

Abstract
Alveolar type II (ATII) respiratory epithelial cells are essential to normal lung function. They may be also central to the pathogenesis of diseases such as acute lung injury, pulmonary fibrosis, and pulmonary adenocarcinoma. Hence, ATII cells are important therapeutic targets. However, effective ATII cell-specific drug delivery in vivo requires carriers of an appropriate size, which can cross the hydrophobic alveolar surfactant film and polar aqueous layer overlying ATII cells, and be taken up without inducing ATII cell dysfunction, pulmonary inflammation, lung damage, or excessive systemic spread and side-effects. We have developed lipoplexes as a versatile nanoparticle carrier system for drug/RNA delivery. To optimize their pulmonary localization and ATII cell specificity, lipoplexes were conjugated to an antibody directed against the ATII cell-specific antigen surfactant protein-C (SP-C) then administered to C57BL/6 mice via the nares. Intranasally-administered, anti-SP-C-conjugated lipoplexes targeted mouse ATII cells with >70% specificity in vivo, were retained within ATII cells for at least 48 hours, and did not accumulate at significant levels in other lung cell types or viscera. 48 hours after treatment with anti-SP-C-conjugated lipoplexes containing the test microRNA miR-486, expression of mature miR-486 was approximately 4-fold higher in ATII cells than whole lung by qRT-PCR, and was undetectable in other viscera. Lipoplexes induced no weight loss, hypoxemia, lung dysfunction, pulmonary edema, or pulmonary inflammation over a 6-day period. These findings indicate that ATII cell-targeted lipoplexes exhibit all the desired characteristics of an effective drug delivery system for treatment of pulmonary diseases that result primarily from ATII cell dysfunction.
Keywords: Lipoplex, alveolar type II cell, surfactant protein-C, microRNA, targeted delivery
Introduction
The primary physiologic function of the lungs is gas exchange, which occurs in the terminal bronchioles and alveoli of the distal lung. The bronchoalveolar epithelium is composed of a single layer of epithelial cells covered with a thin layer of aqueous fluid and an overlying film of surfactant[1]. Thin, squamous alveolar type I cells comprise only ~40% of the cells in the alveolus, but account for more than 90% of the surface area of the alveolar epithelium [2]. The remaining 5–10% is made up of small (~10 μm) cuboidal alveolar type II (ATII) cells [3]. ATII cells synthesize, secrete, and recycle pulmonary surfactant lipids and proteins, which contribute to the maintenance of low intra-alveolar surface tension and thereby facilitate ventilation [4]. ATII cells also regulate the depth of the alveolar lining fluid layer by active ion transport, participate in lung inflammatory responses, and serve as progenitors for alveolar type I cells [5–7]. Hence, ATII cells are essential to normal lung function. Importantly, they may be also central to the pathogenesis of multiple acute and chronic, potentially lethal lung diseases, including neonatal respiratory distress syndrome, acute lung injury, pulmonary fibrosis, pulmonary adenocarcinoma, and severe influenza, for which current treatment options are limited [8–12]. As such, ATII cells are important therapeutic targets.
There are several limitations to current methods for targeted delivery of nanoparticle therapeutics for in vivo applications, such as limited stability in serum, rapid blood clearance, poor cellular uptake, and off-target effects. To overcome these limitations, our group has developed lipoplexes (LPs) as carrier systems for drug/nucleic acid delivery [13–16]. Because hydrophobic therapeutics can be incorporated in the lipid bilayers and hydrophilic therapeutics can be encapsulated in the liquid core of LPs, they are highly versatile. In previous studies, we showed that cationic LPs administered to mice by the intravenous route could target the lungs and be retained therein for at least 48 hours without inducing obvious lung toxicity[13]. However, in addition to lung tissue, we found significant accumulation of LPs in the liver and kidneys of treated mice. The aim of the current study was therefore to develop a universal delivery platform that can specifically deliver drugs/nucleic acids to ATII cells, without off-target deposition in other cell types in the lung and other organs. To achieve this objective, we directly administered LPs to the mouse lung via the nares. We also determined the impact of conjugating LPs to a monoclonal antibody directed against the ATII cell-specific antigen surfactant protein C (SP-C) on targeting of a microRNA (miR) to that specific lung cell type. Synthetic miR-486 was selected as a model drug for LP delivery because miR-486 is one of the most down-regulated miRs in lung cancer [17–19]. Additionally, because the delivered amount of miR-486 can be quantitatively measured by qRT-PCR, drug delivery efficiency could be easily and accurately quantified.
Material and Methods
Materials
1, 2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) was purchased from Avanti Polar Lipids (Alabaster, AL, USA). Linoleic acid, polyethyleneimine (PEI, MW~2000) and ethanol were purchased from Sigma-Aldrich (St Louis, MO, USA). D-α-tocopheryl polyethylene glycol 1000 succinate (vitamin E TPGS) was purchased from Eastman (Kingsport, TN, USA). Cy5 dye-labeled oligodeoxynucleotide (5′-Cy5-TCT-CCC-AGC-GTG-CGC-CAT-3′ [Cy5-ODN]) was custom synthesized by Alpha DNA, Inc. (Montreal, Canada). Rabbit polyclonal IgG anti-SP-C antibody FL-197 was purchased from Santa Cruz Biotechnology, Inc. (sc-13979; Dallas, TX, USA). MirVana™ miR mimic hsa-miR-486-5p (miR-486; UCC-UGU-ACU-GAG-CUG-CCC-CGA-G) and scrambled miR mimic Negative Control #1 (miR-SCR) were purchased from Life Technologies, Inc. (Grand Island, NY, USA).
Preparation of lipoplexes containing nucleic acids
Empty liposomes were first generated by injecting a lipid mixture in ethanol (DOPE:linoleic acid:TPGS at 50:48:2 molar ratio) into 20 mM HEPES buffer (pH = 7.4) to achieve 10% ethanol and 90% aqueous in the final mixture. An equal volume of 0.516 mg/ml PEI solution was added to 0.4 mg/ml nucleic acid solution (ODNCy5, miR-486, or miR-SCR) resulting in an N:P ratio (the ratio of moles of the amine group of PEI to moles of the phosphate groups of nucleic acid) of 10. The PEI/nucleic acid mixture was then sonicated for 5 minutes and incubated at room temperature for 10 minutes. LPs containing nucleic acids (LP-ODNCy5, LP-miR-486, or LP-miR-SCR) were prepared by adding the PEI/nucleic acid mixture to empty liposomes at a lipid:nucleic acid mass ratio of 10. The mixture was sonicated for 5 minutes and incubated at room temperature for 15 minutes.
Incorporation of anti-SP-C antibody onto lipoplexes
Anti-SP-C antibody was incorporated onto LPs by a post-insertion method, as previously described [20]. Briefly, anti-SP-C antibody was thiolated at its N-terminus with 2-iminothiolane (Traut’s reagent) in PBS (pH = 8.0) and purified by gel filtration on a PD-10 column. The thiolated anti-SP-C antibody (anti-SP-C-SH) was then reacted with micelles of Mal–PEG–DSPE at a protein-to-lipid molar ratio of 1:10 for 3 hours at room temperature in PBS (pH = 6.5) to obtain anti-SP-C–PEG–DSPE, which was then post-inserted onto LPs by co-incubation at 37°C for 1 hour. The molar ratio of lipids and anti-SP-C antibody was 2000:1. SP-C targeted LPs (LP-ODNCy5/ANTI-SP-C, LP-miR-486ANTI-SP-C, or LP-miR-SCRANTI-SP-C) were concentrated using Amicon Ultra-15 Centrifugal Filter Units (UFC900308; Millipore, Billerica, MA, USA) so that preparations for in vivo applications contained miRs at a concentration of 0.7 mg/ml. Finally, absence of endotoxin contamination of LP preparations was confirmed by a standard Limulus amebocyte assay.
Lipoplex particle size and surface charge measurement
LP size distributions were measured by dynamic light scattering (BI 200SM; Brookhaven Instruments Corp., Holtsville, NY, USA). The wavelength of the laser was 632.8 nm, and the detection angle was 90°. The size distributions of three batches of LPs prepared independently were measured at 20°C. LP surface charges were measured using a ZetaPALS zeta potential analyzer (Brookhaven Instruments). Three batches of independently prepared LPs were diluted in 20mM HEPES buffer. Three measurements, each consisting of 5 runs, were performed at 20°C. The Smoluchowski model was used to calculate the zeta potential.
Uptake of lipoplexes by A549 cells
A549 human lung adenocarcinoma cell uptake of LPs was evaluated by flow cytometry. 2 × 105 viable A549 cells/well were seeded in 6-well plates and incubated overnight in 2 ml of RPMI 1640 media supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA). The culture medium was then replaced with serum-free RPMI 1640. LP-ODNCy5 and LP-ODNCy5/ANTI-SP-C were added to the cells at an ODNCy5 concentration of 1 μM. 4 hours post-treatment, cells were detached from culture plates using 0.25% trypsin, washed with PBS twice, fixed using 4% paraformaldehyde, and subjected to analysis on a BD LSR Fortessa flow cytometer (Becton Dickinson, San Jose, CA, USA). Cy5 fluorescence was measured in the APC channel. 10,000 events were collected for each sample.
Animals
Pathogen-free, 8 week-old female C57BL/6AnNCr mice (Mus musculus) were purchased from the National Cancer Institute (Frederick, MD, USA). C57BL/6-congenic SP-CGFP mice [21;22] (generously provided by the late Dr. Jo Rae Wight, Duke University, NC, USA) were bred in-house and used at 8 weeks of age. All mice were maintained in sterile caging and provided with food and water ad libitum. All animal procedures were approved by The Ohio State University Institutional Animal Care and Use Committee. Care and handling of all animals was in accordance with the NRC/NIH Guide for the Care and Use of Laboratory Animals.
Lipoplex administration
Mice were anesthetized by i.p. injection of ketamine (8.7 mg/kg)/xylazine (1.3 mg/kg). To facilitate pulse oximetry, the fur over the neck was removed by application of Nair (Church & Dwight, Ewing, NJ, USA) for 2 minutes. Mice were held vertically by the scruff and LPs (1.5 mg/kg, suspended in 50 μl saline) were administered dropwise to both nostrils. Animals were placed on a heat pad in left lateral recumbency then returned to their cages upon recovery. Treated mice were weighed every other day. Data for each experimental animal group were derived from a minimum of two independent experiments.
Whole organ imaging
Immediately prior to imaging, C57BL/6 mice were euthanized by i.p. injection of ketamine (87 mg/kg)/xylazine (13 mg/kg). The lungs, heart, spleen, liver, and both kidneys were removed by careful dissection and fixed in 10% formalin. Organ Cy5 fluorescence was detected using the IVIS-200 Bioluminescent Imaging System (Perkin Elmer, Waltham, MA, USA), in accordance with manufacturer’s instructions.
ATII cell isolation
ATII cells were isolated from C57BL/6 and SP-CGFP mice by a standard lung digestion protocol [3]. Mice were euthanized, the pulmonary artery was cannulated, and the lungs were perfused with normal saline in situ to flush out any remaining blood. The trachea was cannulated, and 2 ml dispase II (5 U/ml in PBS; Life Technologies) was injected into the lungs. Dispase was followed by 0.3 ml warmed low-melting-point agarose (1% in PBS) to prevent the isolation of Clara cells and upper airway epithelial cells. The lungs were cooled on ice, removed, and placed in 5 ml dispase to digest at room temperature for 60 minutes with gentle rocking. Pancreatic DNase (0.01% in DMEM; Sigma-Aldrich) was added for the final 5 minutes of incubation. Lung tissue was teased apart, and the resulting cell suspension was filtered sequentially through 100 μm, 40 μm, and 21 μm sterile nylon meshes. Leukocytes were removed by panning with biotin-labeled rat polyclonal anti-murine CD45 antibody (Becton Dickinson) and biotin-labeled rat polyclonal anti-murine CD16/CD32 antibodies (Becton Dickinson) on polystyrene plates for 2 hours at 37°C. Non-adherent cells were collected, pelleted by centrifugation, resuspended in normal saline, and counted using a hemocytometer. Purity of isolated cell ATII cell preparations was determined by visualization of lamellar bodies (refractile inclusions containing stored surfactant lipids) in modified Papanicolaou-stained cytospins.
Confocal microscopy of ATII cells
Isolated ATII cells were counterstained with DAPI and mounted on glass slides. DAPI and Cy5 fluorescence was detected by laser scanning confocal microscopy (Olympus FV1000, Center Valley, PA, USA) in the DAPI (dichroic mirror 430–470 nm) and Cy5 (dichroic mirror 650–700 nm) channels, respectively.
Flow cytometric analysis of ATII cells
Isolated ATII cells were fixed in 4% paraformaldehyde. GFP and Cy5 fluorescence signals were measured using a BD Calibur flow cytometer (Becton Dickinson). 10,000 events were collected per sample. Data were analyzed using FlowJo v.10.0 (TreeStar, Inc. Ashland, OR, USA).
Quantification of expression of mature miR-486
Following euthanasia, ATII cells and major organs were collected and quickly frozen in liquid nitrogen. Frozen tissues were pulverized and total RNA was extracted using TRIzol. Total RNA was then reverse transcribed into cDNA using the TaqMan microRNA reverse transcription kit (Applied Biosystems, Grand Island, NY, USA). qRT-PCR amplification of cDNA was then performed using TaqMan microRNA assay (Assay ID 001278; Applied Biosystems). The expression of mature miR-486 relative to tissues from untreated mice was determined by the ΔΔCt method and normalized to the endogenous control sno135 (Assay ID 001230; Applied Biosystems).
Measurement of cardiopulmonary function in conscious mice
As in our previous studies [10;23;24], carotid arterial oxygen saturation, heart rate, respiratory rate, and pulse distension were measured in conscious mice every other day using the MouseOx system with a collar clip sensor (Starr Life Sciences Corp., Allison Park, PA, USA), in accordance with manufacturer’s instructions. Data were collected for a minimum of 10 seconds (150 data points) per sample.
Measurement of lung mechanics
Mechanical properties of mouse lungs were assessed using the forced-oscillation technique [25;26] as in our previous studies [27]. Briefly, mice were anesthetized with valium (5 mg/kg, i.p.) followed by ketamine (200 mg/kg, i.p.) 6 minutes later. After tracheal cannulation, mice were mechanically ventilated on a flexiVent piston ventilator (SciReq, Montreal, Canada), with 8 ml/kg tidal volume, at a frequency of 150 breaths/minute, against 2–3 cmH2O PEEP. Following two total lung capacity maneuvers to standardize volume history, static lung compliance values were derived from volume-stepped discontinuous pressure-volume loops. Subsequently, pressure and flow data were collected and used to calculate total lung resistance at baseline using the single-compartment model [25]. As before, maximal airway responsiveness to bronchoconstrictors was measured following exposure to 5 escalating doses of nebulized methacholine (0.1 to 50 mg/ml) [28].
Measurement of total lung water content
Mice were euthanized as above then exsanguinated. The lungs were removed, weighed, and dried in an oven at 55°C for 3 days. After drying, the lungs were weighed again. Wet:dry weight ratio was then calculated as an index of intrapulmonary fluid accumulation. No correction for blood content was made.
Bronchoalveolar lavage
Mice were euthanized as above and the lungs were lavaged in situ with 0.8 ml of sterile saline. BALF was subjected to low-speed centrifugation (1,000 rpm for 10 minutes). Total cell numbers were then determined from the cell pellet. Leukocyte viability was determined via trypan blue exclusion, and cell types were differentiated on Wright-Giemsa-stained cytospin preparations using standard hematological criteria.
Measurement of bronchoalveolar lavage fluid mediators
BALF protein and lactate dehydrogenase were measured by a standard BCA assay and a colorimetric assay (Cayman Chemical, Ann Arbor, MI, USA), respectively. BALF cytokine, chemokine, and growth factor content was determined by Bioplex-Pro Mouse Cytokine 23-plex assay (Bio-Rad Laboratories, Hercules, CA, USA). All assays were performed in accordance with manufacturer’s instructions.
Statistical analyses
Descriptive statistics were calculated using Instat 3.05 (GraphPad Software, San Diego, CA, USA) as in our previous studies [10]. Gaussian data distribution was verified by the method of Kolmogorov and Smirnov. Differences between group means were analyzed by t-test for two-group comparisons and by ANOVA for multi-group comparisons, with a post hoc Tukey-Kramer multiple comparison post-test for ANOVA. P<0.05 was considered statistically significant. All data are presented as mean ± S.D.
Results
Characteristics of lipoplexes
LP-ODNCy5 had a mean diameter of approximately 215 nm, and a zeta potential of approximately −5 mV (Figs. 1A and 1B, respectively). Anti-SP-C antibody incorporation (to generate LP-ODNCy5/ANTI-SP-C) did not significantly alter mean LP diameter or zeta potential.
Fig. 1.
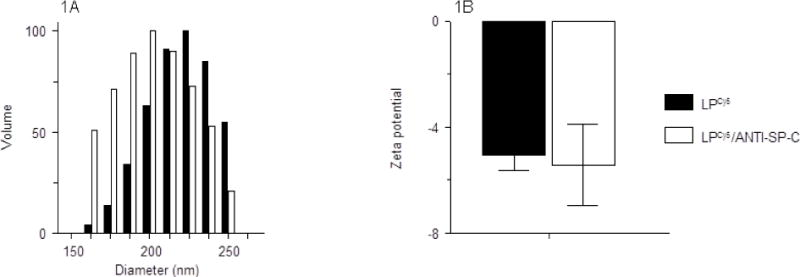
Effect of anti-SP-C antibody conjugation on (A) LP-ODNCy5 diameter (nm); and (B) LP-ODNCy5 surface charge (zeta potential; mV). n=3 LP preparations per group. Data are presented as mean ± S.D.
Incorporation of anti-SP-C antibody significantly improved targeting of lipoplexes to A549 cells in vitro
Before proceeding to animal studies, initial proof-of-concept experiments to demonstrate that conjugation to an anti-SP-C antibody could improve targeting of LP-ODNCy5 to respiratory epithelial cells were preformed using A549 human lung adenocarcinoma cells, which express SP-C [29]. 4 hours after in vitro treatment with LP-ODNCy5 and LP-ODNCy5/ANTI-SP-C containing 1 μM ODNCy5, cellular uptake of LPs by A549 cells was evaluated by flow cytometry. Mean Cy5 fluorescence intensity in A549 cells treated with LP-ODNCy5/ANTI-SP-C was approximately 2-fold higher than in cells treated with LP-ODNCy5, indicating that conjugation to anti-SP-C antibody significantly improved LP delivery efficiency (Fig. 2).
Fig. 2.
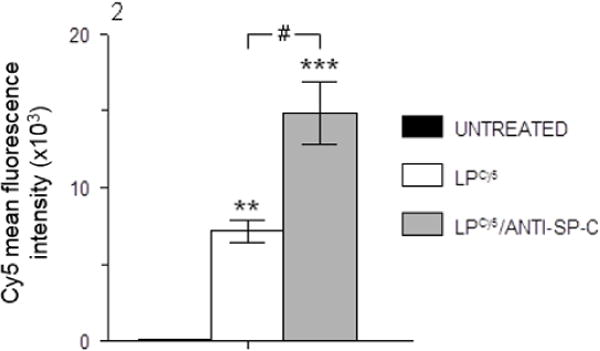
Cy5 mean fluorescence intensity in A549 cells following treatment with LP-ODNCy5 and LP-ODNCy5/ANTI-SP-C containing 1 μM Cy5-ODN for 4 hours, measured by flow cytometry. n=3 cultures per group. **P<0.005, ***P<0.0005, vs. untreated cells. #P<0.0005, vs. LPCy5-treated cells. Data are presented as mean ± S.D.
Biodistribution of lipoplexes in mice
C57BL/6 mice (3 per group) were treated intranasally with 50 μl sterile saline, 50 μl LP-ODNCy5 in saline containing 35 μg ODNCy5 (1.5 mg/kg), or 50 μl LP-ODNCy5/ANTI-SP-C in saline containing 35 μg ODNCy5. At 2 days post-treatment (d.p.t.), mice were euthanized and intact viscera were removed. Relative fluorescence of each organ was measured using the IVIS® imaging system. No fluorescence was detected in lungs, hearts, or spleens isolated from saline-treated animals, although modest autofluorescence was detectable in kidney and liver (Figs. 3A and 3B). In contrast, very high levels of fluorescence were detectable in lungs from mice treated with either LP-ODNCy5 or LP-ODNCy5/ANTI-SP-C. Overall, more than 95% of all Cy5 signal was localized to the lungs, indicating that 95% of administered LPs were retained in this organ at 2 d.p.t.
Fig. 3.
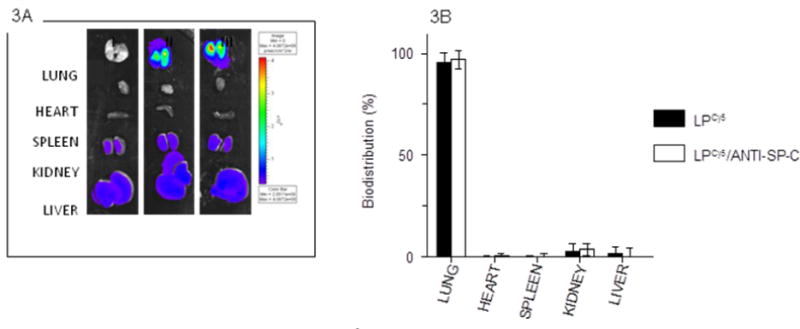
Composite of representative IVIS® images of isolated organs from one saline-treated C57BL/6 mouse (I), one LP-ODNCy5-treated C57BL/6 mouse (II), and one LP-ODNCy5/ANTI-SP-C-treated C57BL/6 mouse (III) at 2 d.p.t.; and (B) Cy5 fluorescence intensity values for lungs shown in panel (A). Organs shown were imaged and quantified at the same time. n=3 mice per group. Data are presented as mean ± S.D.
Incorporation of anti-SP-C antibody significantly improved targeting of lipoplexes to murine ATII cells in vivo
Transgenic, C57BL/6-congenic SP-CGFP mice were used to quantify specific uptake of LPs by ATII cells. These mice express green fluorescent protein (GFP) under the control of the SP-C gene promoter, which is only active in ATII cells [21;22]. Hence, GFP serves as a specific ATII cell marker in these mice. SP-CGFP mice were treated with LP-ODNCy5 or LP-ODNCy5/ANTI-SP-C and ATII cells were isolated by a standard lung digestion protocol at 2 d.p.t. [3]. Papanicolau-positive lamellar bodies (refractile inclusions containing stored surfactant lipids) which are characteristic of ATII cells were visible in cytospins (Fig. 4A, arrows) [30]. When isolated cells were subjected to flow cytometric analysis, the ATII cell population was identified based on its forward- and side-scatter characteristics (Fig. 4B). Cells within a wide gate designed to capture all ATII cells and any other cells of a similar size were selected for further analysis (Fig. 4C). LP-ODNCy5 delivery efficiency was then calculated as the percentage of Cy5-positive/GFP-positive cells among all GFP-positive cells. Conjugation of LP-ODNCy5 with anti-SP-C antibody to generate LP-ODNCy5/ANTI-SP-C increased delivery efficiency of Cy5 to GFP-positive ATII cells by more than 2-fold (Fig. 4D).
Fig. 4.
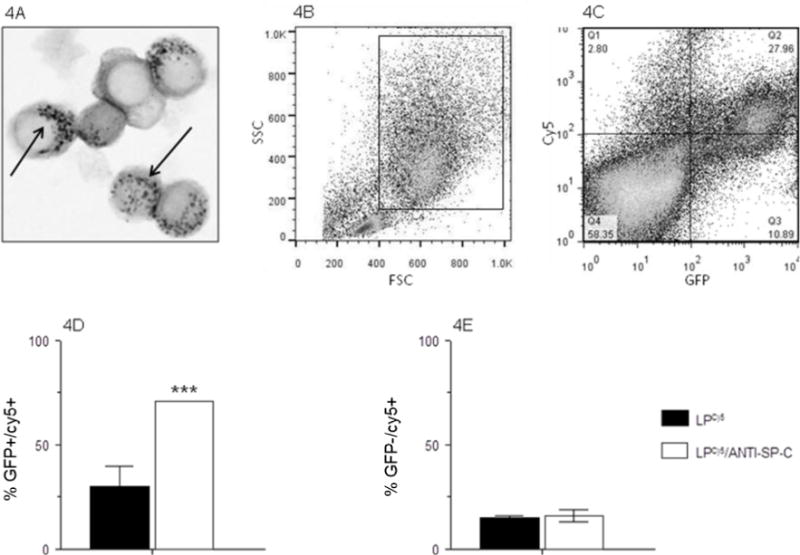
(A) Representative photomicrograph of Papanicolau-stained ATII cells (arrows indicate characteristic Papanicolau-positive lamellar bodies within the cell cytoplasm); (B) Representative flow cytometric plot showing ATII cell gating strategy, based on forward scatter (FSC) and side scatter (SSC) characteristics; (C) Representative flow cytometric quad-plot showing co-localization of green fluorescent protein (GFP) with Cy5 fluorescence in ATII-gated cells isolated from an SP-CGFP mouse treated with LP-ODNCy5/ANTI-SP-C at 2 d.p.t.; (D) Percentage of ATII cells that contain LP-ODNCy5 or LP-ODNCy5/ANTI-SP-C (% GFP+/Cy5+) at 2 d.p.t.; and (E) Percentage of non-ATII cells within the ATII cell gate that contain LP-ODNCy5 or LP-ODNCy5/ANTI-SP-C (% GFP−/Cy5+) at 2 d.p.t. n=4 mice per group. ***P<0.0005, vs. LP-ODNCy5-treated mice. Data are presented as mean ± S.D.
Off-target effects of LP-ODNCy5 treatment were calculated as the percentage of Cy5-positive/GFP-negative cells in all Cy5-positive cells. On average, approximately 15% of Cy5-positive cells were GFP-negative (Fig. 4E). No significant difference was observed between LP-ODNCy5 and LP-ODNCy5/ANTI-SP-C. This indicates that SP-C targeting specifically enhanced the efficiency of LP-ODNCy5 delivery to ATII cells but did not affect uptake of LP-ODNCy5 by non-ATII cells.
Confocal microscopy confirmed that the intensity of Cy5 fluorescence at 2 d.p.t. was higher in ATII cells from SP-CGFP mice that had been treated with LP-ODNCy5/ANTI-SP-C (Fig. 5).
Fig. 5.
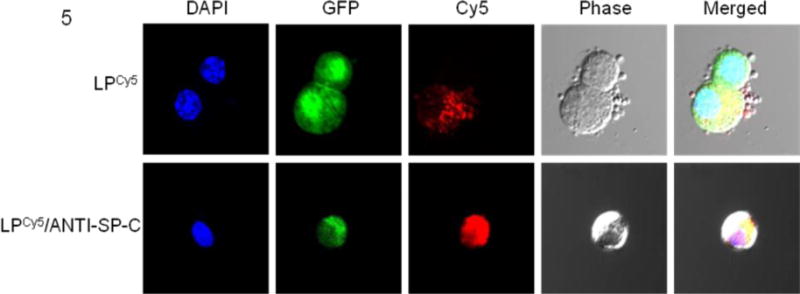
Representative confocal microscopic images of purified ATII cells from SP-CGFP mice treated with LP-ODNCy5 (upper row) or LP-ODNCy5/ANTI-SP-C (lower row) at 2 d.p.t. Cell nuclei were counterstained with DAPI.
Expression of miR-486 delivered by ATII cell-targeted lipoplexes was restricted to ATII cells and lung tissue
Given their superior ATII cell targeting efficiency, subsequent studies used ATII cell-targeted LPs, which were not labeled with Cy5. To evaluate their potential utility for delivery of therapeutics, miRs (miR-486 and miR-SCR) were encapsulated in SP-C targeted LPs. These miRs are 22 nucleotides long and are therefore of comparable length to ODNCy5, which contains 18 nucleotides conjugated to a Cy5 moiety which is approximately the size of two nucleotide residues. When C57BL/6 mice were treated with 50 μl LP-miR-486ANTI-SP-C (containing 35 μg miR-486), expression of mature miR-486 was detectable by qRT-PCR in ATII cells and whole lung homogenates at 2 d.p.t., but was not present in homogenates of tissue harvested from the heart, spleen, kidney, or liver (Fig. 6). Importantly, miR-486 expression was significantly higher in purified ATII cells than in whole lung homogenates. Moreover, miR-486 expression was 8-fold higher in ATII cells and 2-fold higher in lungs from mice treated with LP-miR-486ANTI-SP-C than those treated with untargeted miR-486-complexed LPs. No miR-486 expression was detectable in ATII cells, lungs, or any other organs from mice treated with 50 μl LP-miR-SCRANTI-SP-C containing 35 μg scrambled control miR.
Fig. 6.
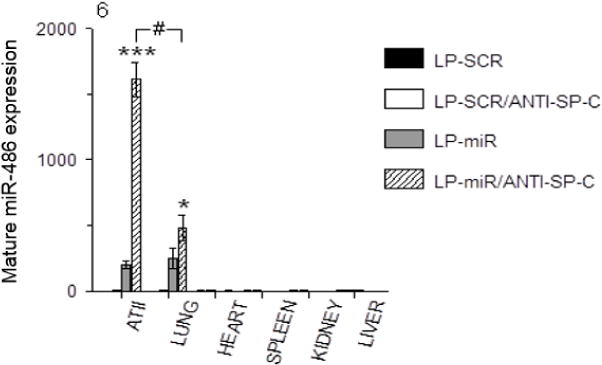
Effects of treatment of C57BL/6 mice with LP-SCR, LP-SCRANTI-SP-C, LP-miR, and LP-miR-486ANTI-SP-C on ATII cell and organ expression of mature miR-486 (by qRT-PCR) at 2 d.p.t. n=3 mice per group. *P<0.05, ***P<0.0005, vs. LP-miR. #P<0.0005, ATII cells vs. whole lung. Data are presented as mean ± S.D.
Treatment with ATII cell-targeted lipoplexes did not alter body weight or cardiopulmonary function in mice
To evaluate potential toxic effects of LPs in vivo, we treated C57BL/6 mice with 50 μl LP-miR-SCRANTI-SP-C containing 35 μg miR-SCR. Neither saline treatment nor LP-miR-SCRANTI-SP-C treatment had any effect on mouse body weights over a 6-day period (Fig. 7A). Likewise, neither treatment resulted in hypoxemia (reduced carotid arterial O2 saturation), bradycardia (reduced resting heart rate), or altered respiratory rate over the same timecourse (Figs. 7B–7D).
Fig. 7.
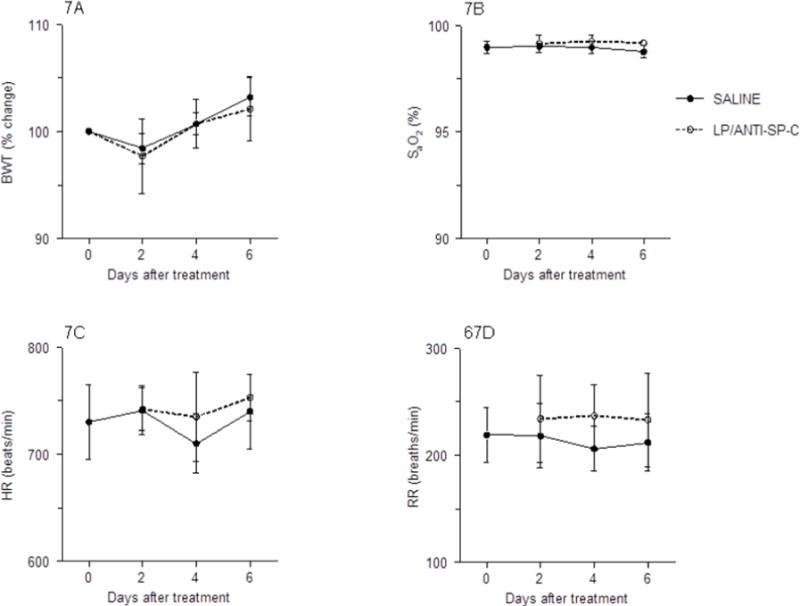
Effects of treatment of C57BL/6 mice with saline or LP-miR-SCRANTI-SP-C on: (A) Body weight (BWT; % change from day 0); (B) Carotid arterial oxygen saturation (% SaO2); (C) Heart rate (HR; beats per minute); and (D) Respiratory rate (RR; breaths per minute). n=8 mice per group. Data are presented as mean ± S.D.
Treatment with ATII cell-targeted lipoplexes did not negatively impact murine lung function
Static lung compliance is an index of lung tissue stiffness and resistance to inflation on inspiration, as measured at a fixed lung volume. Importantly, reduced synthesis of surfactant lipids and proteins by dysfunctional ATII cells results in increased alveolar surface tension and decreased compliance (increased lung stiffness). Likewise, impairment of ATII cell-mediated ion transport results in increased intra-alveolar fluid and dilution of alveolar surfactant proteins, which will also reduce static lung compliance [6]. Hence, compliance is a highly-sensitive measure of ATII cell physiologic function. Treatment with LP-miR-SCRANTI-SP-C had no effect on static lung compliance in mechanically-ventilated live mice at 2 and 6 d.p.t. (Fig. 8A).
Fig. 8.
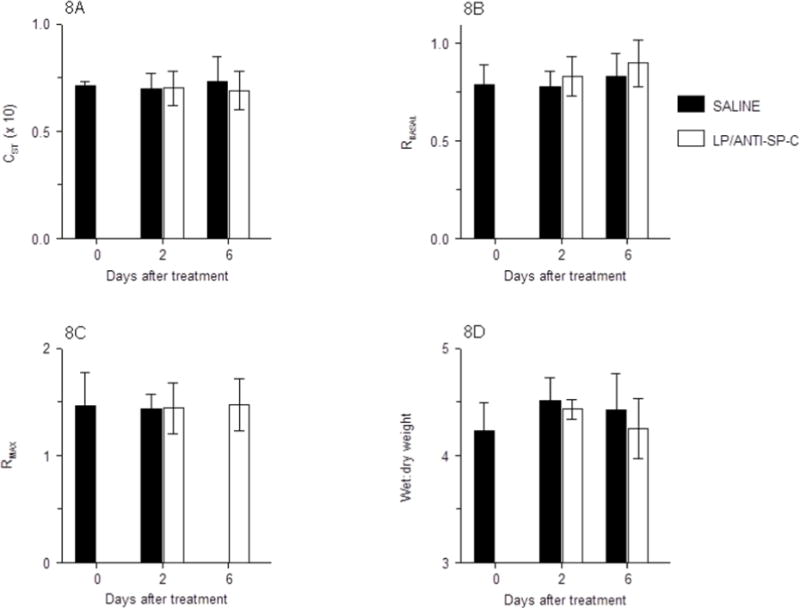
Effects of treatment of C57BL/6 mice with saline or LP-miR-SCRANTI-SP-C on: (A) Static lung compliance (CST; ml/cmH2O, × 10); (B) Baseline total lung resistance (RBASAL, cmH2O.s/ml); (C) Maximal lung resistance following nebulization of 50 mg/ml methacholine (RMAX, cmH2O.s/ml); and (D) Lung water content (wet:dry weight ratio). n=4 mice per group. Data are presented as mean ± S.D.
Total airway resistance is an indicator of airway patency and resistance to airflow during breathing. When ATII cell-mediated alveolar fluid clearance becomes impaired or the alveolar-capillary barrier is damaged by inflammatory processes, fluid accumulation in the alveoli and small conducting airways increases airway resistance. Treatment with LP-miR-SCRANTI-SP-C had no effect on airway resistance at 2 or 6 d.p.t., indicating that these particles have no inherent bronchoconstrictive effect (Fig. 8B). Additionally, neither saline nor LP-miR-SCRANTI-SP-C induced airway hyperresponsiveness to escalating doses of methacholine at either 2 or 6 d.p.t. (Fig. 8C), indicating that LP-miR-SCRANTI-SP-C had no effect on either epithelial permeability or airway smooth muscle function. Finally, neither treatment altered lung water content (Fig. 8D).
Treatment with LP-miR-SCRANTI-SP-C had no effect on bronchoalveolar epithelial integrity (BALF protein content; data not shown). Moreover, we found no increase in BALF lactate dehydrogenase content at either 2 or 6 d.p.t., indicating that LP-miR-SCRANTI-SP-C treatment did not induce significant respiratory epithelial cell death (data not shown).
Treatment with ATII cell-targeted lipoplexes did not induce pulmonary inflammation
Bronchoalveolar lavage fluid (BALF) from untreated mice contained approximately 7 × 105 leukocytes/ml, of which more than 95% were alveolar macrophages (Fig. 9A). BALF from these animals contained very low numbers of neutrophils (<104/ml; Fig. 9B), as well as the occasional lymphocyte, but no eosinophils or basophils (data not shown). Both saline treatment and LP-miR-SCRANTI-SP-C treatment had no effect on BALF total leukocyte content or composition at either 2 or 6 d.p.t.
Fig. 9.
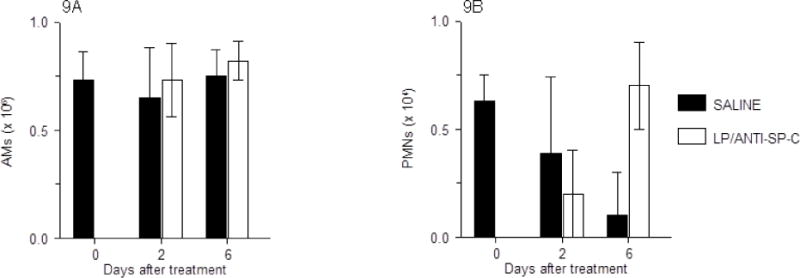
Effects of treatment of C57BL/6 mice with saline or LP-miR-SCRANTI-SP-C on: (A) Bronchoalveolar lavage fluid (BALF) alveolar macrophages (AMs; × 106/ml); and (B) BALF neutrophils (PMNs; × 104/ml). n=4–5 mice per group. Data are presented as mean ± S.D.
In agreement with these findings, levels of inflammatory cytokines (IFN-γ, IL-1α, IL-10, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12[p40], IL-12[p70], IL-13, IL-17, and TNF-α), chemokines (eotaxin, KC, MCP-1, MIP-1α, MIP-1β, and RANTES), and growth factors (G-CSF and GM-CSF) in BALF from saline-treated or LP-miR-SCRANTI-SP-C-treated mice were below the limits of detection at either 2 or 6 d.p.t. (data not shown).
Discussion
Nanoparticles are attractive as novel modalities for systemic and organ-specific delivery of small molecule inhibitors and genetic materials [31]. In addition, the ease of access to the lung makes this organ an attractive site for administration of nanoparticle-associated drugs, oligonucleotides, small interfering RNAs, and miRs by aerosolization [32]. However, successful delivery of nanoparticles to the distal lung presents a set of specific challenges. Firstly, only particles with an aerodynamic diameter less than 5 μm are generally deposited in the distal lung with reasonable efficiency [33]. Secondly, the surfactant layer can act as a physical barrier to nanoparticle uptake by ATII cells, and macrophages in the alveolar lumen can non-specifically phagocytose and degrade inhaled nanoparticles, particularly those of larger size [34;35]. Finally, ATII cell-driven innate immune responses to inhaled nanoparticles may result in pulmonary inflammation and ATII cell dysfunction or even cell death [36;37]. Ideally, therefore, nanoparticle systems for therapeutic delivery of drugs or RNAs to ATII cells need to be of an appropriate size, be able to cross the hydrophobic alveolar surfactant film and polar aqueous layer which overlay the ATII cell, and be taken up without inducing an inflammatory response. Moreover, because nanoparticles can enter cells by receptor-mediated endocytosis into clathrin-coated pits, caveolae-mediated endocytosis, macro-pinocytosis, or phagocytosis, they must be resistant to proteolytic degradation and capable of delivering their contents efficiently to the cell cytoplasm. Finally, high physical stability, ATII cell-specific uptake, prolonged retention within ATII cells, lack of undesired effects on normal ATII cell functions, limited systemic spread, and minimal systemic side-effects would also be highly-desirable characteristics. To date, however, no nanoparticle systems that exhibit most or all of these characteristics have been reported.
We hypothesized that direct intranasal administration of LPs to the mouse lung would improve pulmonary localization and retention. Furthermore, we proposed that conjugating LPs to a monoclonal antibody directed against the ATII cell-specific antigen SP-C would improve targeting to these cells. In our LP formulation, positively charged, low-molecular-weight PEI (PEI2000) was used to condense nucleic acids. PEI2000 is known to be relatively biocompatible [38]. After endocytosis by the cells, the proton sponge effect of PEI helps LPs escape from endosomes and release miRs [39]. DOPE was selected as the building block of our nanocarrier system. DOPE is a phospholipid which can transform to an inverted hexagonal (HII) phase at acidic pH, which destabilizes the endosomal membrane and enhances the release of miRs in the cytosol [40;41]. Linoleic acid was chosen as a helper lipid because its negative charges made the empty liposomes carry negative surface charges, which easily formed LPs with positively charged PEI/miR complexes by electrostatic interactions. In addition, negatively charged linoleic acids do not form strong interactions with miRs, which may also facilitate the dissociation of miRs from the LPs. TPGS, a short poly(ethylene glycol) molecule linked to vitamin E, was added to increase the stability and minimize the non-specific uptake of LPs in vivo. By careful control of the mass ratio of positively charged PEI/miR complexes to negatively charged liposomes, the final LPs were made slightly negatively charged, which reduces the opsonization of LPs by serum proteins, nonspecific uptake, and cytotoxicity in vivo. Anti-SP-C antibody was incorporated on the surface of LPs to significantly improve the targeting efficiency and minimize off-target effects, without significantly altering the physical characteristics of LPs.
Previously, we showed that cationic LPs can efficiently deliver miRs to A549 cells in vitro and in vivo using in a mouse xenograft model [42]. We also demonstrated that LPs can target the lungs and be retained therein for at least 48 hours without inducing obvious lung toxicity when administered to mice by the intravenous route [13]. However, in the latter experiments, only ~30% of intravenously-administered cationic LPs accumulated in lung tissue, and the lung cell type(s) within which these LPs were retained were not determined. Moreover, we also found significant accumulation of cationic LPs in the liver and kidneys of treated mice. In contrast, we found in the current study that intranasally-administered LP-ODNANTI-SP-C localized to the lung and exhibited minimal spread to the liver, kidneys, or spleen. In addition, we found that LP-ODNANTI-SP-C targeted mouse ATII cells with high specificity in vivo and were retained within these cells for at least 48 hours. Furthermore, intranasal administration of LP-miR-486ANTI-SP-C resulted in an ATII cell-specific increase in miR-486 expression at 2 d.p.t., without inducing lung dysfunction, pulmonary edema, or pulmonary inflammation over a 6-day period.
We hypothesize that the hydrophobic nature of the outer shell of the LPs facilitates their movement into and across the surfactant barrier. After intranasal administration, LPs should remain intact during the movement toward the targeted ATII cells because SP-C targeting enhanced the cellular uptake of LPs in vitro and in vivo. If LPs disintegrated prior to being taken up the cells, SP-C targeting should have no effects, which was not we observed. Once anti-SP-C conjugated LPs have entered the subjacent alveolar lining fluid, the anti-SP-C antibody can then bind to SP-C expressed on the apical ATII cell membrane, facilitating the uptake by these cells. SP-C is also co-secreted with surfactant lipids and is essential to correct lipid packing and spreading and stabilization of the surfactant layer [4]. Hence, it is possible that anti-SP-C conjugated LPs also bind to SP-C within the surfactant layer itself. Because surfactant proteins and lipids are continuously recycled by ATII cells via the endosomal pathway, this mechanism would also promote trafficking of LPs to the ATII cell endosomal pool [43]. Recycling may also enhance the ability of anti-SP-C conjugated LPs to persist within ATII cells for a prolonged period. However, further studies will be necessary to determine mechanism(s) of uptake of anti-SP-C conjugated LPs by ATII cells.
MiRs are short (22–25 nucleotides), endogenous, non-coding RNAs that regulate the expression of multiple genes simultaneously at the post-transcriptional level though RNA interference [44;45]. Modulators of miR expression therefore constitute a potent, new class of therapeutics that work through a mechanism that affects multiple regulatory pathways simultaneously, i.e. “one for all” [46;47]. To demonstrate the potential of anti-SP-C antibody-conjugated LPs in delivering therapeutics to ATII cells, we selected miR-486 as a test agent. MiR-486 has been found to be one of the most down-regulated miRs in lung tumor tissues and its suppression contributes to development of non-small cell lung cancer, which is the most common type of lung cancer [17;48]. Delivery of miR-486 via ATII cell-targeted LPs induced its expression at high levels and for at least 48 hours in whole lung homogenates and ATII cells without inducing expression in other tissues. The fact that miR-486 expression was significantly lower in whole lung than in purified ATII cells from LP-miR-486ANTI-SP-C-treated mice further demonstrates that delivery of miR-486 was highly specific to ATII cells: if LP-miR-486ANTI-SP-C had also been taken up by other lung cell types at high levels, then the difference in miR-486 expression between whole lung and purified ATII cells would have been much lower. The absence of a significant difference in miR-486 expression between ATII cells and whole lung tissue from mice treated with untargeted LP-miR-486 further indicates that conjugation to the anti-SP-C antibody is essential to efficient and specific delivery of miR-486 to ATII cells. Taken together, our results suggest that anti-SP-C-antibody-conjugated LPs have potential as an efficient system for delivering miRs to ATII cells undergoing neoplastic transformation.
To determine whether LP-ODNANTI-SP-C treatment had detrimental effects on murine cardiopulmonary function, we performed a battery of physiologically-relevant in vivo assays after nasal delivery of LP-miR-SCRANTI-SP-C to normal C57BL/6 mice. These included measurements of gas exchange and cardiac function in conscious mice by pulse oximetry, analysis of pulmonary mechanics in live, mechanically-ventilated mice, and quantification of pulmonary edema following euthanasia. If LP-miR-SCRANTI-SP-C were to have direct cytotoxic effects on bronchoalveolar epithelial cells and/or induce an inflammatory response, treatment would result in reduced ATII cell ion transport and respiratory epithelial integrity, both of which would promote development of alveolar edema. In turn, alveolar edema would lead to altered lung mechanics, peripheral hypoxemia, and subsequent tachypnea (increased respiratory rate) and bradycardia (decreased heart rate). Importantly, we found no differences in cardiopulmonary function between saline-treated and LP-miR-SCRANTI-SP-C-treated mice at either early (2 d.p.t.) or late (6 d.p.t.) timepoints. Data in these groups were comparable to those we have reported previously for normal C57BL/6 mice [23;24;27]. Likewise, LP-miR-SCRANTI-SP-C treatment did not significantly alter bronchoalveolar permeability and respiratory epithelial cell viability. Finally, we could find no evidence of either cellular or humoral (cytokine/chemokine) inflammatory responses to administration of LP-miR-SCRANTI-SP-C at either 2 or 6 d.p.t. Together, these data indicate that these LPs have very low pulmonary toxicity, if any. To our knowledge, this has not been demonstrated for any other nanoparticle system. Indeed, some nanoparticles, such as fullerene and carbon nanotubes, are actually highly toxic to the lung [49].
In conclusion, we found that intranasally-administered LPs conjugated to an anti-SP-C antibody targeted murine ATII cells with high specificity in vivo, were retained within these cells for a prolonged period without detrimental effects, and could deliver an intact miR in an ATII cell-specific manner. These findings indicate that ATII cell-targeted LPs exhibit all the desired characteristics of an effective drug delivery system for the lung. Hence, they have great promise to enhance treatment of pulmonary diseases, particularly those that result primarily from ATII cell dysfunction or neoplastic transformation: current therapeutic options for many of these diseases are very limited, and they are often intractable. Moreover, we predict that conjugation of LPs to antibodies directed against cell type-specific antigens expressed by bronchial epithelial cells, resident alveolar macrophages, or infiltrating leukocytes would also allow us to target LPs to these cell types with high efficiency and specificity, which would significantly broaden their already widespread therapeutic potential.
Acknowledgments
This work was supported by The Ohio State University Howard Hughes Medical Institute Med-to-Grad Training Program, The Ohio State University Pelotonia Idea Award Program, The National Heart Lung and Blood Institute at the National Institutes of Health (R01-HL102469), and The State University of New York at Buffalo.
The authors would like to acknowledge Lisa Joseph, R.V.T. for her excellent technical support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors report no conflicts of interest.
Contributor Information
Yun Wu, Email: ywu32@buffalo.edu.
Junyu Ma, Email: junyuma@hotmail.com.
Parker S. Woods, Email: Parker.Woods@osumc.edu.
Nicholas M. Chesarino, Email: chesarino.1@buckeyemail.osu.edu.
Chang Liu, Email: cliu36@buffalo.edu.
L. James Lee, Email: lee.31@osu.edu.
Serge P. Nana-Sinkam, Email: Patrick.Nana-Sinkam@osumc.edu.
Ian C. Davis, Email: davis.2448@osu.edu.
References
- 1.Herzog EL, Brody AR, Colby TV, Mason R, Williams MC. Knowns and unknowns of the alveolus. Proceedings of the American Thoracic Society. 2008;5:778–782. doi: 10.1513/pats.200803-028HR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dobbs LG, Johnson MD, Vanderbilt J, Allen L, Gonzalez R. The great big alveolar TI cell: Evolving concepts and paradigms. Cell Physiol Biochem. 2010;25:55–62. doi: 10.1159/000272063. [DOI] [PubMed] [Google Scholar]
- 3.Corti M, Brody AR, Harrison JH. Isolation and primary culture of murine alveolar type II cells. Am J Respir Cell Mol Biol. 1996;14:309–315. doi: 10.1165/ajrcmb.14.4.8600933. [DOI] [PubMed] [Google Scholar]
- 4.Whitsett JA, Wert SE, Weaver TE. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Ann Rev Med. 2010;61:105–119. doi: 10.1146/annurev.med.60.041807.123500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.MASON RJ. Biology of alveolar type II cells. Respirology. 2006;11(Suppl):S12–5. S12–S15. doi: 10.1111/j.1440-1843.2006.00800.x. [DOI] [PubMed] [Google Scholar]
- 6.Davis IC, Matalon S. Epithelial sodium channels in the adult lung–important modulators of pulmonary health and disease. Adv Exp Med Biol. 2007;618:127–140. doi: 10.1007/978-0-387-75434-5_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marconett CN, Zhou B, Rieger ME, Selamat SA, Dubourd M, Fang X, Lynch SK, Stueve TR, Siegmund KD, Berman BP, Borok Z, Laird-Offringa IA. Integrated transcriptomic and epigenomic analysis of primary human lung epithelial cell differentiation. PLoS Genet. 2013;9:e1003513. doi: 10.1371/journal.pgen.1003513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berthiaume Y, Matthay MA. Alveolar edema fluid clearance and acute lung injury. Respir Physiol Neurobiol. 2007;159:350–359. doi: 10.1016/j.resp.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Camelo A, Dunmore R, Sleeman MA, Clarke DL. The epithelium in idiopathic pulmonary fibrosis: breaking the barrier. Front Pharmacol. 2014;4:173. doi: 10.3389/fphar.2013.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolk KE, Lazarowski ER, Traylor ZP, Yu EN, Jewell NA, Durbin RK, Durbin JE, Davis IC. Influenza A virus inhibits alveolar fluid clearance in BALB/c mice. Am J Respir Crit Care Med. 2008;178:969–976. doi: 10.1164/rccm.200803-455OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu X, Rock JR, Lu Y, Futtner C, Schwab B, Guinney J, Hogan BLM, Onaitis MW. Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proc Natl Acad Sci USA. 2012;109:4910–4915. doi: 10.1073/pnas.1112499109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin C, Song H, Huang C, Yao E, Gacayan R, Xu SM, Chuang PT. Alveolar type II cells possess the capability of initiating lung tumor development. PLoS ONE. 2012;7:e53817. doi: 10.1371/journal.pone.0053817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu Y, Crawford M, Yu B, Mao Y, Nana-Sinkam SP, Lee LJ. MicroRNA delivery by cationic lipoplexes for lung cancer therapy. Mol Pharm. 2011;8:1381–1389. doi: 10.1021/mp2002076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X, Yu B, Wu Y, Lee RJ, Lee LJ. Efficient down-regulation of CDK4 by novel lipid nanoparticle-mediated siRNA delivery. Anticancer Res. 2011;31:1619–1626. [PubMed] [Google Scholar]
- 15.Yu B, Hsu SH, Zhou C, Wang X, Terp MC, Wu Y, Teng L, Mao Y, Wang F, Xue W, Jacob ST, Ghoshal K, Lee RJ, Lee LJ. Lipid nanoparticles for hepatic delivery of small interfering RNA. Biomaterials. 2012;33:5924–5934. doi: 10.1016/j.biomaterials.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mao Y, Wang J, Zhao Y, Wu Y, Kwak KJ, Chen CS, Byrd JC, Lee RJ, Phelps MA, Lee LJ, Muthusamy N. A novel liposomal formulation of FTY720 (Fingolimod) for promising enhanced targeted delivery. Nanomed: Nanotech Biol Med. 2014;10:393–400. doi: 10.1016/j.nano.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Võsa U, Vooder T, Kolde R, Vilo J, Metspalu A, Annilo T. Meta-analysis of microRNA expression in lung cancer. Int J Cancer. 2013;132:2884–2893. doi: 10.1002/ijc.27981. [DOI] [PubMed] [Google Scholar]
- 18.Peng Y, Dai Y, Hitchcock C, Yang X, Kassis ES, Liu L, Luo Z, Sun HL, Cui R, Wei H, Kim T, Lee TJ, Jeon YJ, Nuovo GJ, Volinia S, He Q, Yu J, Nana-Sinkam P, Croce CM. Insulin growth factor signaling is regulated by microRNA-486, an underexpressed microRNA in lung cancer. Proc Natl Acad Sci USA. 2013;110:15043–15048. doi: 10.1073/pnas.1307107110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, Tian X, Han R, Zhang X, Wang X, Shen H, Xue L, Liu Y, Yan X, Shen J, Mannoor K, Deepak J, Donahue JM, Stass SA, Xing L, Jiang F. Downregulation of miR-486-5p contributes to tumor progression and metastasis by targeting protumorigenic ARHGAP5 in lung cancer. Oncogene. 2014;33:1181–1189. doi: 10.1038/onc.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allen TM, Sapra P, Moase E. Use of the post-insertion method for the formation of ligand-coupled liposomes. Cell Mol Biol Lett. 2002;7:889–894. [PubMed] [Google Scholar]
- 21.Glasser SW, Korfhagen TR, Wert SE, Bruno MD, McWilliams KM, Vorbroker DK, Whitsett JA. Genetic element from human surfactant protein SP-C gene confers bronchiolar-alveolar cell specificity in transgenic mice. AJP – Lung Cellular and Molecular Physiology. 1991;261:L349–L356. doi: 10.1152/ajplung.1991.261.4.L349. [DOI] [PubMed] [Google Scholar]
- 22.Lo B, Hansen S, Evans K, Heath JK, Wright JR. Alveolar epithelial type II cells induce T cell tolerance to specific antigen. The Journal of Immunology. 2008;180:881–888. doi: 10.4049/jimmunol.180.2.881. [DOI] [PubMed] [Google Scholar]
- 23.Aeffner F, Bratasz A, Flaño E, Powell KA, Davis IC. Post-infection A77-1726 treatment improves cardiopulmonary function in H1N1 influenza-infected mice. Am J Respir Cell Mol Biol. 2012;47:543–551. doi: 10.1165/rcmb.2012-0112OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aeffner F, Abdulrahman B, Hickman-Davis JM, Janssen PM, Amer A, Bedwell DM, Sorscher EJ, Davis IC. Heterozygosity for the F508del mutation in the cystic fibrosis transmembrane conductance regulator anion channel attenuates influenza severity. J Infect Dis. 2013;208:780–789. doi: 10.1093/infdis/jit251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Irvin CG, Bates JH. Measuring the lung function in the mouse: the challenge of size. Respir Res. 2003;4:4–13. doi: 10.1186/rr199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davis IC, Lazarowski ER, Chen FP, Hickman-Davis JM, Sullender WM, Matalon S. Post-infection A77-1726 blocks pathophysiologic sequelae of respiratory syncytial virus infection. Am J Respir Cell Mol Biol. 2007;37:379–386. doi: 10.1165/rcmb.2007-0142OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Traylor ZP, Aeffner F, Davis IC. Influenza A H1N1 induces declines in alveolar gas exchange in mice consistent with rapid post-infection progression from acute lung injury to ARDS. Influenza Other Respir Viruses. 2013;7:472–479. doi: 10.1111/j.1750-2659.2012.00414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis IC, Xu A, Gao Z, Hickman-Davis JM, Factor P, Sullender WM, Matalon S. Respiratory syncytial virus induces insensitivity to b-adrenergic agonists in mouse lung epithelium in vivo. AJP – Lung Cellular and Molecular Physiology. 2007;293:L281–L289. doi: 10.1152/AJPLUNG.00458.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vaporidi K, Tsatsanis C, Georgopoulos D, sichlis PN. Effects of hypoxia and hypercapnia on surfactant protein expression proliferation and apoptosis in A549 alveolar epithelial cells. Life Sci. 2005;78:284. doi: 10.1016/j.lfs.2005.04.070. [DOI] [PubMed] [Google Scholar]
- 30.Dobbs LG. Isolation and culture of alveolar type II cells. AJP – Lung Cellular and Molecular Physiology. 1990;258:L134–L147. doi: 10.1152/ajplung.1990.258.4.L134. [DOI] [PubMed] [Google Scholar]
- 31.Thorley AJ, Tetley TD. New perspectives in nanomedicine. Pharmacol Ther. 2013;140:176–185. doi: 10.1016/j.pharmthera.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 32.Azarmi S, Roa WH, Löbenberg R. Targeted delivery of nanoparticles for the treatment of lung diseases. Adv Drug Delivery Rev. 2008;60:863–875. doi: 10.1016/j.addr.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 33.Bosquillon C, Lombry C, Préat V, Vanbever R. Influence of formulation excipients and physical characteristics of inhalation dry powders on their aerosolization performance. J Control Release. 2001;70:329–339. doi: 10.1016/s0168-3659(00)00362-x. [DOI] [PubMed] [Google Scholar]
- 34.Schleh C, Hohlfeld JM. Interaction of nanoparticles with the pulmonary surfactant system. Inhalation Toxicology. 2009;21:97–103. doi: 10.1080/08958370903005744. [DOI] [PubMed] [Google Scholar]
- 35.Rudt S, Müller RH. In vitro phagocytosis assay of nano- and microparticles by chemiluminescence. I. Effect of analytical parameters, particle size and particle concentration. J Control Release. 1992;22:263–271. [Google Scholar]
- 36.Ali S, Rytting E. Influences of nanomaterials on the barrier function of epithelial cells. In: Capco DG, Chen Y, editors. Nanomaterial. Springer; Netherlands: 2014. pp. 45–54. [DOI] [PubMed] [Google Scholar]
- 37.KENDALL MICH, HOLGATE STEP. Health impact and toxicological effects of nanomaterials in the lung. Respirology. 2012;17:743–758. doi: 10.1111/j.1440-1843.2012.02171.x. [DOI] [PubMed] [Google Scholar]
- 38.Lv H, Zhang S, Wang B, Cui S, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release. 2006;114:100–109. doi: 10.1016/j.jconrel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 39.Godbey WT, Wu KK, Mikos AG. Poly(ethylenimine) and its role in gene delivery. J Control Release. 1999;60:149–160. doi: 10.1016/s0168-3659(99)00090-5. [DOI] [PubMed] [Google Scholar]
- 40.Ma B, Zhang S, Jiang H, Zhao B, Lv H. Lipoplex morphologies and their influences on transfection efficiency in gene delivery. J Control Release. 2007;123:184–194. doi: 10.1016/j.jconrel.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 41.Wasungu L, Hoekstra D. Cationic lipids, lipoplexes and intracellular delivery of genes. J Control Release. 2006;116:255–264. doi: 10.1016/j.jconrel.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 42.Wu Y, Crawford M, Mao Y, Lee RJ, Davis IC, Elton TS, Lee LJ, Nana-Sinkam SP. Therapeutic delivery of microRNA-29b by cationic lipoplexes for lung cancer. Mol Ther Nucleic Acids. 2013;2:e84. doi: 10.1038/mtna.2013.14. e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andreeva AV, Kutuzov MA, Voyno-Yasenetskaya TA. Regulation of surfactant secretion in alveolar type II cells. AJP – Lung Cellular and Molecular Physiology. 2007;293:L259–L271. doi: 10.1152/ajplung.00112.2007. [DOI] [PubMed] [Google Scholar]
- 44.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 45.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009;10:704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9:775–789. doi: 10.1038/nrd3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nana-Sinkam SP, Karsies T, Riscili B, Ezzie M, Piper M. Lung microRNA: from development to disease. Expert Rev Respir Med. 2009;3:373–385. doi: 10.1586/ers.09.30. [DOI] [PubMed] [Google Scholar]
- 48.Hu Z, Chen X, Zhao Y, Tian T, Jin G, Shu Y, Chen Y, Xu L, Zen K, Zhang C, Shen H. Serum microRNA signatures identified in a genome-wide serum microRNA expression profiling predict survival of non-small cell lung cancer. J Clin Oncol. 2010;28:1721–1726. doi: 10.1200/JCO.2009.24.9342. [DOI] [PubMed] [Google Scholar]
- 49.Morimoto Y, Horie M, Kobayashi N, Shinohara N, Shimada M. Inhalation toxicity assessment of carbon-based nanoparticles. Acc Chem Res. 2012;46:770–781. doi: 10.1021/ar200311b. [DOI] [PubMed] [Google Scholar]