

Abstract
Intestinal ischemia and reperfusion (I/R) is encountered in various clinical conditions and contributes to multiorgan failure and mortality as high as 60% to 80%. Intestinal I/R not only injures the intestine, but affects remote organs such as the lung leading to acute lung injury. The development of novel and effective therapies for intestinal I/R are critical for the improvement of patient outcome. AICAR (5-aminoimidazole-4-carboxyamide ribonucleoside) is a cell-permeable compound that has been shown to possess antiinflammatory effects. The objective is to determine that treatment with AICAR attenuates intestinal I/R injury and subsequent acute lung injury (ALI). Male Sprague Dawley rats (275 to 325 g) underwent intestinal I/R injury with blockage of the superior mesenteric artery for 90 min and subsequent reperfusion. At the initiation of reperfusion, vehicle or AICAR (30 mg/kg BW) was given intravenously (IV) for 30 min. At 4 h after reperfusion, blood and tissues were collected for further analyses. Treatment with AICAR significantly decreased the gut damage score and the water content, indicating improvement in histological integrity. The treatment also attenuated tissue injury and proinflammatory cytokines, and reduced bacterial translocation to the gut. AICAR administration after intestinal I/R maintained lung integrity, attenuated neutrophil chemotaxis and infiltration to the lungs and decreased lung levels of tumor necrosis factor (TNF)-α and interleukin (IL)-6. Inflammatory mediators, lung-inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) proteins, were decreased in the lungs and lung apoptosis was significantly reduced after AICAR treatment. These data indicate that AICAR could be developed as an effective and novel therapeutic for intestinal I/R and subsequent ALI.
INTRODUCTION
Mesenteric ischemia remains a critical problem with an overall mortality rate as high as 60% to 80% (1). Intestinal ischemia and subsequent reperfusion are encountered in a variety of clinical conditions including acute mesenteric ischemia, intestinal obstruction, incarcerated hernia, small intestine volvulus and necrotizing colitis. The consequences of mesenteric ischemia are detrimental to the patient and usually result in malabsorption, severe diarrhea, short bowel syndrome and death (2). To date, a limited number of pharmacological agents have been demonstrated to provide some benefit in intestinal ischemia and reperfusion (I/R) injury conditions; however none has been entirely successful (2).
I/R is a clinical condition caused by an initial occlusion of blood supply to a specific organ, for example, the intestine, and subsequent tissue injury due to reperfusion and reoxygenation. The restriction of the arterial blood supply results in tissue hypoxia leading to cellular damage and necrosis. The restoration of blood flow and reoxygenation during reperfusion, however, is associated with tissue injury and an exaggerated inflammatory response (3–11). A wide array of pathological alterations is associated with I/R-induced tissue injury. The initial tissue hypoxia leads to endothelial cell barrier dysfunction and a parallel increase in vascular permeability. The subsequent reperfusion is associated with cell death including necrosis and apoptosis and stimulation of the inflammatory responses (12–14). In particular, during the early phase of reperfusion, innate immune cells such as dendritic cells and monocytes are migrated to the injured tissue to participate in tissue healing (15,16). However, the recruitment of granulocytes should be tightly controlled, as accumulation of too many granulocytes promotes uncontrolled inflammation and tissue injury. Thus, targeting immune activation is a promising therapeutic aspect in the treatment for I/R injury.
AICAR (5-aminoimidazole-4-carboxyamide ribonucleoside) is a revolutionary cell-permeable compound that possesses potent antiinflammatory properties. Recent literature reports that AICAR could inhibit lipopolysaccharide-induced expression of proinflammatory cytokines including tumor necrosis factor (TNF)-α in several immune cells (17,18). In animal models, pretreatment with AICAR attenuated inflammatory responses in dextran sulfate sodium-induced acute and chronic colitis (19). We therefore hypothesize that AICAR with its antiinflammatory feature is able to attenuate systemic inflammation and reduce tissue damage after intestinal I/R. In the current study, we focused on the role of AICAR in the modulation of inflammatory responses caused by intestinal I/R and subsequent acute lung injury.
MATERIALS AND METHODS
Experimental Animals
Male Sprague Dawley rats (275 to 325 g), purchased from Charles River Laboratories (Wilmington, MA, USA) were housed in a temperature-controlled room on a 12-h light-to-dark cycle and fed a standard Purina rat chow diet. All animal experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (20) and were approved by the Institutional Animal Care and Use Committee of the Feinstein Institute for Medical Research.
Animal Model of Intestinal I/R
Rats were fasted for 6 h before surgery. Intestinal I/R was induced by superior mesenteric artery (SMA) occlusion as described previously by us (21). Briefly, rats were anesthetized with isoflurane inhalation and a midline laparotomy was performed. The SMA was isolated and clamped with an atraumatic microvascular clip for 90 min. After 90-min ischemia, the SMA clamp was removed to allow reperfusion. A catheter (PE-50 tubing; BD, Franklin Lakes, NJ, USA) was inserted into the femoral vein to administer drugs. AICAR (30 mg/kg body weight) or vehicle (normal saline) was given IV during a period of 30 min at the beginning of reperfusion. Sham animals underwent the same surgical procedure with the exception of the SMA clamping or AICAR administration. At 4 h after treatment or vehicle administration (that is, 4.5 h after reperfusion), animals were reanesthetized and blood was collected via exsanguination. Afterward, lung tissue and the small intestine were collected. The left upper lobe was used for histologic examination while the lower lobe was used for water-content determination. The right lung was snap frozen in liquid nitrogen and powdered for all other measurements.
Water Content Determination
Lung edema was estimated by comparing wet-to-dry weight ratios as described previously (21).
Intestinal and Lung Histologic Examinations
Morphologic changes in the gut and the lungs were examined by light microscopy at 4 h after treatment. Briefly, tissue samples were fixed in 10% formalin in neutral-buffered solution (Sigma-Aldrich, St. Louis, MO, USA) and paraffin embedded. Tissue blocks were then sectioned at a thickness of 5 μm, transferred to glass slides, and stained with hematoxylin and eosin. The slides were examined by a light microscope for morphologic assessments. Gut injury was scored as described previously (11). The score was based on the following criteria (score: 0–5): preservation of the villi, epithelial degeneration/necrosis, erosions or ulceration and mucin depletion. A scoring system to grade the degree of lung injury was used based on previously described (22) histological features. The injury was graded as absent, mild, moderate or severe with a score of 0 to 3 for each feature. A total score of the three criteria (that is, edema, neutrophil infiltration and intraalveolar hemorrhage and debris) was calculated for each animal.
Granulocyte Myeloperoxidase Assessment
Myeloperoxidase (MPO) activity was determined using a method described by Koike and colleagues (23). Briefly, lung tissues were removed from the euthanized animal, snap frozen in liquid nitrogen and stored at −80°C until assay. Frozen tissue samples were analyzed for MPO activity measurement as described previously by us (21). MPO activity (U/g) = A460 × 13.5/g, where A460 = rate of change in absorbance between 1 and 3 min.
Determination of Blood and Pulmonary Levels of TNF-α and IL-6
Blood and lung homogenates were measured for TNF-α and interleukin (IL)-6 levels using an enzyme-linked immunosorbent assay (ELISA) kit specifically for rat TNF-α or IL-6 (BD Biosciences, San Jose, CA, USA). The assay was carried out according to manufacturer’s instructions. TNF-α and IL-6 levels were normalized to the protein concentration in the sample.
Organ Injury Variables
Serum samples were analyzed for alanine aminotransferase (ALT), aspartate aminotransferase (AST) and lactate dehydrogenase (LDH). Levels were determined using commercial assay kits according to manufacturer’s specifications (Pointe Scientific, Canton, MI, USA).
Measurement of Bacterial Translocation
Mesenteric lymph nodes (MLN) were aseptically removed according to Berg and Garlington (1979) (24), then weighed and homogenized in sterile water. Aliquots of homogenates (1 mL) were plated onto soy agar with 5% sheep blood (Tripticase [TSAII]; BD) for bacterial counts. After 48 h of aerobic incubation at 37°C, the total number of colonies was identified. The bacterial count was expressed as colony-forming units (cfu) per 100 mg of tissue.
Statistical Analysis
All data are expressed as mean ± SE and compared with one-way ANOVA and Student-Newman-Keuls (SNK) test. Differences in values were considered significant if p < 0.05.
RESULTS
AICAR Improved Intestinal Integrity and Reduced Inflammation after Intestinal I/R
In histological sections, severe mucosal damage with denudation of villi and collapse of small vessels were observed in the intestinal tissue after I/R (Figure 1B) as compared with sham group (Figure 1A). The structural appearance and the height of the villi were preserved with AICAR treatment after intestinal I/R (Figure 1C). A significant decrease of 46% in the intestinal injury score also was observed with AICAR treatment (Figure 1D). Intestinal I/R-associated gut edema also was significantly reduced with AICAR treatment (Figure 1E). Intestinal I/R significantly elevated serum markers of tissue damage including ALT (388% increase), AST (281% increase), LDH (238% increase) and lactate (177% increase) compared with sham-operated animals (Figures 2A–D). Treatment with AICAR decreased ALT, AST, LDH and lactate levels by 28%, 44%, 41% and 33%, respectively (see Figures 2A–D). We also investigated whether the serum levels of the cytokines TNF-α and IL-6 were affected by the treatment with AICAR. Intestinal I/R significantly increased serum levels of TNF-α by 595% and IL-6 by 278%, respectively (Figures 3A, B). AICAR administration dramatically reduced the proinflammatory response by 48% and 69%, respectively (see Figures 3A, B). Thus, treatment with AICAR immediate to reperfusion significantly attenuated I/R-induced multiple organ injury.
Figure 1.

Alterations in histological appearances of the gut after intestinal I/R. Representative gut histology (hematoxylin–eosin staining; original magnification 200×) of (A) sham, (B) vehicle, and (C) AICAR treatment in mice at 4 h after intestinal I/R. (D) The gut damage score and (E) the water content of the gut tissue also are shown. Data are expressed as mean ± SE and compared by one-way ANOVA and SNK test. *P < 0.05 versus sham and #P < 0.05 versus vehicle. Scale bar, 100 μm.
Figure 2.

Alterations in serum levels of organ injury indicators. (A) ALT, (B) AST, (C) LDH, (D) lactate in sham-, vehicle- and AICAR-treated mice at 4 h after intestinal I/R. Data are expressed as mean ± SE and compared by one-way ANOVA and SNK test. *P < 0.05 versus sham and #P < 0.05 versus vehicle.
Figure 3.
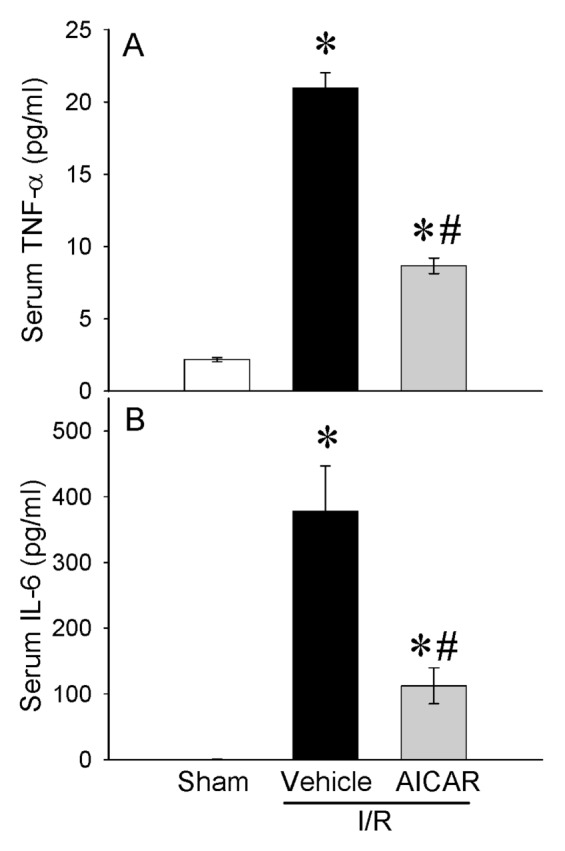
Alterations in serum levels of cytokines. (A) TNF-α and (B) IL-6 in sham-, vehicle- and AICAR-treated mice at 4 h after intestinal I/R. Data are expressed as mean ± SE and compared by one-way ANOVA and SNK test. *P < 0.05 versus sham and #P < 0.05 versus vehicle.
AICAR Reduced Bacterial Translocation into Lymph Nodes after Intestinal I/R
The intestinal barrier function becomes compromised after I/R injury. Bacterial translocation to the mesenteric lymph node (MLN) was minimal in sham (1.6 ± 0.4 colonies/100 mg lymph nodes) and extensive in MLN of rats in the vehicle group subjected to intestinal I/R (104.6 ± 2.3 colonies/100 mg lymph nodes) (Figure 4). Treatment with AICAR at the beginning of reperfusion significantly reduced the bacterial trans location from the gut to the MLN compared with vehicle-treated animals (44.67 ± 4.5 colonies/100 mg lymph nodes) (see Figure 4).
Figure 4.
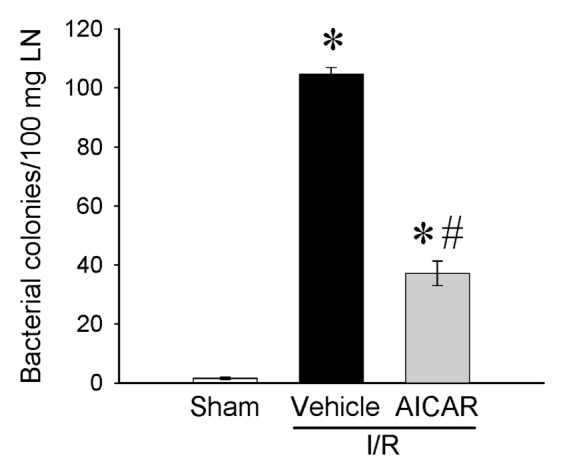
Alterations in bacterial translocation to MLN. MLN homogenates from sham-, vehicle- and AICAR-treated mice at 4 h after intestinal I/R were plated on soy agar plates, incubated at 37°C for 48 h, and the bacterial colonies counted. The number is shown as colony-forming units (cfu) per 100 mg tissue. Data are expressed as mean ± SE and compared by one-way ANOVA and SNK test. *P < 0.05 versus sham and #P < 0.05 versus vehicle.
AICAR Attenuated Lung Injury and Inflammation after Intestinal I/R
Intestinal I/R injury compromises multiple systems where the lungs among other organs are commonly affected (25). In this study, we analyzed lung injury using three parameters: histologic changes, neutrophil infiltration (MPO activity) and lung edema. While lungs of sham-operated animals had a normal microscopic appearance (Figure 5A), the lung specimens from intestinal I/R vehicle group had substantial changes, including alveolar congestion/collapse, hemorrhage and inflammatory cell infiltration (Figure 5B). Histological appearance of the lungs from AICAR-treated animals was similar to that of sham animals (Figure 5C). Lung injury score was increased significantly at 4 h after intestinal I/R and vehicle treatment. Administration of AICAR after gut ischemia significantly reduced the lung injury score by 72% (Figure 5D). In addition, rats subjected to intestinal I/R had a significant increase in lung water content. Lung water content from AICAR-treated animals was reduced significantly with no statistical difference from the sham-operated rats (Figure 5E).
Figure 5.

Alterations in lung morphology after intestinal I/R. Representative lung histology (hematoxylin–eosin staining; original magnification 200×) of (A) sham-, (B) vehicle-, (C) AICAR-treated mice at 4 h after intestinal I/R. The (D) lung damage score calculated as described in Materials and Methods and (E) lung water content are shown. Data are expressed as mean ± SE and compared by one-way ANOVA and SNK test. *P < 0.05 versus sham and #P < 0.05 versus vehicle. Scale bar, 100 μm.
As an indicator of neutrophil infiltration into the lungs caused by intestinal I/R, lung CXCL2 mRNA expression and lung MPO activity were assessed. The lung CXCL2 mRNA was increased by 188% after intestinal I/R and treatment with AICAR decreased it by 82% (Figure 6A). The lung MPO activity showed a significant increase of 241% in the vehicle group after intestinal I/R as compared with sham-operated animals and AICAR treatment decreased it by 53% (Figure 6B). To examine whether AICAR changes tissue cytokine production, we analyzed the cytokine protein levels in the lungs. Similar suppressive effects of AICAR could be found on the lung cytokine levels where TNF-α and IL-6 increased by 254% and 242% respectively at 4 h after intestinal I/R with vehicle treatment, while AICAR treatment decreased tissue TNF-α and IL-6 levels by 67% and 68% respectively (Figure 7), which is similar to those in sham-operated animals. As potential mechanisms of lung inflammation caused by intestinal I/R, (iNOS) and COX-2 protein levels were assessed. The lung iNOS protein was increased significantly by 300% after intestinal I/R with vehicle treatment and the treatment with AICAR reduced it by 70% (Figure 8A). Likewise, the lung COX-2 protein was increased significantly by 211% after intestinal I/R and AICAR treatment reduced it by 72% (Figure 8B).
Figure 6.
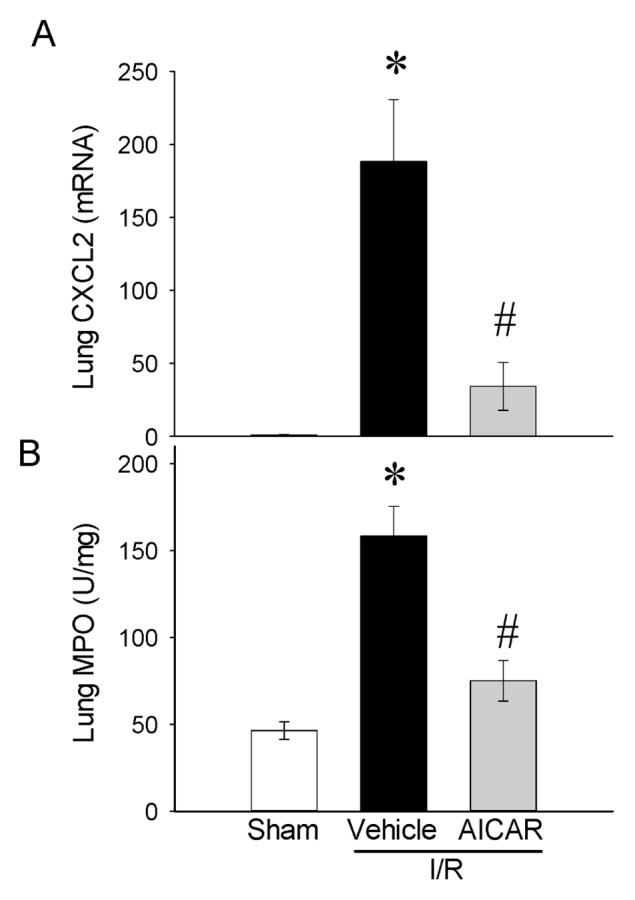
Alterations in lung neutrophil chemotaxis and infiltration. (A) CXCL2 mRNA expression and (B) MPO in sham-, vehicle- and AICAR-treated mice at 4 h after intestinal I/R. Data are expressed as mean ± SE and compared by one-way ANOVA and SNK test. *P < 0.05 versus sham and #P < 0.05 versus vehicle.
Figure 7.
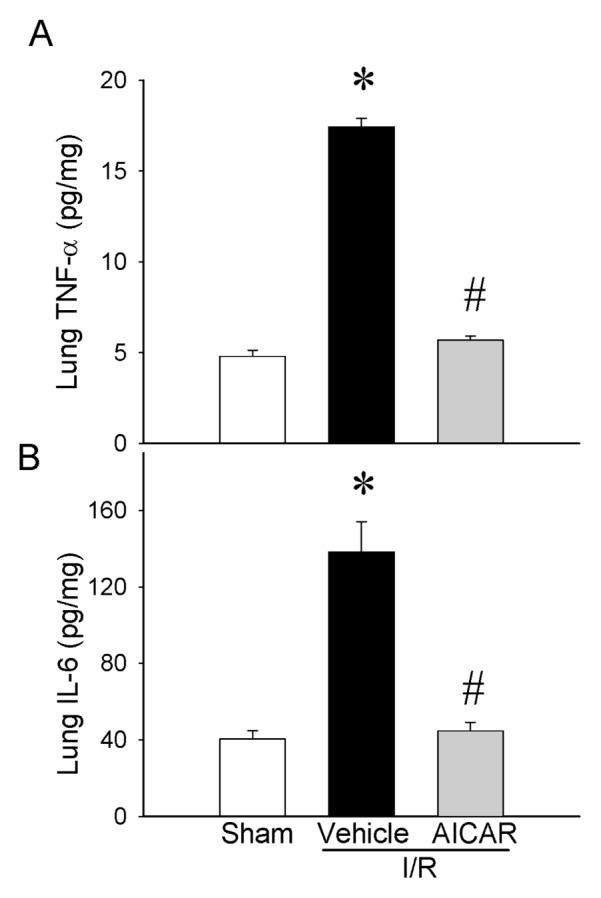
Alterations in lung cytokines. (A) TNF-α, (B) IL-6 in sham-, vehicle- and AICAR-treated mice at 4 h after intestinal I/R. Data are expressed as mean ± SE and compared by one-way ANOVA and SNK test. *P < 0.05 versus sham and #P < 0.05 versus vehicle.
Figure 8.
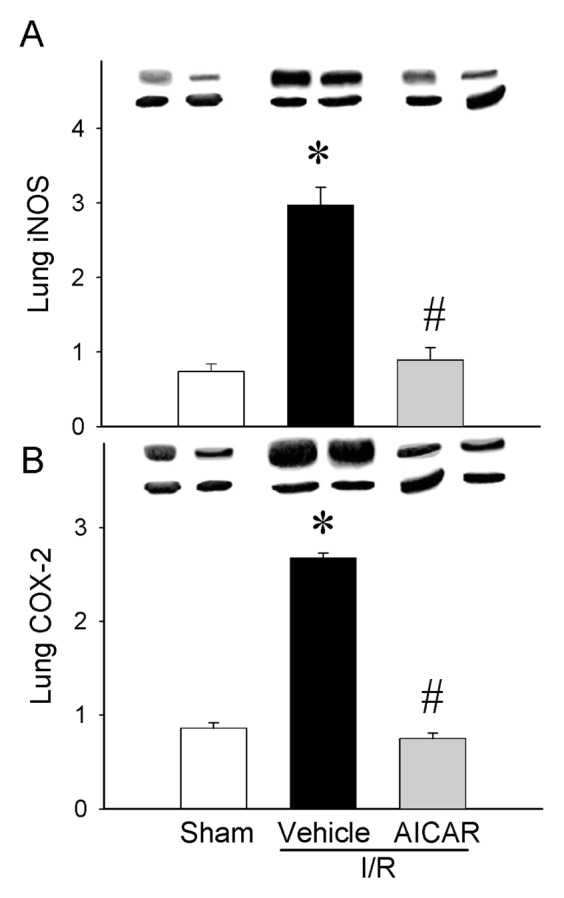
Alterations in lung inflammatory mediators. (A) iNOS, (B) COX-2 in sham-, vehicle- and AICAR-treated mice at 4 h after intestinal I/R. Data are expressed as mean ± SE and compared by one-way ANOVA and SNK test. *P < 0.05 versus sham and #P < 0.05 versus vehicle.
AICAR Attenuated Apoptosis in the Lung and in the Intestine after Intestinal I/R
To determine the effects of AICAR on apoptosis after intestinal I/R, the lungs and the intestine were examined in the sham, vehicle and treatment groups by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining (Figures 9A–C). In the vehicle-treated group, the TUNEL-positive cells in the lungs were increased significantly (see Figure 9B) as compared with sham group (see Figure 9A). Treatment with AICAR significantly decreased it by 59% (Figures 9C, D).
Figure 9.

Apoptosis in the lung after intestinal I/R. Representative TUNEL staining of lung (A) sham, (B) vehicle, (C) AICAR-treated mice at 4 h after intestinal I/R. (D) The number of apoptotic cells in the lung quantified from TUNEL staining. Data are expressed as mean ± SE and compared by one-way ANOVA and SNK test. *P < 0.05 versus sham and #P < 0.05 versus vehicle.
DISCUSSION
I/R injury contributes to a wide range of pathological conditions. Exposure of a single organ to I/R (for example, the intestine), subsequently cause inflammatory activation in other organs (for example, the lung), eventually leading to multiorgan failure. A number of biological processes are associated with I/R-induced tissue injury. The initial tissue ischemia causes impairment in endothelial barrier function and an increase in vascular permeability and leakage. However, I/R leads to, among other things, the activation of cell death programs, including apoptosis and necrosis, and stimulation of the innate and adaptive immune responses. Therefore, targeting the immune response is an emerging therapy for the treatment of I/R injury.
In the current study, we showed that treatment with AICAR at the beginning of reperfusion improved histological integrity of the gut as evidenced by the improvement in the morphological appearance and significant decreases in the gut damage score and the gut water content. The treatment also attenuated tissue injury as indicated by decreases in injury indicators and proinflammatory cytokines and a reduction in bacterial translocation to the gut. In addition to the gut, treatment with AICAR after gut I/R maintained lung integrity, attenuated neutrophil chemotaxis and infiltration to the lungs and decreased lung levels of TNF-α and IL-6. Inflammatory mediators such as iNOS and COX-2 were downregulated in the lungs and both gut and lung apoptosis were attenuated with AICAR treatment. These data clearly demonstrated that AICAR ameliorated gut I/R-associated gut and lung injuries, reduced systemic and tissue inflammation and apoptosis.
The clinical feature of gut ischemia originates from local and systemic responses which are induced by impairment in microcirculation through the activation of the endothelial cells, monocytes, leukocytes and platelets. Damage to the microcirculation leads to intestinal necrosis and/or systemic inflammatory response in the whole body or remote organs such as the lungs. Activated immune cells in the ischemic gut produce inflammatory cytokines, TNF-α and IL-6. In this regard, AICAR treatment significantly decreased systemic levels of TNF-α and IL-6 possibly released from the ischemic gut or other remote organs. Gut I/R injury is one of the most common causes of gut barrier dysfunction (26). The gastrointestinal tract functions as a site for nutrient absorption, but it also acts as a barrier between the gut and the circulation in preventing noxious substances from entering into the circulation. Maintenance of the normal epithelial structure and function is important in preventing trans-movement of bacteria and other large molecules. Increased intestinal permeability due to gut I/R has been associated with increased risk of multiple organ failure and mortality in critically ill patients (27–29). Our study showed that AICAR improved gut histological integrity, indicating the important role it had on the maintenance of the epithelial structure and function.
The organ injury induced by I/R is not limited to the ischemic organ. Gut ischemia due to transient occlusion of the superior mesenteric artery causes tissue injury locally, and the reperfusion of the gut leads to heightened inflammatory response, thereby causing multiple organ failure, including acute lung injury. The systemic inflammatory response after intestinal I/R injury activates neutrophils, which are sequestrated in the pulmonary microcirculation with the consequent increase of endothelial and epithelial permeability, fluid and protein extravasation, leukocyte sequestration and increases in pulmonary capillary endothelial cell injury (3,30–33). Our data show that gut I/R negatively affects lung morphology and increases MPO, and lung neutrophil chemotaxis and local cytokine production. Treatment with AICAR attenuated lung injury and inflammation. These results are consistent with previous reports that AICAR is able to reduce lung injury in a model of hemorrhagic shock (34). Neutrophils play a central role in innate immune and inflammatory responses (35,36). Although neutrophils eradicate microbial infections, excessive neutrophil activation resulting in the release of cytokines and other proinflammatory mediators cause tissue injury and contributes to multiple organ failure including acute lung injury (37). Induction of neutropenia has been shown to diminish severity of pulmonary injury (38–40). Therefore, it is possible that the protective effect of AICAR in gut I/R-associated lung injury resulted from diminished release of proinflammatory mediators from neutrophils.
In the current study, we chose a single time point of 4 h after reperfusion to determine the effect of AICAR in gut I/R injury. This was based on our previous publication where we observed significant increases in organ injury parameters, that is, AST, ALT, lactate and creatinine at 4 h after gut I/R injury. In addition, we also observed significant abnormalities in the gut and the lungs as evidenced by increases in myeloperoxidase activity and cytokines levels, that is, TNF-α and IL-6 in both tissues (41). In the current study, we observed increases in inflammatory mediators such as iNOS and COX-2 protein levels and increases in TUNEL-positive cells in the lungs indicating inflammation and apoptosis as early as 4 h after gut I/R injury. Therefore, amelioration of these early effects systemically and in the gut and the lungs will be highly effective in rendering a compound as therapeutic for gut I/R injury. It is also highly likely that AICAR administered at the beginning of reperfusion will be protective from gut I/R-induced mortality but further experiments are needed for such conclusion.
Our studies also showed that injury indicators, ALT, AST and LDH, are significantly attenuated with AICAR treatment following gut I/R. It is important to recognize that increases in ALT and AST in the circulation are indicative of liver damage and that increases in LDH indicates cellular hypoxia in tissues and as a marker of common injuries. The attenuation of the injury indicators by AICAR treatment suggests that AICAR prevents gut I/R-induced liver damage and other tissue damage caused by cellular hypoxia.
Antiinflammatory effects of AICAR have been reported previously. It has been reported that AICAR treatment reduced proinflammatory cytokine production and decreased histological alterations associated with inflammation caused by experimental autoimmune encephalomyelitis and asthma (42,43). AICAR inhibited release of TNF-α and IL-6 in LPS-stimulated neutrophils (44). Moreover, administration of AICAR in vivo resulted in decreased severity of LPS-induced lung injury, suggesting that the antiinflammatory effects of AICAR is caused by downregulation of the NF-κB pathway and subsequent decreases in TNF-α and IL-6 (44). In this regard, our study showed that AICAR treatment attenuated gut I/R-induced lung levels of TNF-α and IL-6, suggesting a similar molecular mechanism of action of AICAR in gut I/R injury. Inflammatory mediators such as lung iNOS and COX-2 also were attenuated with AICAR treatment, indicating a potential role of these factors in gut I/R-induced lung injury. Impairment in the lung epithelium and endothelium barrier function plays a major role in the development of acute respiratory distress syndrome, and a key feature of this failure is the loss of cells through apoptosis (45). The modulation of apoptosis could prevent lung fibrosis, and the progression into acute lung injury (24,45–47). AICAR attenuated lung apoptosis in gut I/R-associated lung injury as indicated by the reduction of apoptotic cells in the lung parenchyma after treatment. In addition to the lung, AICAR treatment also significantly reduced gut apoptosis.
Although the precise mechanism by which AICAR exerts anti-inflammation in gut I/R injury is not yet clear, AICAR has been shown to have an antiinflammatory effect through inhibition of LPS-induced proinflammatory mediators including TNF-α, IL-1β and IL-6 (17). It has been suggested that the antiinflammatory effects of AICAR could be mediated by systemic changes. LPS stimulation elevates CD14 expression and pretreatment with AICAR significantly suppressed these elevations (48). LPS stimulation decreases AMPK activity in macrophages, and AICAR upregulates its activity and inhibits cytokine production (49). AICAR inhibits NF-κB signaling through AMPK activation, indicating a direct effect of AICAR (48). AICAR is a pharmacological activator of AMPK (50). Some reports have shown that the effects of AICAR are independent of AMPK activation (18,51–53). We have not analyzed AMPK activation in gut I/R injury. It is possible that the protective effect of AICAR in gut I/R injury is mediated by either AMPK independent or dependent action of AICAR or both. Future studies are warranted for such hypothesis.
It is not known whether AICAR acts directly on the leukocytes, endothelial cells and/or epithelial cells to facilitate protection in gut I/R injury. However, it has been reported that AMPK activation with AICAR attenuated LPS-induced endothelial hyperpermeability in vitro and that pretreatment with AICAR reduced LPS-induced lung injury and endothelial hyperpermeability by activation of the Rac/Cdc42/PAK pathway by AMPK activation (54). Furthermore, activation of AMPK attenuated endothelial cell apoptosis by increasing the expression of anti-apoptotic proteins Bcl-2 and survivin (55). In another study, upregulation of AMPK by the cystic fibrosis transmembrane conductance regulator in cystic fibrosis mitigates excessive inflammation in airway epithelial cells (56). Activation of AMPK by AICAR is involved in the regulation of creatine transporter in kidney epithelial cells (57). A recent study showed that activation of AMPK by AICAR enhanced neutrophil chemotaxis and improved phagocytosis and bacterial killing in vitro (58). Thus, there are a myriad of reports available on the molecular mechanism of AMPK activation by AICAR in different cell types, including endothelial cells, epithelial cells and neutrophils. Further studies are needed to examine the exact mechanism of AICAR in gut I/R injury.
Intestinal I/R is encountered in diverse clinical conditions and its consequences are devastating to the patient, eventually leading to death. Ischemia to a specific organ often leads to injuries in other organs and causes multiple organ failure. In this study, a transient occlusion of the superior mesenteric artery caused significant damages to the gut as well as the lung. AICAR improved gut integrity, reduced bacterial translocation and systemic levels of inflammatory cytokines, TNF-α and IL-6. Activated neutrophil migration into the lungs is a key component of the progression of acute lung injury. Intestinal I/R led to neutrophil migration into the lungs as evidenced by increases in MPO, neutrophil chemotaxis and local cytokine production. AICAR treatment attenuated lung injury and inflammation. In summary, AICAR treatment in intestinal I/R protected the ischemic organ as well as the lung. These data indicate that AICAR can be developed as a novel therapeutic for intestinal I/R and subsequent acute lung injury.
ACKNOWLEDGMENTS
This study was supported by the National Institutes of Health grants, R01 HL076179, R01 GM057468, and R01 GM053008 (to P Wang).
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Tendler DA. Acute intestinal ischemia and infarction. Semin Gastrointest Dis. 2003;14:66–76. [PubMed] [Google Scholar]
- 2.Yasuhara H. Acute mesenteric ischemia: the challenge of gastroenterology. Surg Today. 2005;35:185–95. doi: 10.1007/s00595-004-2924-0. [DOI] [PubMed] [Google Scholar]
- 3.Carden DL, Granger DN. Pathophysiology of ischaemia-reperfusion injury. J Pathol. 2000;190:255–66. doi: 10.1002/(SICI)1096-9896(200002)190:3<255::AID-PATH526>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 4.Granger DN, Korthuis RJ. Physiologic mechanisms of postischemic tissue injury. Annu Rev Physiol. 1995;57:311–32. doi: 10.1146/annurev.ph.57.030195.001523. [DOI] [PubMed] [Google Scholar]
- 5.Hassoun HT, et al. Post-injury multiple organ failure: the role of the gut. Shock. 2001;15:1–10. doi: 10.1097/00024382-200115010-00001. [DOI] [PubMed] [Google Scholar]
- 6.Kalia M, Sullivan JM. Brainstem projections of sensory and motor components of the vagus nerve in the rat. J Comp Neurol. 1982;211:248–65. doi: 10.1002/cne.902110304. [DOI] [PubMed] [Google Scholar]
- 7.Kozar RA, et al. Superior mesenteric artery occlusion models shock-induced gut ischemia-reperfusion. J Surg Res. 2004;116:145–50. doi: 10.1016/s0022-4804(03)00301-9. [DOI] [PubMed] [Google Scholar]
- 8.Mallick IH, Yang W, Winslet MC, Seifalian AM. Ischemia-reperfusion injury of the intestine and protective strategies against injury. Dig Dis Sci. 2004;49:1359–77. doi: 10.1023/b:ddas.0000042232.98927.91. [DOI] [PubMed] [Google Scholar]
- 9.Oldenburg WA, Lau LL, Rodenberg TJ, Edmonds HJ, Burger CD. Acute mesenteric ischemia: a clinical review. Arch Intern Med. 2004;164:1054–62. doi: 10.1001/archinte.164.10.1054. [DOI] [PubMed] [Google Scholar]
- 10.Pierro A, Eaton S. Intestinal ischemia reperfusion injury and multisystem organ failure. Semin Pediatr Surg. 2004;13:11–7. doi: 10.1053/j.sempedsurg.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Stallion A, et al. Ischemia/reperfusion: a clinically relevant model of intestinal injury yielding systemic inflammation. J Pediatr Surg. 2005;40:470–7. doi: 10.1016/j.jpedsurg.2004.11.045. [DOI] [PubMed] [Google Scholar]
- 12.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–37. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364:656–65. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–83. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bamboat ZM, et al. Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL-10 secretion. J Clin Invest. 2010;120:559–69. doi: 10.1172/JCI40008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swirski FK, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–6. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giri S, et al. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. J Neurosci. 2004;24:479–87. doi: 10.1523/JNEUROSCI.4288-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jhun BS, et al. 5-Aminoimidazole-4-carboxamide riboside suppresses lipopolysaccharide-induced TNF-alpha production through inhibition of phosphatidylinositol 3-kinase/Akt activation in RAW 264.7 murine macrophages. Biochem Biophys Res Commun. 2004;318:372–80. doi: 10.1016/j.bbrc.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 19.Bai A, et al. Novel anti-inflammatory action of 5-aminoimidazole-4-carboxamide ribonucleoside with protective effect in dextran sulfate sodium-induced acute and chronic colitis. J Pharmacol Exp Ther. 2010;333:717–25. doi: 10.1124/jpet.109.164954. [DOI] [PubMed] [Google Scholar]
- 20.Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Institute for Laboratory Animal Research, Division on Earth and Life Studies, National Research Council of the National Academies. Guide for the Care and Use of Laboratory Animals. 8th edition. Washington (DC): National Academies Press; 2011. [Google Scholar]
- 21.Dwivedi AJ, et al. Adrenomedullin and adrenomedullin binding protein-1 prevent acute lung injury after gut ischemia-reperfusion. J Am Coll Surg. 2007;205:284–93. doi: 10.1016/j.jamcollsurg.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 22.Bachofen M, Weibel ER. Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med. 1982;3:35–56. [PubMed] [Google Scholar]
- 23.Koike K, et al. Gut ischemia/reperfusion produces lung injury independent of endotoxin. Crit Care Med. 1994;22:1438–44. doi: 10.1097/00003246-199409000-00014. [DOI] [PubMed] [Google Scholar]
- 24.Berg RD, Garlington AW. Translocation of certain indigenous bacteria from the gastrointestinal tract to the mesenteric lymph nodes and other organs in a gnotobiotic mouse model. Infect Immun. 1979;23:403–11. doi: 10.1128/iai.23.2.403-411.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–49. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 26.Swank GM, Deitch EA. Role of the gut in multiple organ failure: bacterial translocation and permeability changes. World J Surg. 1996;20:411–7. doi: 10.1007/s002689900065. [DOI] [PubMed] [Google Scholar]
- 27.Ammori BJ, et al. Early increase in intestinal permeability in patients with severe acute pancreatitis: correlation with endotoxemia, organ failure, and mortality. J Gastrointest Surg. 1999;3:252–62. doi: 10.1016/s1091-255x(99)80067-5. [DOI] [PubMed] [Google Scholar]
- 28.Faries PL, Simon RJ, Martella AT, Lee MJ, Machiedo GW. Intestinal permeability correlates with severity of injury in trauma patients. J Trauma. 1998;44:1031–5. doi: 10.1097/00005373-199806000-00016. discussion 1035–6. [DOI] [PubMed] [Google Scholar]
- 29.Doig CJ, et al. Increased intestinal permeability is associated with the development of multiple organ dysfunction syndrome in critically ill ICU patients. Am J Respir Crit Care Med. 1998;158:444–51. doi: 10.1164/ajrccm.158.2.9710092. [DOI] [PubMed] [Google Scholar]
- 30.Simpson R, et al. Neutrophil and nonneutrophil-mediated injury in intestinal ischemia-reperfusion. Ann Surg. 1993;218:444–53. doi: 10.1097/00000658-199310000-00005. discussion 453–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerkin TM, et al. Pulmonary endothelial cell ATP depletion following intestinal ischemia. J Surg Res. 1992;52:642–7. doi: 10.1016/0022-4804(92)90143-n. [DOI] [PubMed] [Google Scholar]
- 32.Schmeling DJ, Caty MG, Oldham KT, Guice KS, Hinshaw DB. Evidence for neutrophil-related acute lung injury after intestinal ischemia-reperfusion. Surgery. 1989;106:195–201. discussion 201–2. [PubMed] [Google Scholar]
- 33.Welbourn CR, et al. Pathophysiology of ischaemia reperfusion injury: central role of the neutrophil. Br J Surg. 1991;78:651–5. doi: 10.1002/bjs.1800780607. [DOI] [PubMed] [Google Scholar]
- 34.Davis KA, et al. Combination therapy that targets secondary pulmonary changes after abdominal trauma. Shock. 2001;15:479–84. doi: 10.1097/00024382-200115060-00012. [DOI] [PubMed] [Google Scholar]
- 35.Strassheim D, et al. Phosphoinositide 3-kinase and Akt occupy central roles in inflammatory responses of Toll-like receptor 2-stimulated neutrophils. J Immunol. 2004;172:5727–33. doi: 10.4049/jimmunol.172.9.5727. [DOI] [PubMed] [Google Scholar]
- 36.Asehnoune K, Strassheim D, Mitra S, Kim JY, Abraham E. Involvement of reactive oxygen species in Toll-like receptor 4-dependent activation of NF-kappa B. J Immunol. 2004;172:2522–9. doi: 10.4049/jimmunol.172.4.2522. [DOI] [PubMed] [Google Scholar]
- 37.Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003;31:S195–9. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- 38.Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1137–45. doi: 10.1152/ajplung.2000.279.6.L1137. [DOI] [PubMed] [Google Scholar]
- 39.Hollingsworth JW, et al. The critical role of hematopoietic cells in lipopolysaccharide-induced airway inflammation. Am J Respir Crit Care Med. 2005;171:806–13. doi: 10.1164/rccm.200407-953OC. [DOI] [PubMed] [Google Scholar]
- 40.Savov JD, Gavett SH, Brass DM, Costa DL, Schwartz DA. Neutrophils play a critical role in development of LPS-induced airway disease. Am J Physiol Lung Cell Mol Physiol. 2002;283:L952–62. doi: 10.1152/ajplung.00420.2001. [DOI] [PubMed] [Google Scholar]
- 41.Zhang F, Wu R, Zhou M, Blau SA, Wang P. Human adrenomedullin combined with human adrenomedullin binding protein-1 is protective in gut ischemia and reperfusion injury in the rat. Regul Pept. 2009;152:82–7. doi: 10.1016/j.regpep.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim TB, et al. Five-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside attenuates poly (I:C)-induced airway inflammation in a murine model of asthma. Clin Exp Allergy. 2007;37:1709–19. doi: 10.1111/j.1365-2222.2007.02812.x. [DOI] [PubMed] [Google Scholar]
- 43.Nath N, et al. 5-aminoimidazole-4-carboxamide ribonucleoside: a novel immunomodulator with therapeutic efficacy in experimental autoimmune encephalomyelitis. J Immunol. 2005;175:566–74. doi: 10.4049/jimmunol.175.1.566. [DOI] [PubMed] [Google Scholar]
- 44.Zhao X, et al. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:L497–504. doi: 10.1152/ajplung.90210.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matthay MA, et al. Future research directions in acute lung injury: summary of a National Heart, Lung, and Blood Institute working group. Am J Respir Crit Care Med. 2003;167:1027–35. doi: 10.1164/rccm.200208-966WS. [DOI] [PubMed] [Google Scholar]
- 46.Perl M, et al. Fas-induced pulmonary apoptosis and inflammation during indirect acute lung injury. Am J Respir Crit Care Med. 2007;176:591–601. doi: 10.1164/rccm.200611-1743OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suzuki J, et al. Inhibitory effect of aminoimidazole carboxamide ribonucleotide (AICAR) on endotoxin-induced uveitis in rats. Invest Ophthalmol Vis Sci. 2011;52:6565–71. doi: 10.1167/iovs.11-7331. [DOI] [PubMed] [Google Scholar]
- 49.Sag D, Carling D, Stout RD, Suttles J. Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008;181:8633–41. doi: 10.4049/jimmunol.181.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sullivan JE, et al. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 1994;353:33–6. doi: 10.1016/0014-5793(94)01006-4. [DOI] [PubMed] [Google Scholar]
- 51.Kuo CL, Ho FM, Chang MY, Prakash E, Lin WW. Inhibition of lipopolysaccharide-induced inducible nitric oxide synthase and cyclooxygenase-2 gene expression by 5-aminoimidazole-4-carboxamide riboside is independent of AMP-activated protein kinase. J Cell Biochem. 2008;103:931–40. doi: 10.1002/jcb.21466. [DOI] [PubMed] [Google Scholar]
- 52.Labuzek K, Liber S, Gabryel B, Okopien B. AICAR (5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside) increases the production of toxic molecules and affects the profile of cytokines release in LPS-stimulated rat primary microglial cultures. Neurotoxicology. 2010;31:134–46. doi: 10.1016/j.neuro.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 53.Qin S, Ni M, De Vries GW. Implication of S-adenosylhomocysteine hydrolase in inhibition of TNF-alpha- and IL-1beta-induced expression of inflammatory mediators by AICAR in RPE cells. Invest Ophthalmol Vis Sci. 2008;49:1274–81. doi: 10.1167/iovs.07-1109. [DOI] [PubMed] [Google Scholar]
- 54.Xing J, et al. Inhibition of AMP-activated protein kinase accentuates lipopolysaccharide-induced lung endothelial barrier dysfunction and lung injury in vivo. Am J Pathol. 2013;182:1021–30. doi: 10.1016/j.ajpath.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu C, Liang B, Wang Q, Wu J, Zou MH. Activation of AMP-activated protein kinase alpha1 alleviates endothelial cell apoptosis by increasing the expression of anti-apoptotic proteins Bcl-2 and survivin. J Biol Chem. 2010;285:15346–55. doi: 10.1074/jbc.M110.102491. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Hallows KR, et al. Up-regulation of AMP-activated kinase by dysfunctional cystic fibrosis transmembrane conductance regulator in cystic fibrosis airway epithelial cells mitigates excessive inflammation. J Biol Chem. 2006;281:4231–41. doi: 10.1074/jbc.M511029200. [DOI] [PubMed] [Google Scholar]
- 57.Li H, et al. Regulation of the creatine transporter by AMP-activated protein kinase in kidney epithelial cells. Am J Physiol Renal Physiol. 2010;299:F167–77. doi: 10.1152/ajprenal.00162.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Park DW, et al. Activation of AMPK enhances neutrophil chemotaxis and bacterial killing. Mol Med. 2013;19:387–98. doi: 10.2119/molmed.2013.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]