

Abstract
The role of stromal cell-derived factor-1 (SDF-1 or CXCL12) and its receptor CXC chemokine receptor-4 (CXCR4) in ischemic liver injury and recovery has not been studied. Some reports suggest that this chemokine may aid in liver regeneration, but others suggest that it may be profibrotic through its activation of hepatic stellate cells. In this study we sought to elucidate the role of SDF-1 and its receptor CXCR4 during liver injury, recovery, and regeneration after ischemia-reperfusion (I/R). A murine model of partial (70%) I/R was used to induce liver injury and study the reparative and regenerative response. CXCR4 was expressed constitutively in the liver, and hepatic levels of SDF-1 peaked 8 h after reperfusion but remained significantly increased for 96 h. Treatment of mice with the CXCR4 antagonist AMD3100 or agonist SDF-1 had no effect on acute liver injury assessed 8 h after I/R. However, treatment with AMD3100 increased hepatocyte proliferation after 72 and 96 h of reperfusion and reduced the amount of liver necrosis. In contrast, treatment with SDF-1 significantly decreased hepatocyte proliferation. These effects appeared to be dependent on the presence of liver injury, as AMD3100 and SDF-1 had no effect on hepatocyte proliferation or liver mass in mice undergoing 70% partial hepatectomy. The data suggest that signaling through CXCR4 is detrimental to liver recovery and regeneration after I/R and that clinical therapy with a CXCR4 antagonist may improve hepatic recovery following acute liver injury.
Keywords: liver regeneration, chemokines, stromal cell-derived factor-1, CXCR4, progenitor cells
hepatic ischemia-reperfusion (I/R) injury, resulting from insufficient oxygen delivery to the liver, followed by the reconstitution of adequate organ perfusion leads to extensive tissue damage and organ dysfunction (11–13). This injury is one of the most common etiologies of liver dysfunction in the surgical patient and can be seen in a variety of clinical scenarios, including liver resection surgery, liver transplantation, and hemorrhagic/septic shock (19). The ischemic insult induces an inflammatory response resulting in acute liver injury that is followed by a reparative phase and subsequent liver regeneration mediated by a complex interaction of growth factors, cytokines, and transcriptional factors (7, 22). We previously showed that specific members of the CXC chemokine family are critical mediators in all aspects of the injury, repair, and regeneration processes (2, 4, 5, 17). Our previous work focused on CXC chemokines that contain a glutamic acid-leucine-arginine (ELR) motif in their amino terminus, which confers binding specificity to the receptors CXCR1 and CXCR2 (2, 17). However, other CXC chemokines may also be involved in the response to hepatic I/R injury.
The physiological importance of stromal cell-derived factor-1 (SDF-1), a CXC chemokine, in embryogenesis and organ development has been well documented, and mice deficient of this chemokine axis uniformly die in the perinatal period (25, 33). In developed organisms, CXCR4 signaling has been implicated in the mobilization and recruitment of progenitor cells to sites of injury (1, 6, 26). The exact role of CXCR4 signaling in normal liver physiology has not been fully elucidated. Previous studies have suggested that SDF-1 binding to CXCR4 may play a role in liver repair through the recruitment of hematopoietic stem cells to the injured liver, and conditional CXCR4 knockout mice have been shown to be more susceptible to chronic liver injury (8, 16, 36). However, CXCR4 signaling has also been implicated as a mediator of liver inflammation and progression to cirrhosis in viral hepatitis through the activation of hepatic stellate cells (10). Consistent with those studies, we previously reported reduced liver injury after ischemic and endotoxic insults in mice depleted of stellate cells (32). Given the seemingly divergent roles for SDF-1/CXCR4 in liver pathobiology, we sought to further elucidate the role of CXCR4 signaling in acute liver injury and liver repair after hepatic I/R.
MATERIALS AND METHODS
Hepatic I/R injury model.
Male wild-type mice on a C57BL/6 background (22–28 g body wt; Jackson Laboratory, Bar Harbor, ME) were used in these experiments. This project was approved by the University of Cincinnati Animal Care and Use Committee and was in compliance with the National Institutes of Health guidelines. The animals underwent sham surgery or I/R. Partial hepatic ischemia was induced as described previously (20). Briefly, mice were anesthetized with pentobarbital sodium (60 mg/kg ip). A midline laparotomy was performed, and an atraumatic clip was used to interrupt blood supply to the left lateral and median lobes of the liver. The caudal lobes retained intact portal and arterial inflow and venous outflow, preventing intestinal venous congestion. After 90 min of partial hepatic ischemia, the clip was removed to initiate hepatic reperfusion. Sham control mice underwent the same protocol without vascular occlusion. For acute injury studies, some mice were injected intraperitoneally with 10 mg/kg AMD3100 (Sigma-Aldrich, St. Louis, MO) or 1 μg of recombinant murine SDF-1α (Peprotech, Rocky Hill, NJ) 1 h prior to initiation of ischemia. For longer studies, some mice were intraperitoneally injected every 12 h with 10 mg/kg AMD3100 or 1 μg of recombinant murine SDF-1α starting at 24 h of reperfusion. An identical volume of sterile phosphate-buffered saline (PBS) was used as a vehicle control for all studies. Mice were euthanized after the indicated periods of reperfusion. Blood and liver tissue samples were taken for analysis.
Partial (70%) hepatectomy model.
Male wild-type C57BL/6 mice (22–28 g body wt; Jackson Laboratory) were used in these experiments. Partial (70%) hepatectomy was performed according to the method of Higgins and Anderson (9). Briefly, mice were anesthetized with pentobarbital sodium (60 mg/kg ip). A midline laparotomy was performed, and 4-0 Vicryl suture (Ethicon Endo-Surgery, Cincinnati, OH) ligatures were secured around the base of the median and left lateral hepatic lobes and the lobes were resected. Some wild-type mice were injected intraperitoneally every 12 h with 10 mg/kg AMD3100 or 1 μg of recombinant murine SDF-1α starting at 24 h after hepatectomy. An identical volume of sterile PBS was used as a vehicle control. Mice were euthanized at the indicated times after hepatectomy, and blood and liver tissue samples were taken for analysis. Liver-to-body weight ratio was determined and normalized to the prehepatectomy liver-to-body weight ratio (30).
Blood and tissue analysis.
Blood was obtained by cardiac puncture for serum analysis. Serum and liver concentrations of SDF-1 were assessed by enzyme-linked immunosorbent assay (ELISA; R & D Systems, Minneapolis, MN). Liver samples were weighed and immediately placed in 10 volumes (wt/vol) of a protease inhibitor cocktail containing 10 nmol/l EDTA, 2 mmol/l PMSF, 0.1 mg/ml soybean trypsin inhibitor, 1.0 mg/ml bovine serum albumin, and 0.002% sodium azide in isotonic PBS, pH 7.0. Tissues were disrupted with a tissue homogenizer, and lysates were incubated at 4°C for 2 h. Samples were clarified by two rounds of centrifugation at 12,500 g for 10 min at 4°C. Liver tissues were fixed in 10% neutral-buffered formalin, processed, and then embedded in paraffin for light microscopy. Sections were stained with hematoxylin and eosin for histological examination.
Western blot analysis.
Liver samples were homogenized in lysis buffer [10 mM HEPES (pH 7.9), 150 mM NaCl, 1 mM EDTA, 0.6% NP-40, 0.5 mM PMSF, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 10 μg/ml soybean trypsin inhibitor, and 1 μg/ml pepstatin]. Samples were sonicated and incubated for 30 min on ice. Samples of equal protein content were separated in a denaturing 4–20% polyacrylamide gel and transferred to a 0.1-μm pore nitrocellulose membrane. Nonspecific binding sites were blocked with Tris-buffered saline containing 5% bovine serum albumin for 1 h at room temperature. Membranes were incubated overnight with antibody to CXCR4 (Abcam, Cambridge, MA) in Tris-buffered saline with 0.1% Tween 20. Membranes were washed and incubated with secondary antibody conjugated to horseradish peroxidase. Immunoreactive proteins were detected by enhanced chemiluminescence.
Proliferating cell nuclear antigen staining.
Immunohistochemical staining for proliferating cell nuclear antigen (PCNA) was performed on paraffin-embedded liver tissue with anti-PCNA antibody using the DakoCytomation ARK kit (Dako, Copenhagen, Denmark). Briefly, a three-step peroxidase method was performed according to the manufacturer's instruction. PC-10 monoclonal antibody (Santa Cruz Biotechnology) was used at a dilution of 1:50 for 15 min at room temperature. The sections were counterstained with hematoxylin. PC-10 immunostaining was evaluated on the basis of the percentage of positive nuclei of 400–600 hepatocytes from the 5 highest positive fields at high power (×400) and expressed as PCNA labeling index.
Flow cytometry.
Single-cell suspensions were prepared from the bone marrow preparations. Cells were suspended in fluorescein-activated cell sorting (FACS) buffer (PBS with 1% bovine albumin and 0.1% sodium azide). Blood was collected via direct cardiac puncture. Whole blood samples were anticoagulated with heparin and transferred to FACS tubes. ACK lysing buffer was added, and the samples were incubated at room temperature. After incubation, the samples were centrifuged at 400 g for 5 min. Cells were suspended in FACS buffer (PBS with 1% bovine albumin and 0.1% sodium azide). Cell counts were determined using a cell counter (AcT 10, Beckman Coulter, Pasadena, CA). Nonspecific binding to cells was controlled by addition of 5% rat serum (Invitrogen) and Fc Block (1 μg/tube; BD Pharmingen) to the FACS buffer. Cells were stained with CD45 phycoerythrin (PE) and CD31 PE/Cy7 antibodies (BioLegend). Cells were surface-labeled as described above and then fixed with 2% paraformaldehyde. Nonviable cells were excluded. All samples were run on an Attune Acoustic Focusing Cytometer and analyzed by FACS Attune software (Applied Sciences).
Hepatocyte isolation and stimulation.
Hepatocytes from male C57BL/6 mice were isolated by nonrecirculating collagenase perfusion through the portal vein as previously described (18). Livers were perfused in situ with 45 ml of GIBCO liver perfusion medium (Invitrogen, Carlsbad, CA) followed by 45 ml of GIBCO liver digestion medium (Invitrogen). The liver was excised, minced, and strained through a steel mesh. The dispersed hepatocytes were collected by centrifugation at 50 g for 2 min at 4°C and washed twice with Williams medium (Invitrogen). Hepatocytes were isolated using Percoll separation and washed twice with Williams medium. The final pellet was resuspended in Williams medium. Hepatocytes were counted and plated in fetal bovine serum-fortified Williams medium, distributed onto 96-well flat-bottomed collagen-coated plates (Life Technologies, Grand Island, NY) at a concentration of 1.5 × 104 cells·200 μl−1·well−1. Plates were incubated for 2 h to allow cell adherence. Medium was removed from adherent hepatocytes, and the cells were reincubated with fetal bovine serum-fortified Williams medium. Some cells were treated with medium alone (control), medium + exogenous SDF-1 (500 ng/ml), medium + exogenous epidermal growth factor (EGF, 50 ng/ml), or medium + SDF-1 (500 ng/ml) and EGF (50 ng/ml). Medium was changed at 48 h. At 96 h, hepatocyte proliferation was determined by measurement of DNA incorporation of 5-bromo-2′-deoxyuridine (BrdU) using the BrdU cell proliferation ELISA kit (Abcam) according to the manufacturer's instructions.
Liver cell production of SDF-1.
Hepatocytes, Kupffer cells, and hepatic stellate cells were isolated from C57BL/6 mice using methods we have previously described (28, 29). Primary liver sinusoidal endothelial cells from C57BL/6 mice were purchased from CellBiologics (Chicago, IL). All cells were cultured for 48 h, at which time culture medium was analyzed by ELISA for SDF-1 concentration according to the manufacturer's directions (R & D Systems).
Liver engraftment of adoptively transferred CD45 cells.
Male C57BL/6.SJL mice, which express CD45.1, and C57BL/6 mice, which express CD45.2, were obtained from Jackson Laboratory. CD45.1 mice underwent 90 min of partial hepatic ischemia and 48 h of reperfusion as described above. Blood was obtained via cardiac puncture, and mononuclear cells were isolated by dextran sedimentation and Histopaque gradient. Cells (1.5 × 105 in PBS) were transferred into C57BL/6 mice via penile vein injection. Control mice were injected with an equivalent volume of PBS. At 1 h after injection, recipient mice were euthanized, and livers were fixed in 10% buffered formalin, processed, embedded, sectioned, and stained for CD45.1 by immunohistochemistry.
Statistical analysis.
Values are means ± SE. Data were analyzed using one-way analysis of variance with subsequent Student-Newman-Keuls test. Differences were considered significant when P < 0.05.
RESULTS
Expression of SDF-1 and CXCR4 are upregulated after I/R.
Previous studies showed increased SDF-1 and CXCR4 expression in fibrotic liver disease (10). To determine its role in acute liver injury and repair after I/R, we first examined the expression of SDF-1. Liver tissue and serum samples were taken from sham mice and injured mice at varying times of reperfusion, and SDF-1 was assessed by ELISA. Liver and serum SDF-1 levels were maximally increased at 8 h of reperfusion (Fig. 1A), which correlates with maximal liver injury after hepatic I/R (21). Serum SDF-1 returned to baseline levels within 24 h, whereas tissue SDF-1 levels remained elevated for up to 96 h after reperfusion. To assess whether liver cells produce SDF-1, primary hepatocytes, Kupffer cells, stellate cells, and sinusoidal endothelial cells were isolated, and their production of SDF-1 was measured after 24 h in culture. As shown in Fig. 1B, all these cell types produced low levels of SDF-1, suggesting that the liver is a source of this chemokine.
Fig. 1.

Expression of stromal cell-derived factor-1 (SDF-1) and CXC chemokine receptor-4 (CXCR4) after hepatic ischemia-reperfusion (I/R) injury. A: liver and serum samples were assessed for SDF-1 expression by enzyme-linked immunosorbent assay at varying times of reperfusion. Values are means ± SE; n = 3–6 per group. *P < 0.05 vs. sham; **P < 0.05 vs. all groups. B: SDF-1 production by primary hepatocytes, Kupffer cells, hepatic stellate cells (HSC), and sinusoidal endothelial cells (SEC). Values are means ± SE; n = 3–6 per group. C: hepatic expression of CXCR4 after I/R. Lysates from whole liver and primary (1°) hepatocytes were assessed for CXCR4 expression by Western blot analysis. Lysate from Jurkat T cells was used as a positive control, and β-actin staining served as a loading control. CXCR4 antibody detects a 43-kDa band, as well as other nonspecific bands. Arrows, 43-kDa band for CXCR4 and band for β-actin.
We next examined hepatic expression of CXCR4 in sham mice and after I/R injury. Liver sections were taken from mice after sham operation and at 4, 8, 24, 48, and 96 h of reperfusion. CXCR4 expression was assessed by Western blot analysis (Fig. 1B). We found CXCR4 to be constitutively expressed in the liver of wild-type mice undergoing sham operation, with several isoforms present (31). CXCR4 expression remained present at all reperfusion times examined, and levels were unchanged compared with sham controls. CXCR4 expression in several primary liver cell lines has been documented, most notably in liver sinusoidal endothelial cells (24) and hepatic stellate cells (10). In addition, we show that CXCR4 is constitutively expressed by primary murine hepatocytes (Fig. 1C). The antibody employed detects a 43-kDa band (Fig. 1C, arrow), as well as other nonspecific bands.
SDF-1/CXCR4 signaling does not alter acute injury after hepatic I/R.
To determine if CXCR4 signaling has an effect on acute I/R injury, mice were treated 1 h prior to ischemia with AMD3100, a CXCR4 antagonist, or recombinant murine SDF-1α. Liver and serum samples were taken from mice at 8 h of reperfusion after I/R injury. Serum alanine aminotransferase (ALT) and tissue myeloperoxidase (MPO) levels were used to determine the extent of liver injury and neutrophil recruitment, respectively (Fig. 2, A and B). At 8 h of reperfusion, there were no differences in serum ALT or liver MPO between the vehicle control and treatment groups. These findings were confirmed by histological examination (Fig. 2C).
Fig. 2.
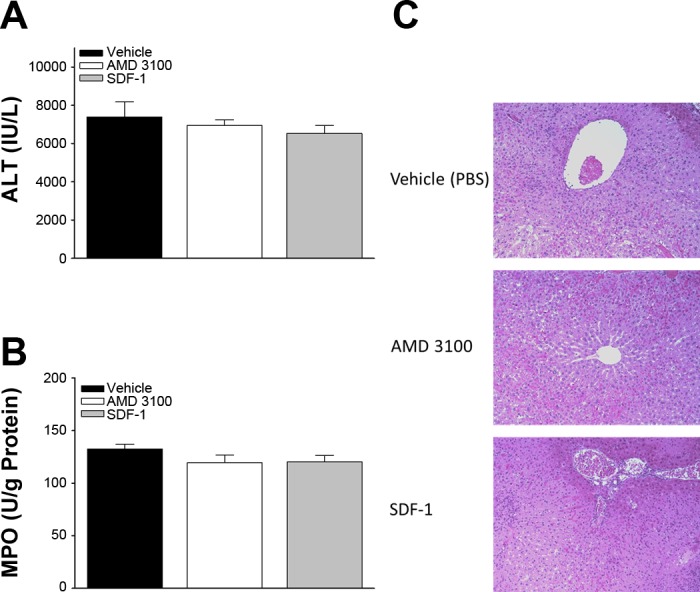
SDF-1/CXCR4 signaling is not involved in acute liver injury induced by hepatic I/R. Liver injury after 8 h of reperfusion was assessed by serum alanine aminotransferase (ALT, A) and liver myeloperoxidase (MPO, B). C: histological analysis of extent of liver injury and tissue destruction. Values are means ± SE; n = 4 per group.
SDF-1/CXCR4 signaling limits hepatocyte proliferation after I/R.
Previous studies suggested that SDF-1/CXCR4 signaling contributes to tissue recovery (8, 14, 35). We evaluated liver recovery after hepatic ischemia and 72, 96, and 168 h of reperfusion. Mice were treated every 12 h with 10 mg/kg AMD3100 or 1 μg of recombinant murine SDF-1α starting at 24 h after reperfusion until euthanization. Hepatocyte proliferation was used to evaluate liver regeneration after hepatic I/R. Liver sections from nonischemic and postischemic tissues were stained for PCNA, and the percentage of PCNA-positive hepatocytes was calculated. PCNA-positive hepatocytes in the nonischemic tissue were increased at 72 and 96 h of reperfusion. The increase in PCNA-positive hepatocytes was consistent and similar in each of the treatment groups (data not shown). The number of PCNA-positive hepatocytes was also increased in postischemic tissues from each of the treatment groups compared with sham controls at 72, 96, and 168 h of reperfusion (Fig. 3). Interestingly, treatment with AMD3100 resulted in increased hepatocyte proliferation at 72 and 96 h of reperfusion compared with vehicle control (Fig. 3). In contrast, treatment with SDF-1 resulted in decreased hepatocyte proliferation at 72 and 96 h.
Fig. 3.

SDF-1/CXCR4 signaling limits hepatocyte proliferation after hepatic I/R injury. Hepatocyte proliferation after I/R was determined by quantitative analysis of immunohistochemical staining for proliferating cell nuclear antigen (PCNA) in mice treated with PBS (vehicle), AMD3100, or recombinant murine SDF-1. Treatment with SDF-1 reduced hepatocyte proliferation, while treatment with the CXCR4 antagonist AMD3100 increased hepatocyte proliferation. Values are means ± SE; n = 6–8 per group. *P < 0.05 vs. control (vehicle).
To examine the degree of liver repair after I/R injury, histological analysis was performed on liver sections from each of the treatment groups at 72, 96, and 168 h of reperfusion. Evaluation of the nonischemic tissue at each time point revealed normal liver architecture without necrosis or signs of hepatic injury (data not shown). In vehicle-treated mice, extensive necrosis of postischemic liver tissue was observed, especially at 72 and 96 h of reperfusion (Fig. 4). Treatment with SDF-1 had no effect on the amount of liver necrosis. However, treatment with AMD3100 resulted in reduced necrosis after 72, 96, and 168 h of reperfusion (Fig. 4), likely due to the increased hepatocyte proliferation in this group (Fig. 3). Interestingly, this effect was not observed after a more modest ischemic insult, i.e., 60 min of ischemia (data not shown).
Fig. 4.

Blockade of CXCR4 improves liver recovery after I/R injury. Mice were injected intraperitoneally with PBS (vehicle), AMD3100 (10 mg/kg), or recombinant murine SDF-1 (1 μg) 24 h after reperfusion and every 12 h thereafter. All postischemic tissues showed extensive destruction and necrosis, consistent with I/R injury. Morphometric quantification of necrotic area showed significantly reduced areas of necrosis in mice treated with AMD3100. Values are means ± SE; n = 6–8 per group. *P < 0.05 vs. SDF-1. **P < 0.05 vs. all other groups.
SDF-1/CXCR4 signaling limits hepatocyte proliferation in injured, but not uninjured, tissue.
Because we found that treatment with AMD3100 or SDF-1 had no effect on hepatocyte proliferation in the nonischemic lobes, which do not undergo significant liver injury, we next assessed whether SDF-1/CXCR4 was important for liver regeneration in the absence of liver injury. Mice underwent partial (70%) hepatectomy and were treated every 12 h with vehicle, 10 mg/kg AMD3100, or 1 μg of recombinant murine SDF-1α beginning 24 h after hepatectomy. We observed no differences in normalized liver weight after partial hepatectomy between the treatment groups (Fig. 5A). Furthermore, the number of proliferating hepatocytes was increased in all groups at 48 h posthepatectomy. However, there were no differences between the treatment groups in the number of PCNA-positive hepatocytes (Fig. 5B).
Fig. 5.

SDF-1/CXCR4 signaling does not regulate hepatic regeneration after partial hepatectomy. Mice were subjected to partial hepatectomy and injected intraperitoneally with PBS (vehicle), AMD3100 (10 mg/kg), or recombinant SDF-1 (1 μg) every 12 h starting 24 h after partial hepatectomy. Recovery of functional liver mass (A) and hepatocyte proliferation (B) was similar in all treatment groups at 48 h posthepatectomy. Values are means ± SE; n = 5 per group.
SDF-1 does not modulate hepatoctye proliferation directly.
To determine if the effects of CXCR4 stimulation or blockade on hepatocyte proliferation in vivo were due to direct effects on hepatocytes, we next evaluated the effects of SDF-1 on EGF-mediated hepatocyte proliferation in primary hepatocytes. Isolated hepatocytes were treated with medium, SDF-1, EGF, or EGF + SDF-1, and proliferation was determined 96 h later by BrdU incorporation. EGF treatment induced significant hepatocyte proliferation, but cotreatment with SDF-1 had no effect (Fig. 6). These data suggest that the in vivo effects of CXCR4 manipulation on hepatocyte proliferation are not mediated by a direct effect on hepatocytes.
Fig. 6.

SDF-1 does not alter epidermal growth factor (EGF)-mediated hepatocyte proliferation in vitro. Primary murine hepatocytes were treated with medium (control), SDF-1 (500 ng/ml), EGF (50 ng/ml), or EGF + SDF-1. Proliferation was determined 96 h later by measurement of 5-bromo-2′-deoxyuridine (BrdU) incorporation. Values are means ± SE; n = 8–16 per group. *P < 0.05 vs. control.
CXCR4 blockade mobilizes vasculogenic cells from the bone marrow to the systemic circulation.
Next, we utilized flow cytometric analysis to evaluate circulating levels of CD45+/CD31+ cells, which have been shown to be a vasculogenic cell population (15). Mice were subjected to hepatic I/R injury or sham operation and treated with twice-daily injections of AMD3100 or SDF-1α starting 24 h after reperfusion. The number of CD45+/CD31+ cells in bone marrow and blood was analyzed by flow cytometry at 48 h (Fig. 7). There was an overall decrease in the number of bone marrow CD45+/CD31+ cells with AMD3100 treatment (Fig. 7A). This decrease was associated with an increase in the number of CD45+/CD31+ cells in peripheral blood (Fig. 7B). To determine if CD45+/CD31+ cells could engraft in the liver, C57BL/6.SJL mice, which express CD45.1 on hematopoietic cells, were subjected to hepatic ischemia and 48 h of reperfusion. Peripheral blood mononuclear cells were isolated, and 1.5 × 105 cells were adoptively transferred into C57BL/6 mice, which express CD45.2 on hematopoietic cells. Liver sections were stained for CD45.1 at 1 h after transfer. As shown in Fig. 7C, CD45.1 cells engrafted in the liver.
Fig. 7.

Blockade of CXCR4 mobilizes CD31+/CD45+ from the bone marrow to the blood after liver I/R. Mice were subjected to hepatic I/R injury or sham procedure and injected intraperitoneally with PBS (vehicle), AMD3100 (10 mg/kg), or recombinant SDF-1 (1 μg) every 12 h starting 24 h after I/R injury. CD31+/CD45+ cells were assessed by flow cytometry in bone marrow (A) and peripheral blood (B). Treatment with AMD3100 resulted in a decrease in the number of CD31+/CD45+ cells in the bone marrow and an increase in the peripheral circulation. Values are means ± SE; n = 4 per group. *P < 0.05 vs. control. **P < 0.05 vs. all groups. C: engraftment of CD45 cells in liver. Liver sections from C57BL/6 mice were obtained 1 h after adoptive transfer of 1.5 × 105 mononuclear cells isolated after I/R injury from C57BL/6 mice, which express CD45.1 on hematopoietic cells. Number of CD45.1 cells per high-powered field (HPF) were counted. Values are means ± SE; n = 5–6 per group. *P < 0.05 vs. control.
DISCUSSION
This study provides new evidence about the contributions of SDF-1/CXCR4 signaling in acute injury, repair, and regeneration after hepatic I/R. While the Western blot for CXCR4 detected multiple bands, detection of the expected 43-kDa band suggests that CXCR4 is constitutively expressed by the liver and that its expression remains consistent throughout the recovery phase after hepatic I/R injury. While maximal expression of SDF-1 in serum and liver tissue occurred simultaneously with maximal liver injury, we found no evidence that SDF-1/CXCR4 signaling has a role in acute liver injury induced by I/R. However, CXCR4 blockade with a receptor antagonist, AMD3100, caused increased hepatocyte proliferation in the postischemic tissue after hepatic I/R, whereas treatment with SDF-1 decreased hepatocyte proliferation. These findings suggest that CXCR4 signaling is a mediator of the reparative and regenerative phases following ischemic injury. Interestingly, stimulation or blockade of CXCR4 did not alter liver regeneration in the nonischemic tissue. Similarly, in a model of liver regeneration induced by partial hepatectomy, CXCR4 stimulation or blockade had no effects on hepatocyte proliferation or liver regeneration, suggesting that the proliferative and regenerative properties relate only to injured liver.
Our in vitro data suggest that the observed effects in vivo with CXCR4 stimulation or blockade are not mediated by direct effects on hepatocytes. Previous studies demonstrated the role of SDF-1 signaling through its receptor CXCR4 in the recruitment of progenitor cells, which would suggest a role for improved recovery after injury (8, 16, 35). Our data suggest that CXCR4 signaling after I/R is detrimental to recovery of injured hepatic tissue and that pharmacological blockade of CXCR4 improved hepatocyte proliferation and reduced tissue damage. In fact, treatment with AMD3100 after hepatic I/R injury resulted in mobilization of a previously described vasculogenic cell population (15). These findings are consistent with recent reports of improved tissue recovery with CXCR4 antagonism after injury in cardiac tissue, lung parenchyma, and skeletal muscle (14, 23, 34). The proposed mechanism for these findings is mobilization of endothelial progenitor cells and resultant improved angiogenesis in the recovery of ischemic tissue (25). This mechanism is consistent with our findings of increased hepatocyte proliferation in postischemic tissue but does not account for attenuated recovery with administration of exogenous SDF-1 after I/R. Treatment with AMD3100 has also been shown to decrease key mediators of asthmatic lung inflammation, suggesting a tissue-specific role for CXCR4 signaling in organ injury (27). Although we did not find differences in our acute markers of injury after hepatic I/R, we did observe differences in the amount of tissue necrosis at later time points, which could result from increased angiogenesis, attenuated inflammation, or both.
Alternatively, the changes to hepatocyte proliferation after CXCR4 stimulation and blockade in postischemic liver could be due to effects on stellate cells. Activation of hepatic stellate cells through CXCR4 signaling has been well documented (10). Additionally, our recent studies have demonstrated reduced hepatic injury after insult in stellate-depleted mice (27, 32). However, since our current data do not support a role for SDF-1/CXCR4 signaling in acute liver injury, it is unlikely that any effects of CXCR4 stimulation or blockade are altering stellate cell contributions to injury. Instead, SDF-1/CXCR4 signaling could be altering stellate cell function in the reparative/regenerative phase. However, the manner in which stellate cells modulate liver regeneration after acute injury has not been thoroughly studied. Additional studies are necessary to determine if SDF-1/CXCR4 signaling in hepatic stellate cells contributes to liver injury.
In summary, this study represents the first examination of hepatic expression of CXCR4 and its role in liver inflammation and recovery after hepatic I/R injury. Our data show that expression of CXCR4 and its ligand SDF-1 is upregulated after hepatic I/R injury and that SDF-1/CXCR4 signaling is associated with decreased hepatocyte proliferation and repair in postischemic, but not nonischemic or uninjured, liver tissue. Furthermore, we demonstrate that these effects are not due to direct signaling in hepatocytes but may be related to altered mobilization of CD31+/CD45+ cells from the bone marrow. These findings shed new light on the regulation of liver repair and regeneration after I/R injury.
GRANTS
This work was supported in part by National Institutes Health Grants DK-56029, AG-025881, and GM-08478 to A. B. Lentsch.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
G.C.W., C.M.F., J.W.K., R.C.Q., H.N., R.S., J.B., and C.C.C. performed the experiments; G.C.W., C.M.F., J.W.K., R.C.Q., H.N., R.S., J.B., M.J.E., C.C.C., and A.B.L. analyzed the data; G.C.W., C.M.F., J.W.K., R.C.Q., H.N., M.J.E., C.C.C., and A.B.L. interpreted the results of the experiments; G.C.W., H.N., and A.B.L. prepared the figures; G.C.W. and A.B.L. drafted the manuscript; G.C.W., C.M.F., J.W.K., R.C.Q., H.N., R.S., J.B., M.J.E., C.C.C., and A.B.L. edited and revised the manuscript; G.C.W., C.M.F., J.W.K., R.C.Q., H.N., R.S., J.B., M.J.E., C.C.C., and A.B.L. approved the final version of the manuscript; A.B.L. developed the concept and designed the research.
REFERENCES
- 1.Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP, Gurtner GC. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med 10: 858–864, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Clarke C, Kuboki S, Sakai N, Kasten KR, Tevar AD, Schuster R, Blanchard J, Caldwell CC, Edwards MJ, Lentsch AB. CXC chemokine receptor-1 is expressed by hepatocytes and regulates liver recovery after hepatic ischemia/reperfusion injury. Hepatology 53: 261–271, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colletti LM, Green M, Burdick MD, Kunkel SL, Strieter RM. Proliferative effects of CXC chemokines in rat hepatocytes in vitro and in vivo. Shock 10: 248–257, 1998. [DOI] [PubMed] [Google Scholar]
- 5.Colletti LM, Green ME, Burdick MD, Strieter RM. The ratio of ELR+ to ELR− CXC chemokines affects the lung and liver injury following hepatic ischemia/reperfusion in the rat. Hepatology 31: 435–445, 2000. [DOI] [PubMed] [Google Scholar]
- 6.Dar A, Goichberg P, Shinder V, Kalinkovich A, Kollet O, Netzer N, Margalit R, Zsak M, Nagler A, Hardan I, Resnick I, Rot A, Lapidot T. Chemokine receptor CXCR4-dependent internalization and resecretion of functional chemokine SDF-1 by bone marrow endothelial and stromal cells. Nat Immunol 6: 1038–1046, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Fausto N, Laird AD, Webber EM. Liver regeneration. 2. Role of growth factors and cytokines in hepatic regeneration. FASEB J 9: 1527–1536, 1995. [DOI] [PubMed] [Google Scholar]
- 8.Hatch HM, Zheng D, Jorgensen ML, Petersen BE. SDF-1α/CXCR4: a mechanism for hepatic oval cell activation and bone marrow stem cell recruitment to the injured liver of rats. Cloning Stem Cells 4: 339–351, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Higgins GM, Anderson RM. Experimental pathology of the liver. I. Restoration of the liver of the white rat following partial surgical removal. Arch Pathol 12: 186–202, 1931. [Google Scholar]
- 10.Hong F, Tuyama A, Lee TF, Loke J, Agarwal R, Cheng X, Garg A, Fiel MI, Schwartz M, Walewski J, Branch A, Schecter AD, Bansal MB. Hepatic stellate cells express functional CXCR4: role in stromal cell-derived factor-1α-mediated stellate cell activation. Hepatology 49: 2055–2067, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaeschke H. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am J Physiol Gastrointest Liver Physiol 284: G15–G26, 2003. [DOI] [PubMed] [Google Scholar]
- 12.Jaeschke H, Bautista AP, Spolarics Z, Spitzer JJ. Superoxide generation by Kupffer cells and priming of neutrophils during reperfusion after hepatic ischemia. Free Radic Res Commun 15: 277–284, 1991. [DOI] [PubMed] [Google Scholar]
- 13.Jaeschke H, Farhood A. Neutrophil and Kupffer cell-induced oxidant stress and ischemia-reperfusion injury in rat liver. Am J Physiol Gastrointest Liver Physiol 260: G355–G362, 1991. [DOI] [PubMed] [Google Scholar]
- 14.Jujo K, Ii M, Sekiguchi H, Klyachko E, Misener S, Tanaka T, Tongers J, Roncalli J, Renault MA, Thorne T, Ito A, Clarke T, Kamide C, Tsurumi Y, Hagiwara N, Qin G, Asahi M, Losordo DW. CXC-chemokine receptor 4 antagonist AMD3100 promotes cardiac functional recovery after ischemia/reperfusion injury via endothelial nitric oxide synthase-dependent mechanism. Circulation 127: 63–73, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim H, Cho HJ, Kim SW, Liu B, Choi YJ, Lee J, Sohn YD, Lee MY, Houge MA, Yoon YS. CD31+ cells represent highly angiogenic and vasculogenic cells in bone marrow: novel role of nonendothelial CD31+ cells in neovascularization and their therapeutic effects on ischemic vascular disease. Circ Res 107: 602–614, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kollet O, Shivtiel S, Chen YQ, Suriawinata J, Thung SN, Dabeva MD, Kahn J, Spiegel A, Dar A, Samira S, Goichberg P, Kalinkovich A, Arenzana-Seisdedos F, Nagler A, Hardan I, Revel M, Shafritz DA, Lapidot T. HGF, SDF-1, and MMP-9 are involved in stress-induced human CD34+ stem cell recruitment to the liver. J Clin Invest 112: 160–169, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuboki S, Shin T, Huber N, Eismann T, Galloway E, Schuster R, Blanchard J, Edwards MJ, Lentsch AB. Hepatocyte signaling through CXC chemokine receptor-2 is detrimental to liver recovery after ischemia/reperfusion in mice. Hepatology 48: 1213–1223, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuboki S, Shin T, Huber N, Eismann T, Galloway E, Schuster R, Blanchard J, Zingarelli B, Lentsch AB. Peroxisome proliferator-activated receptor-γ protects against hepatic ischemia/reperfusion injury in mice. Hepatology 47: 215–224, 2008. [DOI] [PubMed] [Google Scholar]
- 19.Lentsch AB, Kato A, Yoshidome H, McMasters KM, Edwards MJ. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischemia/reperfusion injury. Hepatology 32: 169–173, 2000. [DOI] [PubMed] [Google Scholar]
- 20.Lentsch AB, Yoshidome H, Cheadle WG, Miller FN, Edwards MJ. Chemokine involvement in hepatic ischemia/reperfusion injury in mice: roles for macrophage inflammatory protein-2 and KC. Hepatology 27: 1172–1177, 1998. [DOI] [PubMed] [Google Scholar]
- 21.Lentsch AB, Yoshidome H, Kato A, Warner RL, Cheadle WG, Ward PA, Edwards MJ. Requirement for interleukin-12 in the pathogenesis of warm hepatic ischemia/reperfusion injury in mice. Hepatology 30: 1448–1453, 1999. [DOI] [PubMed] [Google Scholar]
- 22.Luedde T, Trautwein C. Intracellular survival pathways in the liver. Liver Int 26: 1163–1174, 2006. [DOI] [PubMed] [Google Scholar]
- 23.Lukacs NW, Berlin A, Schols D, Skerlj RT, Bridger GJ. AMD3100, a CxCR4 antagonist, attenuates allergic lung inflammation and airway hyperreactivity. Am J Pathol 160: 1353–1360, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mendt M, Cardier JE. Stromal-derived factor-1 and its receptor, CXCR4, are constitutively expressed by mouse liver sinusoidal endothelial cells: implications for the regulation of hematopoietic cell migration to the liver during extramedullary hematopoiesis. Stem Cells Dev 21: 2142–2151, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, Yoshida N, Kikutani H, Kishimoto T. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 382: 635–638, 1996. [DOI] [PubMed] [Google Scholar]
- 26.Petit I, Szyper-Kravitz M, Nagler A, Lahav M, Peled A, Habler L, Ponomaryov T, Taichman RS, Arenzana-Seisdedos F, Fujii N, Sandbank J, Zipori D, Lapidot T. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol 3: 687–694, 2002. [DOI] [PubMed] [Google Scholar]
- 27.Puche JE, Lee YA, Jiao J, Aloman C, Fiel MI, Munoz U, Kraus T, Lee T, Yee HF Jr, Friedman SL. A novel murine model to deplete hepatic stellate cells uncovers their role in amplifying liver damage in mice. Hepatology 57: 339–350, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quillin RC 3rd, Wilson GC, Nojima H, Freeman CM, Wang J, Schuster RM, Blanchard JA, Edwards MJ, Gandhi CR, Gulbins E, Lentsch AB. Inhibition of acidic sphingomyelinase reduces established hepatic fibrosis in mice. Hepatol Res 45: 305–314, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakai N, Van Sweringen HL, Quillin RC, Schuster R, Blanchard J, Burns JM, Tevar AD, Edwards MJ, Lentsch AB. Interleukin-33 is hepatoprotective during liver ischemia/reperfusion in mice. Hepatology 56: 1468–1478, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sekine S, Gutierrez PJ, Lan BY, Feng S, Hebrok M. Liver-specific loss of β-catenin results in delayed hepatocyte proliferation after partial hepatectomy. Hepatology 45: 361–368, 2007. [DOI] [PubMed] [Google Scholar]
- 31.Sloane AJ, Raso V, Dimitrov DS, Xiao X, Deo S, Muljadi N, Restuccia D, Turville S, Kearney C, Broder CC, Zoellner H, Cunningham AL, Bendall L, Lynch GW. Marked structural and functional heterogeneity in CXCR4: separation of HIV-1 and SDF-1α responses. Immunol Cell Biol 83: 129–143, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Stewart RK, Dangi A, Huang C, Murase N, Kimura S, Stolz DB, Wilson GC, Lentsch AB, Gandhi CR. A novel mouse model of depletion of stellate cells clarifies their role in ischemia/reperfusion- and endotoxin-induced acute liver injury. J Hepatol 60: 298–305, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y, Kitamura Y, Matsushima K, Yoshida N, Nishikawa S, Kishimoto T, Nagasawa T. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 393: 591–594, 1998. [DOI] [PubMed] [Google Scholar]
- 34.Tan Y, Li Y, Xiao J, Shao H, Ding C, Arteel GE, Webster KA, Yan J, Yu H, Cai L, Li X. A novel CXCR4 antagonist derived from human SDF-1β enhances angiogenesis in ischaemic mice. Cardiovasc Res 82: 513–521, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Togel F, Isaac J, Hu Z, Weiss K, Westenfelder C. Renal SDF-1 signals mobilization and homing of CXCR4-positive cells to the kidney after ischemic injury. Kidney Int 67: 1772–1784, 2005. [DOI] [PubMed] [Google Scholar]
- 36.Tsuchiya A, Imai M, Kamimura H, Takamura M, Yamagiwa S, Sugiyama T, Nomoto M, Heike T, Nagasawa T, Nakahata T, Aoyagi Y. Increased susceptibility to severe chronic liver damage in CXCR4 conditional knock-out mice. Dig Dis Sci 57: 2892–29002012. [DOI] [PubMed] [Google Scholar]