

Abstract
Our recent work has indicated that the DMP1 locus on 7q21, encoding a haplo-insufficient tumour suppressor, is hemizygously deleted at a high frequency in breast cancer. The locus encodes DMP1α protein, an activator of the p53 pathway leading to cell cycle arrest and senescence, and two other functionally undefined isoforms, DMP1β and DMP1γ. In this study, we show that the DMP1 locus is alternatively spliced in ∼30% of breast cancer cases with relatively decreased DMP1α and increased DMP1β expression. RNA-seq analyses of a publicly available database showed significantly increased DMP1β mRNA in 43–55% of human breast cancers, dependent on histological subtypes. Similarly, DMP1β protein was found to be overexpressed in ∼60% of tumours relative to their surrounding normal tissue. Importantly, alteration of DMP1 splicing and DMP1β overexpression were associated with poor clinical outcomes of the breast cancer patients, indicating that DMP1β may have a biological function. Indeed, DMP1β increased proliferation of non-tumourigenic mammary epithelial cells and knockdown of endogenous DMP1 inhibited breast cancer cell growth. To determine DMP1β's role in vivo, we established MMTV-DMP1β transgenic mouse lines. DMP1β overexpression was sufficient to induce mammary gland hyperplasia and multifocal tumour lesions in mice at 7–18 months of age. The tumours formed were adenosquamous carcinomas with evidence of transdifferentiation and keratinized deposits. Overall, we identify alternative splicing as a mechanism utilized by cancer cells to modulate the DMP1 locus through diminishing DMP1α tumour suppressor expression, while simultaneously up-regulating the tumour-promoting DMP1β isoform.
Keywords: DMP1 (DMP1α and DMP1β), DMTF1, breast cancer, MMTV, Arf, p53, alternative splicing
Introduction
Breast cancer (BC), as the most common tumour type in women, causes a significant health care burden in Western countries [1,2]. Established biomarkers such as hormone receptors (ER/PR) and HER2 play significant roles in the selection of patients for endocrine and trastuzumab therapies [3]. However, these targeted therapies have not produced the anticipated improvement in long-term patient survival. The initial response is often followed by tumour relapse with intrinsic resistance to the first-line therapy [4–6]. In addition, up to 30% of breast cancer patients are overdiagnosed due to the implementation of mammography screening, with minimal reduction in mortality rates [7]. This suggests that many women with early diagnosis of an indolent breast tumour might have been spared psychological stress and therapy-associated side-effects if better prognostic/predictive stratification strategies had existed. Although proliferation markers such as cyclin D1, cyclin E, and p27Kip1 have been proposed for molecular stratification of breast cancer, these have not been routinely used in clinics due to their limited efficacy in deciding therapeutic strategies [3,8,9]. To develop more reliable biomarkers, it is necessary to further delineate oncogenic events that drive the initiation and progression of cancer.
Recently, our laboratory has identified DMP1 (cyclin D-binding Myb-like Protein 1; DMTF1) as a critical tumour suppressor in breast cancer [10–12]. DMP1 is a transcription factor that transactivates p14ARF (p19Arf in mice), leading to p53 stabilization and senescence [10,13,14]. DMP1 also stabilizes p53 by direct protein–protein interaction to block Hdm2-mediated ubiquitination, which is the major mechanism of p53 activation by DMP1 in ARF-null cells [15]. Loss of heterozygosity (LOH) analysis with specific primers demonstrated that the DMP1 locus on 7q21 is hemizygously deleted in ∼42% of breast tumours with mutual exclusiveness to INK4A/ARF or p53 loss. The intact DMP1 allele remained wild type without promoter hypermethylation [11]. Similarly, deletion of Dmp1 in the MMTV-neu mouse model accelerated the development of mammary gland tumours without a significant difference between Dmp1+/− and Dmp1−/− backgrounds, suggesting haploinsufficiency of Dmp1 for tumour suppression [10]. Dmp1 haploinsufficiency was also observed in lymphoma and lung tumour mouse models [16,17]. To date, the molecular mechanisms for Dmp1's haploid insufficiency remain unknown. Moreover, Dmp1−/− females are unable to nurse pups due to poor expansion of luminal cells, suggesting that the Dmp1 locus may possess functions other than tumour suppression [10,18].
The human DMP1 locus encodes three distinct transcripts via alternative splicing of exon 10 [19]. The bonafide tumour suppressor was named DMP1α, while two other transcripts with mostly unknown functions were named DMP1β and DMP1γ. The DMP1β and DMP1γ proteins lack the DNA-binding and C-terminal trans-activation domains found in DMP1α and are therefore unable to transactivate p14ARF or other DMP1α target genes (Supplementary Figures 1A and 1B) [19]. Unlike DMP1γ and DMP1α, DMP1β was found to block differentiation and stimulate monocyte proliferation during PMA-induced differentiation to macrophages [19]. Hence, the DMP1 isoforms may have unique functions, in particular those other than tumour suppression.
Alternative splicing is a mechanism for a single locus to encode multiple functionally distinct proteins that regulates different biological processes [20,21]. Several splicing factors, RNA-binding proteins regulating alternative splicing, have been identified as proto-oncogenes and are frequently overexpressed in human cancer [22,23]. Multiple cancer-associated genes such as PKM, Bcl-x, CD44, cyclin D1, p63, and p73 are alternatively spliced in tumours, compared with matched normal tissues, to produce their tumour-promoting isoforms [21,24,25]. The activities of tumour-associated isoforms vary from regulating novel biological processes to negating the isoforms expressed in normal tissues [26]. Since DMP1 is a critical mediator of breast cancer development in humans and mice, we sought to investigate the involvement of the other DMP1 splice isoforms (DMP1β and DMP1γ) in mammary oncogenesis. Using breast cancer cell lines, clinical samples, and a newly established transgenic mouse model of breast cancer, we demonstrate that DMP1 is aberrantly spliced in breast cancer to increase DMP1β and promote disease progression.
Materials and methods
Details of the human breast cancer samples; the generation of a DMP1β-specific polyclonal antibody in rabbits; the cell lines and mammosphere assays; the DNA and RNA analyses; the western blot analyses; the PCR, qRT-PCR TaqMan, and shRNA sequences; the source of the RNA-seq data; the selection and processing of RNA-seq data; the immunohistochemistry, immunofluorescence, and whole mammary gland mounts; the single staining immunohistochemistry; and the double staining immunohistochemistry are provided in the Supplementary materials and methods.
Establishment of MMTV-DMP1βVH mice
The V5 and 6× His tagged human DMP1β cDNA (a gift from B Torbett) was cloned into a HindIII site of the MMTV-SV40-BSSK vector (from Dr Philip Leder, Harvard Medical School). After DNA sequencing confirmation, pronuclear microinjection of the targeting vector in the FVB/NJ mouse background was carried out by the Transgenic Core Facility at Wake Forest School of Medicine. The founding offspring were identified by PCR. The carrier females of the transgene were bred with pure wild-type FVB/NJ males to expand the colonies. The female mice were monitored daily for palpable tumour development. All of the mice were maintained in accordance with an approved IACUC protocol.
Statistical analyses
Kaplan-Meier graphs for tumour-free survival of MMTV-DMP1β mice and relapse-free survival of breast cancer patients were analysed by MedCalc software, Mariakerke, Belgium. The following statistical analyses were used in other experiments: two-way ANOVA for the cell growth assays; unpaired Student's t-test for the mammosphere assays and RNA-seq analyses; and two-sided chi square tests for the DMP1 LOH versus DMP1β mRNA/protein expression and Supplementary Table 1. A difference was considered statistically significant at p < 0.05.
Results
DMP1 is aberrantly spliced in breast cancer to overexpress DMP1β
To study whether DMP1 is alternatively spliced in human breast cancer, total RNA from the tumours of 20 breast cancer patients and the matched normal tissues was isolated and qRT-PCR was conducted for DMP1α, DMP1β, and DMP1γ transcripts. The expression of DMP1α was used as an internal control to determine the DMP1β to DMP1α (DMP1β/α) and DMP1γ to DMP1α (DMP1γ/α) isoform ratios in each tumour and in its matched normal mammary tissue. To evaluate the relative expression of the two DMP1 isoforms among these tissues, we designated the DMP1β/α ratio in normal tissue 05–173, which was at the median of 20 normal tissues, as 1.0. The DMP1β/α isoform mRNA ratios were significantly higher (ie ≥2.0) in eight breast cancer samples (∼40%) than in their matched normal tissues (Figure 1A), while the DMP1γ/α isoform ratios were higher in only three (∼15%) tumours (Supplementary Figure 2A). In fact, 12 tumours and their normal tissues had no detectable DMP1γ mRNA, suggesting that DMP1γ is unlikely to play a significant role in breast cancer. Hence, we further investigated the alteration of DMP1β expression in breast cancer. qRT-PCR for an additional 26 patients was conducted to evaluate the alteration of DMP1β/α isoform expression. When combined with the patients in Figure 1A, alteration of DMP1 splicing with increased DMP1β/α ratios in the breast tumours versus matched normal tissues was found in 14 out of 46 patients (∼30.4%). Importantly, alteration of DMP1β/α ratios was found in patients with wild-type DMP1 (LOH-negative cases) and in those with hemizygous deletion (LOH-positive cases) (p = 0.1394, χ2 = 2.185). In agreement with human breast cancer, Dmp1β/α ratios were high in ∼53% (8/15) of mammary tumours from the MMTV-neu mouse model, regardless of hemizygous deletion of the Dmp1 gene (Supplementary Figure 2B). Using a publicly available database (GSE58135) for RNA-seq analyses of breast cancers [42 ER+/HER2– breast cancer primary tumours, 30 uninvolved adjacent breast tissues, 42 triple-negative breast cancer (TNBC) primary tumours, 21 uninvolved adjacent breast tissues], we found that the DMP1β levels were higher in breast cancer than in uninvolved tissues, as shown by the Student's t analyses (p = 0.0058) (Supplementary Figure 3). The mode for DMP1β is higher in breast cancer samples than that of uninvolved neighbouring tissue (95 versus 45 in ER+/HER2– BC; 85 versus 60 in TNBC). Overall, significantly increased expression of human DMP1β mRNA (ie 80 hits or higher) was observed in 23/42 (54.8%) cases of ER+/HER2– breast cancer and 18/42 (42.9%) cases of TNBC, which is consistent with the percentage of high DMP1β protein expression in immunohistochemistry.
Figure 1.
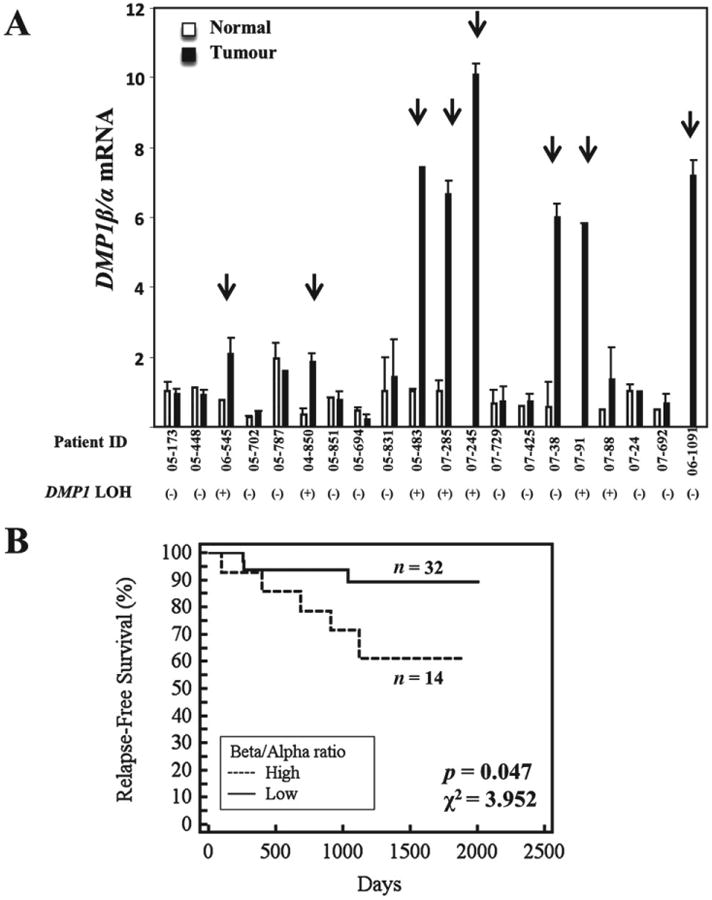
DMP1 alternative splicing in breast cancer leads to increased DMP1β/α ratios. (A) qRT-PCR analysis for DMP1 splicing in 20 breast tumours versus matched normal breast tissues showing increased DMP1β/α mRNA ratios in eight patients. Error bars for normal and tumour tissues of each patient represent experimental variations in the real-time PCR analyses. The arrows indicate patients with significantly altered DMP1β/α splicing in tumours compared with their matched normal tissues. DMP1 LOH (loss of heterozygosity) below each patient indicates breast tumours with hemizygous DMP1 deletion [LOH(+) = one DMP1 allele deletion; LOH(−) = wild-type DMP1 locus]. (B) Kaplan-Meier relapse-free survival analysis of patients with high DMP1β/α ratios (n= 14) versus patients with low DMP1β/α ratios (n = 32) (p = 0.047, χ2 = 3.952)
Using relapse data, we evaluated the correlation between DMP1β/α ratios in tumour and the clinical outcomes of breast cancer patients. Patients with high tumour DMP1β/α ratios were found to relapse significantly faster than patients with low DMP1β/α ratios (p = 0.047, χ2 = 3.952; Figure 1B). We then studied the correlation between DMP1β/α ratios and clinical stages and histological subtypes of breast cancer (Supplementary Table 1). High DMP1β/α ratios tended to associate with stage I and luminal A histological subtypes, but neither was statistically significant. In summary, aberrant DMP1 splicing is present in a significant number of breast cancer cases and carries a biological consequence for the patients.
DMP1β protein is elevated in breast tumour tissues and is associated with poor patient outcomes
Next, we sought to determine whether DMP1β protein is increased in breast cancer. Since our antibodies to Dmp1 (RAX and RAD) [27] detect all the splicing isoforms, we raised a polyclonal antibody to an amino acid epitope found in the C-terminus of DMP1β and DMP1γ, but not in DMP1α protein (Supplementary Figure 1A). To determine the specificity of this new DMP1 antibody (named RAB) to different endogenous DMP1 proteins, all three DMP1 isoforms were simultaneously knocked down to 40% at the RNA level in MDA-MB-231 cells. We found that endogenous DMP1β (∼43kDa) was depleted with each shRNA (Supplementary Figure 4A). The pattern of expression of DMP1β (ie specific to cancer cells) was different from that of DMP1α, which was ubiquitously detectable in both non-transformed and transformed cells (Supplementary Figure 4B). The specificity of RAB was also confirmed in MDA-MB-175VII cells (Supplementary Figure 5A) followed by an immunofluorescent analysis. We further tested the specificity of the RAB antibody to different DMP1 isoforms by individually transfecting each DMP1 isoform into NIH 3 T3 cells followed by western blot analyses. While our previously developed pan-DMP1 antibody (RAX) was able to detect all three DMP1 isoforms, the RAB antibody only detected exogenous DMP1β protein, indicating its high specificity to DMP1β (Supplementary Figures 5B and 5E). Using the RAB antibody, we then carried out immunohistochemistry (IHC) with paraffin-embedded tumour tissues from 63 breast cancer patients. The RAB antibody specificity in the IHC analysis was confirmed by the blocked tumour tissue staining after its pre-incubation with 5μg of the peptide used in the immunization for its production (Supplementary Figure 5C). The staining intensity and percentage of the RAB antibody ranged from low (0–3 in combined scores) to high (4–6) in the tumours compared with surrounding normal breast tissues (Figure 2A and Supplementary Figure 6). Specifically, 35 of 63 (55.6%) breast tumours were highly stained with the RAB antibody relative to the surrounding normal tissues. The staining intensity of the RAB antibody correlated with DMP1β mRNA expression in matched patients (Supplementary Figure 5D). We graphed a Kaplan–Meier relapse-free survival curve based on 4–6 versus 0–3 DMP1β staining intensity. Patients with high DMP1β staining in the tumours relapsed earlier than those with low or absent DMP1β (n = 63, p = 0.0050, χ2 = 7.8653; Figure 2B). There was no correlation between DMP1β protein expression and LOH of the locus, suggesting that these two events are independent (p = 0.4125). Breast cancers with 4–6 DMP1β protein expression were associated with clinical stage I (p = 0.0218, χ2 = 5.26), but were not predominantly of any particular histological subtype (p = 0.7642; Supplementary Table 1). Our data indicate that the DMP1β protein, aside from altered splicing to increase DMP1β/α ratios, is frequently overexpressed in breast tumour tissues and that DMP1β protein expression in breast cancer is associated with significantly shorter survival.
Figure 2.
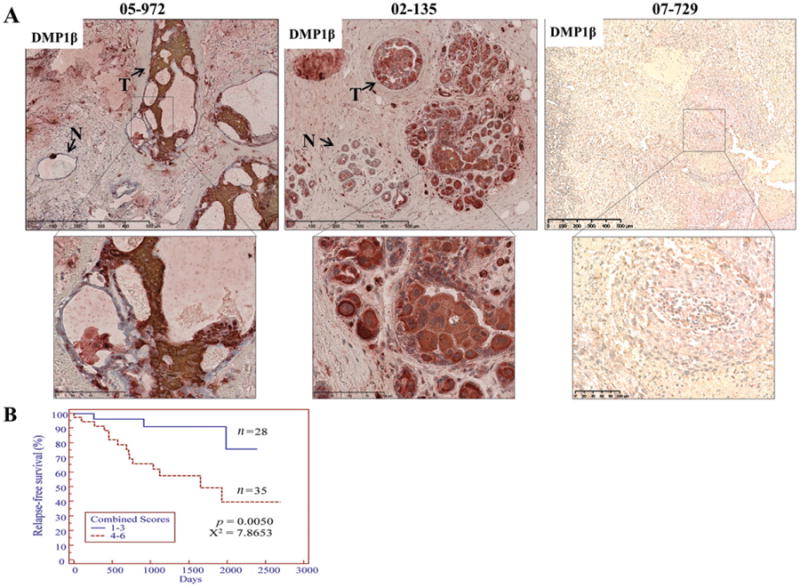
DMP1β immunohistochemistry in human breast tumours. (A) Representative images of DMP1β immunohistochemistry staining from three breast cancer patients (patients 05–972 and 02–135: high DMP1β expression; patient 07–729: low DMP1β expression). A total of 63 human breast tumours were stained with DMP1β-specific antibody (RAB). DMPβ staining was significantly higher in the tumour tissues (05–972 and 02–135) than in the surrounding normal tissues. (B) The immunohistochemical staining sections were scored on both the intensity and the percentage of positive cells with RAB, and Kaplan-Meier relapse-free survival curves were graphed between the high (4–6) and the low (0–3) staining groups. Patients with significantly higher DMP1β scores in tumours had a significantly shorter relapse-free survival than patients with tumours with lower scores (p = 0.0050, χ2 = 7.8653).
Ectopic expression or knockdown of DMP1β in breast epithelial cells modulates proliferation
While the tumour suppressor DMP1α acts as a potent activator of cell cycle arrest and senescence, the DMP1 locus can be aberrantly spliced and DMP1β is overexpressed in breast cancer [11,13,19]. Thus, we set out to determine the biological function of DMP1β in breast non-tumourigenic and cancer cells. MCF10A cells stably expressing DMP1β or vector alone were used to study the effect on cell growth (Figure 3A, right panel). Whereas our previously published work indicated that DMP1α expression in MCF10A cells inhibited proliferation and induced the p53 pathway [11], DMP1β-expressing MCF10A cells grew significantly faster than the cells with the vector alone (Figure 3A, left panel). MCF10A cells expressing DMP1β formed significantly larger mammospheres than those from the control when plated in a 3D Matrigel™ culture system (Figure 3B). To study the consequences of endogenous DMP1β on human breast cancer cell lines, we designed several shRNAs specifically targeting DMP1β and two shRNAs targeting all three DMP1 isoforms. Due to a very limited sequence specific to DMP1β, we encountered difficulty in generating effective shRNAs to specifically silence DMP1β (data not shown). However, the shRNAs (DMP1-1131 [17] and DMP1-465) targeting all three DMP1 isoforms significantly reduced their expression (Figures 4A and 4B). The DMP1-1131 and DMP1-465 shRNAs, independent of mutation or deletion status of the p53 pathway, reduced the proliferation of BT474 and MDA-MB-231 (Figures 4A and 4B), as well as that of MDA-MB-175VII and ZR-75-1 (Supplementary Figure 7B, data not shown). The p53-independent effect of DMP1β on cell proliferation was also con-firmed in SK-BR-3 cells (p53 mutant) (Supplementary Figure 7A). Similarly, when MDA-MB-175VII (p53 wt) cells with the DMP1 shRNA were plated in 3D Matrigel™ culture, they formed significantly smaller mammospheres than the cells expressing a control shRNA (Figure 4C). The growth of non-transformed MCF10A cells was much less affected by DMP1 shRNA (Supplementary Figure 7C). This means that DMP1β plays an essential role in the cell growth of breast cancer cells, but not in non-transformed breast epithelial cells. In summary, our data indicate that DMP1β has a distinct biological role compared with DMP1α; while DMP1α activates the p53 pathway and induces senescence [11,13], the DMP1β isoform increases the proliferation of breast epithelial cells in a p53-independent fashion.
Figure 3.
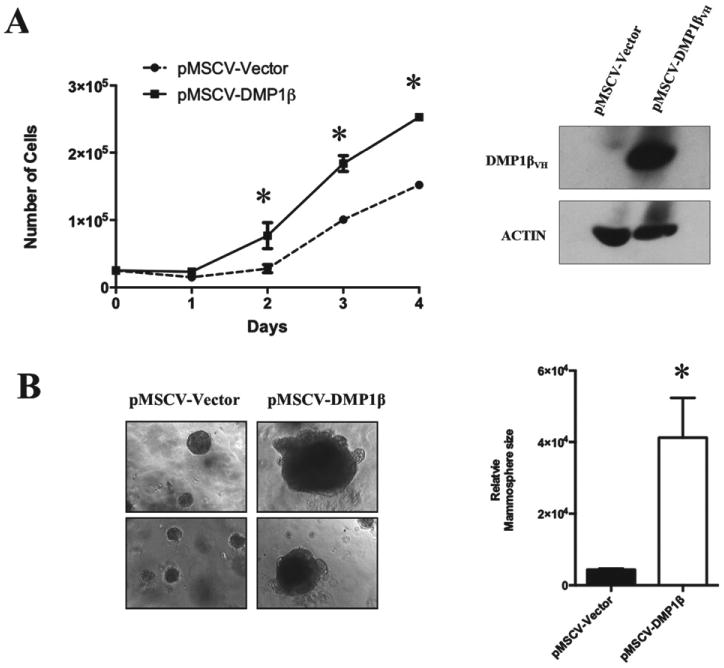
DMP1β expression in non-tumourigenic human breast cell line increases proliferation. (A) Human DMP1β cDNA was stably expressed in MCF10A cells using the pMSCV-puro retroviral vector and growth curves were generated using puromycin-resistant cell pools. DMP1β expression was confirmed by western blot analysis with an antibody to V5. Cells expressing DMP1β grew more rapidly (*p < 0.001). (B) MCF10A cells stably expressing DMP1β were grown as 3D mammospheres in Matrigel™ for over 14 days when images were taken. Relative sizes of mammospheres between vector and DMP1β-expressing MCF10A cells were measured using Image J software (*p < 0.001).
Figure 4.
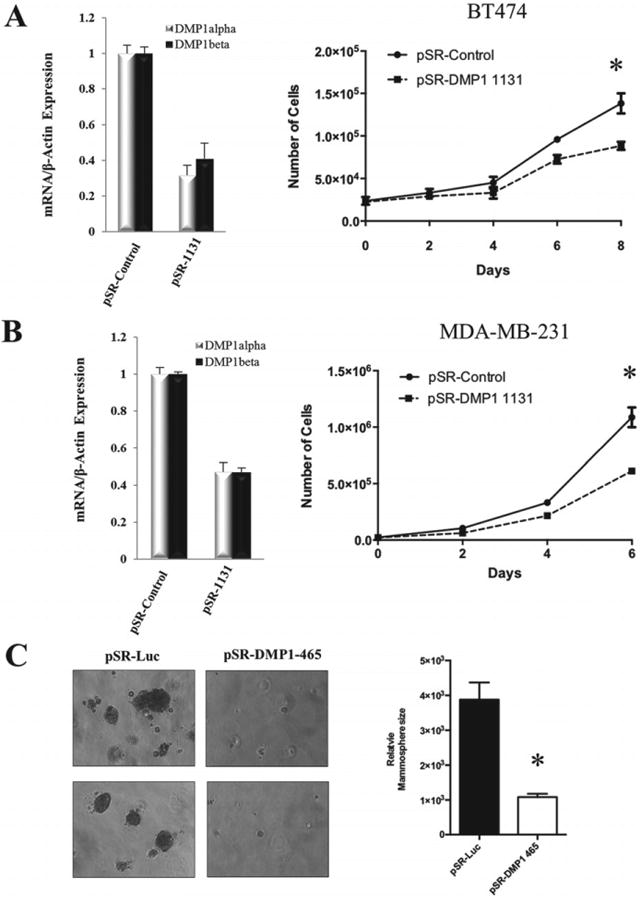
Knockdown of endogenous DMP1 reduces breast cancer cell proliferation. (A, B) BT474 and MDA-MB-231 cells were stably infected with pSUPER.retro.puro retroviral vectors expressing the DMP1-1131 shRNA or a control shRNA. qRT-PCR confirmed the knockdown of endogenous DMP1α and DMP1β isoforms in puromycin-resistant cell pools. Cell proliferation was measured by cell counting over the time period indicated. (C) MDA-MB-175VII cells were infected with retrovirus carrying a shRNA targeting endogenous DMP1 3′UTR (DMP1-465) and puromycin-resistant cells were plated in Matrigel™. The growth inhibitory effect of the DMP1 shRNA on mammospheres was quantitated with Image J software and graphed (*p < 0.001).
DMP1β induces proliferation and mammary gland tumours in vivo
To examine DMP1β function in vivo and whether it has the capacity to induce proliferation of mammary epithelial cells, we set out to establish MMTV-DMP1β transgenic mouse lines. Human V5/6×His-DMP1β cDNA was subcloned into the MMTV-LTR vector for establishment of the transgenic mice (Figure 5A). The founder mice were genotyped by PCR and four transgenic females were identified for the colony expansion (Figure 5A). Western blot and qRT-PCR analyses confirmed the expression of transgenic DMP1β protein and mRNA in the mammary glands of MMTV-DMP1β mice (Figure 5B). In agreement, immunohistochemistry using RAB and pan-DMP1 (RAD [27]) antibodies detected DMP1β protein expression in the luminal cells of transgenic mice (Figure 5C), but not in control tissues (lung, liver; data not shown). Pregnancy in mice significantly alters tumourigenic susceptibility and activity of the MMTV-LTR promoter [28,29]; therefore, we analysed both nulliparous and multiparous female mice. Parous (n = 19 for transgenics; n = 18 for non-transgenics) and nulliparous (n = 26 for transgenics; n = 19 for non-transgenics) females were monitored for mammary lesions/tumour development for 6–20 months. DMP1β-transgenic (42% parous) females developed mammary tumours with a mean latency of 16 months (p < 0.0001, χ2 = 19.7818; Figures 6A and 6B, upper). Multiparous DMP1β-transgenic females developed mammary tumours earlier than non-parous transgenic females (meanlatency 460 versus 545 days, p = 0.0052, χ2 = 7.8233; Figure 6B, lower). Thus, the onset of mammary tumours in MMTV-DMP1β-transgenic females was earlier than that of MMTV-cyclin D1/D3/E, c-rel, but later than MMTV-ErbB2 mice [28]. We also observed a low incidence (>4%) of pituitary prolactinomas, which have been linked to spontaneous tumours in the FVB strain at an average age of 100 weeks and therefore were eliminated from the analysis [30]. As expected, the whole mammary gland mounts and H&E analysis of the mammary glands from the 18-month-old transgenic mice of the four strains showed extensive hyperplasia with multifocal tumour lesions (Supplementary Figures 8 and 9). Immunohistochemical studies demonstrated strong staining of the proliferation markers, Ki67 and cyclin D1, in DMP1β-transgenic mammary glands, which further confirmed the hyperplastic phenotype of the glands (Figure 6C). The observed tumour lesions were highly infiltrated with immune cells (Figure 7) and showed evidence of keratinization and squamous differentiation. The immune infiltrates were mostly T lymphocytes as they were positive for CD3 (Supplementary Figure 10A), but negative for CD20 (not shown). To ascertain which cellular compartment of the mammary gland proliferated within tumours, we double stained these lesions with antibodies for markers of basal/myoepithelial cells (cytokeratin 14; CK14) and luminal cells (cytokeratin 8; CK8). The majority of the tumour cells were positive for CK8, suggesting the luminal phenotype of the tumours; however, some tumour cells were positively stained for both CK8 and CK14 (Figure 7 and Supplementary Figures 10B and 10C). As expected, the keratinized sheets were exclusively positive for CK14, supporting the transdifferentiation phenotype (Supplementary Figure 11 for double staining). We observed low cytoplasmic staining for ER and undetectable staining for PR, indicating that these hormone receptors are not involved in the DMP1β-induced tumour initiation or progression (Figure 7). Hence, the observation that DMP1β has a distinct role from DMP1α in vitro was recapitulated in our in vivo mouse model. Overall, while DMP1α functions as a bona fide tumour suppressor to activate the p53 pathway, we provide evidence that the DMP1β splice isoform induces cell proliferation and mammary tumour formation.
Figure 5.
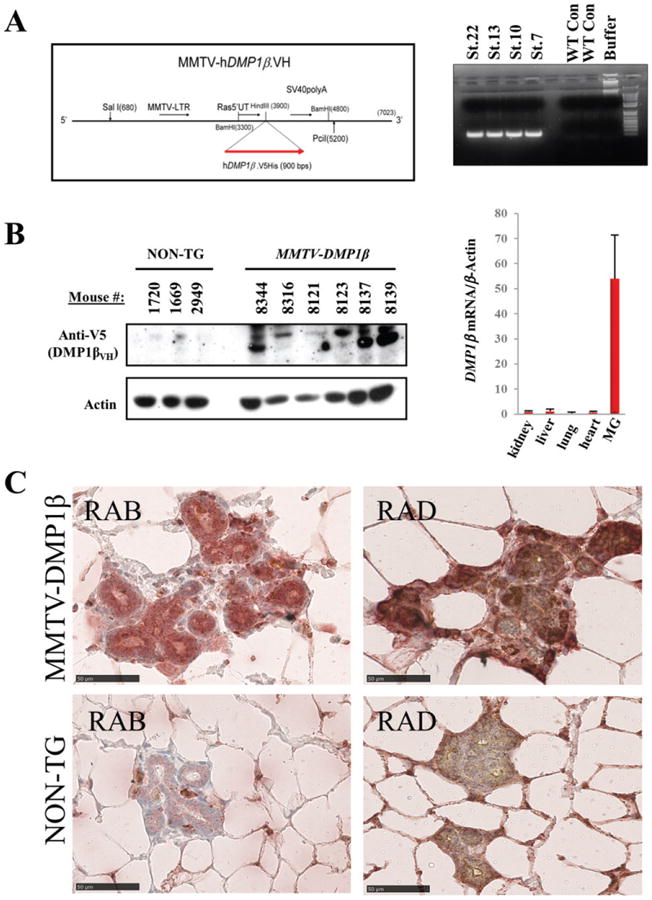
Establishment of MMTV-DMP1βVH mice and expression of the transgene in mammary glands. (A) Schematic map of the MMTV-LTR targeting construct used for expressing of V5/6× His-tagged human DMP1β cDNA. DNA agarose gel analysis of the PCR genotyping results shows four transgenic strains (strains 7, 10, 13, and 22) used for expanding the colonies. (B) Western blot analysis shows the DMP1β protein expression in the mammary glands of DMP1β transgenic mice. qRT-PCR analysis shows specific expression of the DMP1β mRNA in mammary gland versus other tissues from MMTV-DMP1βVH transgenic females (MG = mammary gland). The DMP1βVH proteins migrated as multiple bands at 45-50 kDa dependent on possible post-translational modifications. (C) Immunohistochemical analysis of mammary glands from MMTV-DMP1β and non-transgenic females using DMP1β-specific (RAB) and pan-DMP1 (RAD) antibodies.
Figure 6.
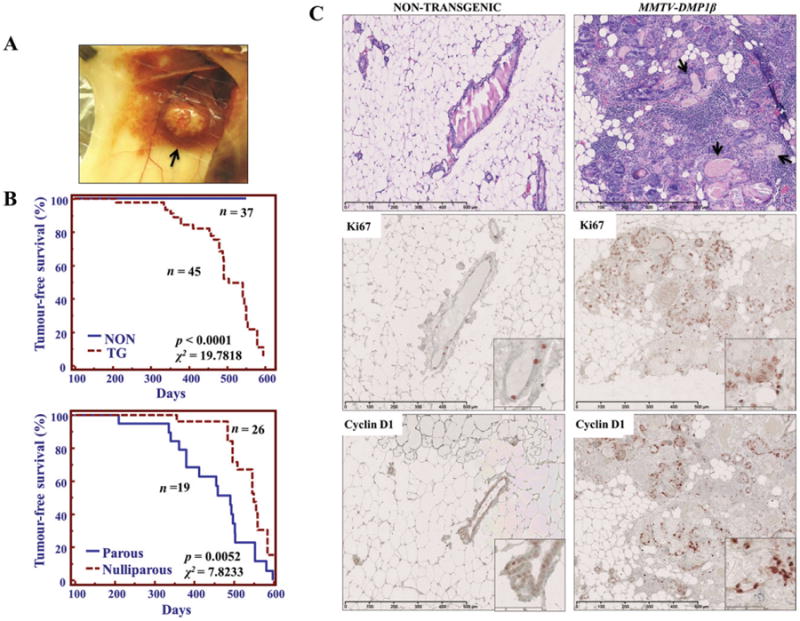
DMP1β induces mammary gland tumours in vivo. (A) Macroscopic image of a palpable mammary gland tumour in an MMTV-DMP1β mouse. The arrow indicates a large tumour mass in the mammary gland. (B) Kaplan-Meier tumour-free survival curves analysing DMP1β-transgenic versus non-transgenic and parous versus nulliparous mice. MMTV-DMP1β mice developed tumours around 16 months of age (upper) (p = 0.0001, χ2 = 19.7818). Since the frequency of hyperplasia was equal in transgenic and non-transgenic cases, only the animals with tumours and without tumours are shown. Tumour development in DMP1β-transgenic mice was accelerated when females went through several rounds of pregnancy (lower) (mean latency, 460 versus 545 days; p = 0.0052, χ2 = 7.8233). (C) Representative histological analysis of mammary glands from MMTV-DMP1β and non-transgenic mice at 18 months of age. The transgenic mammary glands show evidence of hyperplasia with focal tumours, immune cell infiltrate, and keratinized deposits indicative of adenosquamous carcinoma. Black arrows indicate areas of keratinization. Proliferative markers, Ki67 and cyclin D1, were overexpressed in mammary glands from transgenic females.
Figure 7.
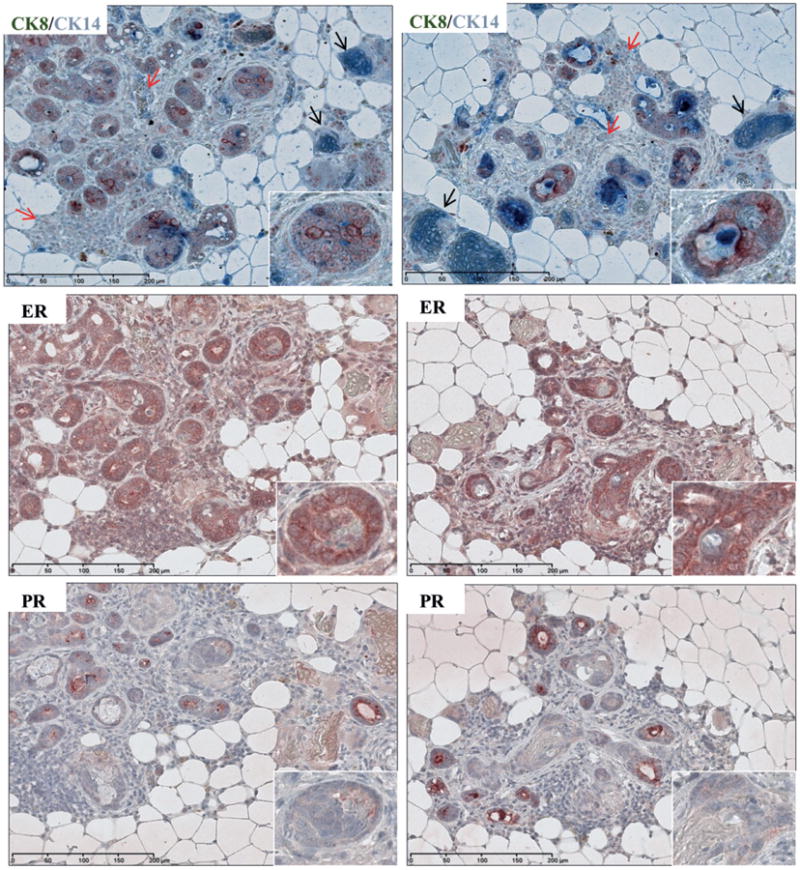
Characterization of mammary tumours from MMTV-DMP1β mice. Immunohistochemical staining of tumour lesions from two transgenic strains for oestrogen receptor (ER) and progesterone receptor (PR), and double staining for cytokeratin 8 (CK8; brown) and cytokeratin 14 (CK14; blue). CK8/CK14 double staining indicates that tumour epithelial cells mostly express luminal marker (CK8), while some cells express the basal/myoepithelial marker (CK14). Double CK8/CK14 staining is indicative of transdifferentiation. Black arrows indicate keratinized sheets strongly stained with basal marker (CK14). Red arrows indicate immune cell infiltrates. ER staining was weak and mostly cytoplasmic, indicating a non-active receptor. PR staining was completely absent in tumour cells.
Discussion
In this study, we have uncovered a novel function of DMP1β, an alternative splicing isoform of DMP1. The DMP1 locus encodes three unique mRNA transcripts [19], which were found alternatively spliced in breast tumours but not in matched normal tissues. The splicing alteration increased the tumour DMP1β/α isoform ratio in ∼30% of breast cancer cases and the DMP1β protein was highly expressed in ∼60% of breast cancers; both were associated with poor clinical outcomes of the patients. The other DMP1 isoform, DMP1γ, was rarely increased and often completely absent in the tumour tissues. Hence, our finding supports a notion for the existence of a selective advantage in breast tumours to overexpress DMP1β but down-regulate DMP1α and perhaps also DMP1γ. Alternative splicing of cancer-associated genes by tumour tissues has been reported previously. For example, prostate tumours express a fetal pyruvate kinase isoform, PKM2, which promotes aerobic glycolysis, a phenomenon known as the Warburg effect, and stimulates tumour progression [31]. DMP1β was previously reported to block PMA-induced differentiation of monocytes to allow continued proliferation, while DMP1γ had little effect in this setting [19]. The evidence that the Dmp1 locus regulates other mammary gland processes aside from the p53 pathway came from Dmp1-null females, as they are unable to nurse offspring due to poor mammary gland development resulting from reduced proliferation of luminal cells [10,18]. In agreement with the role of DMP1β in monocytes, our analysis indicates that patients with high DMP1β/α ratios in their tumours exhibited poor clinical outcomes. By developing a DMP1β-specific antibody, we also show that DMP1β protein is overexpressed in the tumours, while maintained at low levels in the surrounding normal tissues.
We have previously reported that DMP1 is hemizygously deleted in the tumours of ∼42% breast cancer cases, while maintaining the other wild-type allele without promoter hypermethylation [11]. The question why the loss of one DMP1 allele is sufficient to inactivate DMP1α tumour suppressor activity remained unanswered. Here we provide a possible explanation for this phenomenon by showing that increased DMP1β/α ratios occurred in both LOH(+) and LOH(−) cases, indicating that tumours may modulate the wild-type DMP1 allele with or without hemizygous DMP1 deletion. Similarly, MMTV-neu tumours had altered Dmp1 splicing in both Dmp1 wild-type tumours and those with a naturally occurring hemizygously deleted Dmp1 locus. Thus, the DMP1 locus is inactivated by two independent mechanisms: (1) hemizygous deletion of the DMP1α gene that has a tumour-suppressive function (p53-dependent), and (2) altered splicing that increases the DMP1β isoform with tumour-promoting activity (p53-independent). These two mechanisms could have synergistic effects in tumour development, which needs to be addressed in future experiments by crossing Dmp1-knockout mice and DMP1β-transgenic mice.
The generation of oncogenic splicing variants from tumour suppressor loci has been reported for p63 and p73 [24,25]. In both cases, the products of the oncogenic splicing isoforms that lack N-terminal transactivation domains are overexpressed in tumours and act in a dominant-negative fashion to all p53 family proteins (p53, p63, and p73) [25,32]. The function of DMP1α is dependent on its ability to stabilize the wild-type p53 via transactivating ARF expression or directly interacting with p53 [11,13,15–18]. The DMP1β protein lacks the DNA-binding domain and C-terminal transactivation domain of DMP1α, both of which are necessary for activating the Arf promoter and protein-protein interaction with p53 [15,33]. Therefore, it is unlikely that DMP1β directly modulates the Arf-p53 pathway. Our data and previously published work suggest a p53-independent mechanism of tumour promotion by DMP1β. Dmp1-null animals show a distinct mammary gland phenotype from MMTV-DMP1β mice, supporting the notion that DMP1β does not primarily function as a dominant-negative isoform on DMP1α. In fact, Dmp1-null female mice rarely develop mammary tumours even after 2 years of age. Hence, the tumour-promoting function of DMP1β is independent of the DMP1α–p53 axis. The detailed mechanism underlying DMP1β's action deserves further investigation using molecular/genetic approaches and in vivo mouse models.
Our previous work showed that knockdown of endogenous DMP1 using shRNAs targeting all three DMP1 isoforms surprisingly reduced proliferation in two out of three breast cancer cell lines [11]. Due to limited isoform-specific sequences, we were unable to design effective shRNAs unique to each DMP1 isoform. DMP1 splicing occurs on exon 10, where a short sequence containing the TAA stop codon is retained to produce DMP1β and DMP1γ proteins. DMP1α-specific shRNAs cannot be designed because the sequence of this transcript is included in all DMP1β and DMP1γ isoforms. Thus, we used shRNAs targeting all three DMP1 isoforms to show reduced proliferation in multiple breast cancer cell lines. Since overexpression experiments indicate that DMP1β accelerates proliferation while DMP1α induces growth arrest in the same breast epithelial cells, we attribute the attenuated growth of the DMP1-knockdown cells to the effect of DMP1β silencing [11]. The DMP1 -knockdown cells proliferate more slowly regardless of the p53 mutation or deletion status, suggesting a p53-independent mechanism.
A recently developed MMTV-Dmp1α transgenic mouse provided evidence that Dmp1α overexpression was non-tumourigenic but induced the p53 pathway, resulting in impaired mammary glands. Additionally, Dmp1α delayed HER2/neu-induced mammary gland tumour initiation, which further demonstrated its tumour suppressor function [12]. Conversely, mammary glands from our newly established MMTV-DMP1β transgenic mice developed normally without evidence of impairment. Indeed, when analysed at 6–20 months of age, mammary glands from MMTV-DMP1β transgenic mice had developed diffuse hyperplasia and multifocal tumours in four independent transgenic strains. The latency for mammary tumour development was much earlier (460days) in multiparous DMP1β transgenic mice than in nulliparous (545 days) females. The tumours and surrounding glands in the transgenic mice were hyperplasic, as they were strongly stained by the proliferation markers Ki67 and cyclin D1. All of the tumour lesions had evidence of keratinization, suggesting that the tumours are of the adenosqua-mous phenotype. In fact, the tumour epithelial cells were stained with both CK8 and CK14, suggesting transdifferentiation. Although adenosquamous carcinomas are infrequent in human breast cancer, they are induced in the mammary glands of cyclin D1 and cyclin D3-transgenic mice [34–36]. Likewise, tumours from MMTV-DMP1β mice were composed of keratinized sheets with evidence of transdifferentiation. Therefore, it is possible that cyclin D3 and DMP1β converge on the same signalling pathways to induce the transformation of mammary epithelial cells.
In conclusion, we have demonstrated that the DMP1 locus is alternatively spliced to increase the DMP1β isoform during mammary oncogenesis, which was associated with breast cancer progression. Our data strongly support the notion that isoform switching at the DMP1 locus observed in the tumours was not a mere reduction in DMP1α expression, but rather simultaneous inactivation of tumour suppressor activity mediated by DMP1α and an increase in the DMP1β isoform which promotes cell proliferation. The signalling pathways regulating DMP1 splicing in normal and tumour tissues remain unknown. Exon 10 of DMP1 contains multiple consensus sequences for splicing factors such as SF2/ASF, Tra2-β, and SC35, all of which have been implicated in tumourigenesis [23,37–39]. It is possible that HER2/neu signalling is involved since we have observed alteration of Dmp1 splicing in MMTV-neu tumours. Moreover, the mechanism of DMP1β-induced proliferation is still unclear. DMP1β lacks the necessary domains to function as a transcription factor and therefore is most likely acting through protein–protein interactions. Future studies are needed to dissect the upstream signalling that regulates DMP1 splicing in breast cancer and delineate DMP1β-interacting partners necessary for its oncogenic activity.
Supplementary Material
Figure S1. The domain structure of DMP1 splice isoforms.
Figure S2. Alteration of DMP1 splicing in human breast cancer and mouse mammary tumours.
Figure S3. DMP1β expression in human breast cancers from the publically available RNA-seq data.
Figure S4. Detection of the endogenous DMP1α/β proteins in breast cancer cells.
Figure S5. Evaluation of a newly generated DMP1 splice isoform-specific antibody (RAB).
Figure S6. Intensity grading of human breast cancers with the RAB antibody.
Figure S7. Influences of DMP1β overexpression and knockdown on breast epithelial cell growth.
Figure S8. Whole mammary gland mounts from MMTV-DMP1β female mice.
Figure S9. Mammary gland phenotype of 18-month-old MMTV-DMP1β female mice.
Figure S10. High-magnification images of mammary tumours found in MMTV-DMP1β transgenic mice.
Figure S11. Double staining immunohistochemistry of mammary tumours found in an MMTV-DMP1β transgenic mouse.
Table S1. Correlation of DMP1β expression with current histological and molecular classifications of human breast cancer.
Acknowledgments
We thank C Sherr, M Roussel, B Torbett, M Tschan, and P Leder for plasmid DNAs. We are grateful for technical assistance with microscopy received from Kenneth Grant and Brandi Bickford and the Cell and Virus Vector Core Laboratory of the Comprehensive Cancer Center at WFUHS for providing access to cell culture materials. Dr Greg Kucera provided clinical samples in this study. We thank L Miller and J Chou for their advice on RNA-seq analysis and Robert Kendig for his support in the in vitro cell growth assays. KI was supported by ACS RSG-07-207-01-MGO, NIH/NCI 5R01CA106314, and by Director's Challenge Award #20595 from WFUHS. GS was supported by ACS 116403-RSG-09-082-01-MGO and 5R01CA106314. DC was supported by 5R01CA106314. DM was supported by DOD pre-doctoral fellowship BC100907. DBS was supported by NCI training grant 5T32CA079448.
Footnotes
No conflicts of interest were declared.
Author contributions: The authors contributed in the following way: concepts and design for the study: DM, GS, KI; writing the manuscript: DM, AM, GS, KI; Figure 1: DM, EAF, PT, KI; Figure 2: DM, DBS, DC, MCW, KI; Figure 3: DM; Figure 4: DM, GS; Figures 5–7: DM, DBS, MC; Supplementary Figure 1: DM, KI; Supplementary Figure 2: DM, EAF, PT, KI; Supplementary Figure 3: AM, KI; Supplementary Figure 4: DM, DBS, KI; Supplementary Figure 5: DM, EAF, PT; Supplementary Figure 6: DM, DBS, DC, KI; Supplementary Figure 7: DM, GS; Supplementary Figure 8: DM; Supplementary Figure 9: DM, DBS, MC, KI; Supplementary Figure 10: DM, KI; Supplementary Figure 11: DBS, KI; Supplementary Table 1: DM, DC, MCW, KI; care and genotyping of mice: DM, DBS, EAF, GS; immunohistochemistry grading of human breast cancer: DM, DC, MCW, KI.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Mariotto AB, Robin Yabroff K, Shao Y, et al. Projections of the cost of cancer care in the United States: 2010–2020. J Natl Cancer Inst. 2011;103:117–128. doi: 10.1093/jnci/djq495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taneja P, Maglic D, Kai F, et al. Classical and novel prognostic markers for breast cancer and their clinical significance. Clin Med Insights Oncol. 2010;4:15–34. doi: 10.4137/cmo.s4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Higgins MJ, Baselga J. Targeted therapies for breast cancer. J Clin Invest. 2011;121:3797–3803. doi: 10.1172/JCI57152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ring A, Dowsett M. Mechanisms of tamoxifen resistance. Endocr Relat Cancer. 2004;11:643–658. doi: 10.1677/erc.1.00776. [DOI] [PubMed] [Google Scholar]
- 6.Marquette C, Nabell L. Chemotherapy-resistant metastatic breast cancer. Curr Treat Options Oncol. 2012;13:263–275. doi: 10.1007/s11864-012-0184-6. [DOI] [PubMed] [Google Scholar]
- 7.Bleyer A, Welch HG. Effect of three decades of screening mammography on breast-cancer incidence. N Engl J Med. 2012;367:1998–2005. doi: 10.1056/NEJMoa1206809. [DOI] [PubMed] [Google Scholar]
- 8.Keyomarsi K, Tucker SL, Buchholz TA, et al. Cyclin E and survival in patients with breast cancer. N Engl J Med. 2002;347:1566–1575. doi: 10.1056/NEJMoa021153. [DOI] [PubMed] [Google Scholar]
- 9.Esteva FJ, Hortobagyi GN. Prognostic molecular markers in early breast cancer. Breast Cancer Res. 2004;6:109–118. doi: 10.1186/bcr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taneja P, Maglic D, Kai F, et al. Critical roles of DMP1 in human epidermal growth factor receptor 2/neu–Arf–p53 signaling and breast cancer development. Cancer Res. 2010;70:9084–9094. doi: 10.1158/0008-5472.CAN-10-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maglic D, Zhu S, Fry EA, et al. Prognostic value of the hDMP1–ARF–Hdm2–p53 pathway in breast cancer. Oncogene. 2013;32:4120–4129. doi: 10.1038/onc.2012.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fry EA, Taneja P, Maglic D, et al. Dmp1α inhibits HER2/neu-induced mammary tumorigenesis. PLoS One. 2013;8:e77870. doi: 10.1371/journal.pone.0077870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inoue K, Roussel MF, Sherr CJ. Induction of ARF tumor suppressor gene expression and cell cycle arrest by transcription factor DMP1. Proc Natl Acad Sci U S A. 1999;96:3993–3998. doi: 10.1073/pnas.96.7.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sreeramaneni R, Chaudhry A, McMahon M, et al. Ras–Raf–Arf signaling critically depends on the Dmp1 transcription factor. Mol Cell Biol. 2005;25:220–232. doi: 10.1128/MCB.25.1.220-232.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frazier DP, Kendig RD, Kai F, et al. Dmp1 physically interacts with p53 and positively regulates p53's stability, nuclear localization, and function. Cancer Res. 2012;72:1740–1750. doi: 10.1158/0008-5472.CAN-11-2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inoue K, Zindy F, Randle DH, et al. Dmp1 is haplo-insufficient for tumor suppression and modifies the frequencies of Arf and p53 mutations in Myc-induced lymphomas. Genes Dev. 2001;15:2934–2939. doi: 10.1101/gad.929901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mallakin A, Sugiyama T, Taneja P, et al. Mutually exclusive inactivation of DMP1 and ARF/p53 in lung cancer. Cancer Cell. 2007;12:381–394. doi: 10.1016/j.ccr.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inoue K, Wen R, Rehg JE, et al. Disruptionof the ARF transcriptional activator DMP1 facilitates cell immortalization, Ras transformation, and tumorigenesis. Genes Dev. 2000;14:1797–1809. [PMC free article] [PubMed] [Google Scholar]
- 19.Tschan MP, Fischer KM, Fung VS, et al. Alternative splicing of the human cyclin D-binding Myb-like protein (hDMP1) yields a truncated protein isoform that alters macrophage differentiation patterns. J Biol Chem. 2003;278:42750–42760. doi: 10.1074/jbc.M307067200. [DOI] [PubMed] [Google Scholar]
- 20.Kornblihtt AR, Schor IE, Allo M, et al. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nature Rev Mol Cell Biol. 2013;14:153–165. doi: 10.1038/nrm3525. [DOI] [PubMed] [Google Scholar]
- 21.David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010;24:2343–2364. doi: 10.1101/gad.1973010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen-Eliav M, Golan-Gerstl R, Siegfried Z, et al. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J Pathol. 2013;229:630–639. doi: 10.1002/path.4129. [DOI] [PubMed] [Google Scholar]
- 23.Karni R, de Stanchina E, Lowe SW, et al. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nature Struct Mol Biol. 2007;14:185–193. doi: 10.1038/nsmb1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murray-Zmijewski F, Lane DP, Bourdon JC. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ. 2006;13:962–972. doi: 10.1038/sj.cdd.4401914. [DOI] [PubMed] [Google Scholar]
- 25.Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Mol Cancer Res. 2004;2:371–386. [PubMed] [Google Scholar]
- 26.Miura K, Fujibuchi W, Unno M. Splice isoforms as therapeutic targets for colorectal cancer. Carcinogenesis. 2012;33:2311–2319. doi: 10.1093/carcin/bgs347. [DOI] [PubMed] [Google Scholar]
- 27.Mallakin A, Sugiyama T, Kai F, et al. The Arf-inducing transcription factor Dmp1 encodes transcriptional activator of amphiregulin, thrombospondin-1, JunB and Egr1. Int J Cancer. 2010;126:1403–1416. doi: 10.1002/ijc.24938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taneja P, Frazier DP, Kendig RD, et al. MMTV mouse models and the diagnostic values of MMTV-like sequences in human breast cancer. Expert Rev Mol Diagn. 2009;9:423–440. doi: 10.1586/ERM.09.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ross SR. Mouse mammary tumor virus molecular biology and onco-genesis. Viruses. 2010;2:2000–2012. doi: 10.3390/v2092000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radaelli E, Arnold A, Papanikolaou A, et al. Mammary tumor phenotypes in wild-type aging female FVB/N mice with pituitary prolactinomas. Vet Pathol. 2009;46:736–745. doi: 10.1354/vp.08-VP-0280-R-FL. [DOI] [PubMed] [Google Scholar]
- 31.David CJ, Chen M, Assanah M, et al. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364–368. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uramoto H, Sugio K, Oyama T, et al. Expression of deltaNp73 predicts poor prognosis in lung cancer. Clin Cancer Res. 2004;10:6905–6911. doi: 10.1158/1078-0432.CCR-04-0290. [DOI] [PubMed] [Google Scholar]
- 33.Inoue K, Sherr CJ. Gene expression and cell cycle arrest mediated by transcription factor DMP1 is antagonized by D-type cyclins through a cyclin-dependent-kinase-independent mechanism. Mol Cell Biol. 1998;18:1590–1600. doi: 10.1128/mcb.18.3.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soo K, Tan PH. Low-grade adenosquamous carcinoma of the breast. J Clin Pathol. 2013;66:506–511. doi: 10.1136/jclinpath-2012-201084. [DOI] [PubMed] [Google Scholar]
- 35.Pirkmaier A, Dow R, Ganiatsas S, et al. Alternative mammary oncogenic pathways are induced by D-type cyclins; MMTV-cyclin D3 transgenic mice develop squamous cell carcinoma. Oncogene. 2003;22:8. doi: 10.1038/sj.onc.1206488. [DOI] [PubMed] [Google Scholar]
- 36.Wang TC, Cardiff RD, Zukerberg L, et al. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature. 1994;369:669–671. doi: 10.1038/369669a0. [DOI] [PubMed] [Google Scholar]
- 37.Merdzhanova G, Gout S, Keramidas M, et al. The transcription factor E2F1 and the SR protein SC35 control the ratio of pro-angiogenic versus antiangiogenic isoforms of vascular endothelial growth factor-A to inhibit neovascularization in vivo. Oncogene. 2010;29:5392–5403. doi: 10.1038/onc.2010.281. [DOI] [PubMed] [Google Scholar]
- 38.Watermann DO, Tang Y, zur Hausen A, et al. Splicing factor Tra2-β1 is specifically induced in breast cancer and regulates alternative splicing of the CD44 gene. Cancer Res. 2006;66:4774–4780. doi: 10.1158/0008-5472.CAN-04-3294. [DOI] [PubMed] [Google Scholar]
- 39.Xiao R, Sun Y, Ding JH, et al. Splicing regulator SC35 is essential for genomic stability and cell proliferation during mammalian organogenesis. Mol Cell Biol. 2007;27:5393–5402. doi: 10.1128/MCB.00288-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The domain structure of DMP1 splice isoforms.
Figure S2. Alteration of DMP1 splicing in human breast cancer and mouse mammary tumours.
Figure S3. DMP1β expression in human breast cancers from the publically available RNA-seq data.
Figure S4. Detection of the endogenous DMP1α/β proteins in breast cancer cells.
Figure S5. Evaluation of a newly generated DMP1 splice isoform-specific antibody (RAB).
Figure S6. Intensity grading of human breast cancers with the RAB antibody.
Figure S7. Influences of DMP1β overexpression and knockdown on breast epithelial cell growth.
Figure S8. Whole mammary gland mounts from MMTV-DMP1β female mice.
Figure S9. Mammary gland phenotype of 18-month-old MMTV-DMP1β female mice.
Figure S10. High-magnification images of mammary tumours found in MMTV-DMP1β transgenic mice.
Figure S11. Double staining immunohistochemistry of mammary tumours found in an MMTV-DMP1β transgenic mouse.
Table S1. Correlation of DMP1β expression with current histological and molecular classifications of human breast cancer.