

Abstract
Intramyocellular accumulation of lipids is often associated with insulin resistance. Deficiency of comparative gene identification-58 (CGI-58) causes cytosolic deposition of triglyceride (TG)-rich lipid droplets in most cell types, including muscle due to defective TG hydrolysis. It was unclear, however, whether CGI-58 deficiency-induced lipid accumulation in muscle influences insulin sensitivity. Here we show that muscle-specific CGI-58 knockout mice relative to their controls have increased glucose tolerance and insulin sensitivity on a Western-type high-fat diet, despite TG accumulation in both heart and oxidative skeletal muscle and cholesterol deposition in heart. Although the intracardiomyocellular lipid deposition results in cardiac ventricular fibrosis and systolic dysfunction, muscle-specific CGI-58 knockout mice show increased glucose uptake in heart and soleus muscle, improved insulin signaling in insulin-sensitive tissues, and reduced plasma concentrations of glucose, insulin, and cholesterol. Hepatic contents of TG and cholesterol are also decreased in these animals. Cardiac steatosis is attributable, at least in part, to decreases in cardiac TG hydrolase activity and peroxisome proliferator-activated receptor-α/peroxisome proliferator-activated receptor-γ coactivator-1-dependent mitochondrial fatty acid oxidation. In conclusion, muscle CGI-58 deficiency causes cardiac dysfunction and fat deposition in oxidative muscles but induces a series of favorable metabolic changes in mice fed a high-fat diet.
In mammals, excess energy is stored in cytosolic lipid droplets (LDs) as triglycerides (TGs) or in the form of glycogen. During the period of starvation or increased energy demand, this stored energy is mobilized for use. The stored fat is mobilized as free fatty acids (FFAs) and glycerol via a lipolytic process. There are at least 2 important pathways involved in cellular fat lipolysis. One is lipophagy, which delivers some cytosolic LDs to lysosomes for digestion by acidic lipases (1). The other pathway is catalyzed by cytosolic neutral lipases. Adipose TG lipase (ATGL) was identified as a cytosolic neutral lipolytic enzyme that cleaves a fatty acyl chain from a TG molecule stored in LDs, therefore playing a critical role in cellular fat lipolysis (2–4).
Comparative gene identification-58 (CGI-58), also known as α/β-hydrolase domain-containing 5, is the fifth member of α/β-hydrolase fold family (5, 6). It lacks a critical serine residue in the canonical lipase catalytic domain and, thus, possesses no intrinsic lipase activity (6, 7). The protein is ubiquitously expressed in mammals (7–9). In 2004, 3 laboratories independently reported that CGI-58 is a LD-associated protein (8, 10, 11). CGI-58 interacts with several LD-coat proteins and ATGL (7, 8, 11–13). It has been shown that CGI-58 functions as a coactivator of ATGL to robustly increase ATGL's in vitro TG hydrolase activity (7). Recently, it was reported that CGI-58 has lysophosphatidylglycerol acyltransferase activity (14), although the in vivo significance of this interesting in vitro finding has yet to be elucidated. Using cultured hepatoma cells and antisense oligonucleotide-mediated knockdown approach, we have shown that CGI-58 limits cellular TG contents by facilitating hydrolysis, but not by reducing synthesis (9, 15).
Although CGI-58 may function via activating ATGL, in humans CGI-58 mutations cause a neutral lipid storage disease with ichthyosis (also known as Chanarin-Dorfman syndrome [CDS]) (5, 16–19), whereas ATGL mutations cause a neutral lipid storage disease with mild myopathy but without ichthyosis (20). CGI-58-null mice die within 16 hours after birth due to a skin barrier defect, whereas ATGL-deficient mice are viable (21). Deletion of CGI-58 and ATGL in liver or macrophages in mice also causes distinct phenotypes (22–25). Thus CGI-58 must have functions beyond activating ATGL.
Patients with CDS exhibit neutral lipid accumulation in skeletal muscle (26). Recently, Haemmerle laboratory has reported that muscle-specific deletion of CGI-58 causes intramyocellular TG accumulation and cardiac dysfunction in mice on a regular chow diet (27). Chow diets are enriched of fiber and low in fat and cholesterol, which differ from Western-type diets that drive metabolic disorders in humans. Despite this, Haemmerle laboratory was able to show that muscle CGI-58 deficiency reduces blood levels of glucose and insulin in the refed state, increases glucose tolerance and glucose uptake in cardiac muscle, and raises respiratory quotient values (indicative of increased glucose oxidation) in mice on a chow diet (27). These previous findings are interesting because they imply that mice lacking CGI-58 in muscle may have increased insulin sensitivity despite intramyocellular lipid accumulation that often impairs insulin signaling and causes insulin resistance (28). In this study, we generated our own muscle-specific CGI-58 knockout (mCgi58-KO) mice and challenged them with a Western-type high-fat diet (HFD), because mice on a chow diet do not normally develop glucose intolerance and insulin resistance. We found that despite severe lipid accumulation in both soleus and cardiac muscle, mCgi58-KO mice relative to their control counterparts on HFD display many favorable metabolic changes, including increased glucose tolerance and insulin sensitivity, reduced hepatic TG, and decreased plasma concentrations of glucose and insulin. In addition, our study for the first time uncovers an important role of muscle CGI-58 in regulating hepatic and plasma cholesterol levels.
Materials and Methods
Mice and diet
The creation of CGI-58-floxed mice was described in our previous publication (22). To inactivate CGI-58 expression specifically in muscle, CGI-58-floxed mice with more than 75% C57BL/6 background were crossed to muscle creatine kinase-Cre transgenic mice of C57BL/6 background (B6.FVB(129S4)-Tg(Ckmm-cre)5Khan/J, Backcrossed to C57BL/6J for >10 generations, stock number 006475; The Jackson Laboratory) that specifically express Cre recombinase in both skeletal and cardiac muscles. Given that wild-type littermates, muscle creatine kinase-Cre transgenic mice, and homozygous CGI-58-floxed mice express similar amounts of CGI-58 (data not shown), in this study, homozygous CGI-58-floxed mice without Cre (CGI-58f/f or control) were crossed to homozygous CGI-58-floxed mice expressing Cre transgene in one allele (CGI-58f/f/cre or mCgi58-KO) to generate littermate mice for all experiments.
All mice were housed in a specific pathogen-free animal facility in plastic cages at 22°C, with a daylight cycle from 6 am to 6 pm. The mice were provided with water and standard chow diet (Prolab RMH 3000; LabDiet) ad libitum, unless stated otherwise. All animal procedures were approved by the Institutional Animal Care and Use Committee at Wake Forest University Health Sciences and at the University of Maryland. At 6 weeks of age, male mice were fed a synthetic Western-type HFD containing 45% energy from 20.68% (wt/wt) lard (16:0, 23.3%; 18:0, 15.9%; 18:1, 34.8%; and 18:2, 18.7%) and 0.2% (wt/wt) cholesterol (29). Mice were fed HFD for 12 weeks and then killed for sample collection.
Antibodies
A mouse monoclonal antibody against human CGI-58 (ABHD5) (also against mouse CGI-58) was purchased from Novus Biologicals (HL00051099-M01). A rabbit polyclonal antibody against human ATGL amino acids 328–400 was obtained from Cayman Chemical (1006409) and this antibody cross-reacts with human, mouse, and rat ATGL. Rabbit monoclonal antibodies against total Akt (C67E7; 4691), phosphorylated Akt at Ser473 (D9E; 4060), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (14C10; 2118) were purchased from Cell Signaling Technology.
Measurement of TG hydrolase activity in tissues
TG hydrolase activity was measured in the whole tissue homogenate using freshly isolated LDs containing radiolabeled TG as the substrate as we have described previously (15).
Blood biochemistries
Necropsy blood samples (n = 8–12) were collected from mice after a 4-hour fast during the light cycle. Fasted and fed blood samples were collected by submandibular vein puncture at 9 am. Fed blood samples were collected from the mice fed the HFD for 3 weeks. Fasted blood samples were collected from the mice fed the HFD for 4 weeks and fasted for 12 hours (9 pm to 9 am). Plasma concentrations of total cholesterol, free cholesterol, TG, and phospholipids were analyzed by enzymatic assays as we have described previously (15). Blood glucose levels were measured directly by Glucometer (Bayer Contour) after tail nicking. Plasma β-hydroxybutyrate (Stanbio Laboratory), and FFA (NEFA-HR; Wako Diagnostics) were measured using commercially available enzymatic reagents.
Tissue gene expression
Total tissue RNAs were extracted using TRIzol reagents and tissue mRNA levels were determined by quantitative real-time PCR as described previously (30). Primer sequences of genes used for quantitative real-time PCR are available upon request.
Tissue lipid analysis, glucose tolerance test (GTT), insulin tolerance test (ITT), tissue insulin signaling assessment, and tissue FA oxidation assay
All of these procedures were conducted exactly as we have described previously (15, 31).
Determination of tissue glucose uptake
The tissue glucose uptake was determined using 2-[3H]deoxyglucose (DG) under the condition for GTT. DG is transported into tissues and phosphorylated to 2-deoxyglucose 6-phosphate (DGP). 2-[3H] DG (12 μCi/mouse) was injected ip to overnight-fasted mice. After 40 minutes, mice were killed and tissues excised and rinsed in ice-cold PBS containing 1mM EDTA. Tissue samples were homogenized in 0.5% perchloric acid and centrifuged at 3000g for 10 minutes at room temperature. The supernatant was neutralized with 50% KOH and an aliquot of the neutralized supernatant was counted to yield total tissue counts (DG and DGP). A second aliquot was treated with Ba(OH)2 and ZnSO4 to remove DGP and counted to yield 2-[3H] DG counts. Specific 2-[3H] DGP accumulation in each tissue was calculated by subtracting the 2-[3H] DG count from the total tissue count. The protein pellet was solubilized in a buffer containing 0.3N NaOH and 0.1% sodium dodecyl sulfate for measuring tissue protein concentrations.
Mouse echocardiography
Left ventricular (LV) structure and systolic function were determined using a commercially available echocardiograph equipped with a 12-MHz phased array probe (Philips 5500; Philips Medical Systems) by the same investigator (L.G.), who was blinded to the experimental groups as described previously (32).
Statistical analysis
All data are presented as mean ± SEM. The differences between the mean values of CGI-58-floxed control mice and mCgi58-KO mice were tested for statistical significance by the 2-tailed Student's t test. P < .05 was considered to be statistically significant.
Results
Muscle-specific inactivation of CGI-58 causes fatty degeneration in cardiac and soleus muscles
To determine whether CGI-58 was specifically deleted in the muscle of our mice, we did Western blotting with tissue homogenates. As expected, CGI-58 protein levels were decreased only in the cardiac and skeletal muscles (Figure 1A) but not in other tissues examined (Supplemental Figure 1A). The residual CGI-58 protein in muscle likely results from other cell types present in this tissue.
Figure 1.

Muscle-specific inactivation of CGI-58 causes cardiac and soleus steatosis. A, Muscle-specific deletion of CGI-58. Western blotting was performed using homogenates of tissues from adult mCgi58-KO (KO) mice and their littermates (Con). B, The gross appearance of hearts at necropsy showing the normal heart from control mice and the fatty and enlarged heart from mCgi58-KO mice. C, Increases in percentages of heart-to-body weight ratios in mCgi58-KO mice (n = 4–6) (**, P < .0001). D and E, H&E and Oil red-O staining showing LD and neutral lipid deposition in the LV (D) and soleus (E) muscles of mCgi58-KO mice. F, Representative electron micrographs of heart taken at ×2900. G, Representative electron micrographs of heart taken at ×9300, showing cardiac mitochondria and LDs.
mCgi58-KO mice appeared normal grossly. They had similar body weight at birth and gained similar weight over time (Supplemental Figure 1B). At 6 weeks of age, mice were challenged with the HFD. During the 10 weeks of HFD feeding, mCgi58-KO mice and their control littermates gained weight similarly. When body composition was measured in mice fed the HFD for 6 weeks using EchoMRI, no differences were observed in both lean mass and fat mass between the 2 genotypes (Supplemental Figure 1C). Around 8 weeks on HFD, mCgi58-KO mice stopped to gain weight, which may be attributable to the development of heart dysfunction as shown below. mCgi58-KO mice started to die around 13 weeks on HFD, and none of the control mice died at this point. The food intake was not different between mCgi58-KO and control mice fed the HFD for 8 weeks (data not shown).
When mice on HFD for 12 weeks were examined, the hearts of mCgi58-KO mice appeared pale and yellowish and had a substantial increase in size (Figure 1B). There were increases in heart weight (∼2-fold) and the heart-to-body weight ratio (∼2.5-fold) in mCgi58-KO vs control mice (Figure 1C). Hematoxylin and eosin (H&E) staining of heart sections from mCgi58-KO mice displayed accumulation of LDs of varying sizes in cardiac muscle cells (Figure 1D). Oil red-O staining revealed severe neutral lipid deposition in the cardiac muscle (Figure 1D). Histological examinations of mice on the standard chow diet revealed an age-dependent increase of LDs in number and size in cardiomyocytes, and the cardiac microvesicular fatty degeneration was observed even in 3-week-old mCgi58-KO mice (data not shown).
H&E staining of skeletal muscle did not show any differences between mCgi58-KO and control mice fed the HFD for 12 weeks (Figure 1E). However, Oil red-O staining showed that mCgi58-KO mice accumulated neutral lipids-containing LDs in the oxidative slow twitch soleus muscle (Figure 1E), but not in the quadriceps muscle (data not shown) that mainly consists of glycolytic fast-twitch muscle fibers. Some muscle fibers in the gastrocnemius muscle showed Oil red-O-stained LDs, whereas most the gastrocnemius muscle fibers exhibited no LD staining (data not shown), which is consistent with the gastrocnemius muscle being composed of mixed muscle fibers.
Under electron microscope (Figure 1F), CGI-58 KOs, but not control cardiomyocytes, were filled with LDs of varying sizes. Small LDs seemed to fuse to form large LDs in these cells. Mitochondria in CGI-58-deficient cardiomyocytes appeared darker and fewer than those in the control cells. Under high magnification (×9000), mitochondria in CGI-58-deficient cardiomyocyte displayed reduced inner membrane cristae (Figure 1G). LDs and mitochondria were often adjacent to each other, suggesting a close cross talk between these 2 organelles.
Muscle-specific inactivation of CGI-58 increases cardiac and soleus but reduces hepatic contents of TG
To further determine the effect of muscle CGI-58 on intracellular lipid homeostasis, we measured tissue lipids biochemically in mice on HFD for 12 weeks. There was a 40-fold increase in cardiac TG in mCgi58-KO mice (Figure 2A). TG was also significantly increased in soleus but not quadriceps muscles. Interestingly, hepatic TG was decreased by approximately 64% in the KO mice.
Figure 2.

Tissue lipids at necropsy. mCgi58-KO and control mice (n = 4–6) on the HFD for 12 weeks were fasted for 4 hours and then killed for tissue collection. A, Tissue TG content. B, Tissue contents of total cholesterol (TC) and free cholesterol (FC). *, P < .05 and **, P < .01.
Cardiac total cholesterol was also increased, which was associated with a significant decrease in hepatic total cholesterol (Figure 2B). Similar changes were observed for cardiac and hepatic contents of free cholesterol. No alterations were found for cholesterol levels in soleus and quadriceps muscles.
TG hydrolase activity and mitochondrial FA oxidation are reduced in cardiac muscle of mCgi58-KO mice
Because CGI-58 activates the in vitro TG hydrolase activity of ATGL, CGI-58 deficiency may cause cellular TG deposition by reducing TG hydrolysis. Consistently, TG hydrolase activity of the heart homogenate was significantly decreased by 71% in the KO mice (Figure 3A). This was not a result of reduced ATGL expression because ATGL protein was increased in the CGI-58-deficient heart (Figure 3B), despite approximately 50% reduction of ATGL mRNA abundance (Figure 3C).
Figure 3.

Reduced TG hydrolase activity and fatty acid oxidation in the heart of mCgi58-KO mice. A, Cardiac TG hydrolase activity (n = 5). B, Western blottings of ATGL. GAPDH was used as a loading control. C, Relative mRNA levels in the ventricular tissue of control and mCgi58-KO mice (n = 4). D, Cardiac fatty acid oxidation activity (n = 5). E, Cardiac contents of total acyl-carnitine (AC) and AC species (n = 6). All tissue samples were collected from 4-hour-fasted mice fed the HFD for 12 weeks. *, P < .05; **, P < .01.
It was shown that inhibition of ATGL-mediated TG hydrolysis activates peroxisome proliferator-activated receptor (PPAR)α and PPARγ coactivator (PGC)-1 to sustain cardiac mitochondrial biogenesis and function (33). Cardiac levels of mRNAs for PPARα, PGC-1α, and PGC-1β were significantly reduced in mCgi58-KO mice (Figure 3C). Similar reduction was seen for PPARα target genes, including muscle carnitine palmitoyltransferase-1, long-chain acyl-CoA dehydrogenase, and 3-hydroxy-3-methylglutaryl-CoA synthase 2, all of which are involved in mitochondrial FA oxidation. Cardiac mRNA levels of genes encoding cytochrome c oxidase IV and ATP synthase, 2 enzymes critical for ATP production in the mitochondrial respiratory chain, were substantially down-regulated in the CGI-58-deficient heart. Consistently, the in vitro FA oxidation activity was decreased in the homogenate of these hearts (Figure 3D). Cardiac contents of long-chain acyl-carnitines were dramatically increased in the KO mice (Figure 3E), indicative of defective or incomplete FA β-oxidation (34).
Additionally, mCgi58-KO mice showed a significant reduction in cardiac mRNAs for lipoprotein lipase (LPL) and CD36. LPL encodes an enzyme that breaks down circulating lipoprotein-bound TG to release FFAs to cells for energy storage or as an energy source. CD36 encodes a cell membrane-bound FFA transporter. These results suggest that cardiac deposition of TG in mCgi58-KO mice was unlikely a result of increased FA uptake from the circulation.
Despite accumulation of TG and cholesterol in CGI-58-deficient heart, cardiac mRNAs for lipogenic genes sterol regulatory element binding protein-1c and acetyl-CoA carboxylase-1 were unaffected and the mRNA for mitochondrial glycerol-3-phosphate acyltransferase was decreased. Cardiac expression of a cholesterogenic gene 3-hydroxy-3-methylglutaryl-CoA synthase was also decreased. These observations suggest that cardiac lipid accumulation unlikely resulted from increased biosynthesis of TG and cholesterol.
Similar to CGI-58-deficient cardiac muscle, CGI-58-deficient soleus muscle also showed a significant reduction in TG hydrolase activity (Supplemental Figure 2A) and FA oxidation (Supplemental Figure 2B). However, CGI-58-deficient soleus displayed an inconsistent pattern of changes in mRNAs for selected genes involved in lipolysis, FA oxidation, and lipogenesis (Supplemental Figure 2D).
Muscle-specific deletion of CGI-58 improves glucose tolerance and insulin sensitivity in mice on HFD
It has been shown that muscle CGI-58 deficiency promotes glucose use in mice on chow diet (27). This finding implies that deletion of CGI-58 in muscle may protect mice from HFD-induced glucose intolerance and insulin resistance, despite causing intramyocellular lipid accumulation. Indeed, GTT and ITT showed that mCgi58-KO vs control mice on HFD had increased glucose tolerance (Figure 4A) and insulin sensitivity (Figure 4B). When tissue glucose uptake was assessed using DG, mCgi58-KO mice exhibited a significant increase of basal glucose uptake in heart and soleus (Figure 4C). Consistently, the mRNA for glucose transporter (GLUT)1, a GLUT responsible for basal glucose uptake, was significantly elevated in CGI-58-deficient heart. Meanwhile, the mRNA level of insulin-sensitive GLUT4 was down-regulated in this organ (Figure 4D). Soleus mRNAs for these GLUTs were unaltered.
Figure 4.

mCgi58-KO mice have increased glucose tolerance and insulin sensitivity. A, GTT in mCgi58-KO mice (n = 7) and control littermates (n = 6) on HFD for 8 weeks. B, ITTs in mCgi58-KO mice (n = 12) and control littermates (n = 12) on HFD for 10 weeks. C, Basal glucose uptake in heart (n = 5) and soleus (n = 3). D, Cardiac and soleus mRNA levels of GLUTs in 4-hour-fasted mice fed the HFD for 12 weeks (n = 4). E, Western blot analysis (left panel) of Akt phosphorylation at serine 473, total Akt, and GAPDH (as loading controls). Right panel, Densitometry analysis of the blots. *, P < .05 and **, P < .01.
When acute insulin signaling was examined, mCgi58-KO mice displayed a substantial increase in Akt(S473) phosphorylation in heart, soleus, liver, and epididymal fat (Figure 4E). It was noticed that these tissues from the control mice displayed blunted responses to acute insulin-induced phosphorylation of Akt(S473), which was likely a result of 10 weeks of HFD feeding. It was believed that some lipotoxic lipids such as ceramides and diacylglycerols suppress insulin signaling to induce insulin resistance in tissues with “ectopic” lipid deposition (28). We found that the cardiac content of total ceramides tended to increase with the N24:1 and N24:2 ceramide species were significantly elevated in mCgi58-KO mice (Supplemental Table 1). There were no significant differences in other ceramide species detected between the 2 genotypes. Together with improved insulin sensitivity, this result suggests that CGI-58 deficiency-induced lipid accumulation in cardiac and oxidative skeletal muscle is unable to suppress insulin signaling.
To examine how mCgi58-KO mice respond to nutritional fluctuations, we measured plasma concentrations of glucose, insulin, FFA, β-hydroxybutyrate, and TG in nonfasted mice and overnight-fasted mice. A significant reduction in plasma insulin concentrations was observed in nonfasted mCgi58-KO mice relative to controls, although no changes were seen for other plasma parameters examined (Supplemental Figure 3). When necropsy plasma samples from 4-hour-fasted mice that had been fed the HFD for 12 weeks were measured, there were significant decreases in plasma concentrations of glucose, insulin, FFA, β-hydroxybutyrate, total cholesterol, free cholesterol, and cholesterol ester in mCgi58-KO mice (Table 1). Plasma TG concentrations remained unchanged. The reduction in plasma (Table 1) and hepatic (Figure 2B) cholesterol unlikely resulted from altered whole-body cholesterol balance because we did not see any changes in intestinal cholesterol absorption and fecal excretion of neutral sterols (cholesterol and its bacterial metabolites) in mCgi58-KO mice (Supplemental Figure 4). When hepatic mRNA levels were measured using pooled samples from each group (n = 5), we found that expression levels of genes relevant to lipogenesis and cholesterologenesis were decreased (Supplemental Figure 5), implying reduced synthesis of TG and cholesterol in liver of mCgi58-KO mice.
Table 1.
Plasma Parameters in Mice
| Control | mCgi58-KO | |
|---|---|---|
| TG (mg/dL) | 25 ± 4 | 20 ± 6 |
| Total cholesterol (mg/dL) | 246 ± 14 | 172 ± 15b |
| Free cholesterol (mg/dL) | 42 ± 3 | 32 ± 3a |
| Cholesterol ester (mg/dL) | 340 ± 19 | 233 ± 20b |
| β-Hydroxybutyrate (mg/dL) | 7.25 ± 0.96 | 3.92 ± 0.24a |
| FFA (mmol/L) | 0.74 ± 0.02 | 0.63 ± 0.03a |
| Insulin (ng/mL) | 1.19 ± 0.15 | 0.59 ± 0.09a |
| Glucose (mg/dL) | 273 ± 23 | 166 ± 32a |
Shown is mean ± SEM. Mice were fed the HFD for 12 weeks. After a 4-hour fast, plasma samples were collected for assays. n = 8–12.
P < .05 (vs control mice).
P < .01 (vs control mice).
mCgi58-KO mice develop cardiac insufficiency
To determine how muscle CGI-58 deficiency-induced lipid accumulation affects cardiac functions, we performed echocardiography in living control and mCgi58-KO mice fed the HFD for 10 weeks. mCgi58-KO vs control mice exhibited severe impairment in cardiac systolic function (Figure 5A and Table 2). The 2-D-guided M-Mode echocardiography revealed significant increases in LV end systolic and end diastolic dimensions with a concomitant reduction in percent fractional shortening compared with measures obtained from control mice. Moreover, Doppler-derived measures of cardiac output in mCgi58-KO mice declined significantly compared with the control group. There were no alterations in posterior wall thickness (PWT) and peak aortic flow (maximum velocity [Vmax]).
Figure 5.
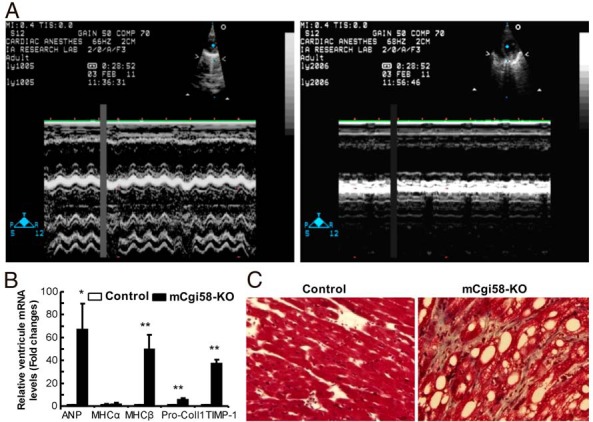
mCgi58-KO mice have cardiac dysfunction and ventricular remodeling. A, Echocardiograph (M-Mode) showing LV systolic function in mice on HFD for 10 weeks. B, Ventricular mRNA levels of genes related to cardiac function and fibrosis in mice on HFD for 12 weeks (n = 4). C, Trichrome staining of ventricular heart tissues from mice on HFD for 12 weeks. *, P < .05 and **, P < .01.
Table 2.
Echocardiography Analysis of Cardiac Function
| Control | mCgi58-KO | |
|---|---|---|
| End systolic dimension (cm) | 0.21 ± 0.02 | 0.34 ± 0.01b |
| End diastolic dimension (cm) | 0.32 ± 0.02 | 0.37 ± 0.01b |
| Fractional shortening (%) | 29.00 ± 2.01 | 7.35 ± 2.57a |
| PWT (cm) | 0.09 ± 0.004 | 0.13 ± 0.02 |
| Relative wall thickness | 0.59 ± 0.04 | 0.69 ± 0.12 |
| Ascending aorta diameter (cm) | 0.12 ± 0.96 | 0.10 ± 0.01a |
| Cross sectional area of ascending aorta (cm) | 0.012 ± 0.0003 | 0.008 ± 0.001a |
| Mean velocity of aortic flow (mmHg) | 3.05 ± 0.33 | 1.64 ± 0.10b |
| LV ejection (s) | 0.066 ± 0.000 | 0.0580 ± 0.0025b |
| Peak aortic flow (cm/s) | 71.33 ± 8.37 | 49.55 ± 5.29a |
| Stroke volume (mL) | 0.03 ± 0.01 | 0.01 ± 0.00a |
| Cardiac output (mL/min) | 13.70 ± 1.79 | 5.00 ± 1.31a |
Shown is mean ± SEM. Mice fed the HFD for 10 weeks were examined by 2-D-guided M-Mode echocardiography. Relative wall thickness = 2 × (PWT/end diastolic dimension).
P < .05 (vs control mice, n = 4).
P < .01 (vs control mice, n = 4).
To further support cardiac dysfunction in mCgi58-KO mice, we measured ventricular mRNAs for several genes sensitive to cardiac insufficiency and remodeling. Atrial natriuretic peptide (ANP) is a protein secreted by cardiomyocytes, and its increase is indicative of hypervolemia in heart chambers. There was a 67-fold increase in cardiac ANP mRNA levels in mCgi58-KO mice relative to their controls (Figure 5B), which was consistent with increased LV end systolic and end diastolic dimensions (indicative of enlarged LV chamber) (Table 2). The reduced end systolic dimension, or weakened contractility, was likely due to chronic fatty degeneration.
It is well known that, as heart failure progresses, the fast contracting myosin α-myosin heavy chain (MHC) is switched to the slow contracting myosin β-MHC (MHC). There was a 50-fold increase in cardiac β-MHC mRNA abundance in mCgi58-KO mice fed the HFD for 12 weeks, strongly supporting a failing heart in these animals. The end-stage failing heart is characterized by cardiac fibrosis as a result of cardiomyocyte loss, collagen accumulation and extracellular matrix remodeling. Consistently, cardiac mRNAs for procollagen type 1 and tissue inhibitor of metalloproteinase-1 increased 6- and 37-fold, respectively, suggesting end-stage heart dysfunction in mCgi58-KO mice. Increased cardiac fibrosis was also revealed by trichrome staining, and its pattern was both interstitial and perivascular (Figure 5C).
Discussion
It has been shown that muscle-specific deletion of CGI-58 in mice on a regular chow diet reduces blood levels of both glucose and insulin in the refed state, increases glucose tolerance and glucose uptake in cardiac muscle, and raises respiratory quotient values (27). These previous findings suggest that muscle CGI-58 deficiency promotes glucose uptake and oxidation. Increased glucose use may protect against HFD-induced glucose intolerance and insulin resistance. Indeed, in the present study we demonstrated that muscle-specific deletion of CGI-58 substantially increases glucose tolerance and insulin sensitivity in mice on HFD, despite ectopic lipid deposition in muscle, which is associated with increased glucose uptake in cardiac and soleus muscles. Additionally, our study uncovered an unidentified role of muscle CGI-58 in regulating tissue and whole-body cholesterol homeostasis.
Dissociation of tissue fat accumulation from insulin resistance was reported in other animal models (35–38) and human subjects (15, 39–41). Multiple mechanisms are implicated in the pathogenesis of insulin resistance due to ectopic fat deposition, including accumulation of insulin signaling-suppressing lipids such as diacylglycerols and ceramides (28). However, cardiac ceramide levels tend to be higher in mCgi58-KO mice than the controls. Although we have not determined muscle levels of diacylglycerols in mCgi58-KO mice, we have previously shown that antisense oligonucleotide-mediated knockdown of CGI-58 in multiple tissues (mainly liver and fat) of adult mice protects against diet-induced insulin resistance and glucose intolerance, despite hepatic steatosis and accumulation of diacylglycerols, ceramides and long-chain fatty acyl-CoAs (15). It was subsequently shown that this is due to sequestration of diacylglycerols and ceramides in cytosolic LDs and endoplasmic reticulum (42). Additionally, it was reported that oleic acid-derived TG protects cultured cells against FA-induced lipotoxicity and that the metabolic fate of a FA molecule may be critical in determining their effects on insulin signaling (43). Likewise, the molecular species relative to the total amount of each type of lipids (diacylglycerols, TG, ceramides, etc) may be more important in modulating insulin signaling.
It has been shown that TG accumulation in different skeletal muscle fiber types leads to distinct metabolic consequences (44–46). When diacylglycerol acyltransferase 2 is overexpressed in glycolytic (type II) muscle, mice accumulate TG in this muscle and become insulin resistant (44). However, when diacylglycerol acyltransferase 1, another TG synthesis enzyme, is overexpressed in muscle, mice accumulate TG in soleus, yet display characteristics of insulin-sensitive myofibers (46). Endurance-trained athletes show increased fat content in skeletal muscle, yet markedly enhanced insulin sensitivity (“athlete paradox”) (47). Here we showed that mCgi58-KO mice accumulate TG in the oxidative muscle only and these animals are more insulin sensitive. These findings collectively support the concept that storing fat in glycolytic muscle may be problematic, whereas storing fat in oxidative muscle may not be detrimental.
There was a mouse model with ATGL disrupted in both cardiac and skeletal muscle, but the researchers did not report the role of ATGL in regulating insulin resistance in these animals, and it is unknown whether intramyocellular TG contents differ among different muscle fibers (33). Recently, Sitnick et al (38) reported that skeletal muscle-specific deletion of ATGL in mice causes intramyocellular TG deposition in both glycolytic and oxidative muscles but does not alter insulin sensitivity. Although this finding may suggest that increasing muscle TG via inhibiting TG hydrolysis neither causes nor prevents insulin resistance, it may also imply that the opposite/different effects of TG accumulation on insulin signaling in the 2 different muscle fiber types are balanced in their animal model. In our study, CGI-58 deletion in muscle causes TG accumulation in the oxidative muscle fibers only, which is consistent with in vitro studies showing that CGI-58 controls oxidative metabolism in muscle (12, 48). Hence it is not too surprising that mCgi58-KO mice display phenotypes different from those seen in skeletal muscle-specific ATGL KO mice. Additionally, phenotypes of mice and humans with mutations in CGI-58 and ATGL are quite different (5, 20, 21, 36), suggesting that CGI-58 may also promote TG hydrolysis via ATGL-independent mechanisms, which may lead to distinct phenotypes. Importantly, it should be emphasized that Sitnick et al (38) deleted ATGL specifically in the skeletal muscle and we deleted CGI-58 in both cardiac and skeletal muscles. Cardiac steatosis and dysfunction in our mice have the potential to confound the establishment of the direct relationship between skeletal muscle TG and insulin sensitivity. Cardiac mRNA levels of ANP are robustly increased in mCgi58-KO mice compared with the controls. It has been shown that cardiac ANP, after release to the circulation, promotes energy expenditure by inducing white adipocyte browning (49), which may reduce body weight and fat, thereby improving glucose tolerance and insulin sensitivity. However, we did not observe any differences in weight gain and fat mass between mCgi58-KO and control mice, suggesting that the ANP effect may be minimal in our animal model, at least within the time window of our experiments. Nonetheless cardiac- or skeletal mCgi58-KO mice are required to clarify this issue.
A striking phenotype seen in mCgi58-KO mice on the HFD is the cardiac steatosis and dysfunction. Muscle-specific deletion of CGI-58 also causes similar cardiac phenotypes in mice on the chow diet (27). In humans, cardiac involvement was noticed only in a couple of CDS patients (18). The rarity of cardiomyopathy in limited CDS patients reported might be explained by variations of multiple factors at the physical examination, such as age, diet composition, and genetic background. Studies are required in the future to identify molecular mediators linking CGI-58 deficiency-induced cardiac steatosis to heart dysfunction.
Cardiac muscle cells import circulating FFA bound to albumin and those released from plasma lipoprotein-TG by LPL (50, 51). Activated FAs may directly enter oxidative pathways, or be reesterified into TG and temporarily stored as cytosolic LDs. Cytosolic LDs are rare in normal cardiomyocytes, suggesting that TG synthesis and hydrolysis are balanced in normal cardiomyocytes. The present study and that by Zierler (27) clearly demonstrate that CGI-58 plays a critical role in regulating this balance. Normal PPARα activity and mitochondrial function are critical in balancing these metabolic pathways (52). Both up-regulation and down-regulation of PPARα are associated with fat deposition in heart (53–55). When PPARα is up-regulated, increased FA uptake may exceed increased FA oxidation, leading to elevation of intracellular fat stores. When PPARα is down-regulated, impaired/attenuated FA oxidation may directly cause fat accumulation. Our results demonstrate that cardiac TG accumulation in mCgi58-KO mice is attributable, at least in part to reduced cellular TG hydrolysis and mitochondrial FA oxidation, although other mechanisms may exist. Deficiency of ATGL, a lipase target of CGI-58, also causes cardiac lipid accumulation and dysfunction by reducing cardiac TG hydrolysis and mitochondrial FA oxidation (33). Cardiac ATGL-dependent TG hydrolysis was shown to sustain mitochondrial functions via activating PPARα-PGC-1 pathway by generating endogenous ligands for PPARα (33). We found that CGI-58-deficient heart accumulates acyl-carnitine, consistent with a defect in FA oxidation (34), a major function of mitochondria. Given the striking similarity in cardiac phenotypes between mCgi58-KO mice and ATGL-deficient mice (33), we speculate that CGI-58 may function through ATGL in murine heart, although additional targets of CGI-58 may exist in other tissues.
In addition to increases of TG in oxidative muscles, mCgi58-KO mice also have a significant increase in cardiac cholesterol. This phenotype was not reported in muscle CGI-58 KO mice on a chow diet (27), or muscle ATGL KO mice on a chow diet or a HFD low in cholesterol (∼0.02%, wt/wt) (33, 38). Because most accumulated cholesterol in heart is in its free, but not esterified form, it is unlikely that a cholesterol esterase is inhibited in the absence of CGI-58, at least, in the mouse heart. Perhaps sequestration of cholesterol at the LDs accounts for increased cholesterol in CGI-58-deficient heart. The use of a low-cholesterol diet in the previous studies (27, 33, 38) may have covered the effects of muscle CGI-58 or ATGL on cholesterol metabolism. A chow diet normally contains 4%–6% fat and only approximately 0.015% (wt/wt) cholesterol. Our diet is a Western-type diet, contains approximately 20% fat (45% energy from lard) and approximately 0.2% (wt/wt) cholesterol. Our diet vs a regular chow diet, or a low-cholesterol HFD, is more relevant to human diets.
Despite increased lipids in cardiac and soleus muscles, mCgi58-KO mice on HFD have reduced hepatic contents of TG and cholesterol. In the studies of chow-fed muscle CGI-58 KO mice, Zierler and associates did not report hepatic cholesterol, but showed that the hepatic TG content was unaltered in the nonfasted state, and reduced in 14-hour-fasted mice (27). Although the molecular mechanism for reduced hepatic lipids has yet to be exploited, improved insulin sensitivity in our mCgi58-KO mice may have played an important role. Insulin resistance is known to drive hepatic lipogenesis (56). Consistently, we found that hepatic mRNA levels of lipogenic and cholesterogenic genes were substantially lower in mCgi58-KO mice relative to control mice. Because intestinal cholesterol absorption and fecal neutral sterol excretion remain unchanged, reduced hepatic lipid synthesis may also account for decreased plasma cholesterol concentrations in mCgi58-KO mice on the HFD. Plasma cholesterol levels remain unaltered in muscle CGI-58 or ATGL KO mice on a chow diet (27, 33). Differences in diet composition and insulin sensitivity may be attributable to distinct plasma cholesterol phenotypes among these animal models. Evidently, detailed studies are required to delineate how muscle CGI-58 regulates cholesterol homeostasis.
In conclusion, simultaneous deletion of CGI-58 in cardiac and skeletal muscles induces a series of favorable metabolic changes, including increased glucose tolerance and insulin sensitivity as well as reduced hepatic and plasma lipids, despite causing muscle steatosis and cardiac dysfunction.
Acknowledgments
We thank Dr Oksana Gavrilova at the Mouse Metabolism Core Laboratory of the National Institute of Diabetes and Digestive and Kidney Diseases for helping with the body composition measurement by EchoMRI. We also thank Dr Jonathan C. Cohen at University of Texas Southwestern Medical Center in Dallas for critical reading of the manuscript.
Present address for P.X.: Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100193, People's Republic of China.
Present address for F.G.: Key Laboratory of Neuroregeneration, Nantong University, Center of Reproductive Medicine, Affiliated Hospital of Nantong University, 20 Xisi Road, Nantong, Jiansu Province 226001, People's Republic of China.
This work was supported in part by intramural funds from Wake Forest University Health Sciences and the University of Maryland (L.Y.); by the National Institutes of Health Award R01DK085176 (to L.Y.), R01DK084172 (to H.S.), and R01HL107500 (to B.X.); and by the China Natural Science Foundation, Overseas, Hong Kong and Macau Scholars Collaborated Research Fund Grant 81228006 (to L.Y. and H.L.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ANP
- atrial natriuretic peptide
- ATGL
- adipose TG lipase
- CDS
- Chanarin-Dorfman syndrome
- CGI-58
- comparative gene identification-58
- DG
- 2-[3H]deoxyglucose
- DGP
- 2-deoxyglucose 6-phosphate
- FFA
- free fatty acid
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- GLUT
- glucose transporter
- GTT
- glucose tolerance test
- HFD
- high-fat diet
- ITT
- insulin tolerance test
- LD
- lipid droplet
- LPL
- lipoprotein lipase
- LV
- left ventricular
- mCgi58-KO
- muscle-specific CGI-58 knockout
- MHC
- myosin heavy chain
- PGC
- PPARγ coactivator
- PPAR
- peroxisome proliferator-activated receptor
- PWT
- posterior wall thickness
- TG
- triglyceride.
References
- 1. Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zimmermann R, Strauss JG, Haemmerle G, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306(5700):1383–1386. [DOI] [PubMed] [Google Scholar]
- 3. Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem. 2004;279(47):48968–48975. [DOI] [PubMed] [Google Scholar]
- 4. Villena JA, Roy S, Sarkadi-Nagy E, Kim KH, Sul HS. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J Biol Chem. 2004;279(45):47066–47075. [DOI] [PubMed] [Google Scholar]
- 5. Lefèvre C, Jobard F, Caux F, et al. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am J Hum Genet. 2001;69(5):1002–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Simon GM, Cravatt BF. Endocannabinoid biosynthesis proceeding through glycerophospho-N-acyl ethanolamine and a role for α/β-hydrolase 4 in this pathway. J Biol Chem. 2006;281(36):26465–26472. [DOI] [PubMed] [Google Scholar]
- 7. Lass A, Zimmermann R, Haemmerle G, et al. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman syndrome. Cell Metab. 2006;3(5):309–319. [DOI] [PubMed] [Google Scholar]
- 8. Subramanian V, Rothenberg A, Gomez C, et al. Perilipin A mediates the reversible binding of CGI-58 to lipid droplets in 3T3-L1 adipocytes. J Biol Chem. 2004;279(40):42062–42071. [DOI] [PubMed] [Google Scholar]
- 9. Brown JM, Chung S, Das A, Shelness GS, Rudel LL, Yu L. CGI-58 facilitates the mobilization of cytoplasmic triglyceride for lipoprotein secretion in hepatoma cells. J Lipid Res. 2007;48(10):2295–2305. [DOI] [PubMed] [Google Scholar]
- 10. Liu P, Ying Y, Zhao Y, Mundy DI, Zhu M, Anderson RG. Chinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane traffic. J Biol Chem. 2004;279(5):3787–3792. [DOI] [PubMed] [Google Scholar]
- 11. Yamaguchi T, Omatsu N, Matsushita S, Osumi T. CGI-58 interacts with perilipin and is localized to lipid droplets. Possible involvement of CGI-58 mislocalization in Chanarin-Dorfman syndrome. J Biol Chem. 2004;279(29):30490–30497. [DOI] [PubMed] [Google Scholar]
- 12. Granneman JG, Moore HP, Mottillo EP, Zhu Z. Functional interactions between Mldp (LSDP5) and Abhd5 in the control of intracellular lipid accumulation. J Biol Chem. 2009;284(5):3049–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Granneman JG, Moore HP, Krishnamoorthy R, Rathod M. Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl). J Biol Chem. 2009;284(50):34538–34544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang J, Xu D, Nie J, Han R, Zhai Y, Shi Y. Comparative gene identification-58 (CGI-58) promotes autophagy as a putative lysophosphatidylglycerol acyltransferase. J Biol Chem. 2014;289(47):33044–33053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brown JM, Betters JL, Lord C, et al. CGI-58 knockdown in mice causes hepatic steatosis but prevents diet-induced obesity and glucose intolerance. J Lipid Res. 2010;51(11):3306–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chanarin I, Patel A, Slavin G, Wills EJ, Andrews TM, Stewart G. Neutral-lipid storage disease: a new disorder of lipid metabolism. Br Med J. 1975;1(5957):553–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dorfman ML, Hershko C, Eisenberg S, Sagher F. Ichthyosiform dermatosis with systemic lipidosis. Arch Dermatol. 1974;110(2):261–266. [PubMed] [Google Scholar]
- 18. Igal RA, Rhoads JM, Coleman RA. Neutral lipid storage disease with fatty liver and cholestasis. J Pediatr Gastroenterol Nutr. 1997;25(5):541–547. [DOI] [PubMed] [Google Scholar]
- 19. Rozenszajn L, Klajman A, Yaffe D, Efrati P. Jordans' anomaly in white blood cells. Report of case. Blood. 1966;28(2):258–265. [PubMed] [Google Scholar]
- 20. Fischer J, Lefèvre C, Morava E, et al. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat Genet. 2007;39(1):28–30. [DOI] [PubMed] [Google Scholar]
- 21. Radner FP, Streith IE, Schoiswohl G, et al. Growth retardation, impaired triacylglycerol catabolism, hepatic steatosis, and lethal skin barrier defect in mice lacking comparative gene identification-58 (CGI-58). J Biol Chem. 2010;285(10):7300–7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guo F, Ma Y, Kadegowda AK, et al. Deficiency of liver comparative gene identification-58 causes steatohepatitis and fibrosis in mice. J Lipid Res. 2013;54(8):2109–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu JW, Wang SP, Alvarez F, et al. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology. 2011;54(1):122–132. [DOI] [PubMed] [Google Scholar]
- 24. Miao H, Ou J, Ma Y, et al. Macrophage CGI-58 deficiency activates ROS-inflammasome pathway to promote insulin resistance in mice. Cell Rep. 2014;7(1):223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goeritzer M, Schlager S, Radovic B, et al. Deletion of CGI-58 or adipose triglyceride lipase differently affects macrophage function and atherosclerosis. J Lipid Res. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miranda A, DiMauro S, Eastwood A, et al. Lipid storage myopathy, ichthyosis, and steatorrhea. Muscle Nerve. 1979;2(1):1–13. [DOI] [PubMed] [Google Scholar]
- 27. Zierler KA, Jaeger D, Pollak NM, et al. Functional cardiac lipolysis in mice critically depends on comparative gene identification-58. J Biol Chem. 2013;288(14):9892–9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148(5):852–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jia L, Ma Y, Liu G, Yu L. Dietary cholesterol reverses resistance to diet-induced weight gain in mice lacking Niemann-Pick C1-Like 1. J Lipid Res. 2010;51(10):3024–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang J, Goldstein JL, Hammer RE, Moon YA, Brown MS, Horton JD. Decreased lipid synthesis in livers of mice with disrupted Site-1 protease gene. Proc Natl Acad Sci USA. 2001;98(24):13607–13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jia L, Ma Y, Rong S, et al. Niemann-Pick C1-Like 1 deletion in mice prevents high-fat diet-induced fatty liver by reducing lipogenesis. J Lipid Res. 2010;51(11):3135–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bharadwaj MS, Strawn WB, Groban L, et al. Angiotensin-converting enzyme 2 deficiency is associated with impaired gestational weight gain and fetal growth restriction. Hypertension. 2011;58(5):852–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haemmerle G, Moustafa T, Woelkart G, et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat Med. 2011;17(9):1076–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koves TR, Ussher JR, Noland RC, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7(1):45–56. [DOI] [PubMed] [Google Scholar]
- 35. Monetti M, Levin MC, Watt MJ, et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 2007;6(1):69–78. [DOI] [PubMed] [Google Scholar]
- 36. Haemmerle G, Lass A, Zimmermann R, et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312(5774):734–737. [DOI] [PubMed] [Google Scholar]
- 37. Sun Z, Miller RA, Patel RT, et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat Med. 2012;18(6):934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sitnick MT, Basantani MK, Cai L, et al. Skeletal muscle triacylglycerol hydrolysis does not influence metabolic complications of obesity. Diabetes. 2013;62(10):3350–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kozlitina J, Boerwinkle E, Cohen JC, Hobbs HH. Dissociation between APOC3 variants, hepatic triglyceride content and insulin resistance. Hepatology. 2011;53(2):467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Amaro A, Fabbrini E, Kars M, et al. Dissociation between intrahepatic triglyceride content and insulin resistance in familial hypobetalipoproteinemia. Gastroenterology. 2010;139(1):149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cantley JL, Yoshimura T, Camporez JP, et al. CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proc Natl Acad Sci USA. 2013;110(5):1869–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Listenberger LL, Han X, Lewis SE, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA. 2003;100(6):3077–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Levin MC, Monetti M, Watt MJ, et al. Increased lipid accumulation and insulin resistance in transgenic mice expressing DGAT2 in glycolytic (type II) muscle. Am J Physiol Endocrinol Metab. 2007;293(6):E1772–E1781. [DOI] [PubMed] [Google Scholar]
- 45. Liu L, Shi X, Choi CS, et al. Paradoxical coupling of triglyceride synthesis and fatty acid oxidation in skeletal muscle overexpressing DGAT1. Diabetes. 2009;58(11):2516–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu L, Shi X, Bharadwaj KG, et al. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem. 2009;284(52):36312–36323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goodpaster BH, He J, Watkins S, Kelley DE. Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab. 2001;86(12):5755–5761. [DOI] [PubMed] [Google Scholar]
- 48. Badin PM, Loubière C, Coonen M, et al. Regulation of skeletal muscle lipolysis and oxidative metabolism by the co-lipase CGI-58. J Lipid Res. 2012;53(5):839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bordicchia M, Liu D, Amri EZ, et al. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J Clin Invest. 2012;122(3):1022–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Goldberg IJ, Eckel RH, Abumrad NA. Regulation of fatty acid uptake into tissues: lipoprotein lipase- and CD36-mediated pathways. J Lipid Res. 2009;50(suppl):S86–S90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Park TS, Yamashita H, Blaner WS, Goldberg IJ. Lipids in the heart: a source of fuel and a source of toxins. Curr Opin Lipidol. 2007;18(3):277–282. [DOI] [PubMed] [Google Scholar]
- 52. Russell LK, Finck BN, Kelly DP. Mouse models of mitochondrial dysfunction and heart failure. J Mol Cell Cardiol. 2005;38(1):81–91. [DOI] [PubMed] [Google Scholar]
- 53. Finck BN, Lehman JJ, Leone TC, et al. The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109(1):121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Duncan JG, Fong JL, Medeiros DM, Finck BN, Kelly DP. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-α/PGC-1α gene regulatory pathway. Circulation. 2007;115(7):909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sharma S, Adrogue JV, Golfman L, et al. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004;18(14):1692–1700. [DOI] [PubMed] [Google Scholar]
- 56. Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 2008;7(2):95–96. [DOI] [PubMed] [Google Scholar]