

Abstract
Recently we described a novel pyranopyridine inhibitor (MBX2319) of RND-type efflux pumps of the Enterobacteriaceae. MBX2319 (3,3-dimethyl-5-cyano-8-morpholino-6-(phenethylthio)-3,4-dihydro-1H-pyrano[3,4-c]pyridine) is structurally distinct from other known Gram-negative efflux pump inhibitors (EPIs), such as 1-(1-naphthylmethyl)-piperazine (NMP), phenylalanylarginine-β-naphthylamide (PAβN), D13-9001, and the pyridopyrimidine derivatives. Here, we report the synthesis and biological evaluation of 60 new analogs of MBX2319 that were designed to probe the structure activity relationships (SARs) of the pyranopyridine scaffold. The results of these studies produced a molecular activity map of the scaffold, which identifies regions that are critical to efflux inhibitory activities and those that can be modified to improve potency, metabolic stability and solubility. Several compounds, such as 22d–f, 22i and 22k, are significantly more effective than MBX2319 at potentiating the antibacterial activity of levofloxacin and piperacillin against Escherichia coli.
Keywords: Pyranopyridine, Efflux pump inhibitor, Enterobacteriaceae, Adjunctive therapy, antibacterial
1. Introduction
Infections caused by Gram-negative bacteria are a growing concern. These bacteria are now responsible for about 30% of hospital-acquired infections and are the major cause of ventilator-associated pneumonia (VAP) and urinary tract infections.1 Among these pathogens, the Enterobacteriaceae have been identified as the most prevalent, followed by the nonfermenting pathogens, e.g. Pseudomonas aeruginosa and Acinetobacter spp.1,2 Infections caused by these pathogens are difficult to treat because they have acquired resistance to multiple antibiotics. Some strains of the Enterobacteriaceae have developed resistance to nearly all antibiotics, including carbapenems, which are often considered the antibiotics of last resort.2 In addition, the challenge of developing new antibiotics against Gram-negative bacteria is made significantly more difficult by the high levels of intrinsic resistance that are caused by a highly protective outer membrane (OM) and efflux pumps.3 The OM acts as a barrier to the diffusion of large lipophilic antibiotics into the cell, and highly efficient efflux pumps recognize a broad range of exogenous molecules (including antibiotics and biocides) and extrude them from the cell.4,5 Overexpression of efflux pumps, especially those of the resistant-nodulation-division (RND) family, is considered one of the key contributors to the bacterial multidrug resistant (MDR) phenotype.6
Because efflux pumps are responsible for much of the intrinsic antibiotic resistance in Gram-negative bacteria,7,8 they are important targets for antimicrobial drug discovery. Inhibition of efflux pumps by genetic mutations or efflux pump inhibitors (EPIs) results in increased antibiotic sensitivity.9 In addition, inhibition of RND pumps in P. aeruginosa by genetic deletion10 or with a potent EPI11 decreases the frequency of resistance to levofloxacin (LVX). RND pumps have been shown to play a role in the virulence of the enteric pathogen Salmonella enterica serovar Typhimuirum,12 and EPIs that target RND pumps have been shown to inhibit biofilm formation in E. coli and K. pneumoniae.13 In principle, by sensitizing bacteria to antibiotics, EPIs would increase the potency or potentially broaden the spectrum of existing classes of antibiotics. EPIs could also prevent the emergence of resistance, inhibit biofilm formation, and decrease the virulence of enteric pathogens. Therefore, the development of potent EPIs could have broad impacts on efforts to control against Gram-negative bacterial infections.
We recently discovered a novel EPI, compound MBX2319 (Figure 1), through a cell-based high-throughput screening campaign designed to identify small molecules that act synergistically with ciprofloxacin (CIP) against Escherichia coli.14 MBX2319 emerged as the most promising hit compound due to its high antibiotic potentiation, drug-like scaffold, and low cytotoxicity. Subsequent characterization of the mechanism of action of MBX 2319 showed that it selectively inhibits AcrB, the major efflux pump of the Enterobacteriaceae. While MBX2319 itself had no antibacterial activity (MIC ≥ 100 µM), it significantly increased the antibacterial activity of fluoroquinolone and β-lactam antibiotics at a concentration of 3.1 µM.14 Structurally, MBX2319 is distinctive from other known Gram-negative EPIs, such as 1-(1-naphthylmethyl)piperazine (NMP),15,16 phenylalanylarginine-β-naphthylamide (PAβN),11 D13-900117 and the related pyridopyrimidine compounds18 (Figure 1). The scaffold of MBX2319 is comprised of a pyranopyridine core with five substituents around the ring. To gain information about the structure-activity relationships (SARs), we initiated a medicinal chemistry exploration in which the substituents were systematically modified. Our goal was to locate sites that could be varied to improve potency, metabolic stability, and solubility. Based on these findings, we have constructed a detailed molecular activity map of the MBX2319 scaffold and identified new compounds with improvements in all three properties.
Figure 1.

Structure of MBX 2319 and some EPIs of Gram-negative bacteria
2. Chemistry
The general strategy for preparing new analogs of MBX2319 is shown in Scheme 1. This method allowed for independent variation of Z and the R1–R4 groups in structures 4 and 5. The synthesis began with a three-component coupling reaction between the hydropyranone 1, malononitrile and carbon disulfide to provide the thione 2.19 Treatment of compound 2 with different amines provided the hydropyranopyridine 3.20 Alkylation of the thiol group with different alkyl halides14 or epoxide provided thioethers 4. In some cases, further derivatization of the nitrile or the thioether moieties provided target compounds 5.
Scheme 1.
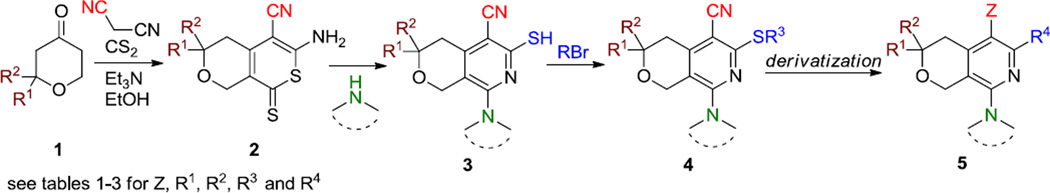
General method for preparation of MBX2319 and analogs
2.1. Modification of the nitrile group and the sulfide
To initiate the project, we treated MBX2319 with acid, base, and oxidizing reagents to prepare new derivatives, as shown in Scheme 2. Hydrolysis of the nitrile group was difficult; either treatment with concentrated mineral acid or alkali hydroxide failed to provide the carboxamide. Specifically, treatment of MBX2319 with concentrated HBr did not provide any hydrolyzed product, while similar treatment with sulfuric acid only provided the sulfonic acid 6, leaving the nitrile group intact. Treatment of MBX2319 with KOH led to removal of the phenethyl group, presumably due to elimination of styrene. We eventually succeeded in preparing the carboxamide 9 and N-hydroxyamidine 8 by treatment of MBX2319 with hydroxylamine hydrochloride and t-BuOK. The difficulty encountered in functionalization of the nitrile is most likely due to the steric hindrance exerted by the sulfide and the isobutyl moieties on both sides. We observed that functionalization of other (unhindered) nitrile groups in the molecule occurred easily and selectively (Scheme 6, conversion c). Lastly, oxidation of the sulfide with m-CPBA provided the sulfoxide 10 and sulfone 11 derivatives.
Scheme 2.

Reagents and conditions: (a) HBr (conc.); (b) H2SO4, TFA; (c) KOH, dioxane, reflux; (d) NH2OH.HCl, tBuOK, DMSO, 100 °C; (e) m-CPBA, CH2Cl2
Scheme 6.

Reagents and conditions: (a) with 7: R6CH2CH2Br, Cs2CO3, DMF, MeCN; (b) with 18e, 18g: R6CH2CH2Br, EtOH (one pot); (c) TMSN3, Bu2SnO, toluene, reflux; (d) TFA, CH2Cl2; (e) Ac2O, Et3N, CH2Cl2; (f) TFAA, Et3N, CH2Cl2; (g) CH3SO2Cl, Et3N, CH2Cl2; (h) acrylic acid, HATU, Et3N, DMF; (i) pivalic acid, HBTU, Et3N, DMF; (j) pyrazole-1-carboxamidine.HCl, pyridine.
2.2. Modification of the alkyl linker
To explore the structural requirements of the alkyl chain (L), thiol 714 was treated with benzyl bromide, 3-phenylpropyl bromide, 2-bromoacetophenone, and styrene oxide to provide analogs 12a–d, respectively, as shown in scheme 3.
Scheme 3.

Reagents and conditions: (a) Cs2CO3, DMF and BnBr for 12a, Ph(CH2)3Br for 12b, PhCOCH2Br for 12c, styrene oxide for 12d.
2.3. Modification of the gem-dimethyl functionality
Two analogs with the gem-dimethyl groups replaced by hydrogens were also prepared (Scheme 4). From tetrahydropyran-4-one, following similar procedures described for the synthesis of MBX2319,14 compounds 16a and 16b were obtained in 3 steps.
Scheme 4.

Reagents and conditions: (a) CS2, CH2(CN)2, Et3N, MeOH; (b) morpholine, EtOH; (c) R3Br, Cs2CO3, DMF, MeCN.
2.4. Modification of the morpholinyl group
Next, we prepared analogs with different amine groups in place of the morpholine (Scheme 5). Thus, the thione 1714 was treated with dimethylamine, diethylamine, pyrolidine, N-methylpiperazine, N-(2-methoxyethyl)piperazine, thiomorpholine, 2,6-dimethylmorpholine, homopyperazine, N-methylhomopiperazine, and homomorpholine to provide the corresponding thiols 18a–18j. These thiols were difficult to purify and isolate because of their air sensitivity. For example, an attempt to purify and isolate compound 18i by preparative HPLC failed because the thiol product completely dimerized into a disulfide after the elution. To minimize the undesired dimerization process, when the conversion of 17 to 18a–j was completed, as judged by LCMS, the reaction mixtures were treated directly with phenethyl bromide in one pot to provide the thioether products 19a–j.
Scheme 5.

Reagents and conditions: (a) R5H, EtOH, reflux; (b) PhCH2CH2Br
2.5. Modification/replacement of the phenyl group
To explore the effects of changes to the phenyl group, thiols 7, 18e and 18g were treated with various alkyl bromides to provide three product series 20a–r, 21a–k and 22a–22k (Scheme 6). The tetrazoles 20b and 20p were prepared from the corresponding nitriles 20a and 20o via treatment with azido(trimethyl)silane and dibutyltin oxide. The cyano group on the pyridine ring did not participate in this reaction. Further modifications of 22d included removal of the Boc group by treatment with trifluoroacetic acid to provide the aniline 22e. Subsequent acylation and sulfonylation of 22e provided the amides 22f–j. Treatment of 22e with 1H-pyrazole-1-carboxamidine provided the guanidine 22k.
3. Results and discussion
We have surveyed modifications of the five substituents around the pyranopyridine core structure. Compounds were evaluated in biological assays that measure potency, cytotoxicity against mammalian cells, and in vitro ADME predictors. The potency of the analogs against efflux pumps is measured by the ability of the analogs to decrease the MICs of levofloxacin (LVX) or piperacillin (PIP) against E. coli and was quantified as the MPC4, which is the minimum potentiation concentration of the analog that decreases the MIC of an antibiotic by 4 fold. Cytotoxicity against mammalian cells (HeLa) is expressed as CC50, the concentration at which cell viability is decreased by 50%. The data from these assays are presented in Tables 1–3. To assess in vitro ADME properties of selected compounds, we measured water solubility, inhibition of CYP450 3A4 and microsomal stability. The data are summarized in Table 4.
Table 1.
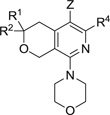
MPC4 values of MBX2319 and analogs with variations in Z, R1, R2 and the alkylsulfide of R4
 | |||||
|---|---|---|---|---|---|
| Compound # |
Z | R1, R2 | R4 | MPC4 (µM)a | |
| LVX | PIP | ||||
| MBX2319 | CN | CH3, CH3 | -SCH2CH2Ph | 3.1 | 3.1 |
| 8 | C(NH)NHOH | CH3, CH3 | -SCH2CH2Ph | ≥100 | ≥100 |
| 9 | C(O)NH2 | CH3, CH3 | -SCH2CH2Ph | ≥100 | ≥100 |
| 10 | CN | CH3, CH3 | -S(O)CH2CH2Ph | ≥100 | ≥100 |
| 11 | CN | CH3, CH3 | -SO2CH2CH2Ph | ≥100 | ≥100 |
| 12a | CN | CH3, CH3 | -SCH2Ph | ≥100 | ≥100 |
| 12b | CN | CH3, CH3 | -SCH2CH2CH2Ph | ≥100 | ≥100 |
| 12c | CN | CH3, CH3 | -SCH2C(O)Ph | ≥100 | ≥100 |
| 12d | CN | CH3, CH3 | -SCH2CH(OH)Ph | ≥100 | ≥100 |
| 16a | CN | H, H | -SCH2CH2Ph | ≥100 | ≥100 |
| 16b | CN | H, H | -SCH2CH2(2-pyridyl) | ≥100 | ≥100 |
MIC values of LVX and PIP against wild-type E. coli (AB1157): 0.063 µM and 4.0 µM, against the the ΔTolC mutant: 0.016 µM and 0.125 µM respectively.
Table 3.
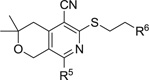
MPC4 and CC50 values of analogs with variations in R5 and R6 groups
 | ||||
|---|---|---|---|---|
| Compound | R6 | MPC4 (µM) | CC50 (µM) |
|
| LVX | PIP | |||
| R5 = morpholinyl | ||||
| 20a | cyano | ≥100 | ≥100 | >100 |
| 20b | tetrazol-5-yl | ≥100 | ≥100 | 30.2 |
| 20c | pyridin-2-yl | 50 | 25 | 78.1 |
| 20d | pyridin-3-yl | 25 | 3.1 | >100 |
| 20e | 2-methoxyphenyl | 3.1 | 3.1 | >100 |
| 20f | 3-methoxyphenyl | 2.4 | 1.6 | >100 |
| 20g | 4-methoxyphenyl | 6.3 | 3.1 | >100 |
| 20h | 3-acetoxyphenyl | 1.6 | 1.6 | 48.3 |
| 20i | 2-fluorophenyl | ≥100 | ≤1.6 | >100 |
| 20j | 3-fluorophenyl | ≥100 | 4.7 | >100 |
| 20k | 4-fluorophenyl | 6.3 | 3.1 | 59.0 |
| 20l | 3-chlorophenyl | 3.1 | 3.1 | >100 |
| 20m | 3-indolyl | 12.5 | 2.4 | 44.8 |
| 20n | 3-benzofuranyl | >100 | 6.3 | >100 |
| 20o | 3-cyanophenyl | 12.5 | 3.9 | >100 |
| 20p | 3- (tetrazol-5-yl)phenyl | ≥100 | 12.5 | 20.7 |
| 20q | 4-(SO3H)phenyl | ≥100 | ≥100 | 17.2 |
| 20r | 3,4-dimethoxyphenyl | 1.3 | 0.8 | 69.4 |
| R5 = 4-(2-methoxyethyl)-piperazinyl | ||||
| 21a | pyridin-2-yl | 12.5 | 3.1 | >100 |
| 21b | pyridin-3-yl | ≥100 | 25 | >100 |
| 21c | pyridine-4-yl | 6.3 | 50 | 95.9 |
| 21d | 1,3-dioxan-2-yl | ≥100 | 25 | >100 |
| 21e | 3-methoxyphenyl | 6.3 | 4.7 | 46.5 |
| 21f | 4-methoxyphenyl | 6.3 | 3.1 | 34.8 |
| 21g | 3,4-dimethoxyphenyl | 1.6 | 0.4 | 38.8 |
| 21h | 2-fluorophenyl | 12.5 | 9.4 | 30.6 |
| 21i | 3-fluorophenyl | ≥100 | 6.3 | 37.9 |
| 21j | 3-bromophenyl | 12.5 | 2.8 | 75.1 |
| 21k | phenoxy | ≥100 | 12.5 | 33.9 |
| R5 = 2,6-dimethyl-morpholinyl | ||||
| 22a | pyridin-3-yl | 12.5 | 3.1 | 42.6 |
| 22b | 3-methoxyphenyl | 9.4 | 1.6 | >100 |
| 22c | 3-fluorophenyl | 1.6 | 1.6 | >100 |
| 22d | 4-BocNHphenyl | 0.8 | 0.4 | >100 |
| 22e | 4-aminophenyl | 0.8 | 0.8 | 52.2 |
| 22f | 4-(CH3CONH)phenyl | 0.1 | 0.1 | 60.5 |
| 22g | 4-(CF3CONH)phenyl | 1.6 | 1.6 | 64.0 |
| 22h | 4-(CH3SO2NH)phenyl | 6.3 | 6.3 | 67.0 |
| 22i | 4-acrylamidophenyl | 0.1 | 0.05 | 62.4 |
| 22j | 4-[(CH3)3CONH]phenyl | ≥100 | ≥100 | >100 |
| 22k | 4-guanidinophenyl | 0.8 | 0.8 | 12.0 |
Table 4.
In vitro PK properties of selected compounds
| Compound# | MLMSa (%) |
HLMSb (%) |
CYP3A4 % INHc |
Solubilityd (µM) |
|---|---|---|---|---|
| morpholinyl compounds | ||||
| MBX 2319 | 0 | 0 | 12 | 12 |
| 20f | 0 | 8 | 29 | 50 |
| 20r | 5 | 21 | 49 | 25 |
| (2-methoxyethyl)piperazinyl compounds | ||||
| 19e | 0 | 0 | 18 | 50 |
| 21a | 3 | 0 | 14 | ≥ 100 |
| 21f | 17 | N.D. | 42 | ≥ 100 |
| 21g | 13 | 20 | 48 | ≥ 100 |
| 2,6-dimethylmorpholinyl compounds | ||||
| 22a | 19 | 53 | 0 | ≥ 100 |
| 22d | 66 | 59 | 0 | 100 |
| 22e | 72 | 76 | 59 | 12 |
| 22f | 0* | 37 | 61 | 50 |
| 22g | 0* | 90 | 66 | 12.5 |
| 22i | 49 | 100 | 90 | 25 |
| 22k | 90 | 90 | 9 | 100 |
MLMS, murine liver microsome stability, % remaining after 60 min at 37 °C in the presence of NADPH.
HLMS, human liver microsome stability, % remaining after 60 min at 37 °C in the presence of NADPH.
CYP3A4 INH, percent inhibition produced by 3 µM compound.
Solubility, maximum solubility in water as determined using a nephelometric assay32
NADPH independent consumption
3.1. Effect of modifications to the nitrile, gem-dimethyl and dimethylenesulfide groups
The effects of modifications to the Z, R1, and R2 groups and to the alkylsulfide portion of the R4 of MBX2319 (see Scheme 1) on potency (MPC4) are summarized in Table 1. None of the analogs potentiated the antibacterial acivity of LVX and PIP (MPC4 ≥100 µM). These results indicate that the Z, R1, R2 groups and the alkylsulfide portion of the R4 group are involved in important interactions with the target and are critical for maintaining the EPI activity of this scaffold. Specifically, oxidation/hydroxylation of the nitrile or the ethylsulfide groups produced inactive compounds (8–11, 12c and 12d). The optimal length of the alkyl chain that connects the pyranopyridine core to the phenyl group is two carbons, which is evident by comparing MBX2319 to 12a and 12b. Consistent with this finding, we observed that addition of an oxygen atom between the ethyl and the phenyl groups was also disfavored (compound 21k, Table 3). Replacement of the gem-dimethyl group with hydrogens resulted in a complete loss of activity, indicating a possibility that this group makes an important hydrophobic interaction with the target. However, it remains to be discovered whether both of the methyl groups are necessary for this interaction, and whether replacing at least one of them with another alkyl group would be advantageous.
3.2. Effect of varying the amino group
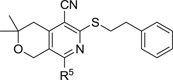
To explore the role of the morpholinyl group of MBX2319 in potency, cytotoxicity and in vitro ADME properties, we synthesized a series of analogs in which the morpholinyl group was replaced with a diverse panel of amino groups (see Scheme 5). The biological activities (MPC4 and CC50 values) of these analogs are summarized in Table 2. All compounds in this series, except the pyrrolidine analog 19c, potentiated the antibacterial activity of PIP (MPC4 ≤25 µM). Notably, four compounds (19e, 19f, 19g and 19j) showed improvements in the MPC4 values with PIP as compared to MBX2319. However, the MPC4 values for LVX are more sensitive to changes in the amino group, as six out of ten analogs (19a–c, 19f, 19g and 19j) exhibited MPC4 ≥100 µM, including the analogs that were potent potentiators of PIP. Although none of the compounds in Table 2 exhibited improved MPC4 values for LVX (comparing to MBX2319), the four compounds that had MPC4 ≤ 12.5 µM all contained the basic piperazinyl or homopiperazinyl groups.
Table 2.
MPC4 and CC50 values of analogs with various amino substituents
 | ||||
|---|---|---|---|---|
| Compound # | R5 | MPC4 (µM) | CC50 (µM) |
|
| LVX | PIP | |||
| MBX2319 | morpholinyl | 3.1 | 3.1 | >100 |
| 19a | dimethylamino | ≥100 | 9.4 | 57.4 |
| 19b | diethylamino | ≥100 | 6.3 | 38.2 |
| 19c | pyrrolidinyl | ≥100 | ≥100 | 20.2 |
| 19d | 4-methylpiperazinyl | 6.3 | 6.3 | 14.4 |
| 19e | 4-(2-methoxyethyl) piperazinyl | 6.3 | 0.4 | 34.2 |
| 19f | thiomorpholinyl | ≥100 | 1.6 | >100 |
| 19g | 2,6-dimethyl morpholinyl | ≥100 | 1.6 | >100 |
| 19h | homopiperazinyl | 12.5 | 12.5 | 20.3 |
| 19i | 4-methylhomo-piperazinyl | 12.5 | 25 | 16.4 |
| 19j | homomorpholinyl | ≥100 | 1.6 | >100 |
The disparity in activities of these analogs when combined with LVX and PIP can be explained by at least two possible mechanisms. First, it is possible that AcrB is sensitive to 19a–c, 19f, 19g and 19j, but another pump that is insensitive to these compounds is responsible for the intrinsic resistance to LVX. While AcrB is the major efflux pump in E. coli that extrudes LVX,21 it is possible that expression of other RND pumps that recognizes LVX is induced in response to the AcrB inhibition. For example, the following efflux pumps in E. coli are able to recognize and transport flouroquinolones: AcrF (RND family), MdfA (major facilitator superfamily(MFS)) YdhE (multidrug and toxic compound extrusion (MATE) family).22 We are currently working on further elucidating the pump specificity of these EPIs. Second, the transport of LVX and PIP could be differentially affected by analogs that exhibit altered interactions with AcrB. Computer docking experiments predict that PIP and LVX bind to different sites in the deep binding pocket of AcrB.23 Molecular dynamic simulations indicate that MBX2319 binds to a “hydrophobic trap” in the deep binding pocket of AcrB,24 which is where the hydrophobic portion of the EPI D13-900117 was shown to bind by X-ray crystallography.25 Interestingly, the hydrophobic trap is not in the main part of the substrate extrusion channel of AcrB. EPIs that bind to this trap, such as MBX2319, are predicted to competitively inhibit the conformational changes of the pump that are required for activity, preventing the binding and/or transport of substrates.26,27 Therefore, differences in the binding interactions of various EPIs to the hydrophobic trap would inhibit the conformational changes of the pump to varying degrees, which could differentially affect the activity of the pump for different antibiotics.
Variations in the morpholinyl group of MBX2319 also affected cytotoxicity against mammalian cells (HeLa). Interestingly, compounds containing the morpholinyl (MBX2319), 2,6-dimethylmorpholinyl (19g), thiomorpholinyl (19f), and homomorpholinyl (19j) groups are not cytotoxic (CC50 > 100 µM). However, seven compounds (19a–e, 19h and 19i) exhibited mild to moderate levels of cytotoxicity. The increased cytotoxicity appeared to be associated with the more basic amino groups. All compounds containing secondary and tertiary amines in the R5 group exhibited CC50 < 50 µM (19d,e,h,i) with compound 19e being the least basic and least cytotoxic.
3.3. The effect of modification/replacement of the phenyl group
To investigate the effects of co-variation of the amino group (R5) and the phenyl group (R6) on the potentiating capacity and cytotoxicity of the compounds, we varied R6 while maintaining R5 as either morpholinyl, 2,6-dimethylmorpholinyl, or 4-(2-methoxyethyl)piperazinyl. The results are summarized in Table 3. When R5 is a morpholinyl group and R6 is a simple cyano group compound 20a was inactive, indicating that a larger group for R6 is necessary for activity. Compounds are less active than MBX2319 when R6 is an aromatic heterocycle, such as pyridine (20c, 20d, 21a–c and 22a), indolyl (20m) and benzofuranyl (20n), and more so when R6 is a cyclic ether such as dioxanyl (21d). Notably, R6 groups containing an acidic functionality seem to be strongly disfavored (20b, 20p and 20q). It is possible that the permeability of these compounds through the OM was impeded by the negative charge of the OM of E. coli.26 Neutral and basic substitutions on the phenyl ring are preferred. Indeed, we identified such compounds with improved potency for both LVX and PIP. Thus, the most preferred substituents for EPI activity are as follows: 3,4-dimethoxy (20r and 21g), 4-amino (22e), 4-BocNH (22d), 4-acetamido (22f), 4-acrylamido (22i) and 4-guanidino (22k). The effect of the amido group on the activity is truly profound, suggesting a possibility that this group forms additional hydrogen bonding interaction(s) with the target, which results in a significant increase in binding affinity. Our computational modeling studies support this hypothesis, and will be published in due time. Using a cell-based assay designed to measure the effects of EPIs on the kinetic parameters of AcrAB-TolC in E. coli (Km and Vmax),27 we found that 22f is at least 20-fold more potent against AcrB than is MBX2319 (manuscript in preparation), which is consistent with a higher binding affinity for 22f. However, it appears that there is a limit to the size of the amide, as demonstrated by the stepwise decrease in potency of the following compounds: acetamido ≈ acrylamido (22i) > trifluoroacetamido (22g) > trimethylacetamido (22j). The weaker activity of the methylsulfonamido compound (22h) compared to the acetamido compound (22f) could be due to its increased electron-withdrawing effect, its size, or both. We will explore other NH containing substituents to further optimize the structure.
Examination of the data in Table 3 revealed that there is a mutual effect of both R5 and R6 on the potentiation activity of the compounds, and the effect is not simply additive. For example, changing R5 from morpholinyl to 2,6-dimethoxymorpholinyl led to increased potency when R6 is 3-fluorophenyl (see 20j and 22c), and the corresponding change caused a decrease in activity when R6 is 3-methoxyphenyl (see 20f and 22b). In terms of cytotoxicity, compounds with acidic or basic groups seem to have lower CC50 values (see 20p, 20q and 22k). Most of the compounds in Table 3 showed CC50 > 50 µM, indicating good tolerance overall of mammalian cells to the scaffold. None of the compounds in Tables 1–3 exhibit growth inhibition against E. coli at 100 µM, consistent with the characterized profile of MBX2319.
3.4. Effects of compounds on bacterial membranes
To rule out the possibility that the new analogs inhibit efflux through an indirect mechanism related to membrane perturbation, we tested compounds 20f, 21f, 21g, 22c–f, 22i and 22k in two assays for membrane activity. To determine whether analogs perturb the transmembrane proton gradient required for AcrAB-TolC activity28, we measured the effect of compounds on the LacY permease-dependent uptake and accumulation of [methyl-3H] β-D-thiogalactopyranoside ([3H]-TMG) in E. coli NCM3722, which requires proton motive force29. Consistent with previous findings for MBX2319,14 none of the analogs significantly inhibited uptake and accumulation of 3H-TMG at concentrations up to 20 µM (see Table S1, supplementary material), indicating that these compounds do not perturb the transmembrane proton gradient. To determine whether MBX2319 analogs affect the integrity of the outer membrane, which could increase the rate of permeation of antibiotics into the periplasm, we measured the influx of nitrocefin in a strain deficient in AcrAB (HN1159). The data, shown in Table S2 (supplementary material), demonstrate that none of the compounds increased the rate of nitrocefin influx, indicating that they had no effect on outer membrane permeability.
3.5. Microsomal stability and CYP inhibition assays for selected compounds
During the SAR study, compounds with good potentiating effects for LVX and PIP were also tested for liver microsome stability, CYP450 3A4 inhibition and aqueous solubility. These parameters were used as indicators of general pharmacokinetic (PK) properties of the compounds.30 The results of these assays are shown in Table 4. Immediately apparent is that MBX2319 and analogs containing the morpholinyl group are unstable in both the mouse and human liver microsome (MLM and HLM, respectively) assays (20f, 20r). Replacing the morpholinyl group with the (2-methoxyethyl)piperazinyl group led to compounds with moderate stability, and significantly improved aqueous solubility (21f, 21g). Most significantly, compounds containing the 2,6-dimethylmorpholinyl group were much more stable in liver microsome preparations (22a–k). These data indicate that the morpholinyl group is the major site of oxidation by liver microsomes. In general, there is a correlation between the stability of compounds in the MLM and HLM assays. Notable exceptions are compounds 22f and 22g, which were unstable in the MLM assay but moderately and highly stable in the HLM assay. It is possible that the acylamido groups of 22f and 22g were hydrolyzed by murine amidase(s), as the compounds were consumed even in the absence of NADPH. The variation in stability of compounds having the same R5 but different R6 groups suggests that modification of the phenyl ring also contributes positively to microsomal stability.
Regarding the CYP450 3A4 inhibition data, it has been suggested that at 3 µM of test compound, inhibition of <15% is low, 15%–50% is moderate and >50% is high.31 Thus, compounds bearing morpholinyl and (2-methoxyethyl)piperazinyl groups exhibited low to moderate levels of CYP inhibition. On the other hand, the inhibitory effects of compounds containing the 2,6-dimethylmorpholinyl group varied greatly, from 0% to 90%. It appears that modification of the phenyl group has a strong influence on CYP450 3A4 inhibition. Interestingly, the amino and amido substituents led to increased inhibition (22e–i) but the carbamate and guanidino substituents led to decreased inhibition (22d, 22k). Thus, we will continue to explore other NH containing substituents to optimize the scaffold. Finally, we do not observe correlations between CYP450 3A4 inhibition, microsomal stability and aqueous solubility. As expected, compounds containing inonizable groups such as piperazinyl, pyridinyl and guanidino are more soluble than the neutral analogs.
4. Conclusions
We have synthesized and tested 60 new analogs of MBX2319. The scaffold is highly responsive to structural changes, suggesting that the binding of the molecule to the target is specific. A molecular activity map has been generated (Figure 2) to guide the next phase of lead optimization. The major findings of this study are: (1) the nitrile, the dimethylenesulfide and the gem-dimethyl groups are important for maintaining potency; (2) non-acidic substituents can be added to the phenyl group to improve potency and in vitro PK properties; and (3) changes to the morpholinyl group lead to improved microsomal stability and solubility. As a trend, modification of the phenyl ring has greater effects on potency and CYP450 inhibition; and modification of the morpholinyl group has greater effects to solubility and microsomal stability. The most potent compounds (22d–f, 22i and 22k) identified in this study exhibited submicromolar MPC4 values, demonstrating significant improvement in potency as compared to MBX2319. Many analogs exhibited aqueous solubility near or greater than 100 µM, which is a 10-fold improvement compared to that of MBX2319. Importantly, while MBX2319 was unstable in the standard liver microsomal assays, replacing the morpholinyl group with the 2,6-dimethylmorpholinyl group markedly improved microsomal stability. We will continue to optimize the scaffold and test the lead compounds in vivo, the result of which will be reported in due course.
Figure 2.

Molecular activity map of the pyranopyridine scaffold
5. Experimental
5.1. Bacterial strains, growth media, and reagents
The bacterial strains and growth media used in this study have been described previously.14 Luria Broth (Miller) and agar were purchased as prepared dehydrated media from Becton Dickenson (Franklin Lakes, NJ). Ciprofloxacin (CIP) was purchased from ICN Biomedicals (Aurora, OH). The following reagents were purchased from Sigma Aldrich (St. Louis, MO): levofloxacin (LVX), piperacillin (PIP), tazobactam, and minocycline (MIN). Mouse (pooled) and human (pooled) liver microsomes were purchased from Xenotech (Lenexa, KS). The P450-Glo™ CYP3A4 assay was purchased from Promega (Madison, WI). [methyl-3H] β-D-thiogalactopyranoside ([3H] TMG; 1 mCi/ml, 7 Ci/mmol) was obtained from Moravek Biochemicals (Brea, CA).
5.2. Antibacterial assays
Assays to measure the Minimum Inhibitory Concentration (MIC) of antibacterial agents were performed as described in the CLSI guidelines33 with modifications described previously.14
Checkerboard assays were used to measure the minimal concentration of an EPI required to decrease the MIC of an antibiotic by 4-fold (MPC4) as described.14
5.3. Membrane activity assays
To estimate the effect of EPIs on the proton motive force in E. coli HN1157, the accumulation of [3H]-TMG by the LacY permease was measured essentially as described.34 The effect of EPIs on the integrity of the outer membrane was measured as described.14
5.4. Cytotoxicity assay
The cytotoxicity of compounds against a mammalian cell line (HeLa, ATCC CCL-2) was determined as described.35
5.5. Liver Microsome Stability Assays36
Compounds were assessed for human or mouse liver microsome stability in an assay mixure (200 µL) containing at final concentration: 25 µM of the test compound, 100 mM potassium phosphate pH 7.4 (Sigma, St. Louis MO), 3 mM MgCl2 (Sigma, St. Louis MO), 0.5 mg/mL human or mouse liver microsomes (Xenotech, Lenexa, KS), and plus or minus (for the control) 2 mM NADPH (Sigma, St. Louis MO). The mixtures were incubated at 37 °C. After one hour, an equal volume of acetonitrile (Med Supply Partners, Atlanta, GA) containing 0.1% trifluouroacetic acid (Sigma, St. Louis MO) was added, the mixture centrifuged, and the supernatant analyzed by HPLC (Agilent 1100 instrument or Gilson 322/215 instrument) equipped with a Grace Ultima C18 column (Grace Davison Discovery Science, Deerfield, IL) using gradients of solvent A (0.1% TFA in water (Med Supply Partners, Atlanta, GA) to Solvent B (0.1% TFA in acetonitrile) for each compound. The reported value was the percent change in peak area of the compound as a result of NADPH dependent consumption (i.e. ratio of plus NADPH/No NADPH control).
5.6. CYP450 3A4 inhibition assay
Compounds were assessed for CYP450 3A4 inhibition in an assay mixture (50 µL) containing at final concentration: 3 µM of the test compound, 100 mM potassium phosphate pH 7.4, 3 mM MgCl2, 0.5 mg/mL mouse liver microsomes, 12 µM Luciferin IPA substrate (from Promega Corporation, Madison, WI) designed to be a specific substrate for CYP450 3A4, and 2.5 mM NADPH. The mixtures were incubated for 10 minutes at 37 °C followed by addition of a Luciferin (with esterase, Promega Corporation, Madison, WI) detection reagent. Assay readout was luminescence signal after 5 minute incubation of the assay mixture in darkness at room temperature comparing to that of the control, in which DMSO (Sigma, St. Louis MO) was used in place of the test compound.
5.7. Aqueous solubility determination
Compound solubility was determined using the nephelometric method as previously described.32
6. General chemical methods
All commercially obtained reagents and solvents were used as received. 1H and 13C NMR spectra were recorded on a Bruker 300 MHz instrument. Chemical shifts are given in δ values referenced to the internal standard tetramethylsilane. LCMS analyses were performed on a Thermo-Finnigan Surveyor LC unit connected to a Thermo LTQ Fleet MS unit. HPLC purification was performed on a Gilson Unipoint instrument equipped with a C18, 10 micron, 150 × 22 mm column. Melting points were taken on EZ-Melt automated melting point apparatus (Stanford Research Systems, Inc.) in manual mode, and are uncorrected. Thin-layer chromatography was performed on silica gel GHLF plates from Analtech (Newark, DE), and the chromatograms were visualized under UV light at 254 nm.
Benzyl bromide; phenethyl bromide; 3-phenylpropyl bromide; 2-bromoacetophenone; styrene oxide; tetrahydro-4H-pyran-4-one; dimethylamine; diethylamine (2.0 M in methanol); pyrrolidine; 1-methylpiperazine; thiomorpholine; 2,6-dimethylmorpholine; homopiperazine; 1-methylhomopiperazine; 3-bromopropionitrile; 2-methoxyphenethyl bromide; 3-methoxyphenethyl bromide; 4-methoxyphenethyl bromide; 3,4-dimethoxyphenethyl bromide; 2-fluorophenethyl bromide; 3-fluorophenethyl bromide; 4-fluorophenethyl bromide; 3-chlorophenethyl bromide; 3-(2-bromoethyl)indole; 2-(2-bromoethyl)-1,3-dioxane; 3-bromophenethyl bromide; beta-bromophenetole; 1H-pyrazole-1-carboxamidine hydrochloride, malononitrile, and carbon disulfide were purchased from Sigma-Aldrich company (St. Louis, MO). 1-(2-methoxyethyl)piperazine was purchased from Goldenbridge Pharma Inc (Arcadia, CA, USA). 2-(2-bromoethyl)pyridine hydrobromide was purchased from AstaTech, Inc. (Bristol, PA, USA). 3-(2-bromoethyl)pyridine; 4-(2-bromoethyl)pyridine were purchased from Beta Pharma Scientific, Inc. (Branford, CT, USA). 3-(2-hydroxyethyl)benzonitrile was purchased from Anichem, Inc. (North Brunswick, NJ, USA). Homomorpholine; 3-(2-hydroxyethyl)benzofuran were purchased from Oakwood Products, Inc. (West Columbia, SC, USA). 4-nitrophenethyl alcohol was purchased from AK Scientific Inc. (Union City, CA, USA).
For the preparation of 7, 17 and MBX2319, see Ref. [14].
6.1. 3-(2-bromoethyl)benzonitrile
To a solution of 3-(2-hydroxyethyl)benzonitrile (0.50 g, 3.40 mmol) and carbon tetrabromide (1.70 g, 5.13 mmol) in dichloromethane (15 mL) was added triphenylphosphine (1.30 g, 4.96 mmol) in portions (exothermic). The reaction mixture was stirred at room temperature for 2 h. Hexane (50 mL) was added, and the precipitates were removed by filtration. The filtrate was concentrated, and the residue was purified by flash chromatography (80 g SiO2; 0–30% ethyl acetate in hexanes) to provide 3-(2-bromoethyl)benzonitrile as a colorless oil, 0.55 g (77%). Rf 0.58 (ethyl acetate/hexanes, 1/3); 1H NMR (CDCl3) δ: 7.55-7.44 (m, 5H), 7.34-7.23 (m, 2H), 3.58 (t, 2H), 3.21 (t, 2H).
6.2. 3-(2-bromoethyl)benzofuran
Following the above procedure, 3-(2-hydroxyethyl)benzofuran (0.50 g, 3.08 mmol) was treated with carbon tetrabromide (1.53 g, 4.61 mmol) and triphenylphosphine (1.21 g, 4.61 mmol) in dichloromethane (20 mL). Purification by flash chromatography (80 g SiO2; 0–30% ethyl acetate in hexanes) provided 3-(2-bromoethyl)benzofuran as colorless oil, 0.66 g (96%). Rf 0.74 (ethyl acetate/hexanes, 1/3); 1H NMR (CDCl3) δ: 7.56-7.47 (m, 3H), 7.34-7.23 (m, 2H), 3.65 (t, 2H), 3.26 (t, 2H).
6.3. tert-Butyl (4-(2-bromoethyl)phenyl)carbamate
Step 1: To a thick-walled 500 mL glass cylinder were sequentially added 4-nitrophenethyl alcohol (5.75g, 34.39 mmol), ethanol (70 mL) and 0.3 g of palladium on carbon (10% by weight). The cylinder was placed in a Parr shaker apparatus. The system was purged three times with hydrogen, sealed at 43 psi. After shaking for 1 h, hydrogen was released, and the cylinder purged with nitrogen. The reaction mixture was filtered to remove solid material. The filtrate was concentrated in a rotary evaporator to ca. 10 mL and allowed to cool to 5 °C. The colorless crystals that formed were collected by filtration to provide 3.86 g (82%) of 4-aminophenethyl alcohol. Rf 0.22 (hexanes/ethyl acetate, 1/1); 1H NMR (CDCl3) δ: 6.84 (d, 2H), 6.47 (d, 2H), 4.83 (br, 2H), 4.56 (t, 1H), 3.47 (td, 2H), 2.52 (t, 2H).
Step 2: To a solution of 4-aminophenethyl alcohol (1.0 g, 7.29 mmol) in dioxane (10 mL) and 20 mL of NaOH aqueous solution (1 N) at 0 °C was added di-tert-butyl dicarbonate (5.0 g, 22.9 mmol) in portions (exothermic). The reaction mixture was allowed to rise to room temperature. After 16 h, water (50 mL) and ether (100 mL) were added. The organic layer was separated, and the aqueous extracted with ether (30 mL). The combined ether solutions were washed with brine, dried over MgSO4 and concentrated in a rotary evaporator. The residue was purified by flash chromatography (80 g SiO2; 0–70% ethyl acetate in hexanes) to provide tert-butyl (4-(2-hydroxyethyl)phenyl)carbamate, 1.4 g (81%). Rf 0.49 (hexanes/ethyl acetate, 1/1); 1H NMR (CDCl3) δ: 7.30 (d, 2H), 7.14 (d, 2H), 6.38 (br s, 1H), 3.82 (dd, 2H), 2.81 (t, 2H), 1.51 (s, 9H).
Step 3: To a solution of tert-butyl (4-(2-hydroxyethyl)phenyl)carbamate (0.55 g, 2.32 mmol) and carbon tetrabromide (0.77 g, 2.32 mmol) in dichloromethane (6 mL) was added triphenylphosphine (0.61 g, 2.32 mmol) in portions (exothermic). The reaction mixture was stirred at room temperature for 16 h and loaded directly on a silica gel column and purified by flash chromatography (80 g SiO2; 0–30% ethyl acetate in hexanes) provided tert-butyl (4-(2-bromoethyl)phenyl)carbamate as white solid, 0.59 g (84%). Rf 0.51 (ethyl acetate/hexanes, 1/3); 1H NMR (CDCl3) δ: 7.30 (d, 2H), 7.14 (d, 2H), 6.38 (br s, 1H), 3.52 (t, 2H), 3.08 (t, 2H), 1.51 (s, 9H).
6.4. N'-hydroxy-3,3-dimethyl-8-morpholino-6-(phenethylthio)-3,4-dihydro-1H-pyrano[3,4-c]pyridine-5-carboximidamide (8) and 3,3-dimethyl-8-morpholino-6-(phenethylthio)-3,4-dihydro-1H-pyrano[3,4-c]pyridine-5-carboxamide (9)
To a solution of MBX2319 (100 mg, 0.24 mmol) in DMSO (5 mL) was added potassium t-butoxide (274 mg, 2.44 mmol) followed by hydroxylamine hydrochloride (170 mg, 2.44 mmol). The mixture was stirred at 100 °C for 24 h, and allowed to cool to room temperature. Crushed ice (ca. 20 g) was added. The suspension was filtered to collect the solid. Purification by preparative HPLC (C18, elution: 10% acetonitrile in water to 100% acetonitrile) provided 22 mg (20%) of 8 as waxy solid, and 27 mg (26%) of 9 as white solid. Characterizations of 8: Rf 0.16 (ethyl acetate/hexane, 1/1); MS(ESI) m/z 443.2 (MH+); 1H NMR (CDCl3) δ: 7.33-7.18 (m, 5H), 6.06 (br s, 2H), 4.65 (s, 2H), 3.83 (t, 4H), 3.45 (t, 2H), 3.25 (t, 4H), 2.99 (t, 2H), 2.73 (br s, 1H), 2.53 (s, 2H), 1.21 (s, 6H). Characterization of 9: mp 171–172 °C; Rf 0.16 (ethyl acetate/hexane, 1/1); MS(ESI) m/z: 428.2 (MH+); 1H NMR (CDCl3) δ: 7.33-7.21 (m, 5H), 5.87 (br s, 1H), 5.76 (br s, 1H), 4.64 (s, 2H), 3.82 (t, 4H), 3.45 (t, 2H), 3.15 (t, 4H), 3.01 (t, 2H), 2.72 (s, 2H), 1.28 (s, 6H).
6.5. 3,3-dimethyl-5-cyano-8-morpholino-6-(phenethylsulfinyl)-3,4-dihydro-1H-pyrano[3,4-c]pyridine (10)
To a solution of MBX2319 (41 mg, 0.10 mmol) in dichloromethane (4 mL) was slowly added 3-chloroperbenzoic acid (22 mg, 77% maximum purity, ca. 0.12 mmol). The mixture was stirred for 1 h at room temperature before additional 3-chloroperbenzoic acid (6 mg, ca. 0.03 mmol) was added. After 2 h, a saturated aqueous solution of sodium bicarbonate (1 mL) was added. After 15 min, the organic layer was collected and concentrated. The product was purified by flash chromatography (12 g SiO2; 0–50% ethyl acetate in dichloromethane) to provide 27 mg (63%) of 10 as white solid. mp 146–147 °C; Rf 0.34 (ethyl acetate/CH2Cl2, 1/1); MS(ESI) m/z: 425.8 (MH+); 1H NMR (CDCl3) δ: 7.29-7.17 (m, 5H), 4.57 (s, 2H), 3.81 (t, 4H), 3.48-3.31(m, 6H), 3.20-3.03 (m, 2H), 2.84 (s, 2H), 1.35 (s, 6H).
6.6. 3,3-dimethyl-5-cyano-8-morpholino-6-(phenethylsulfonyl)-3,4-dihydro-1H-pyrano[3,4-c]pyridine (11)
To a solution of MBX2319 (50 mg, 0.12 mmol) in dichloromethane (4 mL) was added 3-chloroperbenzoic acid (69 mg, 77% max). The mixture was stirred for 1 h at room temperature before a a saturated aqueous solution of sodium bicarbonate (1 mL) was added. After 15 min, the organic layer was collected and concentrated. The product was purified by flashchromatography (12 g SiO2; 0–50% ethyl acetate in hexanes) to provide 40 mg (74%) of 11 as white crystalline solid. mp 162–164 °C; Rf 0.21 (ethyl acetate/hexane, 1/1); MS(ESI) m/z 442.1 (MH+); 1H NMR (CDCl3) δ: 7.26-7.13 (m, 5H), 4.57 (s, 2H), 3.81 (t, 4H), 3.77 (t, 2H), 3.34 (t, 4H), 3.18 (t, 2H), 2.86 (s, 2H), 1.35 (s, 6H).
6.7. General procedure for compounds 12a–c, 20a, 20c–o and 20r
To a stirred solution of compound 7 (0.1 g, 0.33 mmol) in acetonitrile (1.5 mL) and DMF (0.5 mL) was added cesium carbonate (0.2 g, 0.65 mmol), followed by 0.50 mmol of benzyl bromide (for 12a), 3-phenylpropyl bromide (for 12b), 2-bromoacetophenone (for 12c), or R5Br for 20a, 20c–o and 20r (see scheme 6 for structures of R5Br). The mixture was stirred for 16 h, and water (5 mL) was added. Product was extracted with ethyl acetate (3 × 5 mL). The combined organic extracts were washed with brine, dried over Na2SO4 and concentrated by rotary evaporation. The residue was purified by silica gel column chromatography to provide the desired products. Reaction yields and characterization data of 12a–c, 20a, 20c–o and 20r are summarized in Table S3 (see the supporting information).
6.8. 6-(2-hydroxy-2-phenylethylthio)-5-cyano-3,3-dimethyl-8-morpholino-3,4-dihydro-1H-pyrano[3,4-c]pyridine (12d)
To a solution of 7 (210 mg, 0.69 mmol) and triethylamine (0.2 mL) in ethanol (3 mL) was added styrene oxide (0.24 mL, 2.10 mmol). The mixture was stirred for 16 h, and concentrated by rotary evaporation. Purification by flash chromatography (24 g SiO2; 0–100% ethyl acetate in hexanes) provided 135 mg (46%) of 12d as pale yellow solid. mp 142–144 °C; Rf 0.33 (hexanes/ethyl acetate, 1/1); MS(ESI) m/z: 426.0 (MH+); 1H NMR(CDCl3) δ: 7.43-7.29 (m, 5H), 4.97 (dt, 1H), 4.56 (s, 2H), 3.83 (t, 4H), 3.67-3.63 (m, 2H), 3.46 (dd, 1H), 3.32 (m, 4H), 2.78 (s, 2H), 1.33 (s, 6H).
6.9. 8-morpholino-5-cyano-6-(phenethylthio)-3,4-dihydro-1H-pyrano[3,4-c]pyridine (16a)
Step 1: To a solution of tetrahydropyran-4-one (3.0 g, 30.0 mmol) in methanol (3.6 mL) was added carbon disulfide (3.6 mL, 60.0 mmol) and malononitrile (2.0 g, 30.0 mmol) in 4 portions, followed by Et3N (1.5 mL, 81.6 mmol) slowly. The reaction mixture was stirred at room temperature for 24 h. The precipitate was filtered, washed with ether (3 ml × 3) and crystallized from 2-propanol to provide one batch of 14 (0.40 g) as orange solid. The combined filtrates were concentrated and purified by flash chromatography (120 g SiO2; 0–5% methanol in CH2Cl2) to provide a second batch of 14 (1.0 g), 30% total yield. mp 242–247 °C; Rf 0.50 (3% methanol in CH2Cl2); MS(ESI) m/z 224.9 (MH+); 1H NMR (DMSO-d6) δ: 8.93 (s, 2H), 4.73 (s, 2H), 3.80 (t, 2H), 2.71 (t, 2H).
Step 2: A solution of 14 (0.50 g, 2.23 mmol) and morpholine (1.2 mL) in ethanol (2.3 mL) was refluxed under argon for 4 h and then kept at −5 °C over night. The precipitate was filtered, washed with ethanol (2 mL × 2) and dried under vacuum to provide 15, 250 mg (40%). mp 209–216 °C; Rf 0.36 (5% methanol in CH2Cl2); MS(ESI) m/z 278.1 (MH+); 1H NMR (DMSO-d6) δ: 88.41 (s, 2H), 4.36 (s, 2H), 3.84 (t, 2H), 3.73 (t, 4H), 3.66 (t, 4H), 3.05 (t, 4H), 2.98 (t, 4H), 2.62 (t, 2H).
Step 3: To a solution of 15 (100 mg, 0.36 mmol) in MeCN/DMF (1.2 mL/0.3 mL) was added cesium carbonate (235 mg, 0.72 mmol) and phenethyl bromide (0.07 mL, 0.54 mmol). The reaction mixture was heated to 40 °C and stirred overnight at the same temperature. The mixture was cooled to room temperature, and water (10 mL) was added. Product was extracted with ethyl acetate (2 × 10 mL), dried over anhydrous Na2SO4, filtered and concentrated. The yellow residue was purified by flash chromatography (25 g SiO2; 0–50% ethyl acetate in hexanes) to provide 56 mg of 16a (41%) as light yellow solid. mp 132–134 °C; Rf 0.30 (hexanes/ethyl acetate, 1/1); MS(ESI) m/z 382.1 (MH+); 1H NMR (CDCl3) δ: 7.34-7.21 (m, 5H), 4.52 (s, 2H), 4.02 (t, 2H), 3.81 (t, 4H), 3.48 (t, 2H), 3.30 (t, 4H), 3.05 (t, 2H), 2.96 (t, 2H).
6.10. 8-morpholino-5-cyano-6-[[2-(pyridin-2-yl)ethyl]thio]-3,4-dihydro-1H-pyrano[3,4-c]pyridine (16b)
To a solution of 15 (100 mg, 0.36 mmol) in MeCN/DMF (1.2 mL/0.3 mL) was added cesium carbonate (235 mg, 0.72 mmol) and 2-(2-bromoethyl)pyridine hydrobromide (144 mg, 0.54 mmol). The mixture was heated to 40 °C, stirred overnight, and allowed to cool to room temperature. Water (15 mL) was added, and product extracted with ethyl acetate (2 × 25 mL), dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by flash chromatography (25 g SiO2; 0–80% ethyl acetate in hexanes) to provide 55 mg (40%) of 16b as a yellow solid. mp 158–159 °C; Rf 0.20 (hexanes/ethyl acetate, 1/4); MS(ESI) m/z 383.0 (MH+); 1H NMR (CDCl3) δ: 8.48 (d, 1H), 7.56 (td, 1H), 7.11-7.05 (m, 2H), 4.44 (s, 2H), 3.94 (t, 2H), 3.74 (t, 4H), 3.58 (t, 2H), 3.25 (t, 4H), 3.15 (t, 2H), 2.88 (t, 2H).
6.11. General procedures for synthesis of compounds 19a–j, 21a–k and 22a–d
To a suspension of compound 17 (1 mmol) in ethanol (5 mL) was added the amine R5H (10 mmol) (see scheme 5 for structures of R5). The reaction mixture was heated at reflux under nitrogen for 16–32 h, until analysis by LCMS showed a complete conversion of 17, and allowed to cool to room temperature. Phenethyl bromide (for 19a–j) or R6CH2CH2Br (for 21a–k and 22a–d) (1.2–2.0 mmol) was added (see scheme 6 for structures of R6). After 3–16 h (when LCMS analysis showed a complete conversion of the intermediate 18), solvent was evaporated by rotary evaporation. The residue was placed in vacuum for 16 h before being purified by chromatography on silica gel or reversed-phase HPLC. Reaction yields and characterization data of 19a–j, 21a–k and 22a–d are summarized in table S3 (see the supporting information).
6.12. 6-[2-(1H-tetrazol-5-yl)ethylthio]-5-cyano-3,3-dimethyl-8-(4-morpholino)-3,4-dihydro-1Hpyrano[3,4-c]pyridine (20b)
To a solution of 20a (95 mg, 0.27 mmol) in toluene (3 mL) was added di-n-butyltin oxide (11 mg) and azidotrimethylsilane (60 mg). The mixture was refluxed for 16 h, and allowed to cool to room temperature. The solid was collected by filtration, rinsed with ether and dried in vacuum to provide 85 mg of 20b as white solid (80%). mp 230–232 °C; Rf 0.49 (ethyl acetate/methanol/acetic acid, 10/1/0.1); MS(ESI) m/z 402.1 (MH+); 1H NMR (DMSO-d6) δ: 16.08 (br s, 1H), 4.50 (s, 2H), 3.68 (t, 4H), 3.60 (t, 2H), 3.33 (m, 6H, overlapped with water peak), 2.70 (s, 2H), 1.26 (s, 6H).
6.13. 6-[3-(1H-tetrazol-5-yl)phenethylthio]-5-cyano-3,3-dimethyl-8-(4-morpholino)-3,4-dihydro-1Hpyrano[3,4-c]pyridine (20p)
Using the same procedure described for the preparation of 20b, from 128 mg (0.29 mmol) of 20o, 50 mg of 20p was obtained as pale yellow solid (36%). mp 186–188 °C; Rf 0.59 (ethyl acetate/methanol/acetic acid, 10/1/0.1); MS(ESI) m/z 478.2 (MH+); 1H NMR (CDCl3) δ: 7.98-7.94 (m, 2H), 7.45 (t, 1H), 7.37 (d, 1H), 4.55 (s, 2H), 3.85 (t, 4H), 3.49 (t, 2H), 3.30 (t, 4H), 3.06 (t, 2H), 2.75 (s, 2H), 1.33 (s, 6H).
6.14. 3,3-dimethyl-5-cyano-6-(4-sulfophenethylthio)-8-(4-morpholino)-3,4-dihydro-1H-pyrano[3,4-c]pyridine (20q)
MBX 2319 (100 mg, 0.24 mmol) was added to a solution of sulfuric acid and trifluoroacetic acid (1:4, v/v, 1mL). The mixture was stirred for 5 days at room temperature before being poured onto crushed ice (ca. 10 g). Sodium carbonate was added in portions to neutralize the acid. The product was extracted with ethyl acetate (10 mL × 2), concentrated and purified by preparative HPLC (C18, elution: 10% acetonitrile in water to 100% acetonitrile) to provide 14 mg of 20q as yellow wax (12%). MS (ESI) m/z 490.4 (MH+); 1H NMR (DMSO-d6) δ: 7.53 (d, 2H), 7.23 (d, 2H), 4.51 (s, 2H), 3.98 (br s, 1H), 3.70 (t, 4H), 3.45 (t, 2H), 3.33 (t, 4H), 2.97 (t, 2H), 2.69 (s, 2H), 1.26 (s, 6H).
6.15. 6-(4-aminophenethylthio)-5-cyano-3,3-dimethyl-8-(2,6-dimethyl-4-morpholino)-3,4-dihydro-1Hpyrano[3,4-c]pyridine (22e)
Trifluoroacetic acid (0.1 mL) was added to a solution of 22d (50 mg, 0.10 mmol) in dichloromethane (1 mL). The mixture was stirred for 1 h at room temperature then slowly added to 5 mL of aqueous saturated solution of sodium bicarbonate. The product was extracted with dichloromethane, dried over magnesium sulfate and concentrated by rotary evaporation. Purification by flash chromatography (12 g SiO2; 0–100% ethyl acetate in hexanes) provided 37 mg of 22e as light brown solid (90%). mp 81–83 °C; Rf 0.35 (hexanes/ethyl acetate, 1/1); MS(ESI) m/z 453.1 (MH+); 1H NMR (CDCl3) δ: 7.02 (d, 2H), 6.64 (d, 2H), 6.46 (b s, 1H), 4.54 (s, 2H), 3.76 (m, 2H), 3.46-3.34 (m, 4H), 3.08 (br s, 2H), 2.90 (t, 2H), 2.76 (s, 2H), 2.72 (dd, 2H), 1.33 (s, 6H), 1.20 (d, 6H).
6.16. 6-(4-acetamidophenethylthio)-5-cyano-3,3-dimethyl-8-(2,6-dimethyl-4-morpholino)-3,4-dihydro-1H-pyrano[3,4-c]pyridine (22f)
To a solution of 22e (40 mg, 0.09 mmol) and triethylamine (40 µL, 0.29 mmol) in dichloromethane (1 mL) was added acetic acid anhydride (20 µL). The mixture was stirred for 2 h, and purified by flash chromatography (14 g SiO2; 0–80% ethyl acetate in hexanes) to provide 36 mg of 22f as yellow solid (82%). mp 90–91 °C; Rf 0.17 (hexanes/ethyl acetate, 1/1); MS(ESI) m/z 495.1 (MH+); 1H NMR (CDCl3) δ: 7.43 (d, 2H), 7.39 (br s, 1H), 7.18 (d, 2H), 4.54 (s, 2H), 3.76 (m, 2H), 3.46-3.38 (m, 4H), 2.97 (t, 2H), 2.75 (s, 2H), 2.72 (dd, 2H), 2.17 (s, 3H), 1.33 (s, 6H), 1.20 (d, 6H).
6.17 6-[4-(2,2,2-trifluoroacetamido)phenethylthio]-5-cyano-8-(2,6-dimethylmorpholino)-3,3-dimethyl-3,4-dihydro-1H-pyrano[3,4-c]pyridine (22g)
To a solution of 22e (51 mg, 0.12 mmol) and triethylamine (40 µL, 0.29 mmol) in THF (1 mL) was added trifluoroacetic anhydride (16 µL, 0.12 mmol). The mixture was stirred for 16 h. Water (20 µL) was added. After 2 h, the mixture was evaporated to dryness. Purification by flash chromatography (14 g SiO2; 0–100% [10% MeOH/EtOAc] in hexanes) provided 15 mg of 22g as light brown solid (24%). mp 184–185 °C; Rf 0.78 (hexanes/ethyl acetate, 1/3); MS(ESI) m/z 549.2 (MH+); 1H NMR (CDCl3) δ: 7.98 (br s, 1H), 7.49 (d, 2H), 7.26 (d, 2H), 4.53 (s, 2H), 3.75 (m, 2H), 3.46-3.41 (m, 4H), 3.02 (t, 2H), 2.76 (s, 2H), 2.71 (dd, 2H), 1.33 (s, 6H), 1.19 (d, 6H).
6.18. 6-[4-(methylsulfonamido)phenethylthio]-5-cyano-3,3-dimethyl-8-(2,6-dimethyl-4-morpholino)-3,4-dihydro-1H-pyrano[3,4-c]pyridine (22h)
To a solution of 22e (40 mg, 0.09 mmol) and triethylamine (40 µL, 0.29 mmol) in dichloromethane (1 mL) was added methanesulfonyl chloride (20 µL, 0.26 mmol). The mixture was stirred for 3 h, and 1 mL of an aqueous saturated solution of sodium bicarbonate was added. The organic layer was separated and subjected to flash chromatography (14 g SiO2; 0–100% ethyl acetate in hexanes) to provide 40 mg of 22h as white solid (85%). mp 202–203 °C; Rf 0.36 (hexanes/ethyl acetate, 1/1); MS(ESI) m/z 531.2 (MH+); 1H NMR (CDCl3) δ: 7.23 (d, 2H), 7.17 (d, 2H), 6.57 (br s, 1H), 4.54 (s, 2H), 3.76(m, 2H), 3.45-3.40 (m, 4H), 3.03-2.98 (m, 5H), 2.76 (s, 2H), 2.71 (dd, 2H), 1.34 (s, 6H), 1.19 (d, 6H).
6.19. 6-(4-acrylamidophenethylthio)-5-cyano-3,3-dimethyl-8-(2,6-dimethyl-4-morpholino)-3,4-dihydro-1H-pyrano[3,4-c]pyridine (22i)
To a solution of 22e (40 mg, 0.09 mmol) and triethylamine (40 µL, 0.29 mmol) in DMF (1 mL) was added acrylic acid (30 µL, 0.44 mmol) and 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU; 40 mg, 0.11 mmol). The mixture was stirred for 3 h, and subjected to preparative HPLC purification (C18, elution: 10% acetonitrile in water to 100% acetonitrile) to provide 19 mg of 22i as white solid (42%). mp 206–208 °C; Rf 0.39 (hexanes:ethyl acetate, 1:1); MS(ESI) m/z 507.1 (MH+); 1H NMR (CDCl3) δ: 7.51 (br d, 2H), 7.30 (b s, 1H), 7.20 (d, 2H), 6.44 (dd, 1H), 6.24 (dd, 1H), 5.76 (dd, 1H), 4.53 (s, 2H), 3.76 (m, 2H), 3.46-3.39 (m, 4H), 2.99 (t, 2H), 2.76 (s, 2H), 2.71 (dd, 2H), 1.33 (s, 6H), 1.19 (d, 6H).
6.20. 3,3-dimethyl-5-cyano-8-(2,6-dimethylmorpholino)-6-(4-tert-butylamidophenethylthio)-3,4-dihydro-1H-pyrano[3,4-c]pyridine (22j)
To a solution of pivalic acid (15 mg, 0.15 mmol) and N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU, 57 mg, 0.15 mmol) in DMF (1 mL) was added triethylamine (100 µL, 0.72 mmol) and 22e (34 mg, 0.08 mmol). The solution was stirred at rt for 16 h, and directly purified by reversed phase column chromatography (C18, elution: 10% acetonitrile in water to 100% acetonitrile) to provide 30 mg of 22j as light brown crystalline solid (75%). mp 155–157 °C; Rf 0.61 (ethyl acetate:hexanes, 1:1); MS(ESI) m/z 537.3 (MH+); 1H NMR (CDCl3) δ: 7.46 (d, 2H), 7.19 (d, 2H), 4.54 (s, 2H), 3.76 (m, 2H), 3.42 (m, 4H), 2.98 (t, 2H), 2.76 (s, 2H), 2.71 (t, 2H), 1.33 (s, 6H), 1.31 (s, 9H), 1.20 (d, 6H).
6.21. 6-(4-guanidinophenethylthio)-5-cyano-3,3-dimethyl-8-(2,6-dimethyl-4-morpholino)-3,4-dihydro-1H-pyrano[3,4-c]pyridine (22k, TFA salt)
A mixture of 22e (62 mg, 0.15 mmol) and 1H-pyrazole-1-carboxamidine hydrochloride (25 mg, 0.17 mmol) in pyridine (1 mL) was keep at room temperature for 2 days, then heated to 100 °C for 2 h. Pyridine was removed by rotary evaporation at 50 °C. The residue was purified by flash chromatography (25 g SiO2; 0–20% methanol in CH2Cl2). Pure fractions were combined, to which 15 µL of trifluoroacetic acid was added. The solution was evaporated by rotary evaporation and dried in vacuum to provide 40 mg of 22k as white crystalline solid (48%). mp 206–208 °C; MS(ESI) m/z 495.3 (MH+); 1H NMR (CD3CN) δ: 9.54 (br s, 1H), 7.36 (d, 2H), 7.18 (d, 2H), 6.82 (br s, 4H), 4.53 (s, 2H), 3.70 (m, 2H), 3.57-3.45 (m, 4H), 3.04 (t, 2H), 2.71 (s, 2H), 2.65 (t, 2H), 1.28 (s, 6H), 1.11 (d, 6H).
Supplementary Material
Acknowledgments
We thank Dr. Donald T. Moir for his help with the preparation of the manuscript. We thank Ms. Debra M. Mills for her help with the cytotoxicity assay.
The research reported in this article was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (grant R43 AI100332-01). The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Reaction yields and characterization data for compounds 12a–c, 19a–j, 20a, 20c–o, 20r, 21a–k and 22a–d; data on accumulation of [3H]-TMG in E. coli NCM3722 (wild-type) and hydrolysis of nitrocefin in E.coli HN1159 (HN1167, ΔacrAB) in the presence of MBX compounds associated with this article can be found in the online version, at XXX.
Contributor Information
Son T. Nguyen, Email: snguyen@microbiotix.com.
Steven M. Kwasny, Email: skwasny@microbiotix.com.
Xiaoyuan Ding, Email: xding@microbiotix.com.
Steven C. Cardinale, Email: scardinale@microbiotix.com.
Courtney T. McCarthy, Email: cmccarthy@microbiotix.com.
Hong-Suk Kim, Email: hskim@berkeley.edu.
Hiroshi Nikaido, Email: nhiroshi@berkeley.edu.
Norton P. Peet, Email: nppeet@gmail.com.
John D. Williams, Email: jwilliams@microbiotix.com.
Terry L. Bowlin, Email: tbowlin@microbiotix.com.
Timothy J. Opperman, Email: topperman@microbiotix.com.
References
- 1.Peleg AY, Hooper DC. N. Engl. J. Med. 2010;362:1804. doi: 10.1056/NEJMra0904124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kunz AN, Brook I. Chemother. 2010;56:492. doi: 10.1159/000321018. [DOI] [PubMed] [Google Scholar]
- 3.Decad GM, Nikaido H. J. Bacteriol. 1976;128:325. doi: 10.1128/jb.128.1.325-336.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mahamoud A, Chevalier J, Alibert-Franco S, Kern WV, Pages JM. J. Antimicrob. Chemother. 2007;59:1223. doi: 10.1093/jac/dkl493. [DOI] [PubMed] [Google Scholar]
- 5.Vargiu AV, Nikaido H. Proc. Natl. Acad. Sci. USA. 2012;109:20637. doi: 10.1073/pnas.1218348109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nikaido H, Pages JM. FEMS Microbiol. Rev. 2012;36:340. doi: 10.1111/j.1574-6976.2011.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li XZ, Nikaido H. Drugs. 2004;64:159. doi: 10.2165/00003495-200464020-00004. [DOI] [PubMed] [Google Scholar]
- 8.Li XZ, Nikaido H. Drugs. 2009;69:1555. doi: 10.2165/11317030-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poole K. Ann. Med. 2007;39:162. doi: 10.1080/07853890701195262. [DOI] [PubMed] [Google Scholar]
- 10.Lomovskaya O, Lee A, Hoshino K, Ishida H, Mistry A, Warren MS, Boyer E, Chamberland S, Lee VJ. Antimicrob. Agents Chemother. 1999;43:1340. doi: 10.1128/aac.43.6.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lomovskaya O, Warren MS, Lee A, Galazzo J, Fronko R, Lee M, Blais J, Cho D, Chamberland S, Renau T, Leger R, Hecker S, Watkins W, Hoshino K, Ishida H, Lee VJ. Antimicrob. Agents Chemother. 2001;45:105. doi: 10.1128/AAC.45.1.105-116.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishino K, Latifi T, Groisman EA. Mol. Microbiol. 2006;59:126. doi: 10.1111/j.1365-2958.2005.04940.x. [DOI] [PubMed] [Google Scholar]
- 13.Kvist M, Hancock V, Klemm P. Appl. Environ. Microbiol. 2008;74:7376. doi: 10.1128/AEM.01310-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Opperman TJ, Kwasny SM, Kim HS, Nguyen ST, Houseweart C, D'Souza S, Walker GC, Peet NP, Nikaido H, Bowlin TL. Antimicrob. Agents Chemother. 2014;58:722. doi: 10.1128/AAC.01866-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kern WV, Steinke P, Schumacher A, Schuster S, von Baum H, Bohnert JA. J. Antimicrob. Chemother. 2006;57:339. doi: 10.1093/jac/dki445. [DOI] [PubMed] [Google Scholar]
- 16.Schumacher A, Steinke P, Bohnert JA, Akova M, Jonas D, Kern WV. J. Antimicrob. Chemother. 2006;57:344. doi: 10.1093/jac/dki446. [DOI] [PubMed] [Google Scholar]
- 17.Yoshida K, Nakayama K, Ohtsuka M, Kuru N, Yokomizo Y, Sakamoto A, Takemura M, Hoshino K, Kanda H, Nitanai H, Namba K, Yoshida K, Imamura Y, Zhang JZ, Lee VJ, Watkins WJ. Bioorg. Med. Chem. 2007;15:7087. doi: 10.1016/j.bmc.2007.07.039. [DOI] [PubMed] [Google Scholar]
- 18.(a) Nakayama K, Ishida Y, Ohtsuka M, Kawato H, Yoshida K, Yokomizo Y, Hosono S, Ohta T, Hoshino K, Ishida H, Yoshida K, Renau TE, Leger R, Zhang JZ, Lee VJ, Watkins WJ. Bioorg. Med. Chem. Lett. 2003;13:4201. doi: 10.1016/j.bmcl.2003.07.024. [DOI] [PubMed] [Google Scholar]; (b) Nakayama K, Ishida Y, Ohtsuka M, Kawato H, Yoshida K, Yokomizo Y, Ohta T, Hoshino K, Otani T, Kurosaka Y, Yoshida K, Ishida H, Lee VJ, Renau TE, Watkins WJ. Bioorg. Med. Chem. Lett. 2003;13:4205. doi: 10.1016/j.bmcl.2003.07.027. [DOI] [PubMed] [Google Scholar]; (c) Nakayama K, Kawato H, Watanabe J, Ohtsuka M, Yoshida K, Yokomizo Y, Sakamoto A, Kuru N, Ohta T, Hoshino K, Yoshida K, Ishida H, Cho A, Palme MH, Zhang JZ, Lee VJ, Watkins WJ. Bioorg. Med. Chemistry Lett. 2004;14:475. doi: 10.1016/j.bmcl.2003.10.060. [DOI] [PubMed] [Google Scholar]; (d) Nakayama K, Kuru N, Ohtsuka M, Yokomizo Y, Sakamoto A, Kawato H, Yoshida K, Ohta T, Hoshino K, Akimoto K, Itoh J, Ishida H, Cho A, Palme MH, Zhang JZ, Lee VJ, Watkins WJ. Bioorg. Med. Chem. Lett. 2004;14:2493. doi: 10.1016/j.bmcl.2004.03.007. [DOI] [PubMed] [Google Scholar]; (e) Yoshida K, Nakayama K, Kuru N, Kobayashi S, Ohtsuka M, Takemura M, Hoshino K, Kanda H, Zhang JZ, Lee VJ, Watkins WJ. Bioorg. Med. Chem. 2006;14:1993. doi: 10.1016/j.bmc.2005.10.043. [DOI] [PubMed] [Google Scholar]; (f) Yoshida K, Nakayama K, Yokomizo Y, Ohtsuka M, Takemura M, Hoshino K, Kanda H, Namba K, Nitanai H, Zhang JZ, Lee VJ, Watkins WJ. Bioorg. Med. Chem. 2006;14:8506. doi: 10.1016/j.bmc.2006.08.037. [DOI] [PubMed] [Google Scholar]
- 19.Paronikyan EG, Noravyan AS. Chem. Heterocycl. Comp. 1999;35:799. [Google Scholar]
- 20.Hunt JC, Briggs E, Clarke ED, Whittingham WG. Bioorg. Med. Chem. Lett. 2007;17:5222. doi: 10.1016/j.bmcl.2007.06.076. [DOI] [PubMed] [Google Scholar]
- 21.Okusu H, Ma D, Nikaido H. J. Bacteriol. 1996;178:306. doi: 10.1128/jb.178.1.306-308.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poole K. Antimicrob. Agents Chemother. 2000;44:2233. doi: 10.1128/aac.44.9.2233-2241.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takatsuka Y, Chen C, Nikaido H. Proc. Natl. Acad. Sci. USA. 2010;107:6559. doi: 10.1073/pnas.1001460107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vargiu AV, Ruggerone P, Opperman TJ, Nguyen ST, Nikaido H. Antimicrob. Agents Chemother. 2014;58:6224. doi: 10.1128/AAC.03283-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakashima R, Sakurai K, Yamasaki S, Hayashi K, Nagata C, Hoshino K, Onodera Y, Nishino K, Yamaguchi A. Nature. 2013;500:102. doi: 10.1038/nature12300. [DOI] [PubMed] [Google Scholar]
- 26.Nikaido H. Microbiol. Mol. Biol. Rev. 2003;67:593. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagano K, Nikaido H. Proc. Natl. Acad. Sci. USA. 2009;106:5854. doi: 10.1073/pnas.0901695106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zgurskaya HI, Nikaido H. Proc. Natl. Acad. Sci. USA. 1999;96:7190. doi: 10.1073/pnas.96.13.7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel L, Garcia ML, Kaback HR. Biochemistry. 1982;21:5805. doi: 10.1021/bi00266a013. [DOI] [PubMed] [Google Scholar]
- 30.Lin JH, Lu AY. Pharmacol. Rev. 1997;49:403. [PubMed] [Google Scholar]
- 31.Kern EH, Di L. Drug-like properties: concept, structure design and methods from ADME to toxicity optimization. Academic Press; 2008. [Google Scholar]
- 32.Bevan CD, Lloyd RS. Anal. Chem. 2000;72:1781. doi: 10.1021/ac9912247. [DOI] [PubMed] [Google Scholar]
- 33.CLSI. Method for dilution antimicrobial susceptibility testing for bacteria that grow aerobically. Wayne, PA: Clinical and Laboratory Standards Institute; 2006. [Google Scholar]
- 34.Bohnert JA, Karamian B, Nikaido H. Antimicrob. Agents Chemother. 2010;54:3770. doi: 10.1128/AAC.00620-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butler MM, Lamarr WA, Foster KA, Barnes MH, Skow DJ, Lyden PT, Kustigian LM, Zhi C, Brown NC, Wright GE, Bowlin TL. Antimicrob. Agents Chemother. 2007;51:119. doi: 10.1128/AAC.01311-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hill JR. Curr. Prot. Pharmacol. 2003;(Suppl. 23) 7.8.1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.