

Abstract
While urothelial signals, including sonic hedgehog (Shh), drive bladder mesenchyme differentiation, it is unclear which pathways within the mesenchyme are critical for its development. Studies have shown that fibroblast growth factor receptor (Fgfr)2 is necessary for kidney and ureter mesenchymal development. The objective of the present study was to determine the role of Fgfr2 in the bladder mesenchyme. We used Tbx18cre mice to delete Fgfr2 in the bladder mesenchyme (Fgfr2BM−/−). We performed three-dimensional reconstructions, quantitative real-time PCR, in situ hybridization, immunolabeling, ELISAs, immunoblot analysis, void stain on paper, ex vivo bladder sheet assays, and in vivo decerebrated cystometry. Compared with control bladders, embryonic day 16.5 (E16.5) Fgfr2BM−/− bladders had thin muscle layers with less α-smooth muscle actin and thickened lamina propria with increased collagen type Ia and IIIa that intruded into the muscle. The reciprocal changes in mutant layer thicknesses appeared partly due to a cell fate switch. From postnatal days 1 to 30, Fgfr2BM−/− bladders demonstrated progressive muscle loss and increased collagen expression. Postnatal Fgfr2BM−/− bladder sheets exhibited decreased agonist-mediated contractility and increased passive stretch tension versus control bladder sheets. Cystometry revealed high baseline and threshold pressures and shortened intercontractile intervals in Fgfr2BM−/− versus control bladders. Mechanistically, whereas Shh expression appeared normal, mRNA and protein readouts of hedgehog activity were increased in E16.5 Fgfr2BM−/− versus control bladders. Moreover, E16.5 Fgfr2BM−/− bladders exhibited higher levels of Cdo and Boc, hedgehog coreceptors that enhance sensitivity to Shh, compared with control bladders. In conclusion, loss of Fgfr2 in the bladder mesenchyme leads to abnormal bladder morphology and decreased compliance and contractility.
Keywords: bladder development, bladder dysfunction, fibroblast growth factor receptor 2
murine bladder development begins at embryonic day (E)10.5, when the cloaca is subdivided into the urogenital sinus ventrally and the anal canal dorsally (5). At E13.5, the bladder urothelium induces the surrounding mesenchyme to begin differentiating into the inner lamina propria and outer smooth muscle layers (5). Secretion of sonic hedgehog (Shh) by the bladder urothelium is a major inducer for mesenchymal patterning (5, 7, 8, 20). The E16.5 bladder begins receiving urine from the newly developed metanephric nephrons.
While fibroblast growth factor (Fgf) receptor (Fgfr) activity is critical for kidney development (10–12, 21–25, 32, 35), little is known about its role in bladder morphogenesis. Fgfrs are a family of four receptor tyrosine kinases with up to 22 Fgf ligands (15). While Fgf10 affects bladder urothelial cell patterning, no published data exist on Fgfr actions in the developing bladder mesenchyme (9). Although Fgfr2 is expressed in the bladder urothelium and mesenchyme (12), its roles are unclear, as global Fgfr2 deletion results in early embryonic lethality (2).
Using a Tbx18cre line to delete Fgfr2 in E10.5 peri-Wolffian duct stroma, we observed ureteric bud induction defects, abnormal ureteral insertion into the bladder, and high rates of postnatal vesicoureteral reflux (32). Subsequently, we observed that the Tbx18cre line drove cre expression throughout the E13.5 bladder mesenchyme. Closer inspection of embryonic and early postnatal mutants revealed a previously unrecognized expansion in the bladder lamina propria volume and a reduction in the detrusor muscle volume versus littermate controls. Postnatal mice had worsening muscle loss and fibrosis with correlative contractility defects, poor compliance, and voiding dysfunction. Mutant embryos had augmented hedgehog signaling, likely by derepression of Cdo and Boc expression (hedgehog coreceptors).
MATERIALS AND METHODS
Mouse strains.
To visualize Tbx18cre expression in the developing bladder, Tbx18creTg/+ mice (gift from Feng Chen) (33) were bred with Rosa-CAG-LSL-tdTomato-WPRE (CAG) reporter mice (The Jackson Laboratory, Bar Harbor, ME), which express red fluorescent protein (RFP) in all cre-positive derivatives (Tbx18creTg/+CAG mice). For subsequent experiments, Tbx18creTg/+ mice were bred with Fgfr2Lox/Lox mice (gift from David Ornitz) (34) to produce Tbx18creTg/+Fgfr2Lox/Lox mice with conditional deletion of Fgfr2 within the bladder mesenchyme (Fgfr2BM−/− mice). All experiments were carried out with the approval of the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Genotyping.
Genotyping was performed as previously described (32). Briefly, embryonic tissues or tail clippings were digested overnight, after which DNA was precipitated and resuspended. PCR was then performed to detect the various alleles (see Table 1 for the primer pairs used).
Table 1.
Primers used for PCR genotyping
| Primers |
|||
|---|---|---|---|
| Product Size, bp | Forward | Reverse | |
| Tbx18cre | 301 | 5′-CCATCCAACAGCACCTGGGCCAGCTCAACA-3′ | 5-CCACCATCGGTGCGGGAGATGTCCTTCACT-3′ |
| Fgfr2 (wt) | 307 | 5′-TTGACCGGATCTACACACACC-3′ | 5′-GTCAATTCTAAGCCACTGTCTGCC-3′ |
| Fgfr2 (Floxed) | 373 | 5′-TTGACCGGATCTACACACACC-3′ | 5′-CTCCACTGATTACATCTAAAGAGC-3′ |
| tdTomato (+ve) | 196 | 5′-CTGTTCCTGTACGGCATGG-3′ | 5′-GGCATTAAAGCAGCGTATCC-3 |
| tdTomato (−ve) | 297 | 5′-AAGGGAGCTGCAGTGGAGTA-3′ | 5′-CCGAAAATCTGTGGGAAGTC-3′ |
Fgfr2, fibroblast growth factor receptor 2.
Three-dimensional reconstructions.
Three-dimensional (3-D) reconstructions of E13.5, E16.5, and postnatal day (P)1 bladder tissues were performed as previously described (12). Briefly, paraffin-embedded control and Fgfr2BM−/− tissues were serially sectioned (embryonic tissues at 6 μm and postnatal tissues at 10 μm) through the entire bladder and stained with hematoxylin and eosin (H&E). Individual images were projected to a monitor, and bladder layers were traced. Layers were stacked, and a 3-D reconstruction was rendered (Stereo Investigator, MBF Bioscience, Williston, VT). Volumetric analyses were performed on all reconstructed models (n = 4 per genotype, Neurolucida Explorer, MBF Bioscience).
In situ hybridization.
In situ hybridization was performed on 6-μm paraffin cross-sections through the bladder of control and Fgfr2BM−/− embryos. Briefly, digoxigenin UTP-labeled antisense RNA probes were generated using a DIG RNA labeling kit (catalog no. 11175025910, Roche, Basil, Switzerland) against the genes shown in Table 2. Hybridized probes were detected in sections via anti-digoxigenin-AP Fab fragments (catalog no. 11093274910, Roche) and developed in BM purple (catalog no. 11442074001, Roche). Color reactions were stopped with double distilled H20, mounted with aqueous mounting media, and visualized.
Table 2.
List of in situ hybridization probes
| Gene | Accession Number |
|---|---|
| Bmp4 | NM_007554 |
| Boc | NM_172506 |
| Cdo | NM_001243597 |
| Fgfr2 | NM_201601 |
| Gli1 | NM_010296 |
| Hhip | NM_020259 |
| Ptch1 | NM_008957 |
| Shh | NM_009170.3 |
Bmp4, bone morphogenetic protein 4; Hhip, hedgehog-interacting protein; Ptch1, patched homolog 1; Shh, sonic hedgehog.
Quantitative PCR.
Quantitative PCR was performed as previously described (31). Briefly, Fgfr2BM−/− and control bladders (n = 6) were collected, and RNA was extracted using a Mini-prep kit (catalog no. 74704, Qiagen, Valencia, CA). Quantitative PCR primers for α-smooth muscle actin (α-SMA), collagen type Ia (Col1a), collagen type IIIa (Col3a), Shh, patched homolog 1 (Ptch1), Gli1, hedgehog-interacting protein (Hhip), bone morphogenetic protein 4 (Bmp4), growth arrest specific 1 (Gas1), Boc, Cdo, and Gapdh (endogenous control) were designed using Primer3 software (28). SsoAdvanced universal SYBR green supermix master mix (catalog no. 1725272, Bio-Rad, Hercules, CA) was used on an ABI 7900 HT Real Time PCR System (Applied Biosystems, Foster City, CA) to detect mRNA expression levels. n = 6 samples/genotype were used for analysis.
Histology and immunohistochemistry.
For histology, paraffin-embedded tissues were sectioned at 6 μm and stained with H&E or Mason's trichrome. For immunohistochemistry, paraffin sections were dewaxed, antigen retrieved, blocked, and incubated overnight with antibodies against α-SMA (1:250, catalog no. A5528, Sigma-Aldrich, St. Louis, MO), E-cadherin (1:250, catalog no. 13-1900, Life Technologies, Pittsburgh, PA), Col1a (1:250, catalog no. 1310-01, SouthernBiotech, Birmingham, AL), or Col3a (1:250, catalog no. 1330-01, SouthernBiotech) at 4°C. Sections were then washed and incubated with secondary antibodies (rabbit anti-mouse 594: 1:500, catalog no. A-21205; or sheep anti-goat 488: 1:500, catalog no. A-11055, Molecular Probes, Grand Island, NY) and 4′,6-diamidino-2-phenylindole (DAPI; 1:10,000, catalog no. D9542, Sigma-Aldrich). Sections were then washed, mounted, and visualized.
Apoptosis, proliferation, and cell counting.
To identify proliferating cells, immunohistochemistry was performed using primary antibodies against phospho-histone H3 (1:200, catalog no. 9701, Cell Signaling, Beverly, MA), secondary sheep anti-goat 488 antibodies (1:500), and DAPI (1:10,000) as described above. For each assay, immunohistochemistry was performed on cross-sections through the dome, midpoint, and neck regions of each E16.5 bladder. After staining, the mean value for all sections in a single bladder was used to generate a representative value for each bladder per genotype (n = 4 per genotype). To identify apoptotic cells, TUNEL was performed on paraffin-embedded tissue sections via an ApopTag Fluorescein Direct In Situ Apoptosis Detection Kit (catalog no. S7160, EMD Millipore, Darmstadt, Germany) according to the manufacturer's instructions (in similar cross-sections as for proliferation). DAPI-positive nuclei within the lamina propria were also counted in cross-sections through the dome, midpoint, and neck regions of each E16.5 bladder. Quantification of apoptosis, proliferation, and lamina propria cell number (in cells/area) was determined (n = 4 samples per genotype per assay) was performed using ImageJ software (National Institutes of Health, Bethesda, MD).
Western blot analysis.
Protein samples (30 μg each) from whole embryonic and postnatal bladders were resolved in 8% SDS-Tris gels (catalog no. 161-0732, Bio-Rad). Proteins were electrotransferred to nitrocellulose membranes (catalog no. 162-0115, Bio-Rad), blocked with 5% nonfat dry milk in Tris-buffered saline-Tween 20, and incubated overnight at 4°C with primary antibodies (α-SMA: 1:2,000, Sigma-Aldrich; Gli1: 1:1,000, catalog no. 2534, Cell Signaling; α/β-tubulin: 1:1,000, catalog no. 2148, Cell Signaling). Membranes were washed with Tris-buffered saline-Tween 20 and probed with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:2,000, anti-mouse HRP, catalog no. 7074; anti-rabbit HRP, catalog no. 7076, Cell Signaling). Bound antibodies were visualized using the Amersham ECL Prime Western Blotting Detection kit (catalog no. RPN2232GE, Lifesciences, Pittsburgh, PA) according to the manufacturer's instructions.
Total collagen detection.
E16.5 and P1 control and Fgfr2BM−/− bladders were homogenized in 1 mg/ml pepsin in 0.05 M acetic acid and then incubated in the solution for 48 h at 4°C. The supernatant was collected, and collagen concentration was detected with a Sirius Red Collagen Detection Kit (catalog no. 9062, Chondrex, Redmond, WA) in accordance with the manufacturer's instructions. n = 8 samples/genotype were used in each analysis.
Void stain on paper.
P30 control and Fgfr2BM−/− mice (n = 8) were placed individually in metabolic cages lined with Whatman paper. Mice were given ad libitum access to food and water for a 4-h period. After this period, mice were returned to home cages, Whatman papers were collected, and urine stains were photographed using an ultraviolet light box. All images were subsequently analyzed using ImageJ software.
Bladder sheet assays.
Bladder sheet assays were performed as previously described (13). Briefly, bladders were dissected from male control and Fgfr2BM−/− mice (n = 3 per genotype) and incubated at 37°C for 30 min in Ca2+-free Tyrode solution. After incubation, bladders were cut along the dorsal aspect to form a sheet. Sheets were then pinned to the fixed platform (with the mucosal surface facing up) in the recording chamber, with the dome tied to a stainless steel bar connected to a tension transducer. Contractile activity was measured through a tension transducer attached to an oil hydraulic micromanipulator (Narishige, East Meadow, NY) and computer controlled through a stepper motor to allow precise increments of stretch. Recording chambers were placed on a Peltier block (University of Pittsburgh Machine and Electronics shops) to maintain the temperature at 37 ± 0.5°C and superfused with Tyrode solution at a rate of 1 ml/min. Bladder sheets were stretched to 1 g of resting tension, and all preparations were allowed to equilibrate for at least 30 min.
For agonist treatment, α,β-methylene ATP and carbachol were added to equilibrated bladder sheets at physiological concentrations, with the force contraction of the bladder recorded after administration. Bladders were then washed out and returned to equilibrium before subsequent agonist administration. For electrical field stimulation, equilibrated bladder sheets were stimulated at 30-s intervals with increasing electrical frequency ranging from 8 to 20 Hz. Contractile responses of bladders after each stimulation were recorded as previously described in Ref. 13.
In vivo cystometry.
Male P30 control and Fgfr2BM−/− mice were anesthetized with isoflurane and intubated via a small incision using polyethylene-60 tubing. The skull was exposed, and the forepart to the lambdoid suture was removed. The brain stem was sectioned at the supracollicular level, tissues rostral to the section were withdrawn, and anesthesia was discontinued. After supracollicular decerebration, the bladder was exposed via a midline abdominal incision. A flanged polyethylene-50 catheter was passed through a small opening at the apex of the bladder dome and secured with suture. The catheter was connected by a three-way stopcock to an infusion pump (PUMP 33, Harvard Apparatus) and a pressure transducer (World Precision Instruments) joined to a transbridge (4M, World Precision Instruments). Intravesical pressure recordings were made by continuously infusing saline (25°C) at a constant rate of 0.01 ml/min. Mice were observed for voiding, and voiding contractions were marked.
Statistics.
Student's t-tests were conducted where appropriate using GraphPad Prism 5 (GraphPad Software, La Jolla, CA).
RESULTS
Fgfr2 is deleted in the E13.5 Fgfr2BM−/− bladder mesenchyme.
In E13.5 Tbx18creTg/+CAG bladders, RFP was expressed only in the bladder mesenchyme (Fig. 1). Moreover, while in situ hybridization revealed Fgfr2 mRNA expression in both the urothelium and mesenchyme in E13.5 control mice, E13.5 Fgfr2BM−/− mice had selective loss of Fgfr2 in the bladder mesenchyme (Fig. 1). Quantitative PCR revealed an ∼50% reduction in Fgfr2 mRNA in E13.5 Fgfr2BM−/− versus control whole bladders (Fig. 1). In E16.5 control and Fgfr2BM−/− bladders, immunoblot analysis showed reduced phosphorylation of Erk1/2, downstream effectors of Fgfr signaling in Fgfr2BM−/− bladders (likely reflecting the selective loss of Fgfr2 signaling in the mesenchyme; Fig. 1F). Thus, Fgfr2 expression and signaling appear to be deleted in the bladder mesenchyme of Fgfr2BM−/− mice.
Fig. 1.
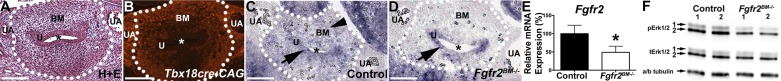
Tbx18cre drives efficient fibroblast growth factor receptor (Fgfr)2 deletion in the embryonic day (E)13.5 bladder mesenchyme (BM). A: hematoxylin and eosin (H&E) stain of a Tbx18cre:CAG red fluorescent protein (RFP) E13.5 bladder cross-section. B: fluorescent image from an adjacent section to that shown in A illustrating RFP-positive cre expression in the bladder mesenchyme but not in the urothelium (U). C and D: in situ hybridization for Fgfr2 showing expression in both bladder mesenchyme (arrowhead) and urothelial tissue layers (arrow) in a control bladder but an absence of Fgfr2 expression in the bladder mesenchyme in a Fgfr2BM−/− bladder. In A–D, the dotted line indicates the outer boundary of the developing bladder. UA, umbilical artery. *Bladder lumen. Scale bars = 150 μm. E: quantitative PCR confirmed the significant reduction in Fgfr2 mRNA expression in Fgfr2BM−/− whole bladders. F: Western blots demonstrating reduced Erk1/2 phosphorylation [phosphorylated (p)Erk1/2] with comparable total (t)Erk1/2 in E16.5 Fgfr2BM−/− bladders compared with age-matched control bladders. a/b Tubulin (Tub), α/β-tubulin (loading control). Values are mean ± SD. *P < 0.05.
E16.5 Fgfr2BM−/− bladders have altered tissue volumes and composition.
Histological analysis and 3-D reconstructions of total bladders revealed no apparent differences between E13.5 control and Fgfr2BM−/− bladders (Fig. 2). Given that bladder muscle development starts at the top of the bladder dome and extends gradually to the neck region, we also performed 3-D analysis of the dome half and neck half of developing bladders separately. As was true for the total bladder, we observed no differences between E13.5 control and Fgfr2BM−/− bladder dome and neck tissue composition or volume (Fig. 2). By E16.5, however, Fgfr2BM−/− bladders exhibited regions of thin detrusor muscle and thickened lamina propria by H&E staining versus age-matched control bladders (Fig. 3). Furthermore, whereas 3-D reconstructions revealed no differences in total bladder and urothelial mean volumes in E16.5 mutants, the mean volume of the lamina propria was increased versus controls (Fig. 3). In addition, E16.5 Fgfr2BM−/− bladder dome regions had a statistical increase in lamina propria tissue volume and neck regions had a significant decrease in smooth muscle volume compared with age-matched control bladder dome regions (Fig. 3). Finally, as a percentage of total, dome, and neck bladder volume, E16.5 mutant muscle was decreased, lamina propria was increased, and the urothelium was unchanged versus controls.
Fig. 2.

Control and Fgfr2BM−/− bladders are comparable at E13.5. A and B: H&E-stained E13.5 sections of control (A) and Fgfr2BM−/− (B) bladders demonstrated similar histology. C and D: three-dimensional (3-D) reconstructions revealed similar lamina propria (LP) volume (red) and muscle (Mus) volume (brown) in E13.5 Fgfr2BM−/− (D) versus control (C) bladders. *Lumen. E: graph of 3D bladder reconstructions confirming comparable total bladder (BV), muscle, lamina propria, and urothelial (Uro) mean volumes in E13.5 controls and mutants. F: graph showing that when normalized to total bladder volumes, mean muscle, lamina propria, and urothelial volume percentages were comparable between Fgfr2BM−/− and age-matched control bladders. G: graph of 3-D bladder “dome” reconstructions confirming comparable bladder, lamina propria, and urothelial mean volumes in E13.5 controls and mutants. H: graph demonstrating that when normalized to bladder dome volume, muscle, lamina propria, and urothelial volume percentages were comparable between Fgfr2BM−/− and control bladders. I: graph of 3-D bladder neck reconstructions confirming comparable bladder, lamina propria, and urothelial mean volumes in E13.5 controls and mutants. J: graph showing that when normalized to bladder neck volume, muscle, lamina propria, and urothelial volume percentages were comparable between Fgfr2BM−/− and age-matched control bladders. Values are means ± SD. Scale bars = 150 μm in A and B.
Fig. 3.

E16.5 Fgfr2BM−/− bladders have reduced muscle and increased lamina propria. A and B: the H&E-stained E16.5 bladder section from an Fgfr2BM−/− embryo (B) appeared to have thinner muscle regions (arrowheads) and an expanded lamina propria compared with that from an age-matched control embryo (A). Scale bars = 300 μm. C and D: 3-D reconstructions revealed an expanded volume of lamina propria (red) and reduced volume of muscle (brown) in E16.5 Fgfr2BM−/− (D) versus control (C) bladders. *Lumen. E: graph of 3-D bladder reconstructions showing comparable total bladder, muscle, and urothelial mean volumes in E16.5 mutants and controls but a significant increase in lamina propria mean volume in Fgfr2BM−/− versus control bladders. F: graph showing that when normalized to total bladder volume, mean muscle volume percentage was reduced, lamina propria percentage was increased, and urothelial percentage was equivalent in E16.5 Fgfr2BM−/− versus control bladders. G: graph of 3-D bladder dome reconstructions showing comparable bladder, muscle, and urothelial mean volumes in E16.5 mutants and controls but a significant increase in lamina propria mean volume in Fgfr2BM−/− versus control bladders. H: graph showing that when normalized to bladder dome volume, mean muscle volume percentage was reduced, lamina propria percentage was increased and urothelial percentage was equivalent in E16.5 Fgfr2BM−/− versus control bladders. I: graph of 3-D bladder neck reconstructions showing comparable bladder, lamina propria, and urothelial mean volumes in E16.5 mutants and controls but a significant decrease in muscle mean volume in Fgfr2BM−/− versus control bladders. J: graph showing that when normalized to bladder neck volume, mean muscle volume percentage was reduced, lamina propria percentage was increased, and urothelial percentage was equivalent in E16.5 Fgfr2BM−/− versus control bladders. Values are means ± SD. *P < 0.05; **P < 0.01.
We then determined if E16.5 mutants had alterations in bladder composition. Immunolabeling revealed robust α-SMA staining in outer mesenchymal layers of control bladders but less intense regional staining in Fgfr2BM−/− bladders (Fig. 4). Quantitative PCR and Western blot analysis confirmed reduced α-SMA mRNA and protein levels in Fgfr2BM−/− versus age-matched control bladders (Fig. 4). Col1a and Col3a immunolabeling revealed more intense signals in E16.5 mutant versus control lamina propria (Fig. 4). Furthermore, Fgfr2BM−/− bladders had collagen fibrils infiltrating into the muscle layer, which were not seen in control bladders (Fig. 4). Quantitative PCR and collagen assays confirmed increases in Fgfr2BM−/− bladder collagen mRNA and protein (Fig. 4). Thus, E16.5 Fgfr2BM−/− bladders have mispatterned muscle and lamina propria versus control bladders.
Fig. 4.

E16.5 Fgfr2BM−/− bladders have decreased smooth muscle and increased collagen levels. A and B: α-smooth muscle actin (α-SMA) immunostaining (red, arrows) revealed regions of thinner muscle in Fgfr2BM−/− (B, arrowheads) versus control (A) bladders. Blue shows 4′,6-diamidino-2-phenylindole (DAPI) nuclear staining. C: quantitative PCR showing significantly reduced α-SMA expression in E16.5 Fgfr2BM−/− versus control bladders. D: Western blots demonstrating reduced total α*SMA protein expression in E16.5 Fgfr2BM−/− compared with age-matched control bladders. α/β-Tubulin was used as a loading control. E and F: collagen type Ia (Col1a) immunohistochemistry revealed more intense staining in the lamina propria (below dotted lines) and regions of collagen infiltration in the smooth muscle layer (arrowheads) of E16.5 Fgfr2BM−/− (F) versus control (E) bladders. G: quantitative PCR showing significantly increased Col1a mRNA expression in E16.5 Fgfr2BM−/− versus control whole bladders. H and I: collagen type IIIa (Col3a) immunostaining showed brighter staining (below dotted lines) and collagen infiltration into the smooth muscle layer (arrowhead) of E16.5 Fgfr2BM−/− (I) versus control (H) bladders. J: quantitative PCR showing significantly increased Col3a mRNA expression in E16.5 Fgfr2BM−/− versus control bladders. K: graph showing increased collagen in whole E16.5 Fgfr2BM−/− bladder lysates compared with age-matched control bladder lysates. Scale bars = 300 μm in A, B, E, F, H, and I. Values are means ± SD in C, G, J, and K. *P < 0.05; **P < 0.01.
E16.5 Fgfr2BM−/− bladders have increased muscle apoptosis, decreased muscle cell proliferation, and increased lamina propria cell numbers.
We then determined whether the changes in E16.5 Fgfr2BM−/− bladder tissue volumes were due to alterations in apoptosis or proliferation. While the mutant lamina propria and urothelium had similar low levels of apoptosis versus controls (not shown), E16.5 Fgfr2BM−/− bladder muscle had a statistical increase in the number of apoptotic cells compared with age-matched control bladder muscle (Fig. 5). Conversely, whereas E16.5 Fgfr2BM−/− bladder lamina propria and urothelial proliferation rates were similar to controls (not shown), mutant muscle did exhibit a statistical decrease in cell proliferation compared with age-matched control muscle (Fig. 5). Although we did not observe changes in lamina propria apoptosis or proliferation between genotypes, Fgfr2BM−/− bladders did have significant increases in both mean lamina propria cell number per cross-section and mean cell density within the lamina propria compared with age-matched control bladders (Fig. 5). Thus, E16.5 Fgfr2BM−/− bladders have perturbed apoptosis and proliferation within the developing muscle layer and not within urothelial or lamina propria tissues. However, the E16.5 Fgfr2BM−/− bladder lamina propria does have increased total cell numbers and cell density versus control bladder lamina propia.
Fig. 5.

E16.5 Fgfr2BM−/− bladder muscle exhibits increased apoptosis and decreased cell proliferation. A and B: representative bladder cross-sections revealed more TUNEL-positive cells (green, arrowheads) in mutant (B) versus control (A) detrusor muscle layers, with a relative lack of staining in both mutant and control lamina propria and urothelium. C and D: representative bladder cross-sections showing decreased phospho-histone H3 positive proliferating cells (red, arrowhead) in muscle layers of E16.5 Fgfr2BM−/− (D) versus control (C) embryos. E: graph showing elevated apoptosis and decreased cell proliferation in Fgfr2BM−/− compared with age-matched control bladders. F and G: graphs demonstrating increased mean cell number per cross section and mean cell density within the lamina propria (in cells/mm2) of Fgfr2BM−/− compared with age-matched control bladders. Scale bars = 300 μm. Values are means ± SD. *P < 0.05.
Postnatal Fgfr2BM−/− mice have perturbations in bladder lamina propria and muscle.
P1 Fgfr2BM−/− bladders appeared smaller with regional muscle thinning (Fig. 6). 3-D reconstructions confirmed that mean Fgfr2BM−/− total bladder and bladder muscle volumes were reduced versus controls (Fig. 6). As a percentage of total bladder volume, P1 mutant muscle was decreased, mutant lamina propria was increased, and the urothelium was unchanged versus control muscle (Fig. 6). Immunostaining, quantitative PCR, and Western blot analysis revealed reduced α-SMA expression in P1 mutant versus control bladder muscle (Fig. 7). Immunostaining revealed more apparent Col1a infiltration in mutant versus control muscle, whereas Col3a staining was not notably different (Fig. 7). By quantitative PCR, Col1a mRNA was significantly increased, and Col3a showed a trend for being increased in P1 Fgfr2BM−/− versus age-matched control bladders. Collagen assays revealed significant increases in P1 Fgfr2BM−/− versus control bladder protein levels (Fig. 7).
Fig. 6.

P1 Fgfr2BM−/− bladders are smaller and have muscle and lamina propria mispatterning. A and B: H&E-stained sections revealed a decrease in bladder size and regions of thin muscle in P1 Fgfr2BM−/− (B) versus control (A) pups. Mus, detrusor muscle. *Lumen. C: graph of 3-D bladder reconstructions demonstrating a significant reduction in total bladder and muscle volumes in Fgfr2BM−/− compared with age-matched control bladders. D: graph showing that when normalized to total bladder volumes, mean muscle percentage was reduced, lamina propria percentage was increased and urothelial percentage was equivalent in P1 Fgfr2BM−/− versus age-matched control bladders. Scale bars = 500 μm. Values are means ± SD. *P < 0.05.
Fig. 7.

P1 Fgfr2BM−/− bladders have decreased smooth muscle and increased collagen. A and B: α*SMA immunostaining (red) in a P1 Fgfr2BM−/− bladder (B) appeared less compact than in a control bladder (A). Blue indicates DAPI nuclear staining. C: quantitative PCR illustrating reduced α-SMA mRNA expression in P1 Fgfr2BM−/− bladders than in control bladders. D: Western blots demonstrating reduced α-SMA protein in P1 Fgfr2BM−/− compared with age-matched control bladders. α/β-Tubulin was used as a loading control. E and F: Col1a labeling revealed regions of increased collagen deposition (arrowhead) in P1 Fgfr2BM−/− (F) versus control (E) bladder muscle layers. G: quantitative PCR showing increased Col1a mRNA expression in P1 Fgfr2BM−/− versus control bladders. H and I: Col3a immunostaining (arrowhead) appeared similar in P1 Fgfr2BM−/− (I) versus control (H) bladder smooth muscle layers. J: quantitative PCR showing a trend for increased Col3a mRNA expression in P1 Fgfr2BM−/− versus control bladders (P = 0.06). K: graph showing increased collagen in whole P1 Fgfr2BM−/− bladder lysates compared with age-matched control bladder lysates. In E, F, H, and I, the dotted line represents the boundary or lamina propria and smooth muscle layer. Scale bars = 300 μm in A, B, E, F, H, and I. Values are means ± SD in C, G, J, and K. *P < 0.05; **P < 0.01.
P30 control and Fgfr2BM−/− bladders were still similar in overall size; however, the mutants had markedly thinner bladder walls, resulting in larger lumens than controls (Fig. 8). Trichrome staining also showed extensive collagen infiltration in the mutant detrusor layer relative to controls (Fig. 8). Quantitative PCR and Western blot analysis revealed less α-SMA in P30 Fgfr2BM−/− versus control bladders (Fig. 8). Finally, quantitative PCR and collagen assays revealed more collagen mRNA and protein in Fgfr2BM−/− versus control bladders(Fig. 8).
Fig. 8.

P30 Fgfr2BM−/− bladders have reduced muscle and increased collagen. A and A′: low-power H&E-stained images of P30 control and Fgfr2BM−/− bladders showing comparable overall sizes but decreased mutant bladder wall thickness versus controls. B and B′: higher-power analysis indicating a thinner and less compact muscle layer in P30 Fgfr2BM−/− (A′, arrowhead) compared with age-matched control (A) bladders. *Lumen. C and C′: trichrome staining showing increased collagen deposition (white arrows) in the detrusor layer of P1 Fgfr2BM−/− (B′) compared with age-matched control (B) bladders. D: quantitative PCR showing significantly reduced α-SMA mRNA expression in P30 Fgfr2BM−/− bladders than in control bladders. E: Western blots demonstrating reduced α-SMA protein in P30 Fgfr2BM−/− compared with age-matched control bladders. α/β- Tubulin was used as a loading control. F and G: quantitative PCR showing increased Col1a and Col3a mRNA expression in P30 Fgfr2BM−/− versus age-matched control bladders. H: graph showing increased collagen in whole P30 Fgfr2BM−/− bladder lysates compared with age-matched control bladder lysates. Scale bar = 700μm in A and A′, 500 μm in B and B′, and 300 μm in C and C′.Values are means ± SD. *P < 0.05; ***P < 0.001.
Postnatal Fgfr2BM−/− mice exhibit bladder dysfunction.
To evaluate functional defects in P1 and P30 Fgfr2BM−/− mice, we first performed assays in isolated bladder sheets. Stimulation with either α,β-methylene ATP (purinurgic agonist) or carbachol (muscarinic agonist) elicited a marked reduction in the Fgfr2BM−/− bladder sheet contractile response versus controls at both ages (Fig. 9). Electrical field stimulation revealed no differences in P1 mutants versus controls (data not shown), whereas P30 mutants had attenuated contractile responses (Fig. 9). With passive stretch, P1 and P30 Fgfr2BM−/− bladder sheets had increased tension (leftward shift of the curves) versus control bladder sheets, indicating decreased compliance (Fig. 9).
Fig. 9.

Fgfr2BM−/− mice have bladder and voiding dysfunction. A: ex vivo bladder sheets from P1 and P30 Fgfr2BM−/− mice exhibited attenuated contractile force generation when stimulated via α,β-methylene ATP (ABMA; purinurgic) or carbachol (muscarinic) agonists compared with age-matched control mice. *P < 0.05; **P < 0.01. B: representative electrical field stimulation profiles of P30 bladder sheets demonstrated an attenuated response to all frequencies in Fgfr2BM−/− compared with age-matched control bladders. C: passive tension profiles in response to stretch showed a leftward shift in Fgfr2BM−/− P1 and P30 compared with age-matched control bladders. D: representative void stain on paper assays from P30 control and Fgfr2BM−/− mice showing the frequency and location of voids (arrows) relative to the source of food and water (diamonds) in the cage. E: representative 20-min in vivo cystometry tracings showing higher baseline and threshold pressures and shortened intercontraction intervals in P30 Fgfr2BM−/− versus control mice (arrows show voiding responses).
We then performed in vivo functional analyses in P30 mice. First, void stain on paper assays revealed that control mice exhibited few void stains (4.14 ± 1.86) distant from the food and water supply, whereas Fgfr2BM−/− mice had multiple void spots (7.83 ± 3.06, P = 0.022) throughout their enclosure. Second, cystometry in decerebrated, live mice showed that Fgfr2BM−/− bladders have higher baseline and threshold pressures and shortened intercontraction intervals versus control bladders. Thus, the bladder mispatterning led to reduced agonist-induced muscle contraction, increased tension/poor compliance, high pressures, and abnormal voiding patterns in Fgfr2BM−/− versus control mice.
Fgfr2BM−/− mice have no evidence of bladder outlet obstruction.
To determine if Fgfr2BM−/− mice had urethral obstruction, we serially sectioned through E14.5 and P1 Fgfr2BM−/− male mice from the bladder to the end of the urethra. The urethra was patent throughout its length at both ages (Fig. 10 and not shown). In addition, Tbx18creTg/+CAG mice had no RFP (cre) expression in the urethral epithelium or surrounding tissue (not shown); thus, Fgfr2 expression would not be perturbed in Fgfr2BM−/− urethras.
Fig. 10.

P1 Fgfr2BM−/− mice have no urethral obstruction. A–X: H&E-stained serial sections from the most distal (A) to proximal portion (X) of the urethra in a P1 Fgfr2BM−/− male mouse showed an observable lumen (arrowhead) at all levels with no evidence of obstruction. Scale bars = 150 μm.
Shh signaling is inappropriately unregulated in embryonic Fgfr2BM−/− bladders.
At E13.5, in situ hybridization and quantitative PCR revealed no changes in Shh or hedgehog target gene expression in Fgfr2BM−/− bladders (not shown). At E16.5, Shh expression still appeared similar in the mutant and control urothelium by in situ hybridization; however, expression of multiple hedgehog readouts appeared to be increased in the Fgfr2BM−/− mesenchyme, including Ptch1, Hhip, and Bmp4 in the lamina propria and Gli1 in the lamina propria and muscle layer (Fig. 11). Moreover, quantitative PCR confirmed no change in Shh but increases in all readouts in E16.5 Fgfr2BM−/− versus control bladders (Fig. 11). Finally, Western blot analysis also showed increased Gli1 protein expression in E16.5 Fgfr2BM−/− versus control bladders (Fig. 11). Thus, while Shh levels appeared equivalent, Shh signaling was inappropriately increased in E16.5 Fgfr2BM−/− bladders.
Fig. 11.
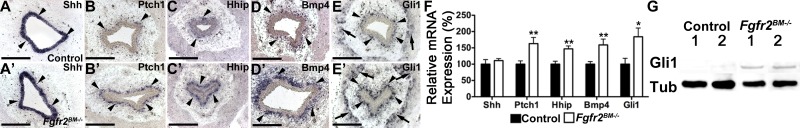
E16.5 Fgfr2BM−/− bladders exhibit augmented sonic hedgehog (Shh) signaling. A: in situ hybridization revealed comparable urothelial Shh mRNA expression (arrowheads) in E16.5 control (A) and Fgfr2BM−/− (A′) bladders. B–D: in situ hybridization revealed that patched homolog 1 (Ptch1), hedgehog-interacting protein (Hhip), and bone morphogenetic protein 4 (Bmp4) mRNA expressions (arrowheads) were more intense in the suburothelial lamina propria in E16.5 Fgfr2BM−/− bladders (B′–E′) than in age-matched control bladders (B–D). E: Gli1 mRNA expression was more robust in both suburothelial (arrowheads) and muscle layers (arrows) in E16.5 Fgfr2BM−/− bladders (E′) than in control bladders (E). F: quantitative PCR confirmed no change in Shh mRNA but increased expression of hedgehog readouts (Ptch1, Hhip, Bmp4, and Gli1) in E16.5 Fgfr2BM−/− versus control bladders. *P < 0.05; **P < 0.01. G: Western blots showing increased Gli1 protein in E16.5 Fgfr2BM−/− compared with age-mated control bladders. α/β-Tubulin was used as a loading control. Values are means ± SD. Scale bars = 300 μm in A–E.
Shh signaling can be enhanced by ectopic expression of cell surface coreceptors Gas1, Boc, and Cdo (1). While quantitative PCR revealed equivalent Gas1 expression in E16.5 Fgfr2BM−/− and control bladders, Fgfr2BM−/− bladders exhibited increased Boc and Cdo levels versus control bladders (Fig. 12). In situ hybridization revealed increased Boc expression in mutant periurothelial lamina propria and in muscle adjacent to the lamina propria versus controls (Fig. 12). Cdo expression in control bladders was restricted to the muscle adjacent to the lamina propria and outer serosa, while Fgfr2BM−/− bladders had stronger and diffuse Cdo expression throughout the smooth muscle layer (Fig. 12). Thus, it is likely that the increases in Shh activity are mediated by increases in Boc and Cdo expression in the E16.5 Fgfr2BM−/− bladder mesenchyme.
Fig. 12.

E16.5 Fgfr2BM−/− bladders have increased Boc and Cdo expression. A: quantitative PCR showing comparable growth arrest specific 1 (Gas1) mRNA expression but elevated Boc and Cdo mRNA expressions in E16.5 Fgfr2BM−/− compared with age-matched control bladders. B and C: in situ hybridization revealed more intense Boc expression in suburothelial (B, arrowheads) and muscle layers adjacent to the lamina propria (C, arrowheads) in E16.5 Fgfr2BM−/− bladders (B′ and C′) than in control bladders (B and C). D and E: in situ hybridization revealed that Cdo mRNA expression was confined to the muscle adjacent to the lamina propria (arrowhead) and outer serosal layer (arrows) of the E16.5 control bladder (D and E), whereas in the E16.5 mutant (D′ and E′), it was strongly expressed throughout the entire muscle layer (arrowheads) up to the serosal layer. Scale bars = 150 μm in B, B′, C, C′, E, and E′ and 300 μm in D and D′. Values are means ± SD. *P < 0.05; **P < 0.01.
DISCUSSION
In a previous publication (32) using the Tbx18cre line to delete Fgfr2, we focused on how loss of the receptor in E10.5 peri-Wolffian duct stroma led to ureteric bud induction defects, abnormal ureteral insertion into the bladder, and high rates of postnatal vesicoureteral reflux. For that study, we only aged mice to P1 and did not systematically examine bladders for structural defects (other than to note that mutants did develop a relatively “normal”-appearing lamina propria and muscle layers based on H&E staining). Subsequent to that study, we noted that mutant mice aged to P30 had obvious bladder histological defects that could not be explained by ureteric induction defects and reflux. Thus, based on the known expression of Tbx18 in the bladder mesenchyme as early as E13.5 (http://www.gudmap.org), we first confirmed that the Tbx18cre line was expressed in and efficiently deleted Fgfr2 in the bladder mesenchyme (Fig. 1). We then began a systematic assessment of structural and functional bladder defects in Tbx18creFgfr2Lox/Lox mice. We ultimately noted that there were indeed changes in mutant E16.5 and early postnatal bladder mesenchyme tissue volumes and composition that had dramatic structural and functional consequences in older postnatal mice.
To our knowledge, Fgfr2 is now only the third known signaling pathway critical for bladder mesenchyme patterning, outside of Shh and the transforming growth factor-β superfamily signaling (5, 7, 8, 14, 20). Interestingly, previous studies (17, 20) have indicated that both Shh and transforming growth factor-β signaling families, in particular Bmp4, are tightly associated (17, 20). While Shh signaling has been shown to modulate smooth muscle differentiation directly, Shh can also regulate the expression of Bmp4, a factor itself capable of inducing smooth muscle patterning (6, 27). In the present study, we demonstrated that Shh signaling activity and Bmp4 expression are concurrently increased in the Fgfr2BM−/− bladder mesenchyme, suggesting two possible mechanisms for altered bladder patterning in our mice.
The alterations in E16.5 Fgfr2BM−/− bladder patterning could be due to a combination of decreased cell survival, reduced proliferation, and altered cell fate determination. Indeed, TUNEL and phospho-histone H3 staining clearly showed that E16.5 Fgfr2BM−/− bladder muscle layers have excessive apoptosis and decreased proliferation compared with age-mated control bladder muscle layers, accounting at least in part for the reduction in muscle. However, the magnitude of these changes may not fully account for the degree of smooth muscle reduction in mutant bladders. Furthermore, Fgfr2BM−/− lamina propria proliferation and apoptosis rates are equivalent to controls, yet total cell number per cross-section and cell density within the lamina propria are significantly increased, indicating that there must be another explanation for the expanded mutant lamina propria, such as a cell fate switch. Indeed, others have shown that hedgehog concentration determines cell fate in the embryonic bladder mesenchyme (4). Cao et al. (4) showed that culture of the fetal urothelium next to intact E13.5 bladders converts the adjacent putative muscle layer to a nonmuscle (i.e., “lamina propria”) fate. Furthermore, coculture of the isolated embryonic bladder mesenchyme with the fetal esophagus, which has very high Shh levels, induces a larger zone of muscle inhibition and a thinner outer muscle layer than with other Shh-expressing epithelia (4). Thus, it follows that the enhanced Shh activity seen in the Fgfr2BM−/− bladder mesenchyme likely contributes to a fate switch of some of the putative muscle cells to lamina propria cells. Together, a cell fate switch and perturbed muscle apoptosis and proliferation likely lead to the observed increases in Fgfr2BM−/− lamina propria and decrease in muscle layer volumes versus controls.
The means by which Fgfr2 represses Shh activity (and Bmp4 expression) in the embryonic bladder appears novel, namely, by dampening Boc and Cdo levels. A recent study (1) has shown that ectopic Boc and/or Cdo expression alone augment Shh activity. Furthermore, Cdo is required for esophageal smooth muscle morphogenesis and orientation (19). Moreover, while a Shh signaling feedback loop may regulate Boc and Cdo levels, there are no published data on other genes or signaling pathways that limit Cdo or Boc expression (1). Thus, directly or indirectly, Fgfr2 appears to repress Cdo and Boc levels in the bladder. This may be relevant in other developing systems in which Fgf and Shh signaling interact.
The reduced bladder muscle volume and expanded lamina propria in Fgfr2BM−/− bladders result in significant structural and functional changes postnatally. The muscle loss likely leads to the decreased contractility, whereas the increased collagen content likely leads to the poor compliance and high pressures seen in functional assays. One previous study (16) showed that an increase in total collagen and a reduction in the smooth muscle-to-connective tissue ratio was strongly associated with bladder dysfunction and a reduction in bladder wall elasticity in humans. Another study (26) found that an increase in the Col1-to-Col3 ratio, as seen in bladders of Col3a-deficient mice, impairs contractility in bladder strips. In Fgfr2BM−/− mice, there appears to be a greater increase in Col1 expression than Col3 (particularly in P1 mice), which may further exacerbate the contractility defects seen in mutants.
Clinical studies have shown that 30–40% of patients (namely children) with bladder dysfunction also exhibit vesicoureteral reflux (VUR) (29, 30). In these patients, the risk of developing chronic kidney disease and reflux nephropathy is greatly increased above those with either VUR or bladder dysfunction alone (3). Despite the apparent association between VUR and bladder dysfunction, there has never been an established genetic link between the two conditions. Our Fgfr2BM−/− mouse model has both high rates of VUR (32) and significant bladder dysfunction, thus providing the first genetic link between the two conditions. It is possible that mutations in the FGF signaling pathway could drive lower urinary tract dysfunction and VUR in some patients with both conditions. Moreover, most children with dysfunctional voiding that present to pediatric urologists or nephrologists are typically thought to have psychosocial defects and not an underlying genetic predisposition. It is possible that permutations in FGF signaling may cause or exacerbate to this clinical condition.
Finally, this study is one of a series from our laboratory on varied (and occasionally opposite) roles of Fgfr2 signaling in the kidney and lower urinary tract mesenchyme. Here, we found that loss of Fgfr2 in the bladder mesenchyme results in embryonic bladder smooth muscle and lamina propria mispatterning and postnatal structural and functional bladder defects. Mechanistically, the embryonic bladder defects appear likely from inappropriate augmented Shh signaling, including increased Bmp4 expression, via derepression of Cdo and Boc. A previous study (18) revealed that Pax3cre-mediated loss of Fgfr2 and concurrent loss of Fgfr1 in the kidney metanephric mesenchyme led to severe defects in metanephric patterning due to excessive apoptosis and aborted ureteric bud induction; moreover, the mice failed to express early (and critical) markers of the metanephric mesenchyme, including Six2, Sall1, and Pax2, and had severely attenuated glial cell line-derived neurotrophic factor levels, likely leading to ureteric induction defects (18). A followup study (23) revealed that while the IIIc isoform of Fgfr2 was most responsible for the early kidney defects seen in Pax3cre-Fgfr1 and Fgfr2 combined mutants, Fgfr2IIIb (the “epithelial” isoform) had an unexpected minor role in mesenchymal development.
More recently, we determined that blockade of Fgfr substrate (Frs)2α-mediated Fgfr2 signaling concurrently with Pax3cre-loss of Fgfr1 in the early kidney mesenchyme resulted in a rescue of the severe renal dysgenesis seen in Pax3cre-Fgfr1 and Fgfr2 mutants; however, the former developed nonautonomous defects in the ureteric lineage characterized by excessive and mispatterned Ret expression, dilated and hyperproliferative ureteric tips, and ultimately renal cystic dysplasia (25). Interestingly, the loss of Fgfr signaling in these Fgfr2-Frs2α/Fgfr1 combined mutants resulted in attenuated Bmp4 expression (unlike the Tbx18cre-Fgfr2 bladder mutants, which had augmented Bmp4 expression). A final set of studies revealed how loss of Fgfr2 from the stromal mesenchyme investing the nephric duct early in kidney development led to ureteric induction defects; this, in turn, caused a range of anomalies, including duplex ureters and kidneys, obstructed hydroureter, renal hypoplasia, renal aplasia, and high rates of vesicoureteral reflux (11, 12, 32). Moreover, as in the kidney mesenchyme, the loss of Fgfr2 expression in the perinephric duct stroma led to a decrease in Bmp4 expression opposite to what occurs with loss of Fgfr2 in the bladder mesenchyme.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants R01-DK-070030 (to C. M. Bates) and DK-093424 (to A. Kanai).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: K.A.W., Y.I., I.Z., C.M.S., D.S.B., W.C.d.G., A.J.K., and C.M.B. conception and design of research; K.A.W., Y.I., I.Z., C.M.S., D.S.B., and A.J.K. performed experiments; K.A.W., Y.I., I.Z., C.M.S., D.S.B., W.C.d.G., A.J.K., and C.M.B. analyzed data; K.A.W., Y.I., I.Z., C.M.S., D.S.B., W.C.d.G., A.J.K., and C.M.B. interpreted results of experiments; K.A.W., Y.I., I.Z., and D.S.B. prepared figures; K.A.W. and C.M.B. drafted manuscript; K.A.W., W.C.d.G., A.J.K., and C.M.B. edited and revised manuscript; K.A.W., C.M.S., W.C.d.G., A.J.K., and C.M.B. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Robert Krauss for Boc and Cdo in situ probe templates.
REFERENCES
- 1.Allen BL, Song JY, Izzi L, Althaus IW, Kang JS, Charron F, Krauss RS, McMahon AP. Overlapping roles and collective requirement for the coreceptors GAS1, CDO, and BOC in SHH pathway function. Dev Cell 20: 775–787, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arman E, Haffner-Krausz R, Chen Y, Heath JK, Lonai P. Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc Natl Acad Sci USA 95: 5082–5087, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avlan D, Gundogdu G, Taskinlar H, Delibas A, Nayci A. Relationships among vesicoureteric reflux, urinary tract infection and renal injury in children with non-neurogenic lower urinary tract dysfunction. J Pediatr Urol 7: 612–615, 2011. [DOI] [PubMed] [Google Scholar]
- 4.Cao M, Liu B, Cunha G, Baskin L. Urothelium patterns bladder smooth muscle location. Pediatr Res 64: 352–357, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao M, Tasian G, Wang MH, Liu B, Cunha G, Baskin L. Urothelium-derived sonic hedgehog promotes mesenchymal proliferation and induces bladder smooth muscle differentiation. Differentiation 79: 244–250, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caubit X, Lye CM, Martin E, Core N, Long DA, Vola C, Jenkins D, Garratt AN, Skaer H, Woolf AS, Fasano L. Teashirt 3 is necessary for ureteral smooth muscle differentiation downstream of SHH and BMP4. Development 135: 3301–3310, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Cheng W, Yeung CK, Ng YK, Zhang JR, Hui CC, Kim PC. Sonic hedgehog mediator Gli2 regulates bladder mesenchymal patterning. J Urol 180: 1543–1550, 2008. [DOI] [PubMed] [Google Scholar]
- 8.DeSouza KR, Saha M, Carpenter AR, Scott M, McHugh KM. Analysis of the sonic hedgehog signaling pathway in normal and abnormal bladder development. PLos One 8: e53675, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eastman R Jr, Leaf EM, Zhang D, True LD, Sweet RM, Seidel K, Siebert JR, Grady R, Mitchell ME, Bassuk JA. Fibroblast growth factor-10 signals development of von Brunn's nests in the exstrophic bladder. Am J Physiol Renal Physiol 299: F1094–F1110, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grieshammer U, Cebrian C, Ilagan R, Meyers E, Herzlinger D, Martin GR. FGF8 is required for cell survival at distinct stages of nephrogenesis and for regulation of gene expression in nascent nephrons. Development 132: 3847–3857, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Hains D, Sims-Lucas S, Kish K, Saha M, McHugh K, Bates CM. Role of fibroblast growth factor receptor 2 in kidney mesenchyme. Pediatr Res 64: 592–598, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hains DS, Sims-Lucas S, Carpenter A, Saha M, Murawski I, Kish K, Gupta I, McHugh K, Bates CM. High incidence of vesicoureteral reflux in mice with Fgfr2 deletion in kidney mesenchyma. J Urol 183: 2077–2084, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ikeda Y, Kanai A. Urotheliogenic modulation of intrinsic activity in spinal cord-transected rat bladders: role of mucosal muscarinic receptors. Am J Physiol Renal Physiol 295: F454–F461, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Islam SS, Mokhtari RB, Kumar S, Maalouf J, Arab S, Yeger H, Farhat WA. Spatio-temporal distribution of Smads and role of Smads/TGF-β/BMP-4 in the regulation of mouse bladder organogenesis. PLos One 8: e61340, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet 20: 563–569, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Landau EH, Jayanthi VR, Churchill BM, Shapiro E, Gilmour RF, Khoury AE, Macarak EJ, McLorie GA, Steckler RE, Kogan BA. Loss of elasticity in dysfunctional bladders: urodynamic and histochemical correlation. J Urol 152: 702–705, 1994. [DOI] [PubMed] [Google Scholar]
- 17.Liu B, Feng D, Lin G, Cao M, Kan YW, Cunha GR, Baskin LS. Signalling molecules involved in mouse bladder smooth muscle cellular differentiation. Int J Dev Biol 54: 175–180, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poladia DP, Kish K, Kutay B, Hains D, Kegg H, Zhao H, Bates CM. Role of fibroblast growth factor receptors 1 and 2 in the metanephric mesenchyme. Dev Biol 291: 325–339, 2006. [DOI] [PubMed] [Google Scholar]
- 19.Romer AI, Singh J, Rattan S, Krauss RS. Smooth muscle fascicular reorientation is required for esophageal morphogenesis and dependent on Cdo. J Cell Biol 201: 309–323, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shiroyanagi Y, Liu B, Cao M, Agras K, Li J, Hsieh MH, Willingham EJ, Baskin LS. Urothelial sonic hedgehog signaling plays an important role in bladder smooth muscle formation. Differentiation 75: 968–977, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Sims-Lucas S, Argyropoulos C, Kish K, McHugh K, Bertram JF, Quigley R, Bates CM. Three-dimensional imaging reveals ureteric and mesenchymal defects in Fgfr2-mutant kidneys. J Am Soc Nephrol 20: 2525–2533, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sims-Lucas S, Cullen-McEwen L, Eswarakumar VP, Hains D, Kish K, Becknell B, Zhang J, Bertram JF, Wang F, Bates CM. Deletion of Frs2α from the ureteric epithelium causes renal hypoplasia. Am J Physiol Renal Physiol 297: F1208–F1219, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sims-Lucas S, Cusack B, Baust J, Eswarakumar VP, Masatoshi H, Takeuchi A, Bates CM. Fgfr1 and the IIIc isoform of Fgfr2 play critical roles in the metanephric mesenchyme mediating early inductive events in kidney development. Dev Dyn 240: 240–249, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sims-Lucas S, Cusack B, Eswarakumar VP, Zhang J, Wang F, Bates CM. Independent roles of Fgfr2 and Frs2α in ureteric epithelium. Development 138: 1275–1280, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sims-Lucas S, Di Giovanni V, Schaefer C, Cusack B, Eswarakumar VP, Bates CM. Ureteric morphogenesis requires Fgfr1 and Fgfr2/Frs2α signaling in the metanephric mesenchyme. J Am Soc Nephrol 23: 607–617, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stevenson K, Kucich U, Whitbeck C, Levin RM, Howard PS. Functional changes in bladder tissue from type III collagen-deficient mice. Mol Cell Biochem 283: 107–114, 2006. [DOI] [PubMed] [Google Scholar]
- 27.Tripathi P, Wang Y, Casey AM, Chen F. Absence of canonical Smad signaling in ureteral and bladder mesenchyme causes ureteropelvic junction obstruction. J Am Soc Nephrol 23: 618–628, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3–new capabilities and interfaces. Nucl Acids Res 40: e115, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ural Z, Ulman I, Avanoglu A. Bladder dynamics and vesicoureteral reflux: factors associated with idiopathic lower urinary tract dysfunction in children. J Urol 179: 1564–1567, 2008. [DOI] [PubMed] [Google Scholar]
- 30.Van Batavia JP, Ahn JJ, Fast AM, Combs AJ, Glassberg KI. Prevalence of urinary tract infection and vesicoureteral reflux in children with lower urinary tract dysfunction. J Urol 190: 1495–1499, 2013. [DOI] [PubMed] [Google Scholar]
- 31.Walker KA, Sims-Lucas S, Caruana G, Cullen-McEwen L, Li J, Sarraj MA, Bertram JF, Stenvers KL. Betaglycan is required for the establishment of nephron endowment in the mouse. PLos One 6: e18723, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walker KA, Sims-Lucas S, Di Giovanni VE, Schaefer C, Sunseri WM, Novitskaya T, de Caestecker MP, Chen F, Bates CM. Deletion of fibroblast growth factor receptor 2 from the peri-wolffian duct stroma leads to ureteric induction abnormalities and vesicoureteral reflux. PLos One 8: e56062, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 33.Wang Y, Tripathi P, Guo Q, Coussens M, Ma L, Chen F. Cre/lox recombination in the lower urinary tract. Genesis 47: 409–413, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130: 3063–3074, 2003. [DOI] [PubMed] [Google Scholar]
- 35.Zhao H, Kegg H, Grady S, Truong HT, Robinson ML, Baum M, Bates CM. Role of fibroblast growth factor receptors 1 and 2 in the ureteric bud. Dev Biol 276: 403–415, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]