

Abstract
We previously demonstrated that in normal glucose (5 mM), methylglyoxal (MG, a model of carbonyl stress) induced brain microvascular endothelial cell (IHEC) dysfunction that was associated with occludin glycation and prevented by N-acetylcysteine (NAC). Herein, we investigated the impact of high glucose and low GSH, conditions that mimicked the diabetic state, on MG-induced IHEC dysfunction. MG-induced loss of transendothelial electrical resistance (TEER) was potentiated in IHECs cultured for 7 or 12 days in 25 mM glucose (hyperglycemia); moreover, barrier function remained disrupted 6 h after cell transfer to normal glucose media (acute glycemic fluctuation). Notably, basal occludin glycation was elevated under these glycemic states. TEER loss was exaggerated by inhibition of glutathione (GSH) synthesis and abrogated by NAC, which corresponded to GSH decreases and increases, respectively. Significantly, glyoxalase II activity was attenuated in hyperglycemic cells. Moreover, hyperglycemia and GSH inhibition increased MG accumulation, consistent with a compromised capacity for MG elimination. α-Oxoaldehydes (MG plus glyoxal) levels were elevated in streptozotocin-induced diabetic rat plasma. Immunohistochemistry revealed a prevalence of MG-positive, but fewer occludin-positive microvessels in the diabetic brain in vivo, and Western analysis confirmed an increase in MG–occludin adducts. These results provide the first evidence that hyperglycemia and acute glucose fluctuation promote MG–occludin formation and exacerbate brain microvascular endothelial dysfunction. Low occludin expression and high glycated-occludin contents in diabetic brain in vivo are factors that would contribute to the dysfunction of the cerebral microvasculature during diabetes.
Keywords: Hyperglycemia & methylglyoxal, Carbonyl stress & endothelial GSH, Occludin glycation & brain endothelial barrier function, Diabetic brain microvascular dysfunction, Streptozotocin & diabetes, N-acetylcysteine & endothelial barrier function
Abbreviations: AGEs, advanced glycation end products; BBB, blood–brain barrier; BSO, L-buthionine-(S,R)-sulfoximine; GSH, glutathione; HPLC, high-performance liquid chromatography; IHEC, immortalized human brain endothelial cell line; MG, methylglyoxal; NAC, N-acetyl-L-cysteine; PCA, perchloric acid; RCS, reactive carbonyl species; RIPA, radio immunoprecipitation assay buffer; SDL, S-D-lactoylglutathione; STZ, streptozotocin; TEER, transendothelial electrical resistance.
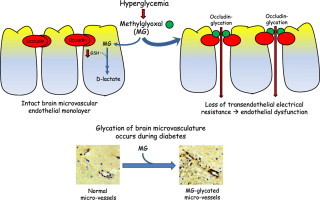
Graphical abstract
Highlights
-
•
Methylglyoxal (MG) induced electrical resistance (TEER)loss in brain microvascular endothelial cells.
-
•
TEER loss was potentiated by hyperglycemia, and low glutathione.
-
•
TEER loss was correlated with occludin-glycation and was attenuated and exacerbated by NAC and BSO, respectively.
-
•
Hyperglycemia decreased glyoxalase II activity and promoted free MG accumulation.
-
•
Diabetic brain in vivo exhibiteda prevalence of MG-positive microvessels and increased occludin–MG adducts.
Introduction
Diabetes is a clinically important risk factor for cardiovascular and cerebrovascular diseases which are underscored by vascular endothelial dysfunction. It is well known that the diabetic condition is characterized by hyperglycemia and elevated plasma levels of reactive carbonyl species (RCS), but the mechanism by which RCS contribute to diabetes-associated cerebrovascular disease is poorly understood. Methylglyoxal (MG) is an RCS dicarbonyl metabolite precursor of advanced glycation endproducts, and is metabolized via a GSH-dependent glyoxalase detoxification pathway. Our recent results demonstrated that occludin glycation induced by MG disrupted barrier function in a human microvascular endothelial cell line (IHEC), and that N-acetylcysteine (NAC) afforded barrier preservation [1]. These results provide a compelling argument that the cerebral microvasculature in diabetes is sensitive to tissue levels of MG and glutathione (GSH).
The integrity of the cerebral microvasculature and microcirculation is maintained by the function of the blood–brain barrier (BBB) which reportedly is disrupted in diabetes [2]. The BBB phenotype is described by the neurovascular unit, comprising of brain capillary endothelial cells on the blood side and perivascular cells on the brain side of microvessels [3,4]. The BBB endothelial monolayer exhibits high transendothelial resistance that is conferred by the intercellular tight junctions between neighboring endothelial cells [5]. Occludin is a member of the tight junctional transmembrane proteins that regulates barrier electrical resistance and paracellular permeability [4]. We recently demonstrated that occludin is a target of MG glycation, and that MG-mediated occludin modification is associated with increased endothelial cell permeability in IHECs [1].
GSH was implicated to play a role in the BBB integrity [6]. The diabetic brain has been associated with decreased tissue GSH [7]; however, the mechanistic relationship between GSH decreases and cerebral microvascular dysfunction is unclear. We have found that GSH attenuated hyperglycemia- or MG-induced endothelial apoptosis [8,9] and barrier permeability [1]. These findings support a role for GSH in endothelial protection. With regards to MG handling, GSH is a rate-limiting cofactor in MG metabolism [11,12]. A reduction in cellular GSH levels leads to compromised glyoxalase function, resulting in the accumulation of free MG, and thereby increasing its glycating potential. Thus, GSH-dependent elimination of MG by glyoxalase I and glyoxalase II could be a major mechanism through which GSH protects against cerebral microvascular dysfunction in diabetes.
Collectively, MG-induced carbonyl stress (protein glycation or carbonylation) and endothelial GSH imbalance would potentiate cerebral microvascular dysfunction, and RCS-mediated dysfunction of the cerebrovascular endothelium and its pathology would be significant in diabetes. Importantly, given that triosephosphates derived from glucose metabolism is an MG source [12], the hyperglycemic state associated with diabetes would have an exacerbating role in brain endothelial injury. The current study addresses new links between hyperglycemia, MG and brain microvascular dysfunction. Using the previously established IHEC cell line, we sought to investigate whether states of high glucose or acute glucose fluctuation exacerbate MG-induced occludin glycation and barrier disruption, and if promoting GSH-dependent MG metabolism via exogenous NAC administration preserves barrier function. We further investigated if glycation of the brain microvasculature in vivo is a significant process during diabetes using a streptozotocin (STZ)-induced diabetic rat model. The results show that hyperglycemia and acute glycemic fluctuation potentiated MG-induced loss of IHEC barrier transendothelial electrical resistance (TEER), an event that was associated with increased occludin-MG adduct formation and prevented by NAC. Importantly, we found that diabetic rat brain microvessels exhibited decreased total occludin expression and elevated glycated–occludin adduct content.
Methods
Reagents
The following reagents were purchased from Sigma (St Louis, MO): d-glucose, methylglyoxal, N-acetyl-l-cysteine, L-buthionine-(S, R)-sulfoximine, insulin-transferrin sodium selenite solution, glutathione, d-lactate, d-lactic dehydrogenase, glutamic-pyruvate transaminase, S-d-lactoylglutathione, Medium 199, o-phenylenediamine, 2-methylquinoxaline, acetonitrile, and HRP-linked goat-anti-rabbit and goat-anti-mouse secondary antibodies. Anti-occludin rabbit polyclonal antibody was obtained from Invitrogen (Carlsbad, CA), anti-MG mouse monoclonal antibody from JcICA (Fukuroi, Japan), anti-actin mouse monoclonal antibody from BD Biosciences (San Jose, CA) and anti-GAPDH mouse monoclonal antibody from Santa Cruz (Santa Cruz, CA). HRP-linked goat-anti-rabbit and goat-anti-mouse antibodies, ECL, and chemi-luminescence detection reagents were purchased from Amersham Biopharmacia (Piscataway, NJ). Fetal bovine serum (FBS) was obtained from Atlanta Biologicals (Atlanta, GA). All other chemicals were of reagent grade and were purchased from Sigma or local sources.
Cell culture and cell incubations
The human brain microvascular endothelial cell line (IHEC) was provided by Dr. Danica Stanimirovic of the National Research Council Canada's Institute for Biological Sciences and was maintained by Dr. Steve Alexander at LSU Health Sciences Center. Routinely, IHECs were cultured in M-199 medium containing 5 mM glucose (normal glucose), 10% FBS, 1% insulin–transferrin–sodium selenite solution, and 1× antibiotic/antimycotic at 37 °C in 5% CO2 on T-75 flask. In all cell studies, IHECs were seeded at specific densities 5 days before the experiment. High glucose-adapted IHECs were achieved by culturing cells in 25 mM glucose for 7 or 12 days. Acute glycemic fluctuation was achieved by transferring 25 mM glucose-adapted cells (12 days) to normal glucose media for 6 h (designated 25→5 mM glucose).
IHECs (1.5×104) were cultured on 8-µm inserts in 24-well plates for measurements of TEER. For assays of GSH, Western blot and glyoxalase I/II activities, IHECs (0.4×106) were grown in 6-well plates. All experiments were conducted on confluent cell monolayers (5 days post-seeding). The experimental glucose concentrations include: 5 mM (normoglycemia), 25 mM (hyperglycemia), or 25→5 mM glucose (acute glycemic fluctuation). To achieve high or low cellular GSH, IHECs were pretreated overnight with 1 mM NAC or 50 µM BSO, respectively. After washing, 2 mM NAC or 300 µM BSO were added to the incubation to maintain elevated or low GSH status in these cells throughout the experiment. A range of MG concentrations were used depending on the type of experiments. MG concentrations of 50 µM to 1 mM were used to examine the time course and dose dependency of TEER responses under normal and high glucose states. These MG levels were previously employed in our recent studies [1]. Since high glucose will contribute to MG production, a lower dose of 300 µM MG was then used in subsequent experiments to test the effect of BSO and NAC under hyperglycemic conditions. An MG level of 50 µM MG was near physiological in diabetic rat plasma (see Fig. 6).
Fig. 6.
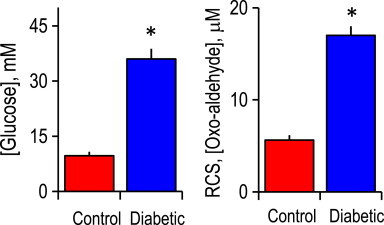
Plasma levels of glucose and reactive carbonyl species (α-oxoaldehydes) in control and streptozotocin (STZ)-induced diabetic rat are elevated. Plasma was from 4-week STZ diabetic rats. Glucose was measured using a glucometer, and oxoaldehydes were quantified by HPLC. Left=glucose content; right=oxoaldehydes content. ⁎p<0.05, diabetic vs control (mean±SEM, n=6). RCS=reactive carbonyl species.
Measurement of transendothelial electrical resistance (TEER)
Studies of TEER were carried out in HBSS containing 25 mM HEPES and 10% FBS (pH 7.35) at 37 °C and 5% CO2. After an initial 1 h adjustment period, changes in electrical resistance at different concentrations of MG without or with NAC or BSO (as above) were recorded for up to 10 h using an epithelial voltohmeter (EVOM, World Precision Instruments, Sarasota, FL). Only inserts with a minimum baseline monolayer resistance of 200 Ω/0.33 cm2 were used. TEER was expressed as the percent of the baseline to account for batch-to-batch variation.
Western analyses of occludin and MG–occludin
Cell extracts
Cells were harvested and lysed in RIPA buffer containing 50 mM Tris, pH 7.4, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100 and a cocktail of protease inhibitors, viz., aprotinin, PMSF, okadaic acid, and leupeptin.
Tissue extracts
Cerebral vessels were isolated as described previously [13]. Briefly, the brain was removed from the skull and pia matter containing large vessels was gently teased off and placed in cold PBS. The brain tissue was then homogenized in ice-cold PBS (polytron, 1 min). The homogenate was passed through an 18G needle 10 times, and centrifuged at 2000g for 10 min at 4 °C. The pellet was suspended in ice-cold PBS, gently layered on top of 30 ml of 15% dextran (MW 38,400) and centrifuged at 17,400g for 45 min at 4 °C. The final pellet represented the microvessel fraction. Micro- and macrovessels were separately homogenized in RIPA buffer by passing through an 18G needle followed by 10 pulses with a polytron. The homogenate was centrifuged at 14,000 rpm (10 min at 4 °C), and the supernatants used for Western blot analyses.
Western blot analyses
Total protein from cell extracts (60 µg), microvessels (30 µg) or macrovessels (50 µg) per sample was resolved on 10% SDS-polyacrylamide gels (110 V, 2 h), and then transferred onto a PVDF membranes at 200 mA at 4 °C for 2 h. The membranes were blocked in 5% non-fat milk in 0.1 M PBS, pH 7.4 at RT for 1 h and then incubated overnight with rabbit anti-occludin polyclonal antibody (1:1000) or with mouse anti-MG monoclonal antibody (1:1000) at 4 °C. The next day, membranes were incubated for 2 h at RT with HRP-conjugated donkey-anti-rabbit or HRP-conjugated sheep-anti-mouse secondary antibody (1:10,000), respectively. Chemiluminescence was detected with ECL reagents per manufacturer's instructions. The membranes were stripped and reprobed for β-actin or GAPDH using mouse monoclonal antibody (1:5000) to verify equal protein loading.
HPLC quantification of GSH and methylglyoxal
GSH determination
Cellular GSH concentrations were determined as we previously described [1,14]. IHECs were harvested by scraping into 5% TCA followed by centrifugation at 14,000 rpm for 5 min. The acid supernatants were derivatized with 6 mM iodoacetic acid and 1% 2,4-dinitrophenyl fluorobenzene to yield the S-carboxymethyl and 2,4-dinitrophenyl derivative of GSH, respectively. GSH derivatives were separated on a 250×4.6 mm2 Alltech Lichrosorb NH2 10 µm column. GSH contents were quantified by comparison to standards derivatized in the same manner and expressed as nmole per milligrams of protein.
Methylglyoxal determination in IHECs and plasma
IHEC cell pellets were washed 3 times with PBS (3000 rpm, 3 min, 4 °C) and sonicated (5 s, 3 times). Blood was taken from the heart of control and diabetic rats using 20G needles, and plasma was collected by centrifugation (5000 rpm for 10 min, 4 °C).
α-Oxoaldehyde (MG plus glyoxal) contents were determined by HPLC as previously described [15]. Cell homogenates or plasma were treated with 0.45 N perchloric acid (PCA) for 24 h at RT. Post 12,000 rpm centrifugation, acid supernatants (500 µl) were incubated with 5 mM o-phenylenediamine for 24 h at RT and then centrifuged and filtered (0.45 µm filter). Separation of MG and glyoxal was performed on a 250×4.6 mm2 Beckman C-18-ODS 5 µm column and quantified using 2-methylquinoline as an external standard. Cellular concentrations were expressed as nmole per milligrams protein and plasma levels as μM.
Assay of cellular glyoxalase I and II activity
IHEC cell pellets were suspended in 10 mM Tris–HCl pH 7.4 containing protease inhibitor cocktail and subjected to 3 freeze–thaw cycles (liquid nitrogen/4 °C), followed by sonication (5 s, 50% amplitude) and centrifugation (12,000 rpm, 20 min at 4 °C). The supernatants were used for assays of glyoxalase I and glyoxalase II activities.
Glyoxalase I activity was determined by S-d-lactoylglutathione (SDLG) formation [16]. The assay solution contained 182 mM imidazole buffer pH 7.0, 14.6 mM magnesium sulfate, 5 mM MG, 1.5 mM GSH and 30 µg/reaction cell lysate. SDLG formation was monitored spectrophoto-metrically at 240 nm at 25 °C, and quantified using the extinction coefficient of 3.37 mM−1 cm−1. Glyoxalase I activity was expressed as nanomol SDLG formed per min/mg protein.
Glyoxalase II function was assessed by d-lactate formation and SDLG hydrolysis. Post-glyoxalase I reaction (above) was stopped by addition of 12% PCA, and the pH was adjusted to 7.4. Samples were centrifuged (14,000 rpm, 10 min at 4 °C) and d-lactate was assayed in 147 mM glycylglycine buffer (pH 10), containing 3 mM NAD+, 30 mM glutamate, 40 U/ml D-LDH, 8 U/ml GPT, and 50 µl supernatant. Reactions were performed at 37 °C for 1 h, and the formation of NADH was determined spectrophotometrically at 340 nm. Glyoxalase II function was expressed as nanomol of d-lactate formed per milligrams of protein.
SDLG hydrolysis was determined in cell extracts by a modified method of Talesa et al. [17] based on SDLG hydrolysis and concomitant GSH regeneration. The reaction mixture consisted of 0.8 mM SDLG, 0.2 mM DTNB, and 150 µl of cell lysate in 100 mM Tris–HCL buffer, pH 7.4. GSH formation was monitored by increases in DTNB absorbance at 412 nm. GSH was quantified using the extinction coefficient of 13.6 mM−1 cm−1, and glyoxalase II activity was expressed as nmole of GSH produced per min/mg protein.
Induction of diabetes
Four-week old male Wistar rats (140–170 g, Harlan Laboratories) were injected with streptozytocin (STZ, 30 mg/kg in sodium citrate buffer, pH 4.5) for 3 consecutive days. Control rats received injections of sodium citrate buffer alone. Animals were housed individually and received standard water and chow diet. At 6 days post-STZ injection and on the day of sacrifice at 8 weeks, blood was obtained via tail vein punctures and glucose was measured using a One Touch Ultra Glucometer (Milpitas, CA). STZ-injected rats with plasma glucose >300 mg/dl were considered diabetic (non-diabetic levels=120–150 mg/dl). Animals were sacrificed by decapitation and brains were quickly removed and processed for immunohistochemistry or Western blot analyses. All animal protocols were approved by the Institutional Animal Care and Use Committee, and were conducted in accordance with the Principles for Use of Animals and Guide to the Care and Use of Experimental Animals.
Immunohistochemistry of occludin and glycated protein expression in rat brain
Rat brains were placed in a brain slicer matrix (Zivic Instruments, Pittsburgh, PA) ventral side up. Two mm coronal sections located 8 mm from the anterior portion of the cerebrum were cut using PBS pre-moistened razor blades. The sections were fixed with ice-cold 4% para-formaldehyde in PBS (4 °C) for at least 24 h, followed by tissue dehydration (graded 70-to-100% ethanol and xylene) and paraffin infusion. Three µm slices were cut and mounted on glass slides, two to three slices per slide. Brain slices were oven-baked at 75 °C, de-paraffinized in xylene and rehydrated by passage through graded series of ethanol solutions (100-to-80%), and distilled water. Tissue slices were then incubated with 1.5 mg/ml serine protease at 37 °C followed by overnight incubation at 4 °C with 5 µg/ml anti-occludin or anti-MG polyclonal antibodies. The slides were washed 3 times with 1× ProHisto washing solution, followed by incubations in 0.3% H2O2 in 50% methanol, and HRP conjugated goat-anti-rabbit or goat-anti-mouse secondary antibody (1:200) at RT. Diaminobenzidine (DAB) solution was applied per manufacturers' recommendation. Brain tissues were counterstained in hematoxylin, mounted and dried. Immunohistochemical staining was visualized using a Nikon Diaphot microscope equipped with a Nikon D90 camera. An average of 3–5 different images per brain slice was photographed and 5–6 microvessels were counted per image to determine the percentage of vessels that were positive for occludin or MG. Coronal sections from 5 control and 5 diabetic rat brains were analyzed.
Protein assay
Protein concentrations were determined using the Bio-Rad Protein Assay Kit (BioRad Laboratories, Irvine, CA) according to the manufacturer's protocol.
Statistical analysis
All data were analyzed by one way ANOVA using Bonferroni's post-test for comparison of sample groups. p<0.05 was considered statistically significant.
Results
Hyperglycemia and acute glycemic fluctuation exacerbates MG-induced IHEC dysfunction
At normal (5 mM) glucose, MG at a pharmacologic dose (1 mM) time-dependently decreased TEER in IHEC monolayers (Fig. 1A), consistent with endothelial dysfunction, and in agreement with our previous study [1]. TEER was next measured in (a) 25 mM glucose-adapted IHECs incubated in media containing 25 mM glucose (hyperglycemia) or (b) 25 mM glucose-adapted IHECs transferred to and incubated in media containing 5 mM glucose (acute glycemic fluctuation) over 6 h. At normal glucose, complete TEER loss occurred at 6 h. Between 1 and 4 h, MG-induced TEER loss was significantly potentiated by high glucose states, suggesting that hyperglycemia did, in fact, exacerbate barrier disruption caused by a pharmacologic MG dose (Fig. 1A). Moreover, TEER at 300 µM MG, a dose that did not elicit barrier dysfunction at normal glucose, was significantly decreased by hyperglycemia and acute glycemic fluctuation (Fig. 1B). These results indicate that IHEC barrier function is sensitive to the media glucose status and that the IHEC cell is a relevant physiological/pathophysiological in vitro model for the cerebral microvasculature.
Fig. 1.
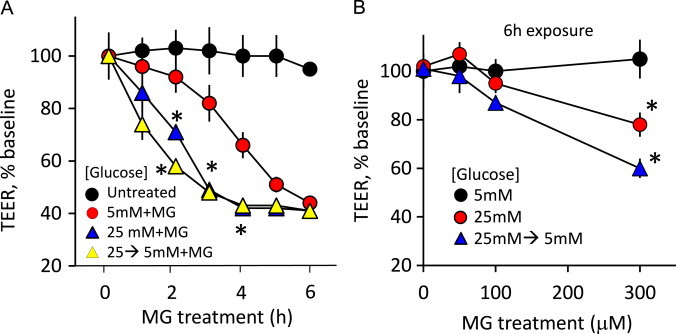
MG induces barrier dysfunction in human brain microvascular endothelial cells (IHECs) under various glucose conditions. (A) Time course of MG-induced transendothelial electrical resistance (TEER) in IHECs adapted to 5 mM glucose (normoglycemia) or to 25 mM glucose (hyperglycemia, 12 days) or to 25 mM glucose followed by transfer to 5 mM glucose (acute glycemic fluctuation, 25→5 mM). MG concentration was 1 mM. TEER in confluent IHEC monolayers on inserts was recorded for 6 h with a voltohmeter. Only inserts with a minimum baseline monolayer resistance of 200 Ω/0.33 cm2 were used in the experiments. TEER was expressed as percent of the baseline to account for batch-to-batch variation. ⁎p<0.05 vs control, n=3. (B) Hyperglycemia and acute glycemic fluctuation exacerbates MG-induced TEER loss. TEER was determined in IHEC monolayers grown in different glucose conditions: 5, 25 mM or 25→5 mM and treated with 0–300 µM MG. ⁎p<0.05 vs 5 mM glucose, n=3.
MG causes occludin glycation
We previously demonstrated that MG-induced IHEC barrier dysfunction was correlated with the glycation of occludin, a tight junctional protein [1]. In the current study, we determined if MG–occludin formation was influenced by glucose status. Fig. 2A shows that the expression of occludin protein per se was unchanged in IHECs grown for 7 or 12 days in 25 mM glucose (HG) or when high glucose-adapted cells were acutely transferred to 5 mM glucose media (GF). However, the basal contents of glycated occludin (i.e., MG–occludin adducts) were significantly elevated in high glucose-adapted IHECs and those subjected to acute glycemic fluctuation (Fig. 2B). MG–occludin levels in these cells were further increased following 8 h treatment with 300 µM MG (Fig. 2B). These results indicate that occludin is a target for MG glycation under conditions of elevated glucose and acute glucose fluctuation, which could explain the exaggerated TEER loss in response to MG (see Fig. 1).
Fig. 2.
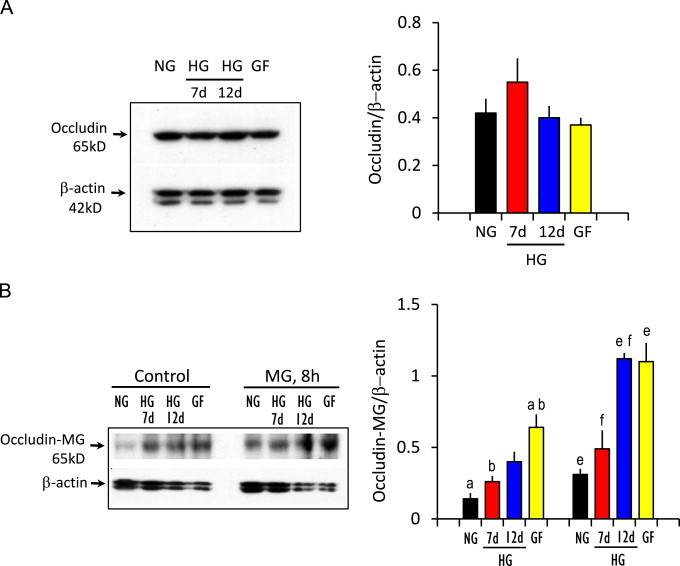
Hyperglycemia and acute glycemic fluctuation enhances MG occludin glycation. IHECs were grown under normal glucose (5 mM) or high glucose (25 mM glucose for 7 days or 12 days) or in cells subjected to acute glycemic fluctuation (25 mM glucose for 12 days, then change from 25→5 mM glucose for 6 h). Total occludin and MG–occludin glycated adducts were determined in whole cell lysates by Western blot analyses. Representative blots were shown. The right panels show the quantification of occludin or MG–occludin band intensities relative to β-actin (mean±SEM) for 6 separate immunoblots. (A) Occludin content and (B) contents of MG–occludin adducts without or after 300 µM MG treatment for 8 h. The bars with similar alphabets are significantly different from one another, p<0.05.
MG-induced IHEC barrier disruption during hyperglycemia and acute glycemic fluctuation is prevented by N-acetylcysteine and exacerbated by buthionine sulfoximine
Our previous study has shown that GSH attenuated MG-induced IHEC barrier dysfunction [1]. Herein, we investigated if GSH protects against IHEC disruption caused by MG under conditions of high glucose and acute glucose decrease. Pretreatment with NAC completely prevented TEER loss induced by 300 or 600 µM MG in IHECs incubated in 25 mM glucose (Fig. 3A) or 25→5 mM glucose (Fig. 3B). MG at 600 µM caused a decrease in cellular GSH at 2 h (Fig. 3C), a time point that preceded significant loss of barrier function at 7 h (see Fig. 3A and B). At 2 h, NAC pretreatment elevated cellular GSH concentrations (Fig. 3C) that correlated with TEER preservation (Fig. 3A and B) regardless of glucose status.
Fig. 3.
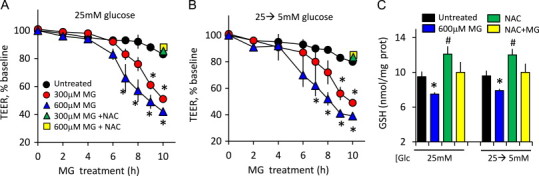
N-acetylcysteine (NAC), a GSH precursor, attenuates MG-induced IHEC dysfunction under conditions of hyperglycemia and acute glycemic fluctuation. TEER loss was induced by 300 or 600 µM MG in IHECs adapted to high glucose (12 days 25 mM, A) or high glucose and then acutely transferred to 5 mM glucose (25→5 mM, B). TEER measurements were performed in the absence or presence of 2 mM NAC as described in Section Methods. ⁎p<0.05 vs MG without NAC, n=3. (C) Effect of NAC on cellular GSH levels in high glucose-adapted IHECs (25 mM) or in cells subjected to acute glycemic fluctuation (25→5 mM). ⁎p<0.05 vs untreated cells; #p<0.05 vs MG alone, n=3.
To confirm the role of GSH, 25 mM glucose-adapted IHECs were pretreated with BSO, a specific inhibitor of GSH synthesis. Fig. 4 shows that TEER loss induced by 300 µM MG was exacerbated by BSO at high glucose status (Fig. 4)A and B that corresponded to significantly decreased cellular GSH beyond the decrease induced by MG alone (Fig. 4C). Between 7 and 10 h, BSO treatment elicited IHEC barrier dysfunction at 50 µM MG, a-near physiological level found in diabetic rat plasma (see Fig. 6). Taken together, these results indicate that patho-physiological MG levels can mediate IHEC barrier dysfunction, a process that is potentiated by low GSH and high glucose, conditions that mimicked the diabetic state.
Fig. 4.
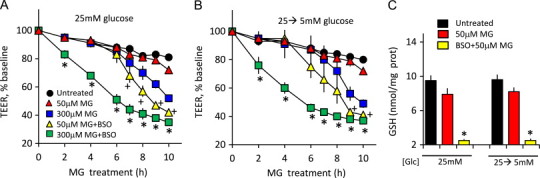
MG-induced IHEC dysfunction is exacerbated by buthionine sulfoximine (BSO), a GSH synthesis inhibitor. TEER loss was induced by 50 or 300 µM MG in IHECs adapted to high glucose (12 days 25 mM, A) or high glucose and then acutely transferred to 5 mM glucose (25→5 mM, B). TEER measurements were performed in the absence or presence of 300 µM BSO as described in Section Methods. ⁎p<0.05 vs MG without BSO, n=3. (C) Effect of BSO on cellular GSH levels in high glucose adapted IHECs (25 mM) or in cells subjected to acute glycemic fluctuation (25→5 mM). ⁎p<0.05 vs untreated or MG-treated cells, n=3.
Glyoxalase II function is compromised by hyperglycemia and acute glycemic fluctuation
Since GSH is an essential co-factor in the glyoxalase pathway in MG metabolism (Fig. 5A) [10,11], we investigated the effect of high glucose and acute glucose change on glyoxalase enzyme activities. Cells adapted to high glucose or subjected to acute glucose changes exhibited decreased d-lactate production (Fig. 5B), consistent with decreased MG metabolism. IHECs treated with MG at 5 mM glucose resulted in significant cellular accumulation of free MG that was further increased by inhibition of GSH synthesis with BSO or in cells grown under conditions of high (25 mM) glucose (Fig. 5C). Cellular levels of MG in 25 mM glucose-adapted IHECs treated with MG and NAC were significantly attenuated (Fig. 5C). These results with BSO and NAC underscore a role of GSH in MG elimination. Interestingly, glyoxalase I activity was unaffected by altered glucose status (Fig. 5D). In contrast, glyoxalase II activity was significantly lower in 25 mM glucose or 25→5 mM glucose cells (Fig. 5E), suggesting that GSH regeneration is compromised by hyperglycemia (Fig. 5A).
Fig. 5.
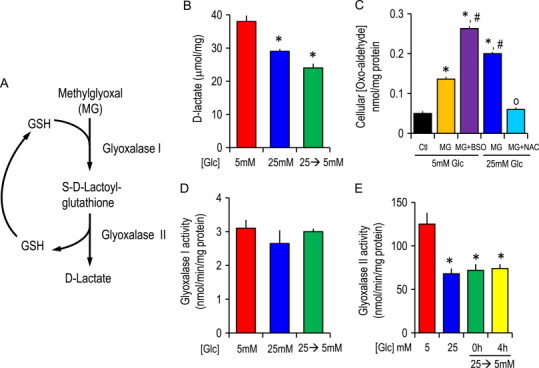
Glyoxalase I and II activities and cellular oxo-aldehyde accumulation in IHECs adapted to normal (5 mM) or high (25 mM) glucose or subjected to acute glycemic fluctuation (25→5 mM glucose). (A) GSH-dependent glyoxalase pathway of MG metabolism: GSH serves as an essential co-factor in glyoxalase I-catalyzed conversion of MG to S-d-lactoylglutathione (SDLG). SDLG is subsequently hydrolyzed to d-lactate by glyoxalase II and GSH is regenerated. (B) The accumulation of d-lactate under conditions of 5, 25 mM or 25→5 mM glucose. d-lactate was determined spectrophotometrically. ⁎p<0.05 vs normal (5 mM) glucose (Glc). (C) The cellular accumulation of α-oxoaldehydes in IHECs cultured in 5 mM normal glucose treated with 300 µM MG without or with BSO or in IHECs adapted to 12 days of 25 mM glucose treated with the same concentration of MG in the absence or presence of NAC. α-Oxoaldehydes levels were determined by HPLC as described in Section Methods. ⁎p<0.05 vs normal (5 mM) glucose; #p<0.05 vs MG treatment in 5 mM glucose; 0p<0.05 vs MG treatment in 25 mM glucose. Glyoxalase I (D) and glyoxalase II (E) activities in IHECs adapted to normal or high glucose or subjected to acute glycemic fluctuation. Glyoxalase I and II activities were determined as the formation of SDLG or d-lactate, respectively. ⁎p<0.05 vs normal (5 mM) glucose. In all studies (B–E), results are mean±SEM for 4 separate IHEC preparations.
Oxo-aldehydes are elevated in diabetic rat plasma, and diabetic brain microvessels are associated with decreased occludin content and increased protein glycation
Using the STZ-induced diabetic rat model, we determined if enhanced protein glycation is a common occurrence in the diabetic brain microvasculature. Fig. 6 shows that total levels of oxo-aldehydes (MG and glyoxal) were elevated in diabetic rat plasma that paralleled the increased in blood glucose, consistent with elevated carbonyl stress during diabetes. These results are in agreement with findings of hyperglycemia and elevated reactive carbonyl species in the plasma of diabetic patients [12].
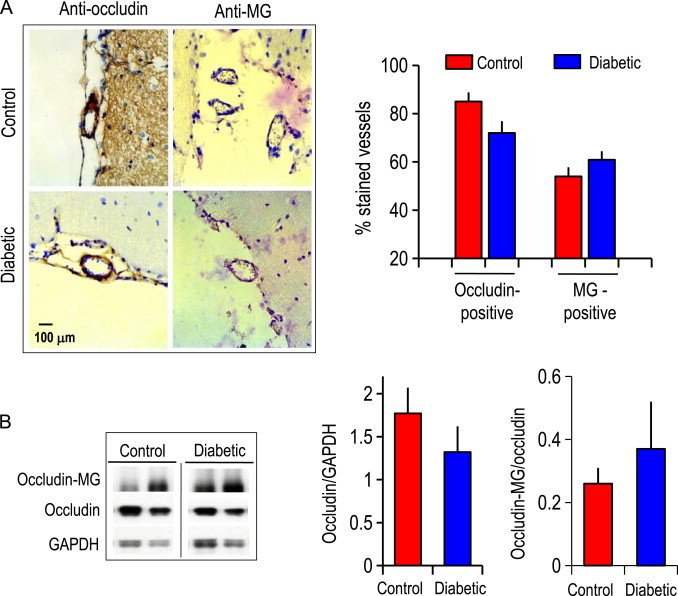
Immunohistochemistry of STZ-induced diabetic rat brain revealed that diabetic brain microvessels displayed 20% less occludin-positive but two-fold higher MG-positive microvessels as compared to non-diabetic brain microvessels (Fig. 7A). Correspondingly, Western blot analyses of brain microvessels confirmed that the expression of occludin was attenuated (40%) while the ratio of glycated-occludin to total occludin was significantly elevated (Fig. 7B). Interestingly, the expression of occludin and glycated protein adducts were not different in macrovessels between normal and diabetic brain (Fig. 8).
Fig. 7.
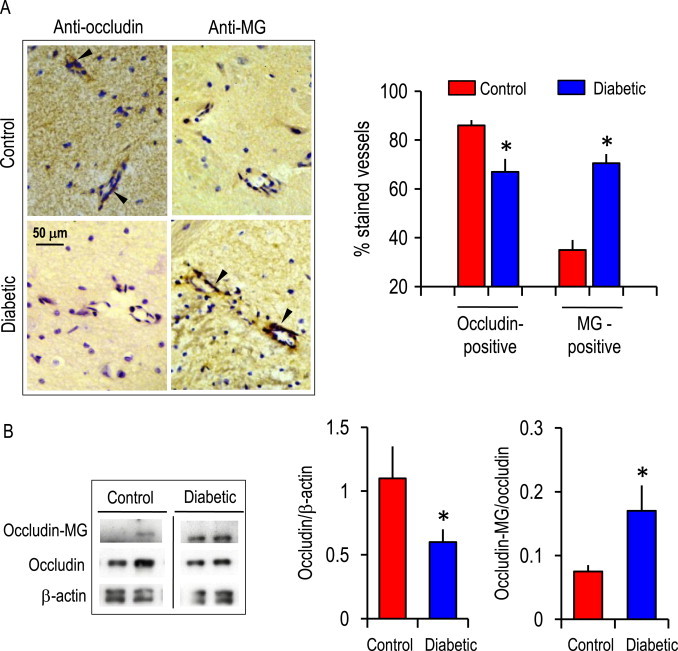
(A) Immunohistochemical staining of occludin and glycated proteins (MG-adducts) in cerebral microvessels in brains from vehicle-treated and diabetic mice at 8 weeks post STZ administration. Arrowheads indicate occludin-positive or MG-positive staining (in dark brown) in representative cerebral microvessels (~50 µm diameter). Cell nuclei are stained blue with DAPI. The right panel shows the number of occludin or MG-positive microvessels, expressed as a percent of total vessels counted (~50–60). ⁎p<0.05 vs control brain. (B) Western blot analysis of occludin and MG–occludin adducts in tissue extracts prepared from brain microvessels. Western blot of two representative brain microvessel preparations of control and diabetic brain are shown. The right panels show the quantification of occludin or MG–occludin band intensities relative to β-actin in brain microvessels (mean±SEM) for 6 separate immunoblots. ⁎p<0.05 vs control brain.
Fig. 8.
(A) Immunohistochemical staining of occludin and glycated proteins (MG-adducts) in cerebral macrovessels in brains from vehicle-treated and diabetic mice at 8 weeks post STZ administration. Occludin-positive or MG-positive staining (in dark brown) of representative cerebral macrovessels (>100 µm diameter) are shown. Cell nuclei are stained blue with DAPI. The right panel shows the number of occludin or MG-positive macrovessels, expressed as a percent of total vessels counted (~20-30). (B) Western blot analysis of occludin and MG–occludin adducts in tissue extracts prepared from brain macrovessels. Western blot of two representative brain macrovessel preparations of control and diabetic brain are shown. The right panels show the quantification of occludin or MG–occludin band intensities relative to GAPDH in brain macrovessels (mean±SEM) for 5 separate immunoblots. There were no statistical differences in contents of occludin or MG-adducts between control and diabetic mice.
Discussion
The current study provides novel evidence that high glucose and acute glucose change potentiate MG-induced brain microvascular endothelial cell barrier dysfunction, a process that was correlated with elevated occludin-MG glycation, decreased glyoxalase II activity, and reduced GSH-dependent cellular MG elimination. Moreover, we demonstrated for the first time that cerebral microvessels in diabetic brain in vivo were highly glycated and exhibited lower expression of the occludin protein than control brain. The significance of low occludin expression and high MG–occludin adduct formation in diabetic brain microvascular dysfunction [18] remains to be determined.
Our results implicate a role for hyperglycemia associated carbonyl stress in the disruption of the cerebral microvasculature. Previously we provided evidence that the brain parenchyma was similarly vulnerable to MG-induced carbonyl stress. We found that MG treatment induced apoptosis in the neuronal cell line, PC12, which was preceded by impaired cellular GSH redox balance, attenuated glucose 6-phosphate dehydrogenase function, and enhanced activator protease factor-1 expression [19]. Significantly, MG-induced PC-12 apoptosis was exacerbated under high glucose conditions [19], consistent with elevated carbonyl stress and enhanced neuronal death. Other investigators have shown that even a physiological level of 5 µM MG combined with high glucose can induce apoptosis and necrosis in human mononuclear cells and human umbilical vein endothelial cells via increases in ROS and alterations in intracellular ATP levels [20,21]. However, we found no evidence of ROS involvement in MG-induced endothelial barrier dysfunction [1], suggesting that carbonyl stress, rather than oxidative stress was the major contributor to the loss of barrier function.
The high resistance of the endothelium of the BBB is determined by the tight junctional complex which restricts paracellular permeability [5,22]. The current results show that the transendothelial electrical resistance of brain microvascular endothelial cell monolayer in culture was disrupted by MG that was correlated with enhanced glycation of the tight junction protein, occludin. Moreover, occludin-glycation was exaggerated in IHECs adapted to high glucose mimicking diabetic states that elevate carbonyl stress. Interestingly, barrier damage persisted during acute glucose normalization. One likely explanation was that at 6-8 h, a significant content of glycated occludin remained (Fig. 2B). The fact that protein carbonylation is an irreversible process and that occludin half-life was around 11 h [23] is consistent with this interpretation. Unfortunately, we encountered problems with maintaining cell viability and stable TEER post 10 h which precluded the examination of the kinetics of barrier function at longer time periods after hyperglycemia. These results suggest that formation of stable protein adducts of MG and occludin is a deleterious process for extended cerebrovascular damage in diabetes.
Notably, MG-induced barrier disruption under these elevated glycemic states was not associated with changes in occludin protein expression, suggesting that, a post-translational process, such as enhanced occludin carbonylation, is likely the major mechanism of hyperglycemia potentiation of endothelial dysfunction. Unlike IHECs, diabetic rat brain exhibited significant decreases in the contents of occludin protein in addition to elevated levels of glycated occludin adducts. In previous studies, we [1] and others [13,24–26] have similarly reported that cerebral occludin content was significantly reduced in STZ-induced diabetic rat. Therefore, the difference in occludin expression in cultured IHECs in high glucose and diabetic brain suggests that other brain cells and/or factors besides hyperglycemia participate in the transcriptional regulation of occludin in vivo. It is noteworthy that the attenuated occludin pool in the diabetic brain appears to be localized to cerebral microvessels, a feature that could contribute to a dysfunctional brain microvasculature during diabetes. Surprisingly, we saw little evidence of glycation of other components of tight junctions, such as ZO-1 or claudin-5 (data not shown) which could be due to inaccessibility of MG to these proteins in the tight junctional complex. Our recent data have demonstrated that pharmacologic levels of MG (1 mM) can markedly induce ZO-1 disorganization in IHEC cells independently of protein carbonylation [1].
Disrupted expressions of ZO-1 and occludin have been described in the brains of various neurological disorders, such as multiple sclerosis [27], HIV-associated dementia [28], and in animal models of Alzheimer's disease [29], Parkinson’s disease [30], and temporal lobe epilepsy [31]. In stroke, hypoxia was shown to relocate claudin-1, ZO-1, and ZO-2 from the plasma membrane to the cytoplasm, while reperfusion elicited MMP-mediated disruption of occludin and claudin-5 [32–35]. It remains to be determined as to whether disrupted components of tight junctions in these pathologies are associated with post-translational oxidative modifications, such as we observed for occludin in the diabetic rat brain. Interesting recent studies revealed that post-translational mechanisms may play a significant role in the regulation of vascular permeability. For instance, occludin phosphorylation and ubiquitination were shown to control tight junction trafficking and vascular endothelial growth factor-induced vascular permeability [36]. Based on the current data, we contend that occludin carbonylation could be a significant contributor to increased permeability of cerebral microvessels in the diabetic brain in vivo. We recently established a mouse model of diabetes and demonstrated that basal BBB permeability, as determined by plasma-to-tissue leakage of Evans Blue, in the diabetic mouse brain at 4 weeks post STZ was 35% higher than WT brains, indicating that the BBB was breached in early diabetes (unpublished).
We have demonstrated that GSH plays an important role in MG elimination and protected against endothelial barrier integrity in brain microvascular endothelial cells grown in normal glucose [1]. In this current study, exogenous NAC afforded similar protection against MG-induced IHEC barrier dysfunction under hyperglycemic conditions. The mechanism of action of NAC in elevated glycemic states is not completely understood. The cysteine residue in NAC could react with MG to yield thiol–aldehyde (thiohemiacetal) adducts [37], but precisely what percent of this no enzymatic MG–thiol adduction was formed and whether NAC was a good nucleophile for MG-adduction are unknown. Our previous finding that BSO ameliorated the effect of NAC in blocking MG-induced TEER loss in IHECs [1] strongly suggests that NAC most likely served as a precursor to maintain endothelial GSH levels to support the glyoxalase pathway in MG metabolism (Fig. 5A). Unexpectedly, we found that glyoxalase II function was compromised by hyperglycemia and glycemic fluctuation. Whether the decreased enzyme activity was due to high glucose-induced decrease in protein expression is unclear; ongoing studies in our laboratory are investigating the influence of hyperglycemia on the transcriptional expression of glyoxalase II. Regardless of mechanism, an attenuated Glo II function means that, besides decreased MG elimination, GSH regeneration from SDLG conversion to d-lactate could also be compromised (Fig. 5A) Consequently, an increase in free MG enhances the potential for protein carbonylation.
It is interesting that we found no change in glyoxalase I activity in high glucose-adapted IHEC cells (Fig. 5D). Overexpression of glyoxalase I was previously shown to reduce hyperglycemia-induced levels of carbonyl stress, AGEs, and oxidative stress in diabetic rats [15]. However, literature evidence is varied on the changes in glyoxalase I and II in diabetes. Glyoxalase I was shown to be increased in insulin-dependent and non-insulin-independent diabetic patients [38], but glyoxalase II activity was increased only in non-insulin-dependent diabetic patients. Therefore, diabetic patients exhibit elevations in both S-d-lactoylglutathione and d-lactate levels. Other studies found that glyoxalase I and II were decreased in the liver but increased in skeletal muscle in STZ diabetic rat. Moreover, glyoxalase I activity in erythrocytes was significantly higher in diabetic patients with microvascular complications (such as nephropathy, retinopathy and neuropathy) than non-diabetic patients [39]. These findings suggest that the expressions or functions of the glyoxalase enzymes are strongly tissue specific and disease-associated.
Plasma α-oxoaldehydes levels were known to be elevated in diabetics [38], which correlates with high hemoglobin A1C contents. Hyperglycemia enhances MG formation, a process that was shown to be potentiated by diabetic ketoacidosis [40], and attenuated by high doses of the anti-glycemic drug, metformin [41]. It is notable that plasma MG levels in diabetic rat (0.75 µmol/l) are at the higher range of those in diabetic patients, ranging from ~200 nmol/l to 1 µmol/l, thus reflecting species-specific differences. In the current study, the cellular MG levels in IHECs were comparable in magnitude to the high values in diabetic rat plasma. If we assume a cell volume of 5 µl/mg protein, the highest concentrations achieved in BSO treated cells (in 5 mM glucose) and cells in adapted to hyperglycemia (25 mM glucose) were estimated to be ~54 and 40 µM. In these cells experiments, a 50 µM MG dose can elicit endothelial barrier damage under high glucose and reduced GSH conditions (Fig. 4), features that characterize the diabetic state.
In summary, this study provides novel insights into a mechanistic basis for diabetes-induced cerebrovascular damage (Fig. 9). The data support the conclusion that carbonyl stress, evidenced by formation of occludin–MG carbonyls, is a likely mechanism of endothelial barrier dysfunction mediated by MG under hyperglycemic states. The study further showed that hyperglycemia compromises GSH-dependent elimination of MG by the glyoxalase pathway and thereby increase the glycating potential of MG. Exogenously administered NAC, likely through GSH production, affords protection against MG-induced endothelial barrier disruption. Taken together, these results will have significant implications for hyperglycemia and carbonyl stress-induced injury to the cerebral microvasculature during diabetes.
Fig. 9.
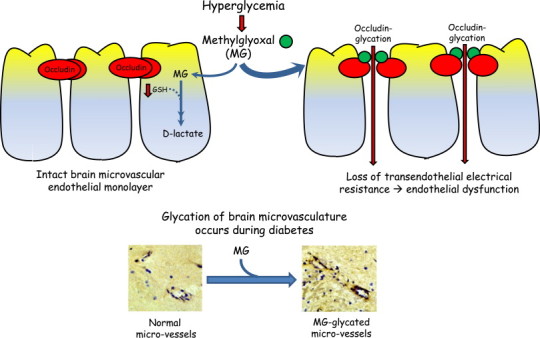
Schematic illustration of (a) MG-induced barrier dysfunction in human brain microvascular endothelial cell monolayer via occludin glycation, a process that is exacerbated by hyperglycemia and decreased GSH (upper panel), and (b) elevated MG-glycation of the cerebral microvasculature in diabetes (lower panel).
Acknowledgments
We thank Christopher Monceaux for help with immunohistochemistry, Dr. Alexander for providing IHEC cells and Dr. Harris for help with diabetes induction in rats. This work was supported by NIH grant DK44510 (TYA) and by an LSUHSC Malcolm Feist Cardiovascular Fellowship (WL).
References
- 1.Li W., Maloney R.E., Circu M.L., Alexander J.S., Aw T.Y. Acute carbonyl stress induces occludin glycation and brain microvascular endothelial barrier dysfunction: role for glutathione-dependent metabolism of methylglyoxal. Free Radical Biology and Medicine. 2013;54:51–61. doi: 10.1016/j.freeradbiomed.2012.10.552. 23108103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huber J.D., VanGilder R.L., Houser K.A. Streptozotocin-induced diabetes progressively increases blood–brain barrier permeability in specific brain regions in rats. American Journal of Physiology—Heart and Circulatory Physiology. 2006;291(6):H2660–H2668. doi: 10.1152/ajpheart.00489.2006. 16951046 [DOI] [PubMed] [Google Scholar]
- 3.Bechmann I., Galea I., Perry V.H. What is the blood–brain barrier (not)? Trends in Immunology. 2007;28(1):5–11. doi: 10.1016/j.it.2006.11.007. 17140851 [DOI] [PubMed] [Google Scholar]
- 4.Shusta E.V. Blood–brain barrier. In: Aird W.C., editor. Endothelial Cells in Health and Disease. Taylor and Francis; New York: 2005. pp. 33–63. [Google Scholar]
- 5.Romero I.A., Radewicz K., Jubin E., Michel C.C., Greenwood J., Couraud P.O. Changes in cytoskeletal and tight junctional proteins correlate with decreased permeability induced by dexamethasone in cultured rat brain endothelial cells. Neuroscience Letters. 2003;344(2):112–116. doi: 10.1016/s0304-3940(03)00348-3. 12782340 [DOI] [PubMed] [Google Scholar]
- 6.Agarwal R., Shukla G.S. Potential role of cerebral glutathione in the maintenance of blood–brain barrier integrity in rat. Neurochemical Research. 1999;24(12):1507–1514. doi: 10.1023/a:1021191729865. 10591399 [DOI] [PubMed] [Google Scholar]
- 7.Ulusu N.N., Sahilli M., Avci A., Canbolat O., Ozansoy G., Ari N. Pentose phosphate pathway, glutathione-dependent enzymes and antioxidant defense during oxidative stress in diabetic rodent brain and peripheral organs: effects of stobadine and vitamin E. Neurochemical Research. 2003;28(6):815–823. doi: 10.1023/a:1023202805255. 12718433 [DOI] [PubMed] [Google Scholar]
- 8.Okouchi M., Okayama N., Alexander J.S., Aw T.Y. NRF2-dependent glutamate-l-cysteine ligase catalytic subunit expression mediates insulin protection against hyperglycemia-induced brain endothelial cell apoptosis. Current Neurovascular Research. 2006;3(4):249–261. doi: 10.2174/156720206778792876. 17109620 [DOI] [PubMed] [Google Scholar]
- 9.Okouchi M., Okayama N., Aw T.Y. Preservation of cellular glutathione status and mitochondrial membrane potential by N-acetylcysteine and insulin sensitizers prevent carbonyl stress-induced human brain endothelial cell apoptosis. Current Neurovascular Research. 2009;6(4):267–278. doi: 10.2174/156720209789630348. 19807652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thornalley P.J. Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems – role in ageing and disease. Drug Metabolism and Drug Interactions. 2008;23(1–2):125–150. doi: 10.1515/dmdi.2008.23.1-2.125. 18533367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thornalley P.J. Glutathione-dependent detoxification of alpha-oxoaldehydes by the glyoxalase system: involvement in disease mechanisms and antiproliferative activity of glyoxalase I inhibitors. Chemico-Biological Interactions. 1998;111–112:137–151. doi: 10.1016/s0009-2797(97)00157-9. 9679550 [DOI] [PubMed] [Google Scholar]
- 12.Stitt A.W., Jenkins A.J., Cooper M.E. Advanced glycation end products and diabetic complications. Expert Opinion on Investigational Drugs. 2002;11(9):1205–1223. doi: 10.1517/13543784.11.9.1205. 12225243 [DOI] [PubMed] [Google Scholar]
- 13.Li W., Prakash R., Kelly-Cobbs A.I., Ogbi S., Kozak A., El-Remessy A.B., Schreihofer D.A., Fagan S.C., Ergul A. Adaptive cerebral neovascularization in a model of type 2 diabetes: relevance to focal cerebral ischemia. Diabetes. 2010;59(1):228–235. doi: 10.2337/db09-0902. 19808897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reed D.J., Babson J.R., Beatty P.W., Brodie A.E., Ellis W.W., Potter D.W. High-performance liquid chromatography analysis of nanomole levels of glutathione, glutathione disulfide, and related thiols and disulfides. Analytical Biochemistry. 1980;106(1):55–62. doi: 10.1016/0003-2697(80)90118-9. 7416469 [DOI] [PubMed] [Google Scholar]
- 15.Dhar A., Desai K., Liu J., Wu L. Methylglyoxal, protein binding and biological samples: are we getting the true measure? Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences. 2009;877(11–12):1093–1100. doi: 10.1016/j.jchromb.2009.02.055. 19299210 [DOI] [PubMed] [Google Scholar]
- 16.Shinohara M., Thornalley P.J., Giardino I., Beisswenger P., Thorpe S.R., Onorato J., Brownlee M. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. Journal of Clinical Investigation. 1998;101(5):1142–1147. doi: 10.1172/JCI119885. 9486985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Talesa V., Uotila L., Koivusalo M., Principato G., Giovannini E., Rosi G. Demonstration of glyoxalase II in rat liver mitochondria. Partial purification and occurrence in multiple forms. Biochimica et Biophysica Acta. 1988;955(1):103–110. doi: 10.1016/0167-4838(88)90183-5. 3382669 [DOI] [PubMed] [Google Scholar]
- 18.Hawkins B.T., Lundeen T.F., Norwood K.M., Brooks H.L., Egleton R.D. Increased blood–brain barrier permeability and altered tight junctions in experimental diabetes in the rat: contribution of hyperglycaemia and matrix metalloproteinases. Diabetologia. 2007;50(1):202–211. doi: 10.1007/s00125-006-0485-z. 17143608 [DOI] [PubMed] [Google Scholar]
- 19.Okouchi M., Okayama N., Aw T.Y. Hyperglycemia potentiates carbonyl stress-induced apoptosis in naïve PC-12 cells: relationship to cellular redox and activator protease factor-1 expression. Current Neurovascular Research. 2005;2(5):375–386. doi: 10.2174/156720205774962665. 16375719 [DOI] [PubMed] [Google Scholar]
- 20.Hsieh M.S., Chan W.H. Impact of methylglyoxal and high glucose co-treatment on human mononuclear cells. International Journal of Molecular Sciences. 2009;10(4):1445–1464. doi: 10.3390/ijms10041445. 19468318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan W.H., Wu H.J. Methylglyoxal and high glucose co-treatment induces apoptosis or necrosis in human umbilical vein endothelial cells. Journal of Cellular Biochemistry. 2008;103(4):1144–1157. doi: 10.1002/jcb.21489. 17721990 [DOI] [PubMed] [Google Scholar]
- 22.Hawkins B.T., Davis T.P. The blood–brain barrier/neurovascular unit in health and disease. Pharmacological Reviews. 2005;57(2):173–185. doi: 10.1124/pr.57.2.4. 15914466 [DOI] [PubMed] [Google Scholar]
- 23.Chen Y.h, Lu Q., Schneeberger E.E., Goodenough D.A. Restoration of tight junction structure and barrier function by down-regulation of the mitogen-activated protein kinase pathway in ras-transformed Madin-Darby canine kidney cells. Molecular Biology of the Cell. 2000;11(3):849–862. doi: 10.1091/mbc.11.3.849. 10712504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li W., Prakash R., Kelly-Cobbs A.I., Ogbi S., Kozak A., El-Remessy A.B. Adaptive cerebral neovascularization in a model of type 2 diabetes: relevance to focal cerebral ischemia. Diabetes. 2010;59(1):228–235. doi: 10.2337/db09-0902. 19808897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antonetti D.A., Barber A.J., Khin S., Lieth E., Tarbell J.M., Gardner T.W. Vascular permeability in experimental diabetes is associated with reduced endothelial occludin content: vascular endothelial growth factor decreases occludin in retinal endothelial cells. Penn State Retina Research Group. Diabetes. 1998;47(12):1953–1959. doi: 10.2337/diabetes.47.12.1953. 9836530 [DOI] [PubMed] [Google Scholar]
- 26.Barber A.J., Antonetti D.A., Gardner T.W. Altered expression of retinal occludin and glial fibrillary acidic protein in experimental diabetes. The Penn State Retina Research Group. Investigative Ophthalmology & Visual Science. 2000;41(11):3561–3568. 11006253 [PubMed] [Google Scholar]
- 27.Bennett J., Basivireddy J., Kollar A., Biron K.E., Reickmann P., Jefferies W.A., McQuaid S. Blood–brain barrier disruption and enhanced vascular permeability in the multiple sclerosis model EAE. Journal of Neuroimmunology. 2010;229(1–2):180–191. doi: 10.1016/j.jneuroim.2010.08.011. 20832870 [DOI] [PubMed] [Google Scholar]
- 28.Kanmogne G.D., Primeaux C., Grammas P. HIV-1 gp120 proteins alter tight junction protein expression and brain endothelial cell permeability: implications for the pathogenesis of HIV-associated dementia. Journal of Neuropathology & Experimental Neurology. 2005;64(6):498–505. doi: 10.1093/jnen/64.6.498. 15977641 [DOI] [PubMed] [Google Scholar]
- 29.Biron K.E., Dickstein D.L., Gopaul R., Jefferies W.A. Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer's disease. PLOS One. 2011;6(8):e23789. doi: 10.1371/journal.pone.0023789. 21909359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen X., Lan X., Roche I., Liu R., Geiger J.D. Caffeine protects against MPTP-induced blood–brain barrier dysfunction in mouse striatum. Journal of Neurochemistry. 2008;107(4):1147–1157. doi: 10.1111/j.1471-4159.2008.05697.x. 18808450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rigau V., Morin M., Rousset M.C., de Bock F., Lebrun A., Coubes P., Picot M.C., Baldy-Moulinier M., Bockaert J., Crespel A., Lerner-Natoli M. Angiogenesis is associated with blood–brain barrier permeability in temporal lobe epilepsy. Brain. 2007;130(7):1942–1956. doi: 10.1093/brain/awm118. 17533168 [DOI] [PubMed] [Google Scholar]
- 32.Fukumoto K., Takagi N., Yamamoto R., Moriyama Y., Takeo S., Tanonaka K. Prostanoid EP1 receptor antagonist reduces blood–brain barrier leakage after cerebral ischemia. European Journal of Pharmacology. 2010;640(1–3):82–86. doi: 10.1016/j.ejphar.2010.05.001. 20470769 [DOI] [PubMed] [Google Scholar]
- 33.Rosenberg G.A., Estrada E.Y., Dencoff J.E. Matrix metalloproteinases and TIMPs are associated with blood–brain barrier opening after reperfusion in rat brain. Stroke. 1998;29(10):2189–2195. doi: 10.1161/01.str.29.10.2189. 9756602 [DOI] [PubMed] [Google Scholar]
- 34.Yang Y., Estrada E.Y., Thompson J.F., Liu W., Rosenberg G.A. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. Journal of Cerebral Blood Flow & Metabolism. 2007;27(4):697–709. doi: 10.1038/sj.jcbfm.9600375. 16850029 [DOI] [PubMed] [Google Scholar]
- 35.Jin R., Song Z., Yu S., Piazza A., Nanda A., Penninger J.M., Granger D.N., Li G. Phosphatidylinositol-3-kinase gamma plays a central role in blood–brain barrier dysfunction in acute experimental stroke. Stroke. 2011;42(7):2033–2044. doi: 10.1161/STROKEAHA.110.601369. 21546487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murakami T., Felinski E.A., Antonetti D.A. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. Journal of Biological Chemistry. 2009;284(31):21036–21046. doi: 10.1074/jbc.M109.016766. 19478092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeng J., Davies M.J. Evidence for the formation of adducts and S-(carboxymethyl)cysteine on reaction of alpha-dicarbonyl compounds with thiol groups on amino acids, peptides, and proteins. Chemical Research in Toxicology. 2005;18(8):1232–1241. doi: 10.1021/tx050074u. 16097796 [DOI] [PubMed] [Google Scholar]
- 38.Rabbani N., Thornalley P.J. Glyoxalase in diabetes, obesity and related disorders. Seminars in Cell and Developmental Biology. 2011;22(3):309–317. doi: 10.1016/j.semcdb.2011.02.015. 21335095 [DOI] [PubMed] [Google Scholar]
- 39.Thornalley P.J. Glyoxalase I − structure, function and a critical role in the enzymatic defence against glycation. Biochemical Society Transactions. 2003;31(6):1343–1348. doi: 10.1042/bst0311343. 14641060 [DOI] [PubMed] [Google Scholar]
- 40.Koc B., Erten V., Yilmaz M.I., Sonmez A., Kocar I.H. The relationship between red blood cell Na/K-ATPase activities and diabetic complications in patients with type 2 diabetes mellitus. Endocrine. 2003;21(3):273–278. doi: 10.1385/ENDO:21:3:273. 14515013 [DOI] [PubMed] [Google Scholar]
- 41.Turk Z., Nemet I., Varga-Defteardarović L., Car N. Elevated level of methylglyoxal during diabetic ketoacidosis and its recovery phase. Diabetes & Metabolism. 2006;32(2):176–180. doi: 10.1016/s1262-3636(07)70266-5. 16735968 [DOI] [PubMed] [Google Scholar]