-coupling factor transporters. Nat Rev Microbiol. 2014;12:79–87. doi: 10.1038/nrmicro3175. [DOI] [PubMed] [Google Scholar]
- Smith DJ, Maggio ET, Kenyon GL. Simple alkanethiol groups for temporary blocking of sulfhydryl groups of enzymes. Biochemistry (Mosc) 1975;14:766–771. doi: 10.1021/bi00675a019. [DOI] [PubMed] [Google Scholar]
- Stauber T, Weinert S, Jentsch TJ. Cell Biology and Physiology of CLC Chloride Channels and Transporters. Compr Physiol. 2012;2:1701–1744. doi: 10.1002/cphy.c110038. [DOI] [PubMed] [Google Scholar]
- Tanford C. Mechanism of free energy coupling in active transport. Annu Rev Biochem. 1983;52:379–409. doi: 10.1146/annurev.bi.52.070183.002115. [DOI] [PubMed] [Google Scholar]
- Tang CY, Chen TY. Physiology and pathophysiology of CLC-1: mechanisms of a chloride channel disease, myotonia. Journal of biomedicine & biotechnology. 2011;2011:685328. doi: 10.1155/2011/685328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong KI, Yamamoto M, Tanaka T. A simple method for amino acid selective isotope labeling of recombinant proteins in E. coli. J Biomol NMR. 2008;42:59–67. doi: 10.1007/s10858-008-9264-0. [DOI] [PubMed] [Google Scholar]
- Tourasse NJ, Li WH. Selective constraints, amino acid composition, and the rate of protein evolution. Mol Biol Evol. 2000;17:656–664. doi: 10.1093/oxfordjournals.molbev.a026344. [DOI] [PubMed] [Google Scholar]
- Verardi R, Traaseth NJ, Masterson LR, Vostrikov VV, Veglia G. Isotope labeling for solution and solid-state NMR spectroscopy of membrane proteins. Adv Exp Med Biol. 2012;992:35–62. doi: 10.1007/978-94-007-4954-2_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdon G, Oh S, Serio RN, Boudker O. Coupled ion binding and structural transitions along the transport cycle of glutamate transporters. Elife. 2014;3:e02283. doi: 10.7554/eLife.02283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walden M, Accardi A, Wu F, Xu C, Williams C, Miller C. Uncoupling and turnover in a Cl-/H+ exchange transporter. J Gen Physiol. 2007;129:317–329. doi: 10.1085/jgp.200709756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter TS, et al. Lysine methylation as a routine rescue strategy for protein crystallization. Structure. 2006;14:1617–1622. doi: 10.1016/j.str.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn R, Richards FM. Unnatural amino acid packing mutants of Escherichia coli thioredoxin produced by combined mutagenesis/chemical modification techniques. Protein Sci. 1993;2:395–403. doi: 10.1002/pro.5560020311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Fulton DB, Andreotti AH. A Selective NMR Probe to Monitor the Conformational Transition from Inactive to Active Kinase. ACS Chem Biol. 2014 doi: 10.1021/cb5004702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Matthews S. TROSY NMR spectroscopy of large soluble proteins. Top Curr Chem. 2013;335:97–119. doi: 10.1007/128_2011_228. [DOI] [PubMed] [Google Scholar]
- Yan N. Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem Sci. 2013;38:151–159. doi: 10.1016/j.tibs.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Yu JX, Hallac RR, Chiguru S, Mason RP. New frontiers and developing applications in 19F NMR. Prog Nucl Magn Reson Spectrosc. 2013;70:25–49. doi: 10.1016/j.pnmrs.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XD, Li Y, Yu WP, Chen TY. Roles of K149, G352, and H401 in the channel functions of ClC-0: testing the predictions from theoretical calculations. J Gen Physiol. 2006;127:435–447. doi: 10.1085/jgp.200509460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Voth GA. The coupled proton transport in the ClC-ec1 Cl(-)/H(+) antiporter. Biophys J. 2011;101:L47–49. doi: 10.1016/j.bpj.2011.10.021. doi:S0006-3495(11)01240-9 [pii] 10.1016/j.bpj.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Terry DS, Shi L, Quick M, Weinstein H, Blanchard SC, Javitch JA. Substrate-modulated gating dynamics in a Na+-coupled neurotransmitter transporter homologue. Nature. 2011;474:109–113. doi: 10.1038/nature09971. doi:nature09971 [pii] 10.1038/nature09971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zifarelli G, Pusch M. CLC chloride channels and transporters: a biophysical and physiological perspective. Rev Physiol Biochem Pharmacol. 2007;158:23–76. doi: 10.1007/112_2006_0605. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.