

Abstract
In this review we summarize the role of inflammasomes in pancreatic physiology and disease with a focus on acute pancreatitis where much recent progress has been made. New findings have identified inducers of and cell specificity of inflammasome component expression in the pancreas, the contribution of inflammasome-regulated effectors to pancreatitis, and metabolic regulation of inflammasome activation, which are strong determinants of injury in pancreatitis. New areas of pancreatic biology will be highlighted in the context of our evolving understanding of gut microbiome- and injury-induced inflammasome priming, pyroptosis, and innate immune-mediated regulation of cell metabolism.
Keywords: G protein-coupled receptor 109a; G protein-coupled receptor 81; mitochondrial oxidation; NACHT, LRR, and PYD domains-containing protein 3
acute and chronic pancreatitis are inflammatory conditions of the pancreas triggered by acinar cell injury with intrapancreatic trypsinogen activation being a hallmark feature (16). However, intrapancreatic trypsinogen activation accounts for only 50% of acinar cell death in experimental acute pancreatitis (16), and trypsinogen and the trypsinogen-activating protease cathepsin B are dispensible for substantial pancreatic injury, inflammation, and fibrosis in experimental chronic pancreatitis (78). Necrotic cell death triggers a robust sterile inflammatory response (37), and there is substantial evidence that this process contributes to pancreatic injury (32). Innate immune sensing components Toll-like receptor 4 (TLR4) (85) and Toll-like receptor 9 (TLR9) (33) in immune cells and nucleotide-binding oligomerization domain-containing protein 1 (NOD1) (89) in pancreatic acinar cells sense damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) in the injured pancreas. These pathways induce activation of NF-κB (25) and the expression of NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome components and effectors (24, 33). The NLRP3 inflammasome is a macroscopic cytosolic protein complex consisting of NLRP3, apoptosis-associated speck-like protein (ASC), and procaspase 1. It functions to proteolytically activate pro-IL-1β and pro-IL-18 (47) and induce release of active IL-1β, IL-18, and high-mobility group protein B1 (HMGB1) (51) in response to a wide range of stimuli, including extracellular ATP, NAD (80), and saturated free fatty acids (93). We discuss regulation of signals that activate the NLRP3 inflammasome, specifically saturated free fatty acids, necrotic acinar cell death pathways, and DAMP release in the context of pancreatitis. Decreasing saturated free fatty acid production with the lipase inhibitor orlistat limits injury in experimental pancreatitis (62). The NLRP3 inflammasome effectors IL-1β, IL-18, and HMGB1 are in turn all major determinants of pancreatic inflammation, parenchymal cell injury, and disease resolution in acute or chronic pancreatitis (66, 73, 76, 79). Recently, cell surface receptors for metabolites induced by TLR stimulation have been found to suppress NLRP3 inflammasome expression, inflammasome activation, and pancreatic injury in experimental pancreatitis. This provides insights into the metabolic regulation of the inflammasome (19, 31). Finally, cell signaling components that regulate inflammasome signaling have been investigated in pancreatic acinar cells in the setting of pancreatic injury. These recent findings contextualize inflammasome biology in acute pancreatitis to guide further investigation, and each will be discussed in further detail.
Cell-Specific Expression of Inflammasome Components
Acinar cells, ductal cells, and endothelial cells in the normal healthy pancreas of mice, rats, and humans do not express caspase 1, IL-1β, or IL-18. Expression of the inflammasome components NLRP3 and ASC in the resting pancreas has not been reported. Alcoholic pancreatitis accounts for up to 45% of pancreatitis (91), and Gu et al. investigated whether chronic ethanol administration or endotoxemia, which occurs with alcohol ingestion (70), could induce expression of inflammasome components in the rat pancreas (24). A single lipopolysaccharide (LPS) injection or ethanol feeding for 14 wk induced expression of caspase 1 and IL-18 in pancreatic acinar cells. In isolated rat pancreatic acinar cell, LPS treatment induced expression of caspase 1 and IL-18, confirming direct regulation of expression in the acinar cell compartment. Confirming a clinical relevance of these findings, IL-1β, caspase 1, and IL-18 were expressed primarily in pancreatic acinar cells in pancreatic specimens from patients with acute or recurrent acute pancreatitis vs. normal pancreas (24). The authors confirm expression of TLR4 in rat and human pancreatic acinar cells and hypothesize that alcohol-mediated sensitization to injury in pancreatitis through endotoxinemia induced TLR4-mediated stimulation of caspase 1, IL-18, and IL-1β in acinar cells. High-fat diets have also been shown to promote low-level endotoxinemia in mice (7) and in humans (71). Whether high-fat diet-derived endotoxemia induces expression of inflammasome components in pancreatic acinar cells is currently unreported. Acute exposure to ethanol with ethanol gavage induces expression of nlrp3, asc, and caspase1 predominantly in liver mononuclear cells as opposed to parenchymal liver cells such as hepatocytes (72). This result is quite different from the immunochemical studies in pancreatic specimens of ethanol-fed mice in which parenchymal cells, specifically the acinar cells, are found to express nlrp3, asc, and caspase1. These differences may reflect differences from acute vs. chronic ethanol toxicity. Steatohepatitis induced by methionine and choline deficiency promotes endotoxemia in mice and expression of nlrp3, asc, pro-IL-1β, and caspase1 in hepatocytes (15). Such findings provide initial insights into potential TLR4-mediated induction of NLRP3 inflammasome components in the pancreatic acinar cell compartment (Fig. 1). Bacterial peptidoglycans from normal gut flora are present in the systemic circulation and are required to prime innate immune responses in neutrophils through NOD1 signaling, demonstrating for the first time a beneficial effect of normal gut flora microbial components on systemic immune responses (12). It is unknown if the leaky gut induced by high-fat diet or ethanol feeding results in increased sensing of bacterial peptidoglycans through NOD1, NOD2, and/or NLRP1. Many parenchymal cell populations, including pancreatic acinar cells, express functional NOD1 (89). It is an intriguing possibility that NOD1 signaling in acute pancreatitis may induce inflammasome components and effectors in acinar cells (Fig. 1). Similarly, TLR3 is also expressed in acinar cells (44), and TLR3 activation plays an important role in high-fat diet-induced dyslipidemia and inflammation in mice fed a high-fat diet (95). The source of TLR3 ligands, either bacterial products or DAMP release from injured host cells (8), remains to be elucidated. TLR3 ligands administered in vivo also strongly induce expression of the inflammatory caspase 11 (45), discussed in detail below. A role for TLR3 priming of pancreatic acinar inflammatory responses remains unexplored.
Fig. 1.
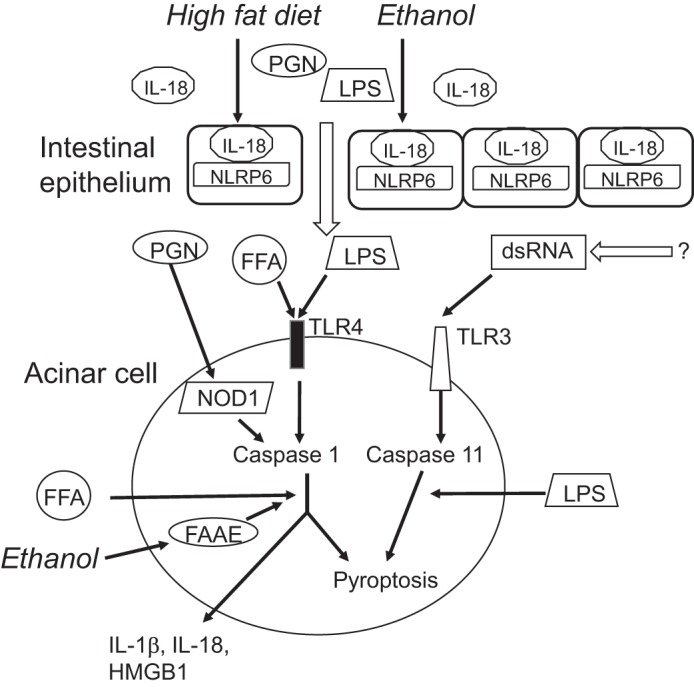
Proposed composite of inflammasome biology in pancreatic acinar cells. NACHT, LRR, and PYD domains-containing protein 6 (NLRP6) and IL-18 maintain intestinal symbiosis and gut barrier function. High-fat diets and ethanol induce a leaky gut and are permissive of peptidoglycan (PGN) and lipopolysaccharide (LPS) translocation, which signal through nucleotide-binding oligomerization domain-containing protein 1 (NOD1) and Toll-like receptor 4 (TLR4), respectively, to prime acinar cell expression of caspase 1 and inflammasome effectors. High-fat diet increases TLR3 signaling with caspase 11 induction, which sensitizes to LPS cytotoxicity through caspase 11-mediated pyroptosis. Ethanol can induce fatty acid acyl ester (FAAE) accumulation in acinar cells, which can be further metabolized to free fatty acids (FFAs). FFAs directly activate TLR4 and also the NLRP3 inflammasome with caspase 1 activation. dsRNA, double-stranded RNA; HMGB1, high-mobility group protein B1.
Inflammasome Activators
Acinar cell necrosis.
The NLRP3 inflammasome is a major sensor of necrotic cell death and required for the sterile inflammatory response to necrosis (37). In many disease states necrosis occurs through a coordinated program termed necroptosis mediated by signaling through receptors harboring a common death domain, suppression of caspase 8 signaling, and signal transduction through the effectors receptor interacting serine/threonine protein kinase 1 and 3 (RIPK1 and RIPK3) (28, 30). The contribution of necroptosis pathways through death domain-containing receptor pathways, including tumor necrosis factor (TNF)-α, Fas, and TNF-related apoptosis-inducing ligand (TRAIL), has been investigated in acute pancreatitis. TNF-α induces necrosis in primary acinar cells (82), and TNF-α deletion or in vivo blockade mitigates pancreatic inflammation and necrosis in experimental acute pancreatitis (17, 55, 65). RIPK3 is required to induce acinar cell necrosis in experimental acute pancreatitis and limits pancreatic inflammation and injury (28). Therefore, acinar cell necrosis in acute pancreatitis occurs predominantly through specific pathways. TRAIL family receptors are expressed de novo in acinar cells in human chronic pancreatitis (27) although the role of TRAIL blockade is unexplored in experimental pancreatitis. Fas is required to limit pancreatic necrosis in experimental acute pancreatitis (38). Fas promotes apoptosis, a noninflammatory form of cell death, in many cell types (1). However, since apoptosis was not identified to be a significant contributor to acinar cell death in murine experimental pancreatitis, the mechanism of Fas-mediated protection in acute pancreatitis remains to be determined (39). CD11c-positive dendritic cells serve a nonredundant function in the phagocytic clearance of injured pancreatic cells in acute pancreatitis and are required to limit pancreatic injury in experimental acute pancreatitis (4). Persistence of necrotic acinar cells therefore promotes an injurious robust sterile inflammatory response in acute pancreatitis. Release of ATP, NAD, HMGB1, and nucleic acids from necrotic cells activates or induces the NLRP3 inflammasome, and these interactions are further discussed below.
P2X7 and CD39.
ATP and NAD released from necrotic cells promotes NLRP3 inflammasome activation in immune cells by stimulating the cell surface receptor P2X7 (80). Genetic deletion of P2X7, enhanced metabolic clearance of extracellular ATP through apyrase injection, or competitive inhibition of extracellular NAD for P2X7 activation through injection of etheno-NAD all suppress pancreatic inflammation, pancreatic caspase 1 activity, and pancreatic injury in experimental acute pancreatitis (33). Pancreatic stellate cells are the major drivers of pancreatic fibrosis. At low concentrations, extracellular ATP promotes cell proliferation in primary pancreatic stellate cells in culture in a P2X7-dependent manner (26); at high concentrations, extracellular ATP induces stellate cell death with P2X7 dependence. Mice deficient in CD39, the major extracellular space ectonucleotidase that hydrolyzes extracellular ATP to ADP and AMP, are predicted to have higher extracellular concentrations of ATP at sites of injury or inflammation and greater P2X7-mediated pancreatic stellate cell death. CD39-deficient mice have decreased pancreatic atrophy and decreased fibrogenesis in experimental pancreatic fibrosis (49), consistent with decreases in fibrogenic pancreatic stellate cell survival. Of note, CD39 and P2X7 interplay is a strong determinant of cell death in other immune cell compartments, including mast cells (48). Finally, the closely related hepatic stellate cell expresses NLRP3 inflammasome and develops a fibrogenic phenotype in response to the NLRP3 activator monosodium urate crystals (92).
Free fatty acids.
Free fatty acids have been implicated as endogenous ligands for TLR4 through binding of the acute phase protein fetuin A (69) and are also direct activators of the NLRP3 inflammasome (93). The unsaturated free fatty acids oleate and linoleate are cytotoxic to isolated pancreatic acinar cells (62). To date, it is unreported if there is enhancement of free fatty acid cytotoxicity to acinar cells in the presence of fetuin A to promote TLR4 activation or in the presence of prior LPS treatment to induce caspase 1 in acinar cells and potentially transduce fatty acid-mediated NLRP3 inflammasome activation. Local free fatty acid production is thought to be mediated by pathological intrapancreatic lipase activity in the inflamed pancreas. Navina et al. identify that regions of pancreatic necrosis colocalize to regions of nonencapsulated adipose tissue in pancreata in human clinical samples in acute pancreatitis, suggesting a significant contribution of lipotoxicity in the human clinical condition. They further demonstrate that in vivo administration of the lipase inhibitor orlistat mitigates experimental pancreatic inflammation and injury (62). Free fatty acids contribute to cytotoxicity in alcoholic pancreatitis through nonoxidative metabolism of ethanol to fatty acid acyl esters (FAAEs), which accumulate in human pancreatitis specimens (99) and are directly cytotoxic to acinar cells (14). FAAEs accumulated in acinar cells with ethanol ingestion are thought to be cleaved to free fatty acids in acinar cells, providing a mechanism for cytosolic free fatty acid accumulation (Fig. 1). The potential for this mechanism to enhance fatty acid-mediated activation of the NLRP3 inflammasome is unexplored. FAAE formation is required to induce injury in alcoholic pancreatitis in vivo in rodent models as evidenced in vivo through use of small molecule inhibitors of FAAE synthase in experimental alcoholic pancreatitis (35). Complementing these findings, genetic deletion or chemical inhibition of alcohol dehydrogenase shifts alcohol metabolism towards fatty acid ethyl ester formation in rodents and human cells, increases pancreatic FAAE production, and induces more severe pancreatic injury in experimental alcoholic pancreatitis (5, 42). A role for TLR4 and NLRP3 activation in experimental alcoholic pancreatitis is currently unreported.
Inflammasome Components NLRP3 and ASC
NLRP3, ASC, and caspase 1 constitute the NLRP3 inflammasome in the setting of NLRP3-activating ligands. Genetic deficiency of NLRP3 or ASC significantly decreases pancreatic inflammation and injury (33). The role of the NLRP3-activating ligands in acute pancreatitis, including extracellular ATP and NAD as well as saturated free fatty acids, are discussed above. Mitochondrial-derived reactive oxygen species are required for NLRP3 inflammasome signaling (29). Recently, hydrogen-infused saline, a scavenger of reactive oxygen species, has been shown to suppress reactive oxygen species, NRLP3 inflammasome activation, inflammation, and cell death in the pancreas when administered postinsult in the caerulein model of experimental pancreatitis in mice (77). A potential role of other NLR family members in caspase 1 activation in pancreatic inflammation has not been reported. The ligands for other well-studied inflammasomes such as NLRP1, NLRC4, and absent in melanoma 2 are of microbial origin, with no clear endogenous ligands detected to date (51). The NLRP6 inflammasome was recently identified as a critical regulator of host defense through production of antimicrobial IL-18 in intestinal epithelial cells (18). Increased gut permeability is an early feature of clinical acute pancreatitis without systemic bacterial translocation (2). NOD1 in acinar cells contributes to inflammation and injury in experimental pancreatitis through recognition of microbial peptidoglycans. Compromise of intestinal epithelial NLRP6 inflammasome-mediated IL-18 release as a mechanism of increased intestinal permeability in acute pancreatitis and increased acinar cell inflammatory responses through NOD1 and TLR4 remain to be investigated (Fig. 1).
Genetic dysregulation of the NLRP3 inflammasome.
Recently IκB kinase α (IKKα) was identified as a negative regulator of inflammasome activation independent of RelA and NF-κB pathway activation. IKKα directly interacts with ASC in resting cells sequestering it away from other inflammasome components. NLRP3-activating signals suppress IKKα kinase activity, dissociate ASC from IKKα, and result in NLRP3-ASC interaction, with NLRP3 inflammasome formation. Moreover, genetic deletion of IKKα results in NLRP3 inflammasome hyperactivation in immune cells, implicating IKKα as an endogenous negative regulator of the NLRP3 inflammasome (57). Specific deletion of IKKα in acinar cell results in spontaneous acinar cell death, pancreatic atrophy, inflammation, and fibrosis in mice (53). A role of inflammasome hyperactivation in acinar cells in this phenotype remains to be explored. Knock-in mice harboring a missense mutation in NLRP3 that induces spontaneous inflammasome activation develop spontaneous neutrophil infiltration in multiple organs, although not in the pancreas (6). The potentially increased susceptibility of such mice to pancreatic injury in experimental pancreatitis models or from ethanol feeding is unexplored.
Caspase 1 and caspase 11.
Caspase 1 activation is required not only for IL-1β, IL-18, and HMGB1 processing and release in response to NLRP3 activation but also for inflammatory cell death. Caspase 1 activity is required for significant injury and inflammation in many experimental models of acute pancreatitis. Genetic deficiency of caspase 1 or injection of small molecule inhibitors of caspase 1 protease activity suppress pancreatic injury and inflammation in experimental acute pancreatitis in mice and rats (33, 76). Independent of NLRP3 inflammasome formation, caspase 1 can mediate an inflammatory form of cell death termed pyroptosis, which requires ASC oligomerization and recruitment of activated caspase 1 into a cytosolic complex independent of NLPR3 termed the pyroptosome. Caspase 11 can form a pyroptosome independent of ASC and NLRP3 with as yet undefined elements (45). Pyroptosis is an inflammatory form of cell death that involves pyroptosome formation, caspase 1 or caspase 11 activation, rapid plasma membrane rupture, and release of proinflammatory cellular contents, including IL-1α and HMGB1 (50). A potential role for caspase 11 in the inflammatory activation in acute pancreatitis derives from experiments in the initially reported caspase 1 null mice. These mice were in fact null for caspase 11 and caspase 1. Recent generation of a caspase 11 null mice has shed light on the important contribution of caspase 11 in innate immunity (45). Curiously, the caspase 1 caspase 11 double knockout mouse had much greater protection from acute pancreatitis than the NLRP3 or ASC mice (33), suggesting either that caspase 11 may have a significant role in acute pancreatitis or that caspase 1 may have NLRP3- and ASC-independent effects, perhaps being directly activated by cathepsin B released from necrotic acinar cells. A direct role of caspase 11 in pancreatic inflammation remains to be explored. TLR3 is a strong inducer of caspase 11 (45), and chronic treatment with TLR3 ligands results in chronic pancreatitis with macrophage and lympohocytic infiltrate in MLR/Mp autoimmune prone mice (74). TLR3 expression has been found in secretory acinar cells (44) and can induce sterile inflammation through recognition of endogenous ligands (8). Whether or not necrotic acinar cells induce TLR3 stimulation and caspase 11 expression in pancreatic acinar cells with functional significance in sterile inflammation is unreported (Fig. 1). Finally, extracellular HMGB1 was identified as a strong inducer of pyroptosis in one report through receptor for advanced glycation end products internalization, cathepsin B activation, early cathepsin B-mediated and NRLP3-independent caspase 1 activation, and complexation with ASC to form a pyroptosome, with resultant cell death from membrane rupture (96). The role of HMGB1 as an inflammatory effector in acute pancreatitis is more fully discussed below.
Inflammasome Effectors
HMGB1.
HMGB1 is described as a ligand for TLR4. TLR4 expression in bone marrow-derived cells is required for full inflammation and injury in experimental acute pancreatitis induced by caerulein hyperstimulation (97). Endotoxin is reported to be undetectable in the pancreas in the caerulein and l-arginine experimental models of acute pancreatitis, and as such TLR4-mediated signaling in immune cells in the inflamed pancreas is thought to occur through recognition of endogenous ligands (85). HMGB1 is strongly implicated as the major endogenous TLR4 ligand in acute pancreatitis. HMGB1 released from immune cells is regulated by caspase 1 activation and is responsible for profound proinflammatory responses in vivo, including late mortality in inflammatory shock models (51). HMGB1 is also passively released from necrotic cells. Serum HMGB1 levels are elevated in clinical pancreatitis and correspond to disease severity (99), consistent with immune cell activation and acinar cell death. The importance of HMGB1 as an effector of pancreatic injury was demonstrated in vivo as HMGB1 masking antibodies mitigate pancreatic inflammation and injury with TLR4 dependence in experimental acute pancreatitis (79). LPS or LPS and chronic ethanol administration promote extracellular release of HMGB1 from pancreatic acinar cells harboring caspase 1 and result in increased serum levels of HMGB1. In this report, acinar cell HMGB1 release occurred in the absence of acinar cell necrosis, suggesting that HMGB1 may be an early effector of pancreatic injury (24). Use of HMGB1 knockout mice to further clarify the proinflammatory functions of this molecule are confounded by its additional functions. Intracellular HMGB1 is required to promote nuclear integrity and to prevent extracellular release of nuclear proteins, including nucleosomes, which are themselves DAMPs released from injured cells. Acinar cell-specific knockout of HMGB1 resulted in more severe experimental pancreatitis consistent with this anti-inflammatory functionality within the cell (41). Extracellular HMGB1 on the other hand complexes with nucleic acid DAMPs released from necrotic cells and promotes TLR9 recognition contributing to innate immune-mediated inflammatory responses (98). TLR9 is identified as a contributor to sterile inflammation in experimental acute pancreatitis (33). Extracellular HMGB1 signaling induces and enhances sterile inflammatory responses through TLR4 and nucleic acid TLRs, such as TLR9, respectively.
IL-1β.
IL-1β is recognized to be a major determinant of sterile inflammation and injury responses in acute pancreatitis. Genetic deficiency of IL-1R or injection of IL-1R antagonist mitigates injury responses in experimental acute pancreatitis (63, 64). Constitutive expression of an IL-1β transgene in acinar cells results in severe chronic pancreatitis with pancreatic atrophy and fibrosis, recapitulating the histopathological changes of human chronic pancreatitis and demonstrating that pancreatic IL-1β expression is sufficient to induce pancreatic damage (56). As noted already, IL-1β is expressed in acinar cells in acute and acute recurrent pancreatitis (24). IL-1β is known to sensitize hepatocytes to TNF-α-mediated cell death in hepatocytes (72). If this effect extends to sensitizing pancreatic acinar cells to TNF-α-mediated cell death is unreported. The role of IL-1β in pancreatic stellate cell-mediated fibrosis remains to be investigated. IL-1β is known to promote fibrogenic responses in the closely related primary hepatic stellate cell, and IL-1R is required for fibrosis in murine experimental nonalcoholic steatohepatitis (60). IL-1β induces hypoxia-inducible factor-1α (HIF-1α) stabilization under normoxia and aerobic glycolysis, both of which are required for stellate cell activation, providing additional potential mechanisms for IL-1β-mediated fibrogenic effects (11).
IL-18.
The role of IL-18 in acute pancreatitis appears more complex. IL-18 is required for gut homeostasis, specifically intestinal epithelial host defense as it is directly bacteriocidal (18). Increased gut permeability is an early feature of clinical pancreatitis (2), and a leaky gut may prime for enhanced systemic innate immune responsiveness (101) and possibly worse sterile inflammatory-mediated injury in remote organs such as the pancreas in acute injury. Genetic deficiency of IL-18 results in greater injury in clinical mild experimental pancreatitis induced by caerulein hyperstimulation, and pretreatment with recombinant IL-18 is dose dependently protective in this model (90). In the interstitial compartment, IL-18 expression promotes proinflammatory responses in the context of other proinflammatory cytokines such as IL-12. Consistent with this model, coadministration of IL-18 with IL-12 induces acute pancreatitis in genetically obese mice (83) or mice fed a high-fat diet (73). Administration of IL-18 alone did not promote pancreatitis in these models. Elevations in serum IL-18 correlate with disease severity in the human clinical condition. IL-18 is expressed in acinar cells and in immune cells in conjunction with other proinflammatory cytokines such as IL-1β and TNF-α in acute and chronic pancreatitis (24, 81).
IL-33.
IL-33 is an IL-1 family member cytokine that is constitutively expressed in endothelial cells and in pancreatic acinar cells but not found in hematopoietic cell lineages (68). Similar to IL-1α, it is passively released from necrotic cells in a full-length biologically active form. IL-33 cleavage and inactivation by caspase 1 has been reported in the literature (9). However, this finding was not reproduced in another extensive investigation that found IL-33 to be a substrate for apoptotic caspases 3 and 7 but not inflammatory caspases 1, 4, or 5 in vitro; caspase 3 seemed to be the major processor of IL-33 in cells in this study (54). Additionally, IL-33 is also a substrate for neutrophil elastase and cathepsin G, resulting in production of hyperactive forms with greater IL-33 receptor activation (52). The IL-33 receptor suppression of tumorigenicity 2 (ST2) and coreceptor IL-1 receptor accessory protein are expressed on mast cells and NK cells, which are implicated in regulation of sterile inflammation. In mast cells, ST2 transduces TH2 proinflammatory cytokine responses with production of IL-5 and IL-13 and promotes mast cell degranulation. ST2 is also expressed on human pancreatic myofibroblasts; IL-33 treatment of these cells induces proliferation and migration and enhances inflammatory cytokine production in response to IL-4 and interferon-γ. Genetic deletion of ST2 increases the severity of pancreatic injury and increases mast cell degranulation in experimental pancreatitis in mice induced by a choline methionine-deficient diet (68). However, exogenous IL-33 administration also increased pancreatic edema and inflammation and mast cell degranulation in experimental pancreatitis induced by bile duct ligation (46). Most recently, ST2 and exogenous IL-33 were found to limit pancreatic injury in coxsackievirus B-induced chronic pancreatitis through an IL-4-dependent induction of M2 macrophages (84). The role of IL-33 in acute pancreatitis is likely to be complex and has only recently been investigated. However, a role for direct caspase 1 functionality in IL-33 processing and release seems increasingly unsupported in the literature.
Inflammasome Regulation of Metabolism
Metabolic pathway regulation.
Caspase 1 activation and NF-kB signaling pathways promote normoxic HIF-1α stabilization, increased expression of glycolytic enzymes, and decreased expression of tricarboxylic acid pathway enzymes with resultant increases in aerobic glycolysis and decreases in oxidative metabolism (Fig. 2). This has been excellently reviewed by O'Neill et al. (67). AMP-activated protein kinase (AMPK) is a ubiquitously expressed cytosolic sensor of energy homeostasis in cells and induces catabolism by promoting glycolysis and β-oxidation as well as by suppressing protein synthesis. It has recently been identified as a key negative regulator of NLRP3 inflammasome activation (93). LPS and free fatty acids suppress AMPK activation in macrophages consistent with AMPK functioning as a negative regulator of TLR and NLRP3 inflammasome signaling (59, 93). Shugrue et al. have recently identified that AMPK suppresses zymogen activation in acinar cells through use of chemical activator and inhibitors (86). A direct role for AMPK in suppression of sterile inflammation in the pancreas is intriguing but remains to be established. The NAD deacetylase sirtuin1 (SIRT1) is a ubiquitously expressed cytosolic protein that also senses cellular energy homeostasis and similarly induces oxidative metabolism. SIRT1 antagonizes NF-kB signaling pathways (43) and is cleaved and inactivated in adipose tissue by caspase 1 in mice fed a high-fat diet (10). The role of suppressed SIRT1 signaling in disease severity in acute pancreatitis remains to be explored.
Fig. 2.
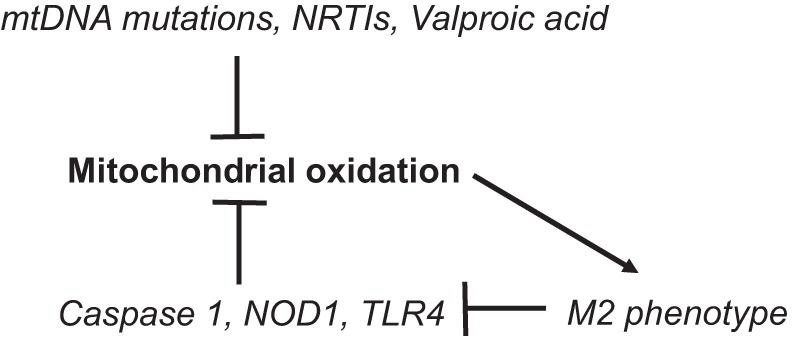
Regulation of mitochondrial oxidation by determinants of pancreatitis. Pancreatitis can be induced by mitochondrial DNA mutations, nucleoside reverse transcriptase inhibitors (NRTIs), and valproic acid, all of which suppress mitochondrial oxidation. Major effectors of pancreatitis severity, caspase 1, NOD1, and TLR4, suppress mitochondrial oxidation. Mitochondrial oxidation promotes an anti-inflammatory macrophage phenotype and suppression of caspase 1 and TLR4 proinflammatory macrophage phenotype signals. mtDNA, mitochondrial DNA.
Macrophage polarization.
Changes in the metabolic phenotype of cells are strong determinants of macrophage polarization (22). Briefly, proinflammatory macrophage polarization requires aerobic glycolysis and decreased oxidative metabolism. This immune phenotype is strongly associated with the acute sterile inflammatory responses in line with TLR4 (22), NOD1 (3), and caspase 1 activation (100). Caspase 1 in particular functions to promote mitochondrial breakdown and suppresses mitophagy, contributing to sustained suppression of oxidative metabolism in cells (100) (Fig. 2). Conversely, anti-inflammatory activated macrophage polarization requires fatty acid oxidation (34), and this immune phenotype is associated with resolution of acute inflammation. Evidence of macrophage polarization effecting pancreatic injury in acute pancreatitis rests largely on investigations of hemin-induced anti-inflammatory macrophage polarization in acute pancreatitis. In these studies, hemin-induced anti-inflammatory macrophages decreased sterile inflammation and tissue injury in experimental acute pancreatitis (61), suggesting that anti-inflammatory macrophage polarization may be protective in pancreatic injury. Metabolic perturbations induced by TLR4, NOD1, or caspase 1 activation in pancreatic acinar cells have not been investigated and may potentially contribute to a metabolic environment conducive to macrophage polarization in the pancreas (Fig. 2).
Evidence of clinical significance.
Drug-induced pancreatitis provides insights into the potential importance of metabolism in acute pancreatitis. Many agents known to induce pancreatitis have mitochondrial toxicity and/or compromise oxidative metabolism. Nucleoside and nucleotide reverse transcriptase inhibitors are well-known causes of drug-induced acute pancreatitis. Nucleoside and nucleotide reverse transcriptase inhibitors induce mitochondrial toxicities in vivo as most commonly documented by type B lactic acidosis and confirmation of pathological ultrastructural changes in mitochondria in human cells induced by such agents in vitro and in clinical specimens (40). Valproic acid is a rare but potentially underreported cause of drug-induced pancreatitis (23). Valproic acid also appears to inhibit fatty acid oxidation at therapeutic doses with carnitine deficiency being the most studied clinical marker of this effect (87). Valproic acid at therapeutic doses decreases the biosynthesis and serum levels of carnitine, a rate-limiting substrate in fatty acid oxidation. Carnitine supplementation is protective from valproic acid-induced hepatotoxicity, suggesting that restoration of β-oxidative function can rescue this phenotype in the clinical setting (20). The most compelling evidence for mitochondrial dysfunction as a key pathological insult precipitating pancreatic disease comes from studies of inherited mitochondrial disorders. Pearson marrow pancreas syndrome is caused by inherited deletions in mitochondrial DNA and exocrine pancreatic insufficiency with pancreatic atrophy, and scarring is the most common manifestation of this syndrome (36). Other inherited mitochondrial disorders have also presented with chronic pancreatitis, including mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes syndromes, for which many mutations in mitochondrial NADH dehydrogenase have been described. Additionally, a patient with inherited deficiency in the β-oxidative pathway component carnitine palmitolytransferase presented with recurrent pancreatitis (88). In aggregate, compromised mitochondrial oxidative metabolism appears to be a common finding in drug-induced and genetic pancreatitis and suggests that drug-induced or genetic-mediated metabolic perturbation may contribute to pancreatitis (Fig. 2). An increased susceptibility to NLRP3 inflammasome activation in these conditions of perturbed oxidative metabolism remains to be demonstrated. Potential mechanisms include alteration of macrophage phenotype favoring the proinflammatory phenotype as discussed above. Alternatively, alterations in metabolic intermediate composition may effect metabolite receptor signaling and regulation of NLRP3 inflammasome activity as discussed in the next section.
Metabolite receptors.
Recent investigation has established that metabolic intermediates such as succinate can regulate NLRP3 inflammasome induction, NF-κB signaling, and inflammatory signaling in immune cells (58). Recently, we identified that TLR-mediated aerobic glycolytic production of lactate negatively regulates TLR4- and TLR9-mediated NF-kB activation, induction of NLRP3 inflammasome components, and NLRP3 inflammasome activation by ATP in LPS-primed macrophages. We further identified that these effects could be reproduced in vivo by lactate injection, which protects from sterile inflammation and pancreatic injury in experimental severe acute pancreatitis. The immune dampening effects of parenteral lactate dosing required the presence of the cell surface lactate receptor G protein-coupled receptor (GPR) 81 in vivo. We further identified that GPR81 is an endogenous limiter of sterile inflammation in experimental acute pancreatitis. Finally, we showed that a small-molecule GPR81 agonist could reproduce these effects in isolated macrophages (31). Additionally, extracellular lactate is required for pancreatic tumor-associated macrophage polarization though HIF-1α-mediated induction of anti-inflammatory macrophage phenotype genes, including arginase 1 in tumor microenvironments; lactate also induced anti-inflammatory macrophage phenotype genes in macrophages in vitro (13). The mechanism of this lactate-mediated effect remains to be more fully characterized. These studies provide a mechanistic basis for the therapeutic benefit of lactated Ringer vs. normal saline infusion in limiting inflammatory organ injury in a randomized clinical trial in acute pancreatitis (94) (Fig. 3). Serum levels of many amino acids increase in acute inflammation (21), and we have recently identified that aspartate levels increase in macrophages in response to TLR4 stimulation in vitro, suggesting that TLR signaling alters aspartate metabolism (19). We further identify that macrophages signaling through the N-methyl-d-aspartic acid (NMDA) receptor component NMDA receptor subunit 2A (NR2A) suppress induction of NLRP3 inflammasome components mediated by TLRs in cell culture and that parenteral aspartate therapy limits pancreatic NLRP3 inflammasome expression, tissue injury, and inflammation in acute pancreatitis. We confirmed that NR2A was required for the therapeutic benefit of aspartate supplementation and had endogenous function in limiting sterile inflammation in a liver injury model. These findings provided one potential mechanism for the well-known immunosuppressive effects of parenteral nutrition (Fig. 3). Whether parenteral amino acid infusion as therapy may be beneficial to limit sterile inflammation early in the course of pancreatitis remains to be investigated.
Fig. 3.
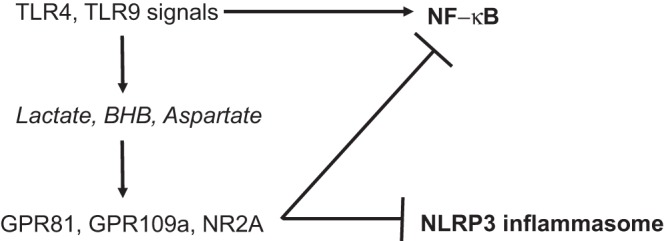
Metabolite receptor feedback suppresses innate immune responses in acute pancreatitis. Macrophage stimulation with TLR4 and TLR9 ligands induces production and release of lactate, β-hydroxybutyrate, and aspartate. These metabolites function as activating ligands for cell surface receptors G protein-coupled receptor (GPR) 81, GPR109a, and NR2A, respectively, and suppress TLR-induced NF-κB activation as well as NLRP3 inflammasome activation.
Finally, we and others have identified that β-hydroxybutyrate, a key intermediate in β-oxidation of fatty acids, suppresses NF-κB signaling in macrophages (75). Recently submitted work from our laboratory identifies the β-hydroxybutyrate production is increased in TLR4 or TLR9 primed primary macrophages and that exogenous β-hydroxybutyrate suppresses NF-κB induction, NLRP3 inflammasome expression, and inflammasome-mediated IL-1β release. Additionally, we show that these effects are mediated by the cell surface β-hydroxybutyrate receptor GPR109a. We further show that β-hydroxybutyrate supplementation can suppress NF-κB activation in tissue macrophages in vivo and protect from experimental acute pancreatitis and drug-induced hepatitis with GPR109a dependence. Moreover GPR109a has endogenous function in limiting sterile inflammation and tissue injury in experimental pancreatitis. In aggregate, these findings suggest that suppression of β-oxidation may promote pancreatic injury in susceptible individuals by limiting production of β-hydroxybutyrate and thereby insufficiently activating a negative regulatory pathway of sterile inflammatory responses (Fig. 3).
Future Directions
The NLRP3 inflammasome has now been recognized for a number of years to be an important determinant of sterile inflammation in many diseases, including pancreatitis. Recent attention has turned to the exact role of individual inflammasome components, and their cellular requirement. Identification of the regulators of the inflammasome machinery has also been a very active area and has revealed multiple connections between inflammasome activation, hypoxia-induced transcription factors, and metabolite receptors. The metabolite receptors are particularly promising therapeutic targets in limiting innate immune-mediated tissue injury in pancreatitis (Fig. 3). The role for inflammasome components and effectors in fibrogenic cell types and in immune cell populations in chronic pancreatitis and in malignant cells and tumor-associated cell populations in pancreatic cancer remain to be extensively interrogated.
GRANTS
This work was supported by National Institutes of Health Grants R01-DK-076674-01A2 and 5U01-AA-021912-02 to W. Z. Mehal, K08-DK-092281 and P30-DK-34989 to R. Hoque and by a Veterans Affairs Merit award to W. Z. Mehal.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: R.H. drafted manuscript; R.H. and W.Z.M. edited and revised manuscript; R.H. and W.Z.M. approved final version of manuscript.
REFERENCES
- 1.Aiba-Masago S, Masago R, Vela-Roch N, Talal N, Dang H. Fas-mediated apoptosis in a rat acinar cell line is dependent on caspase-1 activity. Cell Signal 13: 617–624, 2001. [DOI] [PubMed] [Google Scholar]
- 2.Ammori BJ. Role of the gut in the course of severe acute pancreatitis. Pancreas 26: 122–129, 2003. [DOI] [PubMed] [Google Scholar]
- 3.Bae J, Ricciardi CJ, Esposito D, Komarnytsky S, Hu P, Curry BJ, Brown PL, Gao Z, Biggerstaff JP, Chen J, Zhao L. Activation of pattern recognition receptors in brown adipocytes induces inflammation and suppresses uncoupling protein 1 expression and mitochondrial respiration. Am J Physiol Cell Physiol 306: C918–C930, 2014. [DOI] [PubMed] [Google Scholar]
- 4.Bedrosian AS, Nguyen AH, Hackman M, Connolly MK, Malhotra A, Ibrahim J, Cieza-Rubio NE, Henning JR, Barilla R, Rehman A, Pachter HL, Medina-Zea MV, Cohen SM, Frey AB, Acehan D, Miller G. Dendritic cells promote pancreatic viability in mice with acute pancreatitis. Gastroenterology 141: 1915–1926, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhopale KK, Wu H, Boor PJ, Popov VL, Ansari GA, Kaphalia BS. Metabolic basis of ethanol-induced hepatic and pancreatic injury in hepatic alcohol dehydrogenase deficient deer mice. Alcohol 39: 179–188, 2006. [DOI] [PubMed] [Google Scholar]
- 6.Brydges SD, Broderick L, McGeough MD, Pena CA, Mueller JL, Hoffman HM. Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. J Clin Invest 123: 4695–4705, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, Gibson GR, Delzenne NM. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 50: 2374–2383, 2007. [DOI] [PubMed] [Google Scholar]
- 8.Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, Hogaboam CM, Kunkel SL. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med 205: 2609–2621, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc Natl Acad Sci USA 106: 9021–9026, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chalkiadaki A, Guarente L. High-fat diet triggers inflammation-induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell Metab 16: 180–188, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Choi SS, Michelotti GA, Chan IS, Swiderska-Syn M, Karaca GF, Xie G, Moylan CA, Garibaldi F, Premont R, Suliman HB, Piantadosi CA, Diehl AM. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology 143: 1319–1329, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med 16: 228–231, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, Cline GW, Phillips AJ, Medzhitov R. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513: 559–563, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Criddle DN, Raraty MG, Neoptolemos JP, Tepikin AV, Petersen OH, Sutton R. Ethanol toxicity in pancreatic acinar cells: mediation by nonoxidative fatty acid metabolites. Proc Natl Acad Sci USA 101: 10738–10743, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 54: 133–144, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dawra R, Sah RP, Dudeja V, Rishi L, Talukdar R, Garg P, Saluja AK. Intra-acinar trypsinogen activation mediates early stages of pancreatic injury but not inflammation in mice with acute pancreatitis. Gastroenterology 141: 2210–2217, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Denham W, Yang J, Fink G, Denham D, Carter G, Ward K, Norman J. Gene targeting demonstrates additive detrimental effects of interleukin 1 and tumor necrosis factor during pancreatitis. Gastroenterology 113: 1741–1746, 1997. [DOI] [PubMed] [Google Scholar]
- 18.Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, Flavell RA. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145: 745–757, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farooq A, Hoque R, Ouyang X, Farooq A, Ghani A, Ahsan K, Guerra M, Mehal WZ. Activation of N-methyl-d-aspartate receptor downregulates inflammasome activity and liver inflammation via a β-arrestin-2 pathway. Am J Physiol Gastrointest Liver Physiol 307: G732–G740, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Felker D, Lynn A, Wang S, Johnson DE. Evidence for a potential protective effect of carnitine-pantothenic acid co-treatment on valproic acid-induced hepatotoxicity. Exp Rev Clin Pharmacol 7: 211–218, 2014. [DOI] [PubMed] [Google Scholar]
- 21.Feng B, Wu S, Lv S, Liu F, Chen H, Yan X, Li Y, Dong F, Wei L. Metabolic profiling analysis of a d-galactosamine/lipopolysaccharide-induced mouse model of fulminant hepatic failure. J Proteome Res 6: 2161–2167, 2007. [DOI] [PubMed] [Google Scholar]
- 22.Galvan-Pena S, O'Neill LA. Metabolic reprograming in macrophage polarization (Abstract). Frontiers Immunol 5: 420, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerstner T, Busing D, Bell N, Longin E, Kasper JM, Klostermann W, Hebing B, Hanefeld F, Eckel U, Hoffmann R, Bettendorf U, Weidner B, Wiemer-Kruel A, Brockmann K, Neumann FW, Sandrieser T, Wolff M, Konig S. Valproic acid-induced pancreatitis: 16 new cases and a review of the literature. J Gastroenterol 42: 39–48, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Gu H, Werner J, Bergmann F, Whitcomb DC, Buchler MW, Fortunato F. Necro-inflammatory response of pancreatic acinar cells in the pathogenesis of acute alcoholic pancreatitis (Abstract). Cell Death Dis 4: e816, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gukovsky I, Gukovskaya A. Nuclear factor-kappaB in pancreatitis: Jack-of-all-trades, but which one is more important? Gastroenterology 144: 26–29, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haanes KA, Schwab A, Novak I. The P2X7 receptor supports both life and death in fibrogenic pancreatic stellate cells. PloS one 7: e51164, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hasel C, Durr S, Rau B, Strater J, Schmid RM, Walczak H, Bachem MG, Moller P. In chronic pancreatitis, widespread emergence of TRAIL receptors in epithelia coincides with neoexpression of TRAIL by pancreatic stellate cells of early fibrotic areas. Lab Invest 83: 825–836, 2003. [DOI] [PubMed] [Google Scholar]
- 28.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137: 1100–1111, 2009. [DOI] [PubMed] [Google Scholar]
- 29.Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol 191: 5230–5238, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1: 489–495, 2000. [DOI] [PubMed] [Google Scholar]
- 31.Hoque R, Farooq A, Ghani A, Gorelick F, Mehal WZ. Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology 146: 1763–1774, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoque R, Malik AF, Gorelick F, Mehal WZ. Sterile inflammatory response in acute pancreatitis. Pancreas 41: 353–357, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoque R, Sohail M, Malik A, Sarwar S, Luo Y, Shah A, Barrat F, Flavell R, Gorelick F, Husain S, Mehal W. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology 141: 358–369, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang SC, Everts B, Ivanova Y, O'Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O'Neill CM, Yan C, Du H, Abumrad NA, Urban JF Jr, Artyomov MN, Pearce EL, Pearce EJ. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 15: 846–855, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang W, Booth DM, Cane MC, Chvanov M, Javed MA, Elliott VL, Armstrong JA, Dingsdale H, Cash N, Li Y, Greenhalf W, Mukherjee R, Kaphalia BS, Jaffar M, Petersen OH, Tepikin AV, Sutton R, Criddle DN. Fatty acid ethyl ester synthase inhibition ameliorates ethanol-induced Ca2+-dependent mitochondrial dysfunction and acute pancreatitis. Gut 63: 1313–1324, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishiyama A, Komaki H, Saito T, Saito Y, Nakagawa E, Sugai K, Itagaki Y, Matsuzaki K, Nakura M, Nishino I, Goto Y, Sasaki M. Unusual exocrine complication of pancreatitis in mitochondrial disease. Brain Dev 35: 654–659, 2013. [DOI] [PubMed] [Google Scholar]
- 37.Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, Eisenbarth SC, Florquin S, Flavell RA, Leemans JC, Sutterwala FS. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA 106: 20388–20393, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeyarajah DR, Kielar M, Gokaslan ST, Lindberg G, Lu CY. Fas deficiency exacerbates cerulein-induced pancreatitis. J Invest Surg 16: 325–333, 2003. [DOI] [PubMed] [Google Scholar]
- 39.Kaiser AM, Saluja AK, Sengupta A, Saluja M, Steer ML. Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am J Physiol Cell Physiol 269: C1295–C1304, 1995. [DOI] [PubMed] [Google Scholar]
- 40.Kakuda TN. Pharmacology of nucleoside and nucleotide reverse transcriptase inhibitor-induced mitochondrial toxicity. Clin Ther 22: 685–708, 2000. [DOI] [PubMed] [Google Scholar]
- 41.Kang R, Zhang Q, Hou W, Yan Z, Chen R, Bonaroti J, Bansal P, Billiar TR, Tsung A, Wang Q, Bartlett DL, Whitcomb DC, Chang EB, Zhu X, Wang H, Lu B, Tracey KJ, Cao L, Fan XG, Lotze MT, Zeh HJ 3rd, Tang D. Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology 146: 1097–1107, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaphalia BS, Bhopale KK, Kondraganti S, Wu H, Boor PJ, Ansari GA. Pancreatic injury in hepatic alcohol dehydrogenase-deficient deer mice after subchronic exposure to ethanol. Toxicol Appl Pharmacol 246: 154–162, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal 25: 1939–1948, 2013. [DOI] [PubMed] [Google Scholar]
- 44.Kawakami A, Nakashima K, Tamai M, Nakamura H, Iwanaga N, Fujikawa K, Aramaki T, Arima K, Iwamoto N, Ichinose K, Kamachi M, Ida H, Origuchi T, Eguchi K. Toll-like receptor in salivary glands from patients with Sjogren's syndrome: functional analysis by human salivary gland cell line. J Rheumatol 34: 1019–1026, 2007. [PubMed] [Google Scholar]
- 45.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM. Non-canonical inflammasome activation targets caspase-11. Nature 479: 117–121, 2011. [DOI] [PubMed] [Google Scholar]
- 46.Kempuraj D, Twait EC, Williard DE, Yuan Z, Meyerholz DK, Samuel I. The novel cytokine interleukin-33 activates acinar cell proinflammatory pathways and induces acute pancreatic inflammation in mice. PloS one 8: e56866, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keyel PA. How is inflammation initiated? Individual influences of IL-1, IL-18 and HMGB1. Cytokine 69: 136–145, 2014. [DOI] [PubMed] [Google Scholar]
- 48.Kuhny M, Hochdorfer T, Ayata C, Idzko M, Huber M. CD39 is a negative regulator of P2X7-mediated inflammatory cell death in mast cells (Abstract). Cell Comm Signal 12: 40, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kunzli BM, Nuhn P, Enjyoji K, Banz Y, Smith RN, Csizmadia E, Schuppan D, Berberat PO, Friess H, Robson SC. Disordered pancreatic inflammatory responses and inhibition of fibrosis in CD39-null mice. Gastroenterology 134: 292–305, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell 157: 1013–1022, 2014. [DOI] [PubMed] [Google Scholar]
- 51.Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, Tracey KJ, Kanneganti TD, Dixit VM. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol 185: 4385–4392, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lefrancais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard JP, Cayrol C. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci USA 109: 1673–1678, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li N, Wu X, Holzer RG, Lee JH, Todoric J, Park EJ, Ogata H, Gukovskaya AS, Gukovsky I, Pizzo DP, VandenBerg S, Tarin D, Atay C, Arkan MC, Deerinck TJ, Moscat J, Diaz-Meco M, Dawson D, Erkan M, Kleeff J, Karin M. Loss of acinar cell IKKalpha triggers spontaneous pancreatitis in mice. J Clin Invest 123: 2231–2243, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, Brumatti G, Taylor RC, Kersse K, Vandenabeele P, Lavelle EC, Martin SJ. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 31: 84–98, 2009. [DOI] [PubMed] [Google Scholar]
- 55.Malleo G, Mazzon E, Genovese T, Di Paola R, Muia C, Centorrino T, Siriwardena AK, Cuzzocrea S. Etanercept attenuates the development of cerulein-induced acute pancreatitis in mice: a comparison with TNF-alpha genetic deletion. Shock 27: 542–551, 2007. [DOI] [PubMed] [Google Scholar]
- 56.Marrache F, Tu SP, Bhagat G, Pendyala S, Osterreicher CH, Gordon S, Ramanathan V, Penz-Osterreicher M, Betz KS, Song Z, Wang TC. Overexpression of interleukin-1beta in the murine pancreas results in chronic pancreatitis. Gastroenterology 135: 1277–1287, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martin BN, Wang C, Willette-Brown J, Herjan T, Gulen MF, Zhou H, Bulek K, Franchi L, Sato T, Alnemri ES, Narla G, Zhong XP, Thomas J, Klinman D, Fitzgerald KA, Karin M, Nunez G, Dubyak G, Hu Y, Li X. IKKalpha negatively regulates ASC-dependent inflammasome activation (Abstract). Nat Commun 5: 4977, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McGettrick AF, O'Neill LA. How metabolism generates signals during innate immunity and inflammation. J Biol Chem 288: 22893–22898, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McGettrick AF, O'Neill LA. NLRP3 and IL-1beta in macrophages as critical regulators of metabolic diseases. Diabetes Obesity Metab 15, Suppl 3: 19–25, 2013. [DOI] [PubMed] [Google Scholar]
- 60.Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 139: 323–334, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nakamichi I, Habtezion A, Zhong B, Contag CH, Butcher EC, Omary MB. Hemin-activated macrophages home to the pancreas and protect from acute pancreatitis via heme oxygenase-1 induction. J Clin Invest 115: 3007–3014, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Navina S, Acharya C, DeLany JP, Orlichenko LS, Baty CJ, Shiva SS, Durgampudi C, Karlsson JM, Lee K, Bae KT, Furlan A, Behari J, Liu S, McHale T, Nichols L, Papachristou GI, Yadav D, Singh VP. Lipotoxicity causes multisystem organ failure and exacerbates acute pancreatitis in obesity. Sci Trans Med 3: 107–110, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Norman J, Franz M, Messina J, Riker A, Fabri PJ, Rosemurgy AS, Gower WR Jr. Interleukin-1 receptor antagonist decreases severity of experimental acute pancreatitis. Surgery 117: 648–655, 1995. [DOI] [PubMed] [Google Scholar]
- 64.Norman JG, Fink G, Franz M, Guffey J, Carter G, Davison B, Sexton C, Glaccum M. Active interleukin-1 receptor required for maximal progression of acute pancreatitis. Ann Surg 223: 163–169, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Norman JG, Fink GW, Messina J, Carter G, Franz MG. Timing of tumor necrosis factor antagonism is critical in determining outcome in murine lethal acute pancreatitis. Surgery 120: 515–521, 1996. [DOI] [PubMed] [Google Scholar]
- 66.Norman JG, Fink GW, Sexton C, Carter G. Transgenic animals demonstrate a role for the IL-1 receptor in regulating IL-1beta gene expression at steady-state and during the systemic stress induced by acute pancreatitis. J Surg Res 63: 231–236, 1996. [DOI] [PubMed] [Google Scholar]
- 67.O'Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493: 346–355, 2013. [DOI] [PubMed] [Google Scholar]
- 68.Ouziel R, Gustot T, Moreno C, Arvanitakis M, Degre D, Trepo E, Quertinmont E, Vercruysse V, Demetter P, Le Moine O, McKenzie AN, Delhaye M, Deviere J, Lemmers A. The ST2 pathway is involved in acute pancreatitis: a translational study in humans and mice. Am J Pathol 180: 2330–2339, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, Ray S, Majumdar SS, Bhattacharya S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med 18: 1279–1285, 2012. [DOI] [PubMed] [Google Scholar]
- 70.Parlesak A, Schäfer C, Schütz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol 32: 742–747, 2000. [DOI] [PubMed] [Google Scholar]
- 71.Pendyala S, Walker JM, Holt PR. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology 142: 1100–1101e1102, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, Barrieau M, Min SY, Kurt-Jones EA, Szabo G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest 122: 3476–3489, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pini M, Sennello JA, Cabay RJ, Fantuzzi G. Effect of diet-induced obesity on acute pancreatitis induced by administration of interleukin-12 plus interleukin-18 in mice. Obesity 18: 476–481, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qu WM, Miyazaki T, Terada M, Okada K, Mori S, Kanno H, Nose M. A novel autoimmune pancreatitis model in MRL mice treated with polyinosinic:polycytidylic acid. Clin Exp Immunol 129: 27–34, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rahman M, Muhammad S, Khan MA, Chen H, Ridder DA, Muller-Fielitz H, Pokorna B, Vollbrandt T, Stolting I, Nadrowitz R, Okun JG, Offermanns S, Schwaninger M. The beta-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages (Abstract). Nat Comm 5: 3944, 2014. [DOI] [PubMed] [Google Scholar]
- 76.Rau B, Paszkowski A, Lillich S, Baumgart K, Moller P, Beger HG. Differential effects of caspase-1/interleukin-1beta-converting enzyme on acinar cell necrosis and apoptosis in severe acute experimental pancreatitis. Lab Invest 81: 1001–1013, 2001. [DOI] [PubMed] [Google Scholar]
- 77.Ren JD, Ma J, Hou J, Xiao WJ, Jin WH, Wu J, Fan KH. Hydrogen-rich saline inhibits NLRP3 inflammasome activation and attenuates experimental acute pancreatitis in mice (Abstract). Mediat Inflamm 2014: 930894, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sah RP, Dudeja V, Dawra RK, Saluja AK. Cerulein-induced chronic pancreatitis does not require intra-acinar activation of trypsinogen in mice. Gastroenterology 144: 1076–1085, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sawa H, Ueda T, Takeyama Y, Yasuda T, Shinzeki M, Nakajima T, Kuroda Y. Blockade of high mobility group box-1 protein attenuates experimental severe acute pancreatitis. World J Gastroenterol 12: 7666–7670, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Scheuplein F, Schwarz N, Adriouch S, Krebs C, Bannas P, Rissiek B, Seman M, Haag F, Koch-Nolte F. NAD+ and ATP released from injured cells induce P2X7-dependent shedding of CD62L and externalization of phosphatidylserine by murine T cells. J Immunol 182: 2898–2908, 2009. [DOI] [PubMed] [Google Scholar]
- 81.Schneider A, Haas SL, Hildenbrand R, Siegmund S, Reinhard I, Nakovics H, Singer MV, Feick P. Enhanced expression of interleukin-18 in serum and pancreas of patients with chronic pancreatitis. World J Gastroenterol 12: 6507–6514, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sendler M, Dummer A, Weiss FU, Kruger B, Wartmann T, Scharffetter-Kochanek K, van Rooijen N, Malla SR, Aghdassi A, Halangk W, Lerch MM, Mayerle J. Tumour necrosis factor alpha secretion induces protease activation and acinar cell necrosis in acute experimental pancreatitis in mice. Gut 62: 430–439, 2013. [DOI] [PubMed] [Google Scholar]
- 83.Sennello JA, Fayad R, Pini M, Gove ME, Ponemone V, Cabay RJ, Siegmund B, Dinarello CA, Fantuzzi G. Interleukin-18, together with interleukin-12, induces severe acute pancreatitis in obese but not in nonobese leptin-deficient mice. Proc Natl Acad Sci USA 105: 8085–8090, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sesti-Costa R, Silva GK, Proenca-Modena JL, Carlos D, Silva ML, Alves-Filho JC, Arruda E, Liew FY, Silva JS. The IL-33/ST2 pathway controls coxsackievirus B5-induced experimental pancreatitis. J Immunol 191: 283–292, 2013. [DOI] [PubMed] [Google Scholar]
- 85.Sharif R, Dawra R, Wasiluk K, Phillips P, Dudeja V, Kurt-Jones E, Finberg R, Saluja A. Impact of toll-like receptor 4 on the severity of acute pancreatitis and pancreatitis-associated lung injury in mice. Gut 58: 813–819, 2009. [DOI] [PubMed] [Google Scholar]
- 86.Shugrue CA, Alexandre M, de Villalvilla AD, Kolodecik TR, Young LH, Gorelick FS, Thrower EC. Cerulein hyperstimulation decreases AMP-activated protein kinase levels at the site of maximal zymogen activation. Am J Physiol Gastrointest Liver Physiol 303: G723–G732, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Silva MF, Aires CC, Luis PB, Ruiter JP, LIJ, Duran M, Wanders RJ, Tavares de Almeida I. Valproic acid metabolism and its effects on mitochondrial fatty acid oxidation: a review. J Inher Metab Dis 31: 205–216, 2008. [DOI] [PubMed] [Google Scholar]
- 88.Tein I, Christodoulou J, Donner E, McInnes RR. Carnitine palmitoyltransferase II deficiency: a new cause of recurrent pancreatitis. J Pediatr 124: 938–940, 1994. [DOI] [PubMed] [Google Scholar]
- 89.Tsuji Y, Watanabe T, Kudo M, Arai H, Strober W, Chiba T. Sensing of commensal organisms by the intracellular sensor NOD1 mediates experimental pancreatitis. Immunity 37: 326–338, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ueno N, Kashiwamura S, Ueda H, Okamura H, Tsuji NM, Hosohara K, Kotani J, Marukawa S. Role of interleukin 18 in nitric oxide production and pancreatic damage during acute pancreatitis. Shock 24: 564–570, 2005. [DOI] [PubMed] [Google Scholar]
- 91.Vonlaufen A, Spahr L, Apte MV, Frossard JL. Alcoholic pancreatitis: a tale of spirits and bacteria. World J Gastrointest Pathophysiol 5: 82–90, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Watanabe A, Sohail MA, Gomes DA, Hashmi A, Nagata J, Sutterwala FS, Mahmood S, Jhandier MN, Shi Y, Flavell RA, Mehal WZ. Inflammasome-mediated regulation of hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol 296: G1248–G1257, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 12: 408–415, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu BU, Hwang JQ, Gardner TH, Repas K, Delee R, Yu S, Smith B, Banks PA, Conwell DL. Lactated Ringer's solution reduces systemic inflammation compared with saline in patients with acute pancreatitis. Clin Gastroenterol Hepatol 9: 710–717, 2011. [DOI] [PubMed] [Google Scholar]
- 95.Wu LH, Huang CC, Adhikarakunnathu S, San Mateo LR, Duffy KE, Rafferty P, Bugelski P, Raymond H, Deutsch H, Picha K, Ward CK, Alexoupolou L, Flavell RA, Mbow ML, Susulic VS. Loss of toll-like receptor 3 function improves glucose tolerance and reduces liver steatosis in obese mice. Metab Clin Exp 61: 1633–1645, 2012. [DOI] [PubMed] [Google Scholar]
- 96.Xu J, Jiang Y, Wang J, Shi X, Liu Q, Liu Z, Li Y, Scott MJ, Xiao G, Li S, Fan L, Billiar TR, Wilson MA, Fan J. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ 21: 1229–1239, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xue J, Habtezion A. Carbon monoxide-based therapy ameliorates acute pancreatitis via TLR4 inhibition. J Clin Invest 124: 437–447, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, Nakasato M, Lu Y, Hangai S, Koshiba R, Savitsky D, Ronfani L, Akira S, Bianchi ME, Honda K, Tamura T, Kodama T, Taniguchi T. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 462: 99–103, 2009. [DOI] [PubMed] [Google Scholar]
- 99.Yasuda T, Ueda T, Takeyama Y, Shinzeki M, Sawa H, Nakajima T, Ajiki T, Fujino Y, Suzuki Y, Kuroda Y. Significant increase of serum high-mobility group box chromosomal protein 1 levels in patients with severe acute pancreatitis. Pancreas 33: 359–363, 2006. [DOI] [PubMed] [Google Scholar]
- 100.Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, Horng T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci USA 111: 15514–15519, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zuhl M, Dokladny K, Mermier C, Schneider S, Salgado R, Moseley P. The effects of acute oral glutamine supplementation on exercise-induced gastrointestinal permeability and heat shock protein expression in peripheral blood mononuclear cells. Cell Stress Chaperones 20: 85–93, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]