

Abstract
The NaCl cotransporter (NCC) of the renal distal convoluted tubule is stimulated by low-K+ diet by an unknown mechanism. Since recent work has shown that the STE20/SPS-1-related proline-alanine-rich protein kinase (SPAK) can function to stimulate NCC by phosphorylation of specific N-terminal sites, we investigated whether the NCC response to low-K+ diet is mediated by SPAK. Using phospho-specific antibodies in Western blot and immunolocalization studies of wild-type and SPAK knockout (SPAK−/−) mice fed a low-K+ or control diet for 4 days, we found that low-K+ diet strongly increased total NCC expression and phosphorylation of NCC. This was associated with an increase in total SPAK expression in cortical homogenates and an increase in phosphorylation of SPAK at the S383 activation site. The increased pNCC in response to low-K+ diet was blunted but not completely inhibited in SPAK−/− mice. These findings reveal that SPAK is an important mediator of the increased NCC activation by phosphorylation that occurs in the distal convoluted tubule in response to a low-K+ diet, but other low-potassium-activated kinases are likely to be involved.
Keywords: sodium chloride cotransporter, renal distal convoluted tubule, low-K+ diet, SPAK
understanding how the kidney maintains plasma potassium (K+) homeostasis over a wide range of dietary K+ intake is a long-standing problem in renal physiology. Extensive study has demonstrated a strong impact of changes in dietary K+ on sodium chloride cotransporter (NCC) activity in the distal convoluted tubule (DCT) (2, 9, 10). Aldosterone has long been recognized as a key regulatory factor since aldosterone levels are affected by K+ diet as well as sodium (Na+) diet. While high-K+ and low-Na+ diets both increase aldosterone, the kidney has the remarkable ability to initiate different responses. If aldosterone is increased due to volume depletion such as occurs on a low-Na+ diet, the NCC present in the DCT is activated as well as epithelial Na+ channel (ENaC) in more downstream connecting tubule (CNT) and cortical collecting duct (CCD) segments without disturbing K+ balance. Alternatively, a high-K+ diet elevates aldosterone which acts to promote K+ excretion. But NCC in the DCT is inhibited by high-K+ diet with the physiologically important result that excess K+ can be excreted without disturbing body Na+ balance.
A low-K+ diet is known to increase the abundance and phosphorylation of NCC (2, 10, 35) despite low-aldosterone levels by a so far mysterious mechanism (2, 35). A significant decrease in the abundance of ENaC (10, 21) and the secretory K channel ROMK also occurs with low-K diet (6, 10, 39, 40). This results in reduced Na+ reabsorption and K+ secretion by the CNT/CCD, thereby conserving body K+. Functionally, this aldosterone-independent increase in NCC activity acts to maintain overall Na+ balance even in the face of limited Na+ reabsorption by the CNT/CCD due to the low aldosterone. Although this is an adaptive mechanism for conserving K+ balance, activation of NCC by low-K+ diet may contribute to the rise in blood pressure that is observed in the Na+-rich and K+-deficient Western diet (1).
Multiple studies have identified the STE20/SPS-1-related proline-alanine-rich protein kinase (SPAK) and the related kinase oxidative stress-related kinase (OSR1) as regulators of NCC and NKCC cotransporters (5, 17, 19, 27, 29, 30, 41), serving as terminal kinases in signaling pathways that regulate renal salt transport. Both SPAK and OSR1 can directly phosphorylate NCC and NKCC, and both kinases can be activated by the With No Lysine Kinases (WNKs) (3, 14, 27, 33, 37, 41), which appear to act as intermediate kinases in signaling cascades that couple activation of pressor-activated G protein receptors (4, 31, 34) to activation of NCC and NKCC2. Localization studies have established that both SPAK and OSR1 are expressed in the DCT as well as the thick ascending limb (TAL) of the loop of Henle (19, 41). Despite the apparent functional redundancy of these sister kinases and their overlapping expression pattern in the TAL and DCT, recent studies have illuminated subtle differences between SPAK and OSR1 that suggest they play distinctive physiological roles. Work in genetically modified mice has shown that while OSR1 alone is sufficient to maintain proper TAL function, SPAK is essential for basal activation of NCC and proper DCT function (5, 13, 19, 41). SPAK is primarily responsible for proper activation of NCC in states of sodium deficiency (13, 19), presumably involving activation of the ATR1 receptor by angiotensin (15, 36), while OSR1 has also been found to be important in mediating activation of NCC in response to vasopressin (31) and norepinephrine (34). Here, we explore how the signaling system is affected by dietary potassium.
While SPAK abundance in the cortex is increased during dietary K+ restriction (19, 21), the nature of the K+-sensitive signaling system is still largely unknown. Here, we focus on the most likely downstream signal by testing whether SPAK is responsible for mediating NCC responses to a low-K+ diet.
METHODS
Animals.
The University of Maryland School of Medicine Institutional Animal Care and Use Committee approved all procedures. The generation of SPAK knockout (KO) mice has been described in detail previously (8). The final determination of genotype was assessed by DNA extraction from mouse tails with Extract-N-Amp PCR Mix for SPAK wild-type (WT), and Western blotting of kidney lysate following animal death for SPAK KO. Experimental cohorts were arranged such that WT and KO littermates were paired and equally represented.
Dietary manipulation.
After the genotype (∼8 wk old) was determined, the animals were given a control diet (1% K+, 0.3% Na+, 0.9% Cl−; TD.88238 from Harlan Teklad) for 3 days, and then animals were assigned to a dietary regimen that consisted of either: 1) the control diet or 2) a potassium-deficient diet matched for equal caloric intake to the control diet (15–30 ppm K+, 0.3% Na+, 0.45% Cl−; TD.88239 from Harlan Teklad). The animals were studied after 4 days on this low-K+ or the matched control diet. Both groups consumed similar amounts of the diets.
Antibodies.
The following antibodies were used for both immunoblotting and immunolocalization studies (in all cases sequence numbers refer to that in mouse): rabbit anti-NCC, AA 104–122, chicken anti-pNCC T58, AA 54–66, rabbit anti-SPAK (24), rabbit anti-OSR1 (23). The following antibodies raised in sheep were obtained from the Division of Signal Transduction Therapy at the University of Dundee, Dundee, UK: total SPAK, amino acids: 196–210 (#S150C); total OSR1, amino acids: 333–352 (#S149C); pSPAK/OSR1, pS383/325 (#S670B), pNCC, pT58 (#S995B). Immunolocalization studies used not only the above antibodies but guinea pig anti-NCC (7).
Immunolocalizations.
Anesthetized mice were fixed by perfusion with 2% paraformaldehyde in PBS via the left ventricle for 5 min at room temperature. The kidneys were then removed and fixed an additional 24 h at 4°C in the same fixative, rinsed in PBS, and embedded in paraffin. Cross-sections 3 μm thick, cut at the level of the papilla, were picked up on chrome-alum gelatin-coated glass coverslips and dried on a warming plate. The sections were then deparaffinized in two xylene baths and two absolute ethanol baths, 5 min each, and rehydrated in a graded ethanol series to distilled water. For epitope retrieval, the coverslips were placed in pH 8 aqueous solutions of Tris (1 mM), EDTA (0.5 mM), and SDS (0.02%). The retrieval solution was heated to boiling in a microwave oven, transferred to a conventional boiling water bath for 15 min, and then allowed to cool to room temperature before the sections were thoroughly washed in distilled water to remove the SDS. Sections were preincubated for 30 min with Image-iT blocking solution (Invitrogen), rinsed in PBS, then incubated an additional 30 min in a solution of 2% BSA, 0.2% fish gelatin, 5% normal donkey serum, and 0.2% sodium azide in PBS.
Incubations with specific antibodies (as described above), diluted in PBS containing 1% BSA, 0.2% fish gelatin, 0.1% Tween 20, and 0.2% sodium azide, took place overnight in a humid chamber at 4°C. After thorough washing in high-salt wash (incubation medium plus added sodium chloride at 0.5 M), antibodies were detected with Alexa Fluor-conjugated secondary antibodies. Unconjugated secondary antibodies from Jackson Laboratories were coupled to the respective fluorophores using kits from Invitrogen.
Image analysis.
Images were obtained with Zeiss 410 or 510 laser-scanning confocal microscopes. For quantification and presentation of representative micrographs in the figures, the images from different groups were acquired with identical laser power, pinhole, and contrast settings and within the linear range of the photo detector. Quantitative analysis of antibody-labeling confocal images from four different mice for differences in labeling intensity was performed using Improvision Volocity 5 by a trained investigator who was blind as to the identity of the groups. At least six different tubules sampled per mouse from four WT (≥24 tubules) and four SPAK KO (≥24 tubules) mice were evaluated.
Sample preparation for Western blotting.
Mouse kidney tissue (cortex/medulla) was sonicated in HEENG buffer [20 mM HEPES (pH 7.6), 125 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10% glycerol] containing 1% Triton and 0.5% SDS with protein and phosphatase inhibitor. Samples were sonicated three times with 10-s pulses. The samples were gently agitated at 4°C for 1 h on a rocking shaker and then spun down at high speed (13,600 g) for 10 min to pellet insoluble material. The supernatant was collected and quantified for protein yield using a bicinchoninic acid protein assay reagent kit (Pierce). Equal amounts of kidney protein were suspended in Laemmli buffer (room temperature for 45 min) and resolved on 8% (NKCC2 and NCC) or 10% (SPAK and OSR1) SDS-PAGE gels, and transferred to Amersham Hybond-ECL membranes and blocked in Tris-buffered saline with 0.1% Tween 20 (TBS-T) containing 5% nonfat dry milk (NFDM) for 1 h at room temperature. Membranes were then incubated in 5% NFDM containing primary antibody (4°C, overnight), washed in TBS-T, incubated in 4% NFDM containing horseradish peroxidase-conjugated secondary antibody, and then washed for 10 min (6×) in TBS-T. Bound antibodies were then revealed using enhanced chemiluminescence reagent (Pierce) and fluorography. Quantification of Western blots was performed by scanning autofluorograms using multiple time exposures. Exposures chosen for quantitation were far from saturation for all groups assessed. ImageJ analysis (NIH) was used to quantify differences in protein abundance and phosphorylation.
Statistical analysis.
Data are presented as means ± SE. Statistical analysis was performed using GraphPad PRISM version 5. Statistical significance was determined by t-test when comparing two groups and by two-way ANOVA when comparing multiple groups with two variables (e.g., diet and genotype). For post hoc analysis of multiple group comparisons, Newman-Keuls and Tukey tests were used with identical results. P < 0.05 was considered significant.
RESULTS
Role of SPAK in NCC and pNCC responses to low-K+ diet.
To assess the role of SPAK in elevating NCC phosphorylation in response to low-K+ diet, the SPAK-null mice previously characterized (13) and genetically matched WT controls were placed on a low-K+ diet for 4 days. As shown in Fig. 1, A and B, WT mice showed more than a doubling of NCC abundance and a strong increase in NCC phosphorylation in response to low-K+ diet, corroborating previous observations (35). This response was not detectable in the SPAK-null mice. Since the abundance of NCC is severely reduced in the SPAK-null mice (13), it is possible that the failure to detect an effect of low-K+ diet on phosphorylation of NCC was due to the low amounts of NCC present in SPAK-null mice. To test this possibility, levels of pNCC from SPAK-null mice were analyzed in immunoblots after loading comparable amounts of NCC as observed in WT mice (5 times the levels of protein used in the blots shown in Fig. 1B). Quantitation of these blots shows a statistically significant increase in pNCC levels in SPAK-null mice on a low-K+ diet (Fig. 1C), but smaller than the response in the WT. Indeed, the magnitude of the change in pNCC in the SPAK-null (47%) is statistically less in magnitude than the response to low-K+ diet measured in WT mice (87%). Although the Western blot analysis of this type is semiquantitative, these data suggest that SPAK is an important determinant of NCC phosphorylation in response to low-K+ diet, but that other kinases are also likely to be involved.
Fig. 1.

A: wild-type (WT) mice on a low-K+ diet for 4 days show an increase in total NaCl cotransporter (NCC) expression in cortical homogenates compared with WT mice on a control diet. Increases in response to low-K+ diet were blocked in STE20/SPS-1-related proline-alanine-rich protein kinase knockout (SPAK KO) mice (n = 6, *P < 0.05). B: WT mice on a low-K+ diet for 4 days also show increased pNCC@T58. Increases in pNCC in response to low-K+ diet were not detectable in SPAK KO mice when equal protein was loaded from WT and KO mice (n = 6, *P < 0.05). C: response of WT with normal (1×) loading is compared with the response of KO mice to low-K+ diet when 5× the normal protein load was used to compensate for the reduction of NCC abundance by one-fifth in the SPAK KO mice. Relative abundance (relative to basal phosphorylation of WT) is plotted for genotype and diet. In this case, a 47% increase in pNCC could be detected in KO mice compared with the 87% increase observed in WT mice (n = 10, *P < 0.001). Note: exposure time of blots in B was extended so that low levels of pNCC in the SPAK KO mice could be detected. At these longer exposure times, the signal of the pNCC from WT (Low-K group) is approaching saturation, and thus the change in the WT signal in B is expected to be different (somewhat lower) than seen in C.
Changes in NCC abundance and phosphorylation in response to low-K+ diet were also assessed in immunolocalization studies (Fig. 2) to precisely evaluate and quantitate NCC and pNCC in the same tubules. Low-K+ diet increased pNCC in the DCT of WT mice but this response was significantly impaired in SPAK-null mice (Fig. 2A). Levels of apical NCC labeling and phosphorylation were also assessed in both the DCT1 and DCT2 regions, using calbindin D immunolabeling to identify the DCT2 region (Fig. 2B). To obtain a more direct evaluation of NCC phosphorylation relative to the abundance of NCC, labeling levels were evaluated quantitatively by an observer blinded to the treatment variables (see methods). These studies showed that while levels of NCC phosphorylation in WT mice were strongly increased by low-K+ diet in both regions, no significant effect of low-K+ diet was detected in SPAK KO mice in DCT1 (Fig. 2C). However, in the DCT2 region the level of NCC phosphorylation was very low in WT mice but was increased strongly when mice consumed a low-K+ diet. Interestingly, low-K+ diet significantly increased phosphorylation of NCC in the DCT2 region of SPAK KO mice, although the level of phosphorylation was very blunted (only ∼30% of the WT response) compared with the response to low-K+ diet of WT mice (Fig. 2C).
Fig. 2.

A: immunolocalization studies indicate that low-K+ diet increases both total NCC and pNCC@T58 at the apical surface of distal convoluted tubule (DCT) and that this effect of low-K+ diet is blocked in SPAK KO mice. Bar = 10 μm. B: immunolocalization with calbindin D (a marker to the DCT2 region of the DCT) shows that pNCC abundance is strong in the DCT1 and drops sharply in the adjacent DCT2 region. Bar = 10 μm. C: quantitation of pNCC immunolocalizations shows that low-K+ diet strongly increases apical pNCC in both DCT1 and DCT2 regions in WT mice (n = 4, **P < 0.001). But in SPAK KO mice no significant effect of low-K+ diet could be detected in DCT1 while a small but statistically significant effect was detected in DCT2 (n = 4, *P < 0.05). D: quantitation of pNCC immunolocalizations relative to total NCC shows that DCT1 in WT mice shows a strong increase in relative NCC phosphorylation in response to a low-K+ diet while the DCT2 of WT increases from a much lower level resulting in a stronger relative increase, although the relative levels of the 2 regions become similar following low-K diet. In SPAK KO mice, however, there is only a modest increase in relative labeling in the DCT1 while a strong fractional increase occurs in the DCT2. Nevertheless, relative phosphorylation of NCC remains much lower in SPAK KO than measured in WT mice on a low-K diet (n = 4, *P < 0.05, **P < 0.001).
By measuring the intensity of apical NCC and pNCC in the same tubules, it was possible to directly assess phosphorylation efficiency. This analysis revealed a detectable increase in the proportion of NCC that becomes phosphorylated in response to low-K+ diet in the DCT1 of SPAK-null mice [Δ(pNCC/NCC) = 0.11 ± 0.02], but the response is much smaller than observed in WT mice [Fig. 2D; Δ(pNCC/NCC) = 0.20 ± 0.02 (46% of WT response)]. A similar blunted response was observed in the DCT2 of SPAK-null mice (ΔpNCC/NCC = 0.16 ± 0.04 in SPAK-null vs. ΔpNCC/NCC = 0.40 ± 0.02 in WT, or 40% of WT response, P < 0.0001), but the level of NCC phosphorylation relative to total NCC increased slightly more than in DCT1 (Fig. 2D).
Thus, the phosphorylation of NCC in response to low-K+ diet is blunted in the absence of SPAK. Importantly, however, it is not completely eliminated, especially in DCT2. Taken together, these observations suggest that OSR1 or another kinase is able to phosphorylate NCC even in the absence of SPAK and that this kinase may be more abundant in the DCT2.
SPAK and OSR1 responses to low-K+ diet.
The abundance of SPAK is strongly increased in WT animals on a low-K+ diet as shown both by immunolocalizations (Fig. 3A) and immunoblots (Fig. 3B). Immunoblots reveal that the response involves the higher molecular weight forms of SPAK (50–60 kDa) (13, 19) which are thought to be catalytically active. A similar response was previously reported in rats by Nguyen et al. (21), but not by Castañeda-Bueno et al. (2) (see discussion). Immunoblots also indicate that low-K+ diet enhances phosphorylation of SPAK at its S383 activation site (Fig. 4A). This activation site is also shared by OSR1 which accounts for the low molecular weight band observed in SPAK-null mice in blots with this antibody, and phosphorylation levels of the OSR1 band are not affected by low-K+ diet (Fig. 4A). Immunolabeling studies with the same pSPAK/OSR1 antibody showed a striking increase in pSPAK/OSR1 apical labeling in DCTs of WT mice on low-K+ diet that is not observed in KO mice (Fig. 4B), indicating that SPAK rather that OSR1 is primarily activated by low-K+ diet.
Fig. 3.
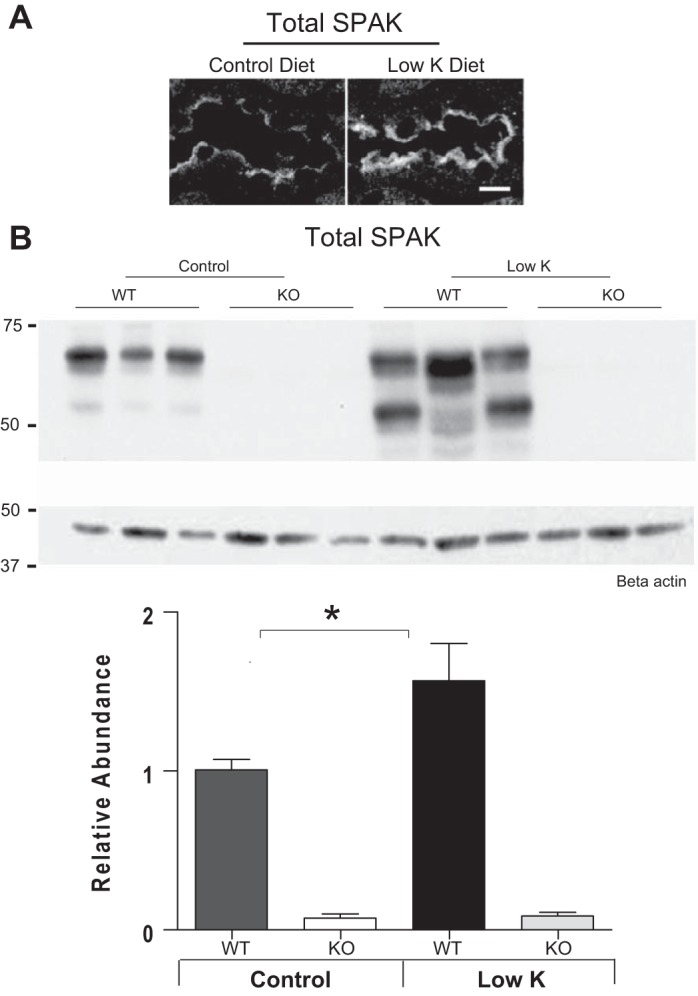
A: immunolocalization studies show that low-K+ diet increases total SPAK in the DCT of WT mice. Bar = 10 μm. B: immunoblots of WT mice on a low-K+ diet for 4 days also show a strong increase in SPAK in the renal cortex (n = 6, *P < 0.05).
Fig. 4.
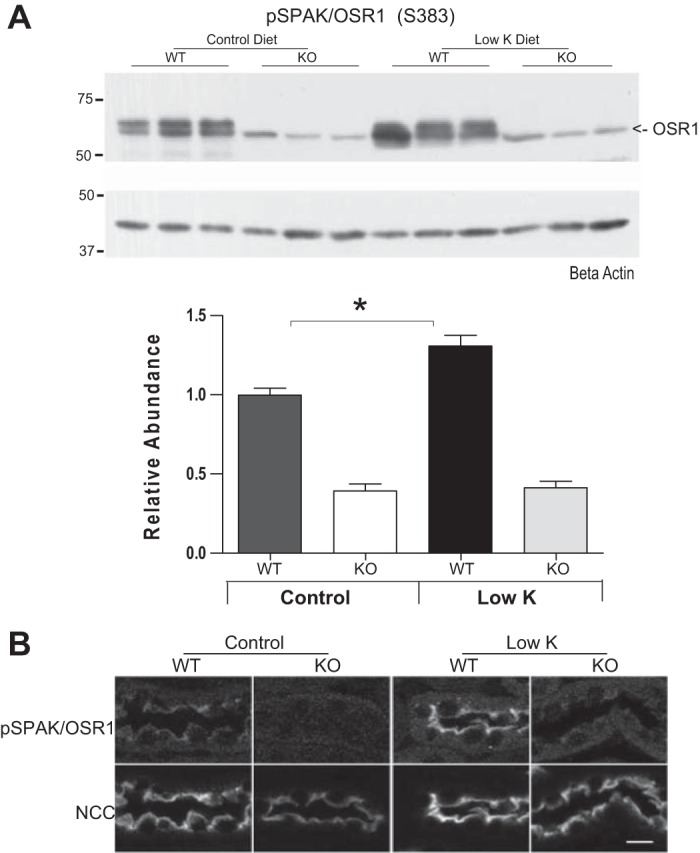
A: WT mice on a low-K+ diet for 4 days show a strong increase in SPAK/oxidative stress-related kinase (OSR1) phosphorylation at their S383/S269 activation sites in the renal cortex (n = 6, *P < 0.05 by ANOVA). Note that the lower molecular weight band detected by this antibody represents phosphorylated OSR1 present in the SPAK KO mice but that its phosphorylation is not significantly changed by low-K+ diet in SPAK KO mice. B: immunolocalizations of pSPAK/OSR1 in WT mice on a low-K+ diet for 4 days show an increase in apical phosphorylation at the apical membrane of DCT segments that is absent in SPAK KO mice. Bar = 10 μm.
To further test the possible role of OSR1 in the NCC response to low-K+ diet, we also assessed total OSR1 abundance in immunoblots of cortex samples. We observed no significant effect of low-K+ diet on OSR1 abundance in either WT or SPAK-null mice (Fig. 5A). However, since OSR1 is highly expressed in the TAL, it is possible that specific changes in OSR1 abundance in the DCT might not be detected in immunoblot assessments. To directly assess OSR1 changes in the DCT, we carried out immunolocalizations of OSR1 that indicated that apical OSR1 increases with low-K+ diet (Fig. 5B). We then carried out quantitative evaluations of the effect of low-K+ diet on OSR1 abundance along the distal tubule using NCC and calbindin D colabeling to distinguish the DCT2 from the DCT1 region. Additionally, since previous studies had shown that the apical localization of OSR1 in the DCT is destabilized in SPAK-null mice (13), we also evaluated quantitatively whether low-K+ diet affects OSR1 localization along the DCT differently in WT and SPAK-null mice. When WT mice were placed on a low-K+ diet for 4 days, there was an increase in apical OSR1 in both the DCT1 and DCT2 regions of the distal tubule (Fig. 5C). By contrast, when SPAK-null mice were placed on a low-K+ diet, the change in apical OSR1 abundance was reduced slightly in DCT1 and DCT2 (Fig. 5C). There was not a consistent effect of low-K+ diet on cytoplasmic OSR1 puncta in these mice (data not shown). Thus, it is possible that apical OSR1 can explain the modest increase in pNCC detected in blots (Fig. 1C) and by immunolocalization (Fig. 2D). However, because the increased apical localization is not accompanied by a detectable phosphorylation-dependent activation of OSR1, it seems more probable that another kinase carries out the muted increase in NCC phosphorylation in SPAK-null mice on a low-K+ diet.
Fig. 5.

A: immunoblots of total cortical homogenates show no significant change in overall OSR1 expression in response to a low-K+ diet. B: immunolocalization of OSR1 showing that low-K+ diet increases apical OSR1 in the DCT. C: quantitation of apical OSR1 immunolocalizations along the DCT shows that low-K+ diet strongly increases apical OSR1 in both DCT1 and DCT2 regions in WT mice (n = 4, *P < 0.01 by ANOVA) but there was no significant effect on apical OSR1 levels in either DCT1 or DCT2 in SPAK KO mice.
DISCUSSION
These studies evaluate the role of SPAK signaling in the stimulation of NCC known to occur in response to a low-K diet (2, 10, 35). We found that while WT mice fed a low-K diet showed strongly increased NCC phosphorylation, this response was blunted but not completely inhibited in the well-characterized SPAK knockout mouse model (13).
A recent study (2) examined the NCC response to low-K+ diet in a knock-in mouse model expressing a mutated SPAK (SPAKT243A/T243A) that cannot be efficiently activated (27). Those studies showed a significant increase in phosphorylation of NCC in these mice in response to low-K diet compared with mice of the same genotype on a control diet. However, a direct quantitative comparison of the response of these mice to WT mice exposed to low-K diet was not performed. Our studies were designed to directly compare the magnitude of the response to low-K+ diet in SPAK KO to what occurs in WT mice. The comparison of responses between WT and SPAK KO mice is difficult because the length of DCT is reduced in SPAK KO mice and thus the amount of NCC is significantly reduced (13). To more fairly assess NCC phosphorylation in response to low-K+ diet, we evaluated comparable amounts of NCC in immunoblot tests of NCC phosphorylation (Fig. 1C). These blots showed a significant increase in phosphorylation due to low-K diet in SPAK KO mice, consistent with the immunoblot findings of Castaneda-Bueno et al. (2). However, we found that the magnitude of the increase in NCC phosphorylation was significantly blunted in SPAK KO mice compared with WT mice. In addition, quantitation of immunolabeling of pNCC and total NCC in the same cells along the DCT revealed that relative NCC phosphorylation at the apical membrane (pNCC/total NCC) was significantly increased by low-K diet in both DCT1 and DCT2 regions of the distal tubule by a much greater extent in WT than in SPAK KO mice (Fig. 2D). Thus, these studies indicate that SPAK plays a role in mediating the increased activity of NCC in the DCT produced by a low-K+ diet.
Consistent with an important role for SPAK in NCC phosphorylation (13, 19, 41), we also found a significant increase in SPAK levels both in immunolocalization and immunoblot studies of WT mice on low-K+ diet (Fig. 3). Analysis of SPAK has also been complicated by the presence of truncated forms of SPAK (2, 13, 27), which have variable catalytic activity. Recent findings indicate that many of the higher molecular weight forms (50–60 kDa) arise from cleavage by the protease aspartyl aminopeptidase-mediated cleavage of the N-terminal PAPA box (18). We found that the higher molecular weight forms of SPAK (50–60 kDa), which are predicted to have catalytic activity (18), are predominately increased when animals are fed a low-potassium diet, consistent with an important role of SPAK in stimulating NCC phosphorylation. Also consistent with the involvement of SPAK in the low-K+ response, we could detect increased phosphorylation at position 383, which reports kinase activation (38). Since the phosphopeptide sequence used to produce this antibody is nearly identical in SPAK and OSR1, the antibody should also be able to detect the presence of OSR1 phosphorylated at this site in SPAK KO mice. However, the amount of pOSR1 in SPAK-null mice was not significantly affected by low-K+ diet (Fig. 4A). Similarly, while we could detect apical labeling of pSPAK/OSR1 in DCTs of WT mice that was increased by low-K+ diet, pOSR1 was very weak and unchanged in SPAK-null mice irrespective of K+ diet (Fig. 4B).
Similar regulation of SPAK abundance and phosphorylation was observed in rats (21), but our results were different from Castañeda-Bueno et al. (2). These investigators reported that potassium depletion caused an increase in levels of pSPAK2 and pKS-SPAK but no changes in total expression of any SPAK forms. Unfortunately, the smaller SPAK forms were not shown on a gel, and it is not clear how SPAK2 and KS-SPAK were defined in these studies. Future studies will be required to elucidate the reason for the variable responses observed, but there are two obvious differences in the experimental design. In contrast to our study with kidney cortex, Castañeda-Bueno et al. (2) examined total kidney, which will contain more of the smaller SPAK forms because these are most abundant in the renal medulla (13, 19). The dietary potassium-depletion regiment used by Castañeda-Bueno et al. (2), of removing potassium citrate, was also different from our potassium chloride depletion protocol.
Importantly, Castañeda-Bueno et al. (2) found that shifting the anion composition of the diet can strongly impact the NCC phosphorylation response to changes in K+ diet (2). Thus, a potential limitation of our study is that changes in intracellular chloride rather than K+ may mediate the activation of SPAK by low-K+ diet. Also of possible relevance to the intracellular signaling mechanism are recent structural studies supporting the concept that WNK kinases may function as chloride sensors (22). It remains to be determined whether intracellular chloride changes in response to dietary K+ restriction.
Our data suggest that OSR1 may normally have a role in the low-potassium response, however. Apical OSR1 labeling along the DCT was significantly increased by low-K+ diet in both the DCT1 and DCT2 of WT mice. A similar effect was observed in SPAK-null mice, but it was blunted compared with the WT (Fig. 5). Taken together, these observations indicate that SPAK is not necessary for apical localization of OSR1, but SPAK may help potentate trafficking or retention of OSR1 in response to low-potassium diet. This is somewhat consistent with evidence showing that long-term dDAVP treatment (31) or acute sympathetic stimulation (34) can produce activation of OSR1 and increased apical pNCC in SPAK-null mice. But unlike the effects of dDAVP or adrenergic stimulation, low-potassium diet does not provoke OSR1 activation, as measured by S325 phosphorylation, in SPAK KO mice. These observations suggest low potassium may work through different signaling systems to phospho-activate NCC than other physiological stimuli. It has been generally assumed that WNK kinases (possibly WNK4) (25) act as the key upstream kinases that phosphorylate OSR1 and SPAK at residue S383/S325 in response to physiological activators. All WNK kinases are capable of phospho-activating either OSR1 or SPAK in vitro systems (11, 12). Our observations suggest that low potassium activates SPAK through different upstream kinases than are involved in activating OSR1 by dDAVP or norepinephrine, possibly through different WNKs.
Kinases other than SPAK and OSR1 may also play a role in the low-K+ diet response, especially in the late DCT. The phosphorylation of NCC was evaluated along the DCT in immunolocalization studies allowing us to compare the response of the early DCT (DCT1), which lacks calbindin D, to the later portion of the DCT (DCT2), where calbindin D is abundant. Interestingly, while pNCC levels in WT animals on control K+ diet were very low in the DCT2 compared with the DCT1, both regions showed a strong increase in pNCC in response to low-K+ diet. In SPAK-null mice the response was blunted, but a significant increase in NCC phosphorylation was still observed with a slightly bigger response DCT2 compared with DCT1. In the absence of OSR1 activation, as discussed above, it seems likely that another kinase increases NCC phosphorylation in the absence of SPAK.
SPAK-mediated NCC phosphorylation in response to low-K+ diet likely plays a role in limiting urinary potassium loss in states of dietary potassium deprivation. SPAK KO mice exhibit a phenotype similar to NCC-null mice with a tendency to develop hypokalemia. Frank hypokalemia develops when SPAK KO (19) or NCC KO mice (20) are placed on a potassium-deficient diet. By activating NCC-mediated sodium reabsorption in the DCT in states of dietary potassium deprivation, less sodium is delivered downstream to the major site of regulated potassium secretion, the CNT/CCD, where potassium is secreted in exchange for sodium. It remains an important question to determine how the SPAK signaling system senses K+. Both intrarenal (28, 32) and gastrointestinal-based (16, 26) potassium-sensing mechanisms have been implicated.
In summary, phosphorylation-dependent activation of NCC is modulated by dietary potassium. Our data indicate that SPAK and possibly OSR1 or another kinase contribute to the aldosterone-independent activation of NCC that occurs in response to low-K+ diet. It will be interesting to learn how K+ is sensed to activate SPAK.
GRANTS
This study was supported by National Institutes of Health Grants DK54231 and DK63049 to P. A. Welling, DK32839 to J. B. Wade, GM074771 and DK093501 to E. Delpire, and 5T32HL072751 to P. R. Grimm.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.B.W., P.R.G., E.D., and P.A.W. conception and design of research; J.B.W., J.L., R.A.C., and P.A.W. analyzed data; J.B.W., J.L., R.A.C., P.R.G., E.D., and P.A.W. interpreted results of experiments; J.B.W., E.D., and P.A.W. drafted manuscript; J.B.W., R.A.C., P.R.G., E.D., and P.A.W. edited and revised manuscript; J.B.W., J.L., R.A.C., P.R.G., E.D., and P.A.W. approved final version of manuscript; J.L. and R.A.C. performed experiments; J.L., R.A.C., and P.A.W. prepared figures.
REFERENCES
- 1.Appel LJ, Brands MW, Daniels SR, Karanja N, Elmer PJ, Sacks FM, American Heart Association. Dietary approaches to prevent and treat hypertension: a scientific statement from the American Heart Association. Hypertension 47: 296–308, 2006. [DOI] [PubMed] [Google Scholar]
- 2.Castaneda-Bueno M, Cervantes-Perez LG, Rojas-Vega L, Arroyo-Garza I, Vazquez N, Moreno E, Gamba G. Modulation of NCC activity by low and high K+ intake: insights into the signaling pathways involved. Am J Physiol Renal Physiol 306: F1507–F1519, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Castaneda-Bueno M, Cervantes-Perez LG, Vazquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G. Activation of the renal Na+:Cl− cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci USA 109: 7929–7934, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castaneda-Bueno M, Gamba G. Mechanisms of sodium-chloride cotransporter modulation by angiotensin II. Curr Opin Nephrol Hypertens 21: 516–522, 2012. [DOI] [PubMed] [Google Scholar]
- 5.Chiga M, Rai T, Yang SS, Ohta A, Takizawa T, Sasaki S, Uchida S. Dietary salt regulates the phosphorylation of OSR1/SPAK kinases and the sodium chloride cotransporter through aldosterone. Kidney Int 74: 1403–1409, 2008. [DOI] [PubMed] [Google Scholar]
- 6.Chu PY, Quigley R, Babich V, Huang CL. Dietary potassium restriction stimulates endocytosis of ROMK channel in rat cortical collecting duct. Am J Physiol Renal Physiol 285: F1179–F1187, 2003. [DOI] [PubMed] [Google Scholar]
- 7.Coleman RA, Wu DC, Liu J, Wade JB. Expression of aquaporins in the renal connecting tubule. Am J Physiol Renal Physiol 279: F874–F883, 2000. [DOI] [PubMed] [Google Scholar]
- 8.Delpire E, Gagnon KB. SPAK and OSR1: STE20 kinases involved in the regulation of ion homoeostasis and volume control in mammalian cells. Biochem J 409: 321–331, 2008. [DOI] [PubMed] [Google Scholar]
- 9.Frindt G, Houde V, Palmer LG. Conservation of Na+ vs. K+ by the rat cortical collecting duct. Am J Physiol Renal Physiol 301: F14–F20, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frindt G, Palmer LG. Effects of dietary K on cell-surface expression of renal ion channels and transporters. Am J Physiol Renal Physiol 299: F890–F897, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gagnon KB, England R, Delpire E. Characterization of SPAK and OSR1, regulatory kinases of the Na-K-2Cl cotransporter. Mol Cell Biol 26: 689–698, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gagnon KB, England R, Delpire E. Volume sensitivity of cation-Cl− cotransporters is modulated by the interaction of two kinases: Ste20-related proline-alanine-rich kinase and WNK4. Am J Physiol Cell Physiol 290: C134–C142, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Grimm PR, Taneja TK, Liu J, Coleman R, Chen YY, Delpire E, Wade JB, Welling PA. SPAK isoforms and OSR1 regulate sodium-chloride cotransporters in a nephron-specific manner. J Biol Chem 287: 37673–37690, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, Nelson-Williams C, Ellison DH, Flavell R, Booth CJ, Lu Y, Geller DS, Lifton RP. Wnk4 controls blood pressure and potassium homeostasis via regulation of mass and activity of the distal convoluted tubule. Nat Genet 38: 1124–1132, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Lee DH, Maunsbach A, Riquier-Brison A, Nguyen M, Fenton R, Bachmann S, Yu A, McDonough AA. Effects of ACE inhibition and ANG II stimulation on renal Na-Cl cotransporter distribution, phosphorylation, and membrane complex properties. Am J Physiol Cell Physiol 304: C147–C163, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee F, Oh G, McDonough A, Youn J. Evidence for a gut factor in K+ homeostasis. Am J Physiol Renal Physiol 293: F541–F547, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Lin SH, Yu IS, Jiang ST, Lin SW, Chu P, Chen A, Sytwu HK, Sohara E, Uchida S, Sasaki S, Yang SS. Impaired phosphorylation of Na+-K+-2Cl− cotransporter by oxidative stress-responsive kinase-1 deficiency manifests hypotension and Bartter-like syndrome. Proc Natl Acad Sci USA 108: 17538–17543, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Markadieu N, Rios K, Spiller BW, McDonald WH, Welling PA, Delpire E. Short forms of SPAK in the kidney are created by Dnpep-mediated proteolytic cleavage. J Biol Chem 289: 29273–29284, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang CL, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab 14: 352–364, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morris RG, Hoorn EJ, Knepper MA. Hypokalemia in a mouse model of Gitelman's syndrome. Am J Physiol Renal Physiol 290: F1416–F1420, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen MT, Yang LE, Fletcher NK, Lee DH, Kocinsky H, Bachmann S, Delpire E, McDonough AA. Effects of K+-deficient diets with and without NaCl supplementation on Na+, K+, and H2O transporters' abundance along the nephron. Am J Physiol Renal Physiol 303: F92–F104, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal 7: ra41, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piechotta K, Garbarini N, England R, Delpire E. Characterization of the interaction of the stress kinase SPAK with the Na+-K+-2Cl− cotransporter in the nervous system: evidence for a scaffolding role of the kinase. J Biol Chem 278: 52848–52856, 2003. [DOI] [PubMed] [Google Scholar]
- 24.Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J Biol Chem 277: 50812–50819, 2002. [DOI] [PubMed] [Google Scholar]
- 25.Ponce-Coria J, Markadieu N, Austin TM, Flammang L, Rios K, Welling PA, Delpire E. A novel Ste20-related proline/alanine-rich kinase (SPAK)-independent pathway involving calcium-binding protein 39 (Cab39) and serine threonine kinase with no lysine member 4 (WNK4) in the activation of Na-K-Cl cotransporters. J Biol Chem 289: 17680–17688, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rabinowitz L. Aldosterone and potassium homeostasis. Kidney Int 49: 1738–1742, 1996. [DOI] [PubMed] [Google Scholar]
- 27.Rafiqi FH, Zuber AM, Glover M, Richardson C, Fleming S, Jovanovic S, Jovanovic A, O'Shaughnessy KM, Alessi DR. Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol Med 2: 63–75, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rengarajan S, Lee DH, Oh YT, Delpire E, Youn JH, McDonough AA. Increasing plasma K+ by intravenous potassium infusion reduces NCC phosphorylation and drives kaliuresis and natriuresis. Am J Physiol Renal Physiol 306: F1059–F1068, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richardson C, Rafiqi FH, Karlsson HK, Moleleki N, Vandewalle A, Campbell DG, Morrice NA, Alessi DR. Activation of the thiazide-sensitive Na+-Cl− cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci 121: 675–684, 2008. [DOI] [PubMed] [Google Scholar]
- 30.Richardson C, Sakamoto K, de los Heros P, Deak M, Campbell DG, Prescott AR, Alessi DR. Regulation of the NKCC2 ion cotransporter by SPAK-OSR1-dependent and -independent pathways. J Cell Sci 124: 789–800, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saritas T, Borschewski A, McCormick JA, Paliege A, Dathe C, Uchida S, Terker A, Himmerkus N, Bleich M, Demaretz S, Laghmani K, Delpire E, Ellison DH, Bachmann S, Mutig K. SPAK differentially mediates vasopressin effects on sodium cotransporters. J Am Soc Nephrol 24: 407–418, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int 83: 811–824, 2013. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi D, Mori T, Nomura N, Khan MZ, Araki Y, Zeniya M, Sohara E, Rai T, Sasaki S, Uchida S. WNK4 is the major WNK kinase positively regulating NCC in the mouse kidney. Biosci Rep 34: 195–205, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terker AS, Yang CL, McCormick JA, Meermeier NP, Rogers SL, Grossmann S, Trompf K, Delpire E, Loffing J, Ellison DH. Sympathetic stimulation of thiazide-sensitive sodium chloride cotransport in the generation of salt-sensitive hypertension. Hypertension 64: 178–184, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vallon V, Schroth J, Lang F, Kuhl D, Uchida S. Expression and phosphorylation of the Na+-Cl− cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol 297: F704–F712, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van der Lubbe N, Lim CH, Fenton RA, Meima ME, Danser AH, Zietse R, Hoorn EJ. Angiotensin II induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter independent of aldosterone. Kidney Int 79: 66–76, 2011. [DOI] [PubMed] [Google Scholar]
- 37.van der Lubbe N, Lim CH, Meima ME, van Veghel R, Rosenbaek LL, Mutig K, Danser AH, Fenton RA, Zietse R, Hoorn EJ. Aldosterone does not require angiotensin II to activate NCC through a WNK4-SPAK-dependent pathway. Pflügers Arch 463: 853–863, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon's hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J 391: 17–24, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wade JB, Fang L, Coleman RA, Liu J, Grimm PR, Wang T, Welling PA. Differential regulation of ROMK (Kir1.1) in distal nephron segments by dietrary potassium. Am J Physiol Renal Physiol 300: F1385–F1393, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei Y, Bloom P, Lin D, Gu R, Wang WH. Effect of dietary K intake on apical small-conductance K channel in CCD: role of protein tyrosine kinase. Am J Physiol Renal Physiol 281: F206–F212, 2001. [DOI] [PubMed] [Google Scholar]
- 41.Yang SS, Lo YF, Wu CC, Lin SW, Yeh CJ, Chu P, Sytwu HK, Uchida S, Sasaki S, Lin SH. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol 21: 1868–1877, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]