

Abstract
Pulmonary arterial hypertension (PAH) is a progressive disease that, if left untreated, eventually leads to right heart failure and death. Elevated pulmonary arterial pressure (PAP) in patients with PAH is mainly caused by an increase in pulmonary vascular resistance (PVR). Sustained vasoconstriction and excessive pulmonary vascular remodeling are two major causes for elevated PVR in patients with PAH. Excessive pulmonary vascular remodeling is mediated by increased proliferation of pulmonary arterial smooth muscle cells (PASMC) due to PASMC dedifferentiation from a contractile or quiescent phenotype to a proliferative or synthetic phenotype. Increased cytosolic Ca2+ concentration ([Ca2+]cyt) in PASMC is a key stimulus for cell proliferation and this phenotypic transition. Voltage-dependent Ca2+ entry (VDCE) and store-operated Ca2+ entry (SOCE) are important mechanisms for controlling [Ca2+]cyt. Stromal interacting molecule proteins (e.g., STIM2) and Orai2 both contribute to SOCE and we have previously shown that STIM2 and Orai2, specifically, are upregulated in PASMC from patients with idiopathic PAH and from animals with experimental pulmonary hypertension in comparison to normal controls. In this study, we show that STIM2 and Orai2 are upregulated in proliferating PASMC compared with contractile phenotype of PASMC. Additionally, a switch in Ca2+ regulation is observed in correlation with a phenotypic transition from contractile PASMC to proliferative PASMC. PASMC in a contractile phenotype or state have increased VDCE, while in the proliferative phenotype or state PASMC have increased SOCE. The data from this study indicate that upregulation of STIM2 and Orai2 is involved in the phenotypic transition of PASMC from a contractile state to a proliferative state; the enhanced SOCE due to upregulation of STIM2 and Orai2 plays an important role in PASMC proliferation.
Keywords: store-operated calcium channel, pulmonary artery, PASMC, phenotype
pulmonary arterial hypertension (PAH) is a progressive and fatal disease that predominantly affects women. In patients with PAH, the elevated pulmonary arterial pressure (PAP) is mainly caused by increased pulmonary vascular resistance (PVR), which results in an increase in the afterload for the right ventricle and, if untreated, leads to right heart failure and eventually death. Elevated PVR in patients with PAH is primarily caused by sustained pulmonary vasoconstriction and excessive pulmonary vascular remodeling (including pulmonary vascular wall thickening, small vessel obliteration, and formation of plexiform lesions). In pediatric patients, such as those with persistent pulmonary hypertension in the newborn (PPHN) or hypoxia-induced pulmonary hypertension, inhalation of nitric oxide (NO) and infusion of vasodilator (e.g., adenosine, prostacyclin) significantly reduces PAP and PVR (2). In adult patients with idiopathic PAH, however, only a minority of the patients respond to vasodilators, i.e., acute treatment of the patients with inhaled NO or infused vasodilators can decrease PAP and PVR to the normal level in ∼15% patients (34). These data indicate that, at early stage, sustained pulmonary vasoconstriction plays an important role in the initiation and development of pulmonary hypertension in patients with PAH; while at late stage, a gradual transition from sustained vasoconstriction to vascular remodeling may play a critical role in the progression of pulmonary hypertension. Then, at the stage when patients develop physiological right ventricular hypertrophy, excessive pulmonary vascular remodeling characterized by the adventitial, medial, and intimal hypertrophy of the pulmonary arteries, the intraluminal obliteration and occlusion of small pulmonary arteries and arterioles, the neointimal and plexiform lesions in the pulmonary vasculature are the predominant causes for maintaining the increase in PVR and PAP. Sustained increase in PVR and PAP, or the elevated afterload, would eventually cause pathological right ventricular hypertrophy and, if untreated, right heart failure and death.
These assumptions and observations are also in good agreement with the clinical manifestation of the disease. In pediatric patients, for example, intravenous administration of vasodilators or inhalation of NO are very effective to reduce PVR and PAP, while in adult patients there are only a few patients (15%) who are defined as “responders” (23, 31, 33, 34). Administration of vasodilators (e.g., adenosine, prostacyclin) or inhalation of NO cause significant reduction of PVR and PAP only in “responders.” Most of the adult patients with PAH, however, are defined as “nonresponders”; conventional treatment with Ca2+ channel blockers like nifedipine and verapamil or vasodilators are no longer effective and useful for the patients. Other newly developed drugs which have antiproliferative effects are commonly used to treat nonresponder patients (11, 12).
Both basic and clinical research data indicate that, at the early stage of PAH, sustained pulmonary vasoconstriction is an important contributor to the development or initiation of the disease manifestation such as the elevated PVR and PAP (24, 29, 40). However, at the progression and/or late stage of the disease, the transition from a sustained pulmonary vasoconstrictive phenotype to an excessive pulmonary vascular remodeling phenotype is an important pathogenic process in the progression of PAH.
Smooth muscle cells including pulmonary arterial smooth muscle cells (PASMC) are extremely plastic and can dedifferentiate in response to various environmental stimuli from a contractile or quiescent phenotype to a proliferative or synthetic phenotype (28, 30). In addition, a phenotypic switch of PASMC from a contractile to proliferative phenotype is inevitable for any pathological and physiological vascular remodeling process to occur. Contractile PASMC represent the majority of PASMC in the in vivo functional pulmonary vessels, and they are responsible for maintenance and regulation of vascular tone that is required for maintaining PVR to the blood flow. Normal pulmonary vasoconstriction and vasodilatation, due primarily to PASMC contraction and relaxation, respectively, play an important role in regulating and maintaining a normal pulmonary arterial pressure and blood flow.
Enhanced PASMC proliferation has been demonstrated to play an important role in the development and progression of pulmonary vascular remodeling in patients with idiopathic PAH and animals with experimental pulmonary hypertension. The increased PASMC proliferation is due at least in part to an increased cytosolic Ca2+ concentration ([Ca2+]cyt) resulting from augmented store-operated Ca2+ entry (SOCE) and upregulated Orai and canonical transient receptor potential (TRPC) channel expression (45, 47). There have been some studies suggesting that Orai2, in addition to forming Orai channels, also directly interacts with TRPC6 to form heteromeric channels to allow SOCE (3). Furthermore, it was demonstrated that clustered aggregates of stromal interacting molecule (STIM)-Orai associate with activated TRPC6 and stabilize their conformation in a state in which it operates as a Ca2+ release-activated channel (CRAC) (19). Therefore, we aimed to examine whether Ca2+-permeable channels (e.g., Orai and TRPC6) and their regulatory proteins (e.g., STIM) are involved in the transition of PASMC from a contractile phenotype to a highly proliferative phenotype. Specifically, we examined 1) whether upregulated expression of STIM2 correlated with the transition of PASMC from a contractile to a proliferative phenotype; and 2) whether upregulated expression of store-operated Ca2+ channels (SOC) was involved in the development of PAH. Our results provide strong evidence that the transition of PASMC from a contractile phenotype to a proliferative phenotype is associated with a significantly enhanced SOCE and that upregulated expression of STIM1/STIM2, Orai1/Orai2, and TRPC6 is required for the enhanced SOCE.
MATERIALS AND METHODS
Isolation of rat pulmonary artery smooth muscle tissue.
Protocols involving the use of experimental animals for all experiments were reviewed and approved by the Ethics/Animal Care Committee of the University of Illinois at Chicago and The University of Arizona. Sprague-Dawley male rats (150–200 g) were decapitated, and the whole lung and heart were removed en bloc and placed in warm Hanks' balanced salt solution (HBSS, Life Technologies, Carlsbad, CA) supplemented with 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES; Sigma Aldrich, St. Louis, MO). The right and left branches of the main pulmonary as well as the intrapulmonary arteries were first isolated from the whole lung with fine forceps under a dissecting microscope. The fat and connective tissues were then removed gently from the isolated pulmonary artery (PA) under sterile conditions. The isolated PA was incubated in HBSS containing 1.7 mg/ml collagenase type II (Worthington Biochemical; Lakewood Township, NJ) for 20 min at 37°C. Then, the shortly digested PA ring was rinsed with HBSS to remove residual collagenase, the adventitia of the PA ring was carefully stripped off with fine forceps, and the endothelium was denuded with a sterile cotton swab. The remaining PA smooth muscle tissue was then used to prepare single PASMC and to extract total protein for Western blot experiments.
Isolation and preparation of rat PASMC.
Following removal of the adventitia and endothelium, the rat PA was further digested in HBSS 1.7 mg/ml collagenase type II, 0.5 mg/ml elastase (Sigma), and 1 mg/ml bovine serum albumin (BSA, Sigma) at 37°C for 50 min. The PA tissue was agitated every 15–20 min to speed digestion. The dispersed PA tissue was then triturated approximately 8–10 times with a fire-polished Pasteur pipette, to further dissociate the cells. Ten milliliters of Dulbecco's modified Eagle's medium (DMEM; Corning, Herndon, VA) supplemented with 7 mM NaH2CO3, 10 mM HEPES (pH 7.2), 20% fetal bovine serum (FBS, Corning), and 1% penicillin and streptomycin (Pen/Strep, Corning) was then added to the enzymatic solution to stop digestion. The cell suspension was centrifuged for 5 min at 1,500 rpm at room temperature (22–24°C). The supernatant was aspirated off and the resulting pellet was resuspended in 2 ml of fresh 10% FBS-DMEM and triturated to separate the cells.
For experiments using freshly dissociated PASMC, aliquots of the cell suspension were plated directly onto glass coverslips coated with 5% gelatin (porcine, Sigma Aldrich) or in six-well dishes (Corning) with 2.5 ml of 10% FBS-DMEM. These freshly dissociated rat PASMC were allowed to attach to the coverslips for 3–4 h before loading with fura-2 acetoxymethyl ester (fura-2 AM) for measurement of cytosolic free Ca2+ concentration ([Ca2+]cyt). The freshly dissociated cells plated directly into the six-well dishes were used for immunocytochemistry and immunoblotting experiments to detect protein expression levels of various ion channels.
To prepare primary cultured rat PASMC, aliquots of the cell suspension were plated onto gelatin-coated coverslips in 25-mm petri dishes or directly onto 10-cm petri dishes with 10% FBS-DMEM and incubated in a humidified atmosphere of 5% CO2 in air at 37°C. Twenty-four hours later, the culture media were changed to Medium 199 (M199) supplemented with 10% FBS, 100 μg/ml cell growth supplement, and antibiotics (penicillin and streptomycin). The medium was changed 24 h after initial seeding and every 48 h subsequently. When the cells reached 80–90% confluency, they were gently washed with phosphate-buffered saline (PBS), incubated briefly with 1 ml of 0.025% trypsin-EDTA solution until detachment (3–5 min), and then 9 ml 10% FBS-M199 was added to the plate. The cell suspension was then transferred to a sterile 15-ml round-bottom tube, centrifuged at room temperature for 5 min at 200 × g (1,500 rpm), resuspended in the appropriate growth media and seeded onto coverslips or petri dishes, and used for Western blot and [Ca2+]cyt measurement experiments.
Mouse PASMC isolation.
PASMC were isolated from mouse lungs, as described previously (37). Briefly, a mixture of 5 ml of M199 growth medium containing 5 g/l low-melting-point agarose type VII (Sigma), 5 g/l iron beads (diameter <10 μM; Sigma), and antibiotics (penicillin and streptomycin) was slowly injected over a period of 60 s through the right ventricle, thereby perfusing the PA. M199 growth medium (1 ml) containing 5 g/l agarose type VII was injected in airways through the trachea. The lungs were plunged in cold PBS to cause the agarose to gel. Because of the rapidly solidifying nature of the agarose and the size of the iron particles, the likelihood of traversing the capillary space is minimized. All the lobes were then isolated and finely minced in a petri dish. The tissue was further disrupted by passing through a 16-gauge followed by an 18-gauge needle approximately five times. The suspension was then mixed in M199 growth medium containing 80 U/ml type IV collagenase (Sigma) and incubated at 37°C for 90 min. With the use of a magnetic column (Invitrogen), the arteries containing the iron beads were collected. The supernatant was aspirated and the arteries were washed and suspended in 5 ml M199 containing 20% FBS. Aliquots of the suspension were transferred to T25 culture flasks. Smooth muscle cell purity was determined by immunostaining with smooth muscle specific actin antibody.
Measurement of [Ca2+]cyt in PASMC.
[Ca2+]cyt was measured using fura-2 AM (Invitrogen-Molecular Probes, Eugene, OR), a membrane-permeable Ca2+-sensitive fluorescent indicator, and a Nikon digital imaging fluorescent microscopy system. Cells on 25-mm coverslips were loaded with 4 μM fura-2 AM in normal physiological salt solution (PSS) for 60 min at room temperature (22–24°C) in the dark. The PSS solution contained (in mM) 137 NaCl, 5.4 KCl, 1.8 CaCl2, 1.2 MgCl2, 10 HEPES, and 10 glucose (pH was adjusted to 7.4 with 1 N NaOH). The fura-2 AM-loaded cells were then placed in a recording chamber on the stage of an inverted fluorescent microscope (Eclipse Ti-E; Nikon, Tokyo, Japan) equipped with an objective lens (S. Plan Fluor ×20/0.45 ELWD; Nikon) and Em-CCD camera (Evolve; Photometrics, Tucson, AZ). The recording chamber was continuously perfused with PSS at a flow rate of 2 ml/min using a mini pump (model 3385; Control, Friendswood, TX). The fura-2 AM-loaded cells were then washed by perfusion with normal PSS for 20 min to remove excess extracellular fura-2 AM and allow sufficient time for intracellular esterase to cleave fura-2 AM to active fura-2. The cells were excited at 340- and 380-nm wavelengths (D340xv2 and D380xv2 filters, respectively; Chroma Technology, Bellows Falls, VT) by a xenon arc lamp (Lambda LS; Sutter Instrument, Novato, CA) and an optical filter changer (Lambda 10-B). Emission of fura-2 fluorescence was collected through a dichroic mirror (400DCLP) and a wide band emission filter (D510/80m). [Ca2+]cyt within the region of interest (4 × 4 μm), which was placed or positioned at the peripheral region of each cell, was measured as the ratio of fluorescence intensities (F340/F380) every 2 s. [Ca2+]cyt measurements were carried out at 32°C using an automatic temperature controller (TC-344B, Warner Instruments, Hamden, CT), as increased fura-2 compartmentalization in intracellular organelles and cell dye leakage have been reported to occur at the physiological temperature (37°C) (32). PASMC were isolated from Trpc6−/− mice and wild-type (WT) littermates for [Ca2+]cyt measurements. Trpc6−/− mice breeding pair was initially obtained from National Institute of Environmental Health Sciences (Research Triangle Park, NC).
Measurement of isometric tension in the isolated mice PA rings.
The intrapulmonary artery was dissected from the mice lungs, and the isolated mice PA ring was cut into 2-mm long segments. Two tungsten hooks (with a diameter of 100 μm) were carefully inserted into the lumen of the PA ring under a dissecting microscope. One hook was then connected to the bottom of a perfusion chamber, and the other was attached to an isometric force transducer (Harvard Apparatus). The resting passive tension was set and maintained at an optimal tension of 300 mg (43), and the rings were allowed to stabilize at resting tension for ∼1 h before experimentation. The isometric tension was continuously measured and recorded, and data were acquired using DATAQ software (DATAQ Instruments). Isolated PA rings were perfused with modified Krebs solution (MKS: at 37°C) consisting of the following chemicals (in mM): 138 NaCl, 1.8 CaCl2, 4.7 KCl, 1.2 MgSO4, 1.2 NaH2PO4, 5 HEPES, and 10 glucose (pH 7.4). The active tension (or the absolute amount of force) relative to the basal tension was measured and expressed as the net increase in tension (mg). The endothelium was removed by repeatedly passing a rough-surface stainless steel wire through the intralumen of the PA ring; functional removal of the endothelium was confirmed by the loss of acetylcholine-mediated dilatation of the isolated PA ring. To stabilize the vessel and obtain a stable contractile response, the isolated PA rings were challenged by superfusion with 60 mM K+-containing solution (60K) three times before experimentation. The amplitude of the 60K-induced PA contraction (or the 60K-induced increase in active tension in isolated PA rings) and the baseline tension of the isolated PA rings are usually stable after three times of challenges with 60K-containing solution.
Measurement of pulmonary arterial pressure in isolated and perfused/ventilated lungs.
Experiments using the mice in this study were approved by the Institutional Animal Care and Use Committee at the University of Illinois at Chicago and The University of Arizona. C57BL/6 mice (22–25 g) were anesthetized with ketamine (100 mg/kg)-xylazine (26 mg/kg) via intra-abdominal injection. After a tracheostomy was performed, mice were ventilated with a gas mixture of 21% O2, 5% CO2 via a rodent ventilator (minivent type 845, Harvard Apparatus). Respiratory rate was maintained at 80 breaths/min and tidal volume was 10 ml/kg (∼250 ml). Positive end-expiratory pressure was maintained at 2 mmH2O. End-inspiratory plateau pressure (EIP) was measured with a pressure transducer (MPX type 399/2, Hugo Sachs Elektronik-Harvard Apparatus, Germany). To prevent blood coagulation, 20 IU heparin was injected into the right ventricle. A stainless steel catheter was inserted into the main pulmonary artery (PA) after a right ventriculotomy was performed, and the PA and ascending aorta were tied together. Pulmonary arterial pressure (PAP) was measured using a pressure sensor (P75 Type 379, Hugo Sachs Elektronik-Harvard Apparatus, Germany), which was connected to the PA catheter. The PAP, left atrial pressure, and EIP were monitored continuously. For data acquisition and data storage, Powerlab 8/30, Quad Bridge Amp, and LabChart (AD Instruments, Australia) were used. After basal PAP was stabilized for 40–60 min, the experiments were performed. The physiological salt solution (PSS) used for the perfusate consisted of the following composition (mM): 120 NaCl 120, 4.3 KCl, 19 NaHCO3, 1.1 KH2PO4, 10 glucose, 1.8 CaCl2, and 1.2 MgCl2 (pH 7.4). To protect prostaglandin synthesis, 3.1 mM sodium meclofenamate was added to the perfusate. For isotonic high-K+ solutions (40 mM), NaCl was replaced by an equimolar amount of KCl. To demonstrate the role of Ca2+ on contraction, voltage-dependent Ca2+ channel blocker (1 μM nifedipine) was added to the perfusate. Full methods were described previously by Yoo et al. (44).
Western blot experiments.
PA smooth muscle tissue was placed in an Eppendorf tube on ice and lysed with cold RIPA lysis buffer (Millipore, Billerica, MA) supplemented with protease inhibitor cocktail (Roche; Basel, Switzerland) by sonication three times for 30 s each time. Cultured rat PASMC and HEK cells were washed with ice-cold PBS, scraped, placed into an Eppendorf tube, and centrifuged. The pelleted cells were resuspended in 20–50 μl of RIPA buffer supplemented with protease inhibitor cocktail. Lysed tissues and cells were incubated in lysis buffer for 15 min on ice. The lysates were then centrifuged at 13,300 rpm for 15 min at 4°C. The pellet was discarded and from the supernatant, protein concentration was determined by Bradford Protein Assay (Bio-Rad, Hercules, CA) with BSA as a standard. Proteins (10–20 μg) were mixed and boiled in Laemmli sample buffer supplemented with 2-mercaptoethanol (BME, Sigma) reducing agent. Protein lysates were resolved on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto 0.45-μm nitrocellulose membranes (Bio-Rad). Membranes were incubated for 1 h at 22–24°C in a blocking buffer [0.1% Tween 20 in TBS (TBST)] containing 5% nonfat dry milk powder. The membranes were then incubated with primary antibodies diluted in TBST containing 5% BSA, with shaking overnight at 4°C. Membranes were washed three times in TBST for 5 min each, followed by incubation in secondary antibody conjugated to horseradish peroxidase for 2 h at room temperature in TBST containing 5% milk. Membranes were washed three times for 5 min each, and peroxidase activity was visualized with enhanced chemiluminescence substrate (Pierce, Rockford, IL). Primary antibodies included anti-calponin (1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA), myosin heavy chain (MYH, 1:1,200, Santa Cruz), smooth muscle 22-α (SM22α; 1:1,000, Santa Cruz), proliferating cellular nuclear antigen (PCNA, 1:1,000, Santa Cruz), STIM1 (1:1,000, Pro-Sci), STIM2 (1:1,000, Sigma), Orai1 (1:500, Pro-Sci), Orai2 (1:500, Alomone, Israel), Orai3 (1:500, Alomone), TRPC6 (1:500, Sigma), and β-actin (1:2,000, Santa Cruz). Band intensity was quantified with ImageJ (National Institutes of Health, Bethesda, MD), normalized to β-actin control, and is expressed as arbitrary units.
Transfection of PASMC.
Twenty-four hours after seeding, HEK293 cells at 60–80% confluency were transiently transfected with STIM2 and Orai2 expression constructs using Lipofectamine 2000 transfection reagent, based on the manufacturer's protocol. Transfection was performed at 37°C in serum-free Opti-MEM medium (Gibco) with 0.5 μg/ml DNA and transfection reagent (μl). After 5–6 h of incubation at 37°C with transfection medium, the medium was changed to serum-containing DMEM and cells were incubated for 24–72 h before experiments. Rat PASMC were transiently transfected with STIM2 and Orai2 expression constructs and siRNA targeting STIM2 and Orai2 using Amaxa Basic Nucleofector kit (Lonza, Walkersville, MD) via electroporation according to the manufacturer's instructions. Serum-free Opti-MEM medium (Gibco) was used to dilute cells, transfection reagent, DNA, and siRNA. After 5–6 h of incubation at 37°C with transfection medium, the medium was changed to serum-containing M199 and cells were incubated for 24–72 h. [Ca2+]cyt measurements and Western blotting using DNA- and siRNA-transfected cells were performed 48–72 h after transient transfection. cDNA vectors used for upregulation were as follows: STIM2 [plasmid 18868: pEX-CMV-SP-STIM2 (15-746)], Orai2 (plasmid 16369: pcDNA3.1 Orai2). Small interfering RNA agents used for downregulation were as follows: STIM2 (Ambion Life Technology, Carlsbad, CA), Orai2 (siGENOME Thermo Scientific), and scrambled siRNA (siGENOME Thermo Scientific).
Cell proliferation assays.
To determine rat PASMC proliferation rate in vitro, we used bromodeoxyuridine (BrdU) incorporation assay and measured the changes in cell number using a cell counter. BrdU incorporation assay (Millipore) was used according to the manufacturer's protocol. Confluent primary cultured cells were detached with 0.05% trypsin-EDTA, seeded into 96-well plates (2.5 × 103 cells/well) in M199 containing 10% FBS, and allowed to attach onto the plates overnight. PASMC were then incubated in serum-free M199 for 24 h and then incubated with vehicle control or with 1, 2, or 3 ng/ml of transforming growth factor-β (TGF-β) for 24, 72, or 96 h, respectively. In addition, cells were incubated with 0.3%, 1.0%, or 10% FBS-M199 for 24, 72, or 96 h. Cells were also incubated in 10% FBS-M199 with 500 nM Ca2+ or 1.2 mM Ca2+ for 24 or 72 h. During the final 4 h of incubation, BrdU was incorporated into actively dividing cells. Cells were fixed/denatured in room temperature fixative solution. Anti-BrdU antibody was added, followed by incubation with secondary anti-mouse IgG peroxidase-conjugate. Fluorescence was measured using a spectrophotometer microplate reader at 450/540 nm.
For cell counting experiments, PASMC growth curves were examined by cell counting with the Bio-Rad TC10 automated cell counter. Subculture cells were growth arrested overnight, after which they were collected, counted, and equally seeded into eight-well multidishes (Nunclon Δ, 10.5 cm2 of culture area per well; Thermo Scientific), with 1 × 105 cells/well (0.95 × 104 cells/cm2, 3 wells/sample); this number was used as baseline (0 h). Treatment cells were counted 24, 72, and 96 h after the start of experiments. Each count was an average of three repeats, and each data point was the average of four experiments.
PCR analysis.
Genomic DNA from a tail biopsy was extracted by standard procedures and subjected to PCR for genotyping. The primer sequences and anticipated band sizes are as follows: TRPC6-WT (378 bp): forward (F)-TCT TTA TGC AAT CGC TGT GG; reverse (R)-GCT AGT CTT CCT GCA ATC CA; TRPC6-Mut (175 bp): F-TCT ATT AAC ACT CAA CTG GCA CCT; R-GCC AGA GGC CAC TTG TGT AG.
The cycling parameters for the PCR were as follows: 1 cycle at 94°C for 3 min, 33 cycles of 94°C for 15 s, then 15 s of 60°C, and 72°C for 18 s. The PCR ended with 5 min at 72°C.
Statistical analysis.
Data are expressed as means ± SE and were analyzed for statistical significance by the unpaired Student's t-test or one-way ANOVA for multiple groups using SigmaPlot software. Differences were considered to be significant at P < 0.05. Significant difference is expressed in the figures or figure legends as P < 0.05, P < 0.01, and P < 0.001.
RESULTS
Upregulated expression of STIM2, TRPC6, and Orai2 in the proliferative phenotype of PASMC in culture compared with the contractile phenotype of PASMC in isolated PA.
To determine the potential differences in the expression levels of STIM1, TRPC6, and Orai2 between proliferative phenotype and contractile phenotype of PASMC, we used Western blot analysis to compare protein levels of STIM1, TRPC6, and Orai2 in isolated PA rings with denuded endothelium and stripped-off adventitia (contractile PASMC phenotype) and in primary cultured PASMC (proliferative PASMC phenotype). To confirm the contractility of PASMC in isolated PA rings, we first examined the response of isolated PA rings from rat to high K+-containing solution (40 mM and 60 mM) in the presence and absence of extracellular Ca2+ or in the absence or presence of nifedipine (1 μM), a dihydropyridine Ca2+ channel blocker that potently blocks the L-type and T-type voltage-dependent Ca2+ channels in PASMC.
As shown in Fig. 1, in isolated pulmonary artery (PA) rings from rats with denuded endothelium and stripped-off adventitia (Fig. 1A), raising extracellular K+ concentration ([K+]o) from 4.7 to 60 mM caused a large vasoconstriction determined by the increase in isometric tension (Fig. 1Ba, left). Increasing [K+]o, by changing the K+ equilibrium potential, causes membrane depolarization, opens L-type voltage-dependent Ca2+ channels (VDCC), and increases [Ca2+]cyt (39, 41, 43). The high K+ (60 or 40 mM K+)-mediated PA contraction shown in Fig. 1Ba (left) was obviously due to membrane depolarization-mediated Ca2+ influx through VDCC. Removal of extracellular Ca2+ almost abolished the 60K-induced increase of tension (Fig. 1Ba, left, and Bb, left), while application of 1 μM nifedipine, a dihydropyridine L-type VDCC blocker, significantly reduced the 60 mM K+-mediated PA contraction (Fig. 1Ba, right and Bb, right) (41). In isolated perfused/ventilated mouse lung, raising [K+]o from 4.7 mM to 40 mM also significantly increased pulmonary arterial pressure (PAP) by causing pulmonary vasoconstriction or contraction of PASMC in contractile phenotype. Extracellular application of the VDCC blocker nifedipine (Fig. 1, Ca and Cb) significantly and reversibly inhibited 40K-induced increase in PAP. These results obtained from rat and mouse lung PA demonstrate that 1) PASMC in isolated PA rings (used in these contraction experiments and the following Western blot experiments) are in contractile phenotype, and 2) high K+-mediated pulmonary vasoconstriction in the contractile phenotype is significantly dependent on the rise in cytosolic [Ca2+]cyt due to Ca2+ influx through VDCC.
Fig. 1.

Stromal interacting molecule protein 2 (STIM2), canonical transient receptor potential channel 6 (TRPC6), and Orai2 are upregulated in proliferative pulmonary arterial smooth muscle cells (PASMC) in comparison to contractile PASMC. A: pulmonary artery (PA) isolated from normal male rat for the contraction experiments. B: representative tracing (a) of isometric tension measured in isolated PA rings constricted by 60-mM K+ (60K) before, during, and after application of Ca2+-free (0Ca) modified Krebs solution (MKS) (left) or 1 μM nifedipine (Nif)-containing MKS (right). Summarized data (means ± SE, b) showing 60K-induced active tension before (Cont), during (0Ca or Nif), and after (Rec) application of 0Ca-MKS (0Ca) or Nif-containing MKS (Nif). ***P < 0.001 vs. Cont and Rec bars. C: representative tracing (a) of pulmonary arterial pressure (PAP) in isolated perfused/ventilated lung preparation showing 40 mM K+ (40K)-mediated changes in PAP before, during, and after application of nifedipine. Summarized data (means ± SE, b) showing 40K-induced increase in PAP before (Cont), during (Nif), and after (Rec) application of 1 μM Nif. ***P < 0.001 vs. Cont and Rec bars. D: primary cultured PASMC (a) isolated from rat PA stained with antibody against smooth muscle α-actin (green fluorescence) and DAPI (blue fluorescence). Bromodeoxyuridine (BrdU) incorporation (b) of PASMC cultured in 0.3% FBS-DMEM (0.3% FBS) and 10% FBS-DMEM in the normal Ca2+ (1.8 mM Ca2+) and low Ca2+ (0.5 mM Ca2+) was measured 24 or 72 h after cells were plated (means ± SE). ***P < 0.001 vs. 10% FBS-1.8 mM Ca2+ bars (72 h). E and F: representative Western blot images (E) and summarized data (means ± SE, F) for myosin heavy chain (MYH), smooth muscle 22-α (SM22α), and calponin (Ea) as well as STIM2, TRPC6, and Orai2 (Eb) in isolated PA or contractile PA tissues (PA) and cultured PASMC or proliferative PASMC (PASMC). β-Actin was used as a loading control. *P < 0.05, **P < 0.01 vs. PA.
To determine the role of STIM and SOC proteins in the phenotypic transition of PASMC, we first compared the protein expression levels of STIM2, Orai2, and TRPC6 between PASMC in contractile phenotype and proliferative phenotype. In primary cultured PASMC derived from rat PA (Fig. 1Da), addition of 10% FBS significantly increased the number of cells as determined by measuring BrdU incorporation (Fig. 1Db). Reducing extracellular Ca2+ concentration in the culture media from 1.8 mM to 0.5 μM (by adding the Ca2+ chelator, EGTA) completely abolished the 10% FBS-mediated PASMC proliferation (Fig. 1Db). The purity of PASMC in culture was determined by the positive staining with smooth muscle cell α-actin (SMαA) and the filament structure of SMαA (Fig. 1Da). These data indicate that primary cultured PASMC (used in the cell proliferation or BrdU incorporation experiments and the following Western blot experiments) are all in a proliferative phenotype.
To further confirm the phenotype of PASMC in isolated PA and in primary culture, we compared the expression levels of myosin heavy chain (MYH), smooth muscle 22-α (SM22α), and calponin. As shown in Fig. 1, the expression level of these differentiation markers was significantly higher in the contractile phenotype of PASMC in PA rings than in the proliferative PASMC cultured in media with 10% FBS (Fig. 1, Ea and F, top). Importantly, the protein expression of STIM2, TRPC6, and Orai2 were all significantly upregulated in the proliferative phenotype of PASMC cultured in growth media in comparison to the contractile phenotype of PASMC in isolated PA rings (Fig. 1, Eb and F, bottom). These data indicate that, when contractile-phenotype PASMC are dissociated from isolated PA rings and cultured in growth media, the cells undergo phenotypical changes to become proliferative-phenotype cells, and the loss of the differentiation markers (e.g., MYH, SM22α, and calponin) is associated with a significant upregulation of STIM2, TRPC6, and Orai2.
Enhanced store-operated Ca2+ entry (SOCE) in the proliferative phenotype of PASMC compared with the contractile phenotype of PASMC (freshly dissociated PASMC).
To determine whether the amplitude of [Ca2+]cyt increase due to SOCE is different between proliferative and contractile phenotypes of PASMC, we compared cyclopiazonic acid (CPA)-mediated increase in [Ca2+]cyt in freshly dissociated PASMC (contractile phenotype) and primary cultured PASMC (proliferative phenotype) in media containing 10% FBS and growth factors (Fig. 2A). As shown in Fig. 2, extracellular application of 10 μM CPA, a SERCA inhibitor that induces Ca2+ influx due to passive depletion of Ca2+ in the intracellular Ca2+ stores or the sarcoplasmic reticulum (SR), caused a slow increase in [Ca2+]cyt due to Ca2+ mobilization from the SR in freshly dissociated (contractile phenotype) PASMC bathed in Ca2+-free (0Ca) solution. Approximately 10 min after treatment with CPA in the absence of extracellular Ca2+, restoration of extracellular Ca2+ (to 1.8 mM) induced a rapid increase in [Ca2+]cyt due apparently to SOCE (Fig. 2, B and C, top). The CPA-mediated increase in [Ca2+]cyt due to SOCE was significantly enhanced in proliferative PASMC compared with freshly dissociated (contractile phenotype) PASMC (Fig. 2, B and C, top), while 60 mM K+-mediated increase in [Ca2+]cyt was significantly reduced in proliferative PASMC compared with freshly dissociated contractile PASMC (Fig. 2, B and C, bottom). The enhanced SOCE in proliferative PASMC was associated with 1) downregulated MYH, a differentiation marker, and upregulated PCNA, a proliferation marker (4), and 2) upregulated TRPC6, STIM2, Orai2, and Orai3 (Fig. 2, D and E). These data indicate that 1) SOCE is enhanced as a result of upregulated expression of proteins that participate in forming store-operated Ca2+ channels (SOC), such as TRPC6, Orai2/3, and STIM2, in proliferative PASMC (in which PCNA expression is upregulated) compared with contractile or differentiated PASMC (in which MYH expression is much higher), and 2) Ca2+ entry through voltage-dependent Ca2+ channels (VDCC) induced by 60 mM K+-mediated membrane depolarization is significantly reduced in proliferative PASMC compared with contractile PASMC.
Fig. 2.

Store-operated Ca2+ entry (SOCE) is enhanced and voltage-dependent Ca2+ entry (VDCE) is attenuated in freshly dissociated (contractile) PASMC and primary cultured (proliferative) PASMC. A: Fura 2 fluorescence (F360) images showing freshly dissociated contractile PASMC (top) and primary cultured proliferative PASMC (bottom) from which cytosolic Ca2+ concentration ([Ca2+]cyt) was measured. B: representative records showing cyclopiazonic acid (CPA)-induced changes in [Ca2+]cyt in the absence (0Ca) and presence of extracellular (1.8 mM) Ca2+ (top) and 60K-induced changes in [Ca2+]cyt in the presence of extracellular (1.8 mM) Ca2+ (bottom) in contractile (left) and proliferative (right) PASMC. C: summarized data (means ± SE) showing the amplitudes of CPA-induced increases (top) in [Ca2+]cyt due to Ca2+ mobilization (Release) and store-operated Ca2+ entry (SOCE), and the 60K-induced increase (bottom) in [Ca2+]cyt due to voltage-dependent Ca2+ entry (60K) in contractile (open bars) and proliferative (closed bars) PASMC. **P < 0.01, ***P < 0.001 vs. contractile PASMC. D: representative Western analyses on MYH, proliferating cellular nuclear antigen (PCNA), TRPC6, STIM2, Orai2, and Orai3 in contractile and proliferative PASMC. β-Actin was used as a loading control. E: summarized data (means ± SE) showing protein expression levels of MYH, PCNA, STIM2, TRPC6, Orai2, and Orai3, in contractile PASMC (open bars) and proliferative PASMC (closed bars). *P < 0.05 vs. contractile PASMC. The upregulation of PCNA is associated with significant increase in protein expression of STIM2, TRPC6, Orai2, and Orai3 and marked decrease in protein expression of MYH in proliferative PASMC compared with contractile PASMC.
Expression level of STIM2 and TRPC6 is positively correlated with expression of PCNA, a cell proliferation marker, in proliferative phenotype of PASMC.
Reduction of serum concentration and removal of growth factors from culture media lead to growth arrest of PASMC (47). Addition of serum or increase of serum concentration (from 1% to 10%, for example) stimulated PASMC proliferation and significantly increased protein expression level of PCNA, a cell proliferation marker. As shown in Fig. 3, A and B, the protein expression level of PCNA in PASMC cultured in media containing 10% FBS was significantly greater than in PASMC cultured in 0.1% FBS-containing media (Fig. 3, A and B). When PASMC were cultured in 10% FBS-containing media, the number of cells was increased by 2.5 times after 72 h (Fig. 3C, solid triangles); when PASMC were cultured in 0.1% FBS-containing media, however, the number of cells was not significantly changed at 72 h (Fig. 3C, open circles). While the increased PCNA expression in PASMC cultured in 10% FBS-containing media was correlated well with the increased cell proliferation (Fig. 3), the protein expression levels of the differentiation markers calponin and SM22α were actually decreased in PASMC cultured in 10% FBS-containing media compared with cells cultured in 0.1% FBS-containing media.
Fig. 3.

Upregulation of STIM2, TRPC6, and Orai2 is associated with increased expression of PCNA and decreased expression of calponin and SM22α in proliferative PASMC cultured in high concentration of serum (10% FBS). A: Western blot analyses on PCNA, calponin, and SM22α (top) as well as STIM2, TRPC6, and Orai2 (bottom) in PASMC cultured in media containing 1%, 3%, or 10% FBS. β-Actin was used as a loading control. B: summarized data (means ± SE) showing the protein expression levels of PCNA, calponin, and SM22α (top) as well as STIM2, TRPC6, and Orai2 (bottom) in rat PASMC cultured in media supplemented with 1% (open bars), 3% (gray bars), or 10% (solid bars) FBS. *P < 0.05, **P < 0.01 vs. 1% FBS bars. C: time course of cell growth (or changes in cell numbers) in rat PASMC cultured in media containing 1%, 3%, or 10% FBS at 24, 48, or 72 h. ***P < 0.001 vs. 1% FBS and 3% FBS curves.
The increased proliferation and upregulated PCNA expression in proliferating phenotype of PASMC cultured in 10% FBS-containing media were positively associated with upregulation of STIM2, TRPC6, and Orai2 (Fig. 3, A and B). These data indicate that the change from quiescent state to proliferative state is associated with an upregulation of STIM2, TRPC6, and Orai2 in PASMC. In other words, the higher proliferation rate of PASMC is closely correlated with the higher expression level of STIM2, TRPC6, and Orai2. Given the fact that Ca2+ influx through store-operated Ca2+ channels (SOC) is required for the enhanced PASMC proliferation (8, 26, 35, 47), it is possible that gradually upregulated STIM2, TRPC6, and Orai2 are involved in the transition of PASMC from quiescent state to the proliferative state. The upregulated STIM2, TRPC6, and Orai2 may also play an important role in maintaining a high proliferation rate in proliferating PASMC to, for example, repair vascular injury and cause pulmonary vascular medial hypertrophy.
Treatment of proliferative PASMC with TGF-β upregulates expression of smooth muscle cell differentiation markers and downregulates expression of TRPC6 and STIM1.
TGF-β is a growth factor that enhances smooth muscle progenitor cell differentiation (6, 17) by activating SMAD signaling cascades (15, 38). In proliferative PASMC cultured in media containing 10% FBS, treatment of the cells with TGF-β (1 to 2 ng/ml) significantly decreased expression of PCNA, a cell proliferation marker, and inhibited cell proliferation, but significantly increased expression levels of the differentiation markers, such as calponin and SM22α (Fig. 4A). Furthermore, TGF-β-mediated PASMC differentiation was associated with a significant downregulation of TRPC6 and STIM2 (Fig. 4B) as well as Orai1 and Orai2 (Fig. 4C). In PASMC cultured in 10% FBS-containing media, TGF-β at concentrations of 1 ng/ml, 2 ng/ml, and 3 ng/ml (for 24–72 h) significantly inhibited the cell growth (Fig. 4D). These observations are consistent with the data showing that STIM2, Orai2, and TRPC6 are upregulated when PASMC underwent the transition from the quiescent (more differentiated state) to the highly proliferative state (see Fig. 3). The results shown in Fig. 4 also imply that PASMC in highly proliferative state (e.g., cultured in media containing 10% FBS and growth factors) tend to have higher expression of STIM2, Orai1/2, and TRPC6 than PASMC in more differentiated state (e.g., treated with TGF-β or cultured in media without serum and growth factors).
Fig. 4.

Transforming growth factor-β (TGF-β) upregulates the cell differentiation markers, downregulates the cell proliferation markers, and downregulates protein expression of TRPC6, STIM2, and Orai1/2 in proliferative PASMC. A: Western blot analyses on PCNA, calponin, and SM22α in control proliferative PASMC (Control) and PASMC treated with 1 and 2 ng/ml TGF-β (left). β-Actin was used as a loading control. Summarized data (means ± SE, right) showing the protein expression levels of PCNA, calponin, and SM22α in control rat PASMC (Control, treated with vehicle) and PASMC treated with 1 ng/ml (1 ng) and 2 ng/ml (2 ng). *P < 0.05, **P < 0.01, and ***P < 0.001 vs. control. B and C: Western blot analyses on TRPC6, STIM1, and STIM2 (B) as well as Orai1 and Orai2 (C) in control proliferative PASMC (Control) and PASMC treated with 1 and 2 ng/ml TGF-β (left). β-Actin was used as a loading control. Summarized data (means ± SE, right) showing the protein expression levels of TRPC6, STIM1, and STIM2 (B) as well as Orai1 and Orai2 (C) in control rat PASMC (Control, treated with vehicle) and PASMC treated with 1 ng/ml (1 ng) and 2 ng/ml (2 ng). *P < 0.05 vs. control. D: time course of cell growth (or changes in cell numbers) in PASMC cultured in media (10% FBS-M199) containing vehicle (○) and TGF-β (1 ng/ml, ■; 2 ng/ml, ▲; or 3 ng/ml, ▼). *P < 0.05 vs. TGF-β-treated groups.
Treatment of proliferative PASMC with heparin induces cell differentiation (by upregulating smooth muscle cell differentiation markers) and downregulates expression of STIM, TRPC6, and Orai.
To further investigate the relationship between the phenotypical state of PASMC and the expression level of STIM, TRPC, and Orai, we examined the effect of heparin, an anticoagulant that has been shown to inhibit PASMC proliferation and enhance PASMC differentiation (13, 14, 25), on the protein expression levels of STIM1/2, TRPC6, and Orai1/2/3. As shown in Fig. 5, incubation of proliferative PASMC in media containing 10% FBS and heparin (30 μg/ml) for 72 h significantly downregulated the cell proliferation marker PCNA but markedly upregulated the smooth muscle cell differentiation markers calponin and SM22α (Fig. 5A). In PASMC treated with heparin, the protein expression levels of STIM (STIM1 and STIM2), TRPC6, and Orai (Orai1-3) were all significantly downregulated in comparison to control proliferative PASMC (Fig. 5, B and C). Again, these results indicate that heparin-mediated PASMC differentiation is associated with a significant downregulation of STIM1/2, TRPC6, and Orai1/2/3.
Fig. 5.
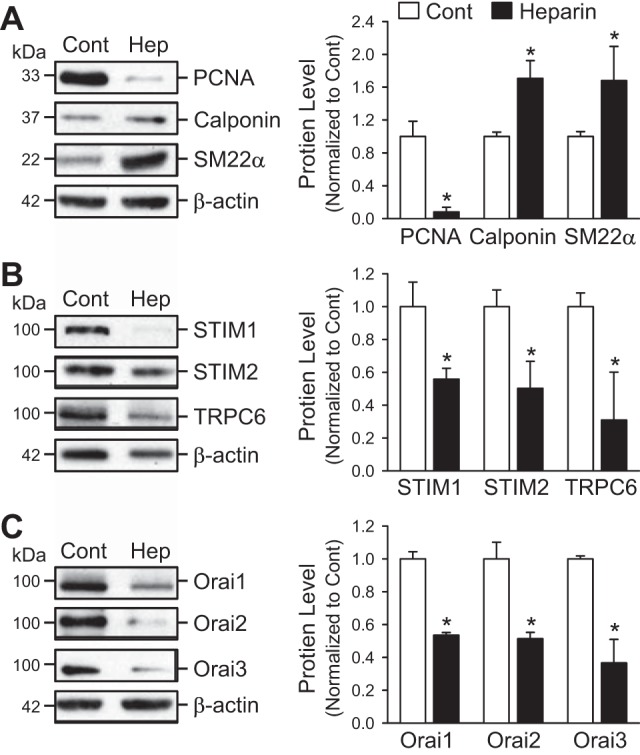
Heparin downregulates the cell proliferation markers, upregulates the cell differentiation markers, and downregulates protein expression of STIM1/2, TRPC6, and Orai1/2/3 in proliferating PASMC. A: Western blot analyses on PCNA, calponin, and SM22α in control proliferative PASMC (Cont) and PASMC treated with 30 μg/ml heparin (Hep) (left). β-Actin was used as a loading control. Summarized data (means ± SE, right) showing the protein levels of PCNA, calponin, and SM22α in control PASMC (Cont) and PASMC treated with heparin. B and C: Western blot analyses on STIM1, STIM2, and TRPC6 (B) as well as Orai1, Orai2, and Orai3 (C) in control proliferative PASMC (Cont) and PASMC treated with 30 μg/ml of heparin (Hep, left). β-Actin was used as a loading control. Summarized data (means ± SE, right) showing the protein expression levels of STIM1, STIM2, and TRPC6 (B) as well as Orai1, Orai2, and Orai3 (C) in control rat PASMC (Cont) and PASMC treated with heparin (Hep). *P < 0.05 vs. control (Cont) bars.
Overexpression of STIM2 enhances the increase in [Ca2+]cyt due to SOCE in PASMC.
To test whether STIM2 is sufficient to enhance SOCE, we compared the CPA-induced increases in [Ca2+]cyt in control PASMC (transfected with an empty vector) and PASMC transfected with STIM2. Overexpression of STIM2 in human PASMC (hPASMC) resulted in a marked increase in protein expression of STIM2 (Fig. 6A) and a significant increase in the amplitude of CPA-mediated increase in [Ca2+]cyt due to SOCE (Fig. 6B). In the absence of extracellular Ca2+, extracellular application of 10 μM CPA, a SERCA inhibitor that depletes Ca2+ from the SR, caused a small and slow increase in [Ca2+]cyt due to Ca2+ leakage from the SR to the cytosol in proliferative (Fig. 6B, top). When the CPA-mediated increase in [Ca2+]cyt in the absence of extracellular Ca2+ returns to the baseline level, restoration of extracellular Ca2+ caused a large and rapid increase in [Ca2+]cyt due obviously to SOCE. Overexpression of STIM2 in proliferative human PASMC (Fig. 6A) significantly enhanced 1) the CPA-induced increase in [Ca2+]cyt due to Ca2+ leakage from the SR to the cytosol and 2) the CPA-induced increase in [Ca2+]cyt due to Ca2+ influx through store-operated Ca2+ channels activated by the passive depletion of Ca2+ from the SR (Fig. 6B). These data demonstrate that increased expression of STIM2 is sufficient to enhance SOCE in PASMC.
Fig. 6.
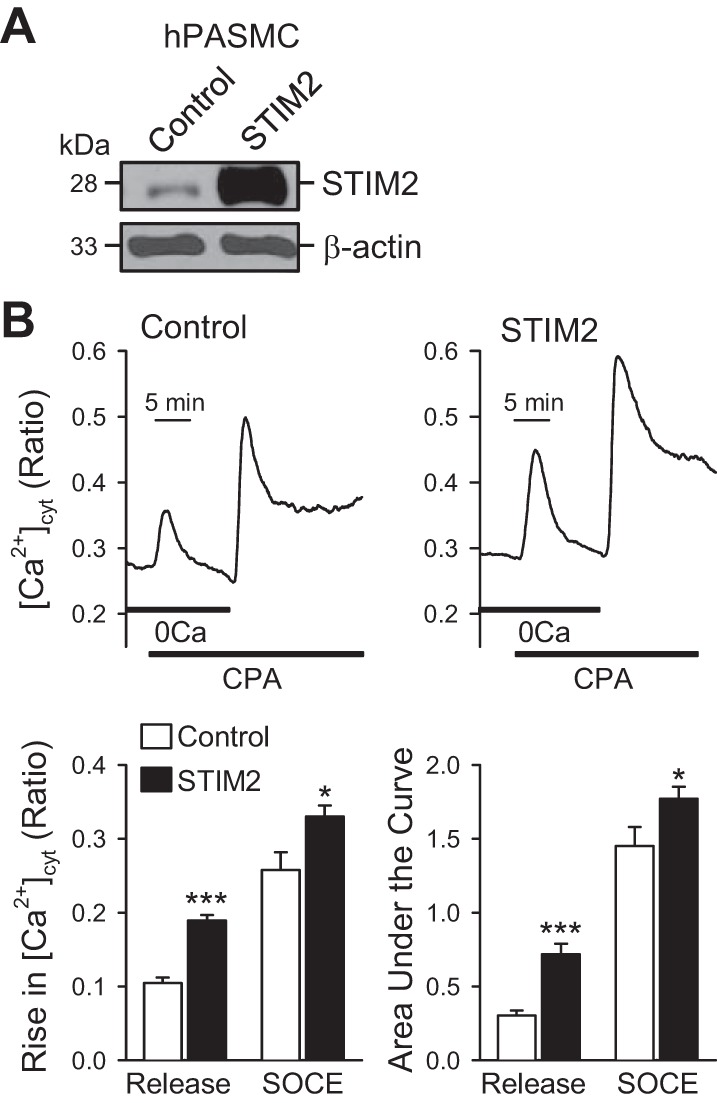
Overexpression of STIM2 enhances SOCE in proliferative human PASMC (hPASMC). A: Western blot analyses on STIM2 in control PASMC (Cont) and PASMC transiently transfected with STIM2 (STIM2). β-Actin was used as a loading control. B: representative records (top) of the 10 μM CPA-induced changes in [Ca2+]cyt in control PASMC (Control, transfected with empty vector) and STIM2-transfected PASMC (STIM2) superfused with Ca2+-free (0Ca) or 1.8 mM Ca2+-containing solution. Summarized data (means ± SE, bottom) showing the 10 μM CPA-induced increases in [Ca2+]cyt (amplitude and area under the curve) due to Ca2+ mobilization (Release) form the sarcoplasmic reticulum (SR) to the cytosol and Ca2+ influx through store-operated Ca2+ channels (SOCE) in control and STIM2-transfected PASMC. *P < 0.05, ***P < 0.001 vs. control (cells treated with empty vector).
Downregulation of Orai2 with siRNA attenuates the amplitude of [Ca2+]cyt increase due to SOCE in proliferative PASMC.
To investigate whether Orai2 is necessary for enhanced SOCE in proliferative PASMC, we compared the rise in [Ca2+]cyt due to SOCE in control PASMC (treated with scrambled siRNA) and PASMC treated with siRNA specifically targeted against Orai2. Downregulation of Orai2 with siRNA significantly decreased the protein expression level of Orai2 in proliferative PASMC cultured in 10% FBS-containing media (Fig. 7A). When proliferative PASMC were bathed in Ca2+-free solution, inhibition of SERCA by extracellular application of CPA induced a small increase in [Ca2+]cyt as a result of Ca2+ mobilization from the SR to the cytosol. Restoration of extracellular Ca2+ (to 1.8 mM) in the presence of CPA then induced a large increase in [Ca2+]cyt due to SOCE (Fig. 7B, top). In proliferating PASMC treated with siRNA for Orai2, the amplitude of the CPA-mediated increase in [Ca2+]cyt due to SOCE was significantly attenuated in comparison to PASMC treated with scrambled (Scram) siRNA, whereas the CPA-mediated increase in [Ca2+]cyt due to Ca2+ mobilization was not significantly changed (Fig. 7B). Downregulation of Orai2 also significantly decreased expression of PCNA (Fig. 7A), suggesting that Orai2 expression also contributes to PASMC proliferation. These results indicate that Orai2 is necessary for the enhanced SOCE in proliferative PASMC. It is possible that upregulated STIM2 and Orai2 functionally interact with each other to enhance SOCE in proliferative PASMC.
Fig. 7.

Downregulation of Orai2 attenuates SOCE in proliferative PASMC. A: Western blot analyses on Orai2 and PCNA in proliferative PASMC treated with scrambled siRNA (Scram-siRNA) and Orai2-targeted siRNA (Orai2-siRNA). β-Actin was used as a loading control. B: representative records (top) of the 10 μM CPA-induced changes in [Ca2+]cyt in Scram-siRNA-treated PASMC and Orai2-siRNA-treated PASMC superfused with Ca2+-free (0Ca) or 1.8 mM Ca2+-containing solution. Summarized data (means ± SE, bottom) showing the 10 μM CPA-induced increases in [Ca2+]cyt (amplitude and area under the curve) due to Ca2+ mobilization (Release) form the SR to the cytosol and Ca2+ influx through store-operated Ca2+ channels (SOCE) in PASMC treated with Scram-siRNA or Orai2-siRNA. *P < 0.05 vs. Scram-siRNA.
Deletion of TRPC6 significantly attenuates the increase in [Ca2+]cyt due to SOCE in proliferative PASMC.
Many TRPC channels contribute to the formation of receptor-operated Ca2+ channels (ROC) and store-operated Ca2+ channels (SOC) in vascular smooth muscle cells including PASMC (45L–47). To confirm that TRPC6 is involved in forming SOC responsible for enhanced SOCE in proliferative PASMC, we isolated PASMC from wild-type (WT) mice and Trpc6−/− mice (Fig. 8A) and cultured the cells in media containing 10% FBS and growth factors. The mice were genotyped using the standard PCR procedure and confirmed downregulation and absence of Trpc6 mRNA expression in the Trpc6+/− and Trpc6−/− mice, respectively (Fig. 8A). In proliferative PASMC isolated and prepared from Trpc6−/− mice, the CPA-mediated increase in [Ca2+]cyt due to SOCE was significantly lower than that in proliferative PASMC isolated from WT mice (Fig. 8, B and C). In human proliferative PASMC, we previously reported that downregulation of TRPC6 with antisense oligonucleotides (46) significantly reduced SOCE, while upregulated TRPC6 was associated with enhanced SOCE in PASMC isolated from patients with idiopathic PAH and animals with experimental pulmonary hypertension (42, 47). These results indicate that upregulated TRPC6 is involved in the enhanced SOCE in proliferative PASMC compared with PASMC in a contractile or differentiated phenotype.
Fig. 8.

Deletion of TRPC6 significantly attenuates CPA-induced increase in [Ca2+]cyt due to store-operated Ca2+ entry (SOCE). A: genotyping characterization of Trpc6+/−, Trpc6−/−, and wild-type (WT) mice demonstrates the presence of only knockout alleles in the homozygous mice, while heterozygotes have both knockout (KO) and WT alleles. PASMC were isolated and prepared from Trpc6−/− and WT mice for the fluorescence microscopy experiments in B. B: representative records of the 10 μM CPA-induced changes in [Ca2+]cyt in WT-PASMC and Trpc6−/−-PASMC superfused with Ca2+-free (0Ca) or 1.8 mM Ca2+-containing solution. C: summarized data (means ± SE) showing the 10 μM CPA-induced increase in [Ca2+]cyt due to Ca2+ influx through store-operated Ca2+ channels (SOCE) in PASMC isolated from WT and Trpc6−/− mice. *P < 0.05 vs. WT.
DISCUSSION
In this study, we demonstrate that 1) upregulated expression of STIM2, TRPC6, and Orai2 is associated with a proliferative phenotype of PASMC and may play an important role in the transition of PASMC from the contractile phenotype to the proliferative phenotype, 2) SOCE is enhanced in the proliferative phenotype of PASMC compared with the contractile phenotype of PASMC, 3) overexpression of STIM2 is sufficient to enhance SOCE in proliferative PASMC, 4) differentiation of proliferative PASMC (by treatment with TGF-β or heparin) results in downregulation of STIM2 and TRPC6, and 5) downregulation of Orai2 or TRPC6 attenuates SOCE in the proliferative phenotype of PASMC. Our data indicate that the transition of PASMC from a contractile phenotype to a proliferative phenotype is associated with enhanced SOCE which requires upregulation of STIM2, TRPC6, and Orai2. This study identifies a potential benefit for targeting STIM2, TRPC6, and/or Orai2 to prevent PASMC proliferation in the development of pulmonary hypertension.
Unlike skeletal or cardiac muscle cells, PASMC are amazingly plastic and can undergo extreme and reversible changes in phenotype in response to local environmental cues that regulate phenotype (27). In response to vascular injury, for example, smooth muscle cells exhibit a phenotypic change characterized by a dramatic increase in rate of proliferation, migration, and synthetic capacity (28). This “synthetic” or “highly proliferative” phenotype plays an active role in repair of vascular damage. Unfortunately, the significant level of plasticity of smooth muscle cells predisposes the cell to abnormal environmental signals that can lead to adverse phenotypic switching and acquisition of characteristics that can contribute to development and/or progression of vascular disease (28). Smooth muscle cell phenotype modulation contributes to the pathogenesis of numerous vascular disorders, including PAH. There is a spectrum of different subtypes of smooth muscle cells that are present in the medial layer, which ranges from a contractile phenotype and a proliferative phenotype. The majority of healthy PASMC show a contractile phenotype, characterized by high contractile ability and low proliferation rate.
The molecular mechanisms driving smooth muscle cell differentiation/dedifferentiation in the PA media layer are not completely understood. Many studies have established that smooth muscle cells express multiple markers that are indicative of their phenotype; however, no single marker exclusively identifies a specific phenotype to the exclusion of other (28). The genes specific for smooth muscle cell differentiation include those that encode for smooth muscle α-actin (SM α-actin), smooth muscle 22-α (SM22α), smooth muscle myosin heavy chain (MYH), smooth muscle myosin light chain kinase (MLCK) and calponin, which all contain CArG elements in their promoter regions. Previous studies have shown that mutations of the conserved CArG elements in these genes promoter enhancer's results in abolished expression of SM22α and MYH (18, 20). Furthermore, these smooth muscle cell markers are useful for assessing smooth muscle cell differentiation with a particular importance on identifying and assessing the degree of phenotypic switching in smooth muscle cells. Loss of contractile markers (MYH, SM22α, and calponin) and increased proliferation is seen in dedifferentiated/proliferative smooth muscle cells (7). Myocardin has been shown to selectively induce the expression of all CArG-dependent smooth muscle cell marker genes (5). It has been also demonstrated that myocardin is both necessary and sufficient to activate various smooth muscle cell contractile markers.
A commonly used smooth muscle cell marker is SM α-actin, in part because it was the first known protein expressed during differentiation of smooth muscle cells during development and it is very specifically selective for smooth muscle cells (10). Additionally, SM α-actin is required for force contraction in fully differentiated smooth muscle cells, and it is the most abundant protein in differentiated smooth muscle cells making up 40% of total cell proteins (9). However, SM α-actin should not be used as a definitive smooth muscle cell lineage marker, since its expression varies in many non-smooth muscle cell types under specific situations (including, e.g., myofibroblasts and endothelial cells in response to TGF-β stimulation), which is why it was not used in these experiments to characterize smooth muscle cell differentiation.
Regardless of the methods of differentiating PASMC, our data presented here demonstrate that STIM2, TRPC6, and Orai2 are all upregulated in highly proliferative PASMC, suggesting that this upregulation is required for phenotypic switching. Indeed, it has been previously confirmed that depletion of STIM2 reduces the proliferative capabilities of dedifferentiated PASMC (35). Additionally, it has been shown that in STIM1 knockout cells there was a failure to progress to S phase of the cell cycle due to upregulation of p21 (CDK inhibitor) and reduction in Rb phosphorylation (Rb is one of the major modulators of the G1/S transition in mammalian cells) (16), as well as a reduction in cAMP response element binding protein (CREB) phosphorylation (36), and nuclear factor of activated T cells (NFAT) transcriptional activity (1), suggesting that multiple signaling pathways may be regulated by STIM2 in PASMC. Furthermore, the roles of various ion channels, specifically Ca2+ channels and K+ channels, have been established to play a role in the development of pulmonary hypertension (21, 48), and they too can potentially contribute to the development of pulmonary hypertension by increasing intracellular Ca2+ and thus cause PASMC differentiation. Alternatively, it was recently published that the Akt signaling cascade is involved in regulating cell proliferation and pulmonary arterial remodeling, specifically through PTEN/Akt1/mTOR signaling, suggesting a potential role of Akt within the pulmonary vasculature along with STIM proteins potentially (37). However, these suggestions have yet to be confirmed and would be necessary to complete this story. On the basis of previous sequence analysis, the promoter of TRPC1 gene contains binding sequences for many transcription factors (such as AP-1, signal transducer and activator of transcription, Smad, and c-myc) that are involved in progression of the cell cycle and regulation of cell proliferation, differentiation, and apoptosis. It would be interesting to investigate whether inflammation mediators (such as cytokines, chemokines, and histamine) and growth factors, which are upregulated in pulmonary hypertension, affect not only TRPC expression in normal PASMC by regulating these transcription factors and their DNA binding activity with the TRPC genes, but as well for the STIM2 and Orai2 genes.
In summary, vascular smooth muscle cells, including PASMC, are extremely plastic and can dedifferentiate in response to various environmental stimuli from a contractile/quiescent to a proliferative/synthetic phenotype (28, 30). Under pathological conditions or vascular injury, PASMC undergo a change in phenotype, characterized by an increase in proliferation rate, migration, and vascular repair, due to increases in cytosolic Ca2+ concentration ([Ca2+]cyt) (22). Importantly, an increase in [Ca2+]cyt in PASMC is a major trigger for pulmonary vasoconstriction and an important stimulus for cell proliferation (24, 29). Here, we demonstrate that the transition from quiescent/contractile PASMC to proliferative/synthetic PASMC is associated with enhanced SOCE due to upregulation of STIM2, TRPC6, and Orai2 (Fig. 9). Understanding the molecular mechanisms that regulate phenotypic switching of PASMC and increased PASMC proliferation is critical to elucidation of the pathogenesis of PAH. STIM2, TRPC6, and Orai2 are potentially good targets for the development of novel therapies for the treatment of PAH.
Fig. 9.

Schematic diagram of the proposed mechanisms in the transition of PASMC from the contractile phenotype to proliferative phenotype and its potential role in the development of pulmonary vascular remodeling. In differentiated or contractile PASMC, the protein expression level of STIM2, TRPC6, and Orai2 and the store-operated Ca2+ entry (SOCE) are both low. When contractile PASMC under phenotypic changes toward proliferative phenotype, the upregulated STIM2, TRPC6, and Orai2 contribute to enhancing SOCE, increasing [Ca2+]cyt, promoting Ca2+ refilling into intracellular Ca2+ stores, maintaining a sustained high level of [Ca2+]cyt, and stimulating PASMC proliferation. The enhanced PASMC proliferation may ultimately contribute to the development and progression of pulmonary vascular remodeling, a major cause of the elevated pulmonary vascular resistance and pulmonary arterial pressure in patients with pulmonary arterial hypertension.
GRANTS
This work was supported, in part, by grants from the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL-110543, HL-066012, HL-125208, and HL-098053).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.A.F., K.A.S., A.M., D.M., and J.X.-J.Y. conception and design of research; R.A.F., J.W., S.S., Y.G., M.T., and H.T. performed experiments; R.A.F., J.W., S.S., K.A.S., Y.G., M.T., H.T., A.M., and J.X.-J.Y. analyzed data; R.A.F., J.W., S.S., K.A.S., H.T., A.M., and J.X.-J.Y. interpreted results of experiments; R.A.F., J.W., S.S., K.A.S., Y.G., M.T., H.T., A.M., D.M., and J.X.-J.Y. prepared figures; R.A.F., K.A.S., A.M., D.M., and J.X.-J.Y. drafted manuscript; R.A.F., J.W., S.S., K.A.S., Y.G., M.T., H.T., A.M., D.M., and J.X.-J.Y. edited and revised manuscript; R.A.F., J.W., S.S., K.A.S., M.T., A.M., D.M., and J.X.-J.Y. approved final version of manuscript.
REFERENCES
- 1.Aubart FC, Sassi Y, Coulombe A, Mougenot N, Vrignaud C, Leprince P, Lechat P, Lompre AM, Hulot JS. RNA interference targeting STIM1 suppresses vascular smooth muscle cell proliferation and neointima formation in the rat. Mol Ther 17: 455–462, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barst RJ, Agnoletti G, Fraisse A, Baldassarre J, Wessel DL. Vasodilator testing with nitric oxide and/or oxygen in pediatric pulmonary hypertension. Pediatr Cardiol 31: 598–606, 2010. [DOI] [PubMed] [Google Scholar]
- 3.Berna-Erro A, Galan C, Dionisio N, Gomez LJ, Salido GM, Rosado JA. Capacitative and non-capacitative signaling complexes in human platelets. Biochim Biophys Acta 1823: 1242–1251, 2012. [DOI] [PubMed] [Google Scholar]
- 4.Bravo R, Macdonald-Bravo H. Existence of two populations of cyclin/proliferating cell nuclear antigen during the cell cycle: association with DNA replication sites. J Cell Biol 105: 1549–1554, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J, Kitchen CM, Streb JW, Miano JM. Myocardin: a component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol 34: 1345–1356, 2002. [DOI] [PubMed] [Google Scholar]
- 6.Davis-Dusenbery BN, Wu C, Hata A. Micromanaging vascular smooth muscle cell differentiation and phenotypic modulation. Arterioscler Thromb Vasc Biol 31: 2370–2377, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duband JL, Gimona M, Scatena M, Sartore S, Small JV. Calponin and SM 22 as differentiation markers of smooth muscle: spatiotemporal distribution during avian embryonic development. Differentiation 55: 1–11, 1993. [DOI] [PubMed] [Google Scholar]
- 8.Fantozzi I, Zhang S, Platoshyn O, Remillard CV, Cowling RT, Yuan JX. Hypoxia increases AP-1 binding activity by enhancing capacitative Ca2+ entry in human pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 285: L1233–L1245, 2003. [DOI] [PubMed] [Google Scholar]
- 9.Fatigati V, Murphy RA. Actin and tropomyosin variants in smooth muscles. Dependence on tissue type. J Biol Chem 259: 14383–14388, 1984. [PubMed] [Google Scholar]
- 10.Frid MG, Shekhonin BV, Koteliansky VE, Glukhova MA. Phenotypic changes of human smooth muscle cells during development: late expression of heavy caldesmon and calponin. Dev Biol 153: 185–193, 1992. [DOI] [PubMed] [Google Scholar]
- 11.Galie N, Corris PA, Frost A, Girgis RE, Granton J, Jing ZC, Klepetko W, McGoon MD, McLaughlin VV, Preston IR, Rubin LJ, Sandoval J, Seeger W, Keogh A. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol 62: D60–D72, 2013. [DOI] [PubMed] [Google Scholar]
- 12.Galie N, Simonneau G. The Fifth World Symposium on Pulmonary Hypertension. J Am Coll Cardiol 62: D1–D3, 2013. [DOI] [PubMed] [Google Scholar]
- 13.Garg HG, Thompson BT, Hales CA. Structural determinants of antiproliferative activity of heparin on pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 279: L779–L789, 2000. [DOI] [PubMed] [Google Scholar]
- 14.Garg HG, Yu L, Hales CA, Toida T, Islam T, Linhardt RJ. Sulfation patterns in heparin and heparan sulfate: effects on the proliferation of bovine pulmonary artery smooth muscle cells. Biochim Biophys Acta 1639: 225–231, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Goumans MJ, Mummery C. Functional analysis of the TGFβ receptor/Smad pathway through gene ablation in mice. Int J Dev Biol 44: 253–265, 2000. [PubMed] [Google Scholar]
- 16.Guo RW, Wang H, Gao P, Li MQ, Zeng CY, Yu Y, Chen JF, Song MB, Shi YK, Huang L. An essential role for stromal interaction molecule 1 in neointima formation following arterial injury. Cardiovasc Res 81: 660–668, 2009. [DOI] [PubMed] [Google Scholar]
- 17.Hautmann MB, Madsen CS, Owens GK. A transforming growth factor β (TGFβ) control element drives TGFβ-induced stimulation of smooth muscle alpha-actin gene expression in concert with two CArG elements. J Biol Chem 272: 10948–10956, 1997. [DOI] [PubMed] [Google Scholar]
- 18.Kim S, Ip HS, Lu MM, Clendenin C, Parmacek MS. A serum response factor-dependent transcriptional regulatory program identifies distinct smooth muscle cell sublineages. Mol Cell Biol 17: 2266–2278, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao Y, Erxleben C, Abramowitz J, Flockerzi V, Zhu MX, Armstrong DL, Birnbaumer L. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc Natl Acad Sci USA 105: 2895–2900, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manabe I, Owens GK. CArG elements control smooth muscle subtype-specific expression of smooth muscle myosin in vivo. J Clin Invest 107: 823–834, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mandegar M, Remillard CV, Yuan JX. Ion channels in pulmonary arterial hypertension. Prog Cardiovasc Dis 45: 81–114, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Marchand A, Abi-Gerges A, Saliba Y, Merlet E, Lompre AM. Calcium signaling in vascular smooth muscle cells: from physiology to pathology. Adv Exp Med Biol 740: 795–810, 2012. [DOI] [PubMed] [Google Scholar]
- 23.Montani D, Savale L, Natali D, Jais X, Herve P, Garcia G, Humbert M, Simonneau G, Sitbon O. Long-term response to calcium-channel blockers in non-idiopathic pulmonary arterial hypertension. Eur Heart J 31: 1898–1907, 2010. [DOI] [PubMed] [Google Scholar]
- 24.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, McMurtry IF, Stenmark KR, Thistlethwaite PA, Weissmann N, Yuan JX, Weir EK. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol 54: S20–S31, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mrabat H, Garg HG, Hales CA. Growth inhibition of bovine pulmonary artery smooth muscle cells following long-term heparin treatment. J Cell Physiol 221: 603–608, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogawa A, Firth AL, Smith KA, Maliakal MV, Yuan JX. PDGF enhances store-operated Ca2+ entry by upregulating STIM1/Orai1 via activation of Akt/mTOR in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 302: C405–C411, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev 75: 487–517, 1995. [DOI] [PubMed] [Google Scholar]
- 28.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84: 767–801, 2004. [DOI] [PubMed] [Google Scholar]
- 29.Reid L. Vascular remodeling. In: The Pulmonary Circulation: Normal and Abnormal: Mechanisms, Management, and the National Registry, edited by Fishman A. Philadelphia, PA: Univ. of Pennsylvania Press, 1990, p. 264. [Google Scholar]
- 30.Rensen SS, Doevendans PA, van Eys GJ. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J 15: 100–108, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rich S, Kaufmann E, Levy PS. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N Engl J Med 327: 76–81, 1992. [DOI] [PubMed] [Google Scholar]
- 32.Roe MW, Lemasters JJ, Herman B. Assessment of Fura-2 for measurements of cytosolic free calcium. Cell Calcium 11: 63–73, 1990. [DOI] [PubMed] [Google Scholar]
- 33.Sitbon O, Humbert M, Jais X, Ioos V, Hamid AM, Provencher S, Garcia G, Parent F, Herve P, Simonneau G. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 111: 3105–3111, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Solik P, Lesny P, Luknar M, Varga I, Goncalvesova E. The long-term response to treatment with calcium channel blockers in a patient with idiopathic pulmonary arterial hypertension. Bratisl Lek Listy 114: 283–286, 2013. [DOI] [PubMed] [Google Scholar]
- 35.Song MY, Makino A, Yuan JX. STIM2 contributes to enhanced store-operated Ca2+ entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Pulm Circ 1: 84–94, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takahashi Y, Watanabe H, Murakami M, Ono K, Munehisa Y, Koyama T, Nobori K, Iijima T, Ito H. Functional role of stromal interaction molecule 1 (STIM1) in vascular smooth muscle cells. Biochem Biophys Res Commun 361: 934–940, 2007. [DOI] [PubMed] [Google Scholar]
- 37.Tang H, Chen J, Fraidenburg DR, Song S, Sysol JR, Drennan AR, Offermanns S, Ye RD, Bonini MG, Minshall RD, Garcia JG, Machado RF, Makino A, Yuan JX. Deficiency of Akt1, but not Akt2, attenuates the development of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L208–L220, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.ten Dijke P, Arthur HM. Extracellular control of TGFβ signalling in vascular development and disease. Nat Rev Mol Cell Biol 8: 857–869, 2007. [DOI] [PubMed] [Google Scholar]
- 39.Thorneloe KS, Nelson MT. Ion channels in smooth muscle: regulators of intracellular calcium and contractility. Can J Physiol Pharmacol 83: 215–242, 2005. [DOI] [PubMed] [Google Scholar]
- 40.Tuder RM, Archer SL, Dorfmuller P, Erzurum SC, Guignabert C, Michelakis E, Rabinovitch M, Schermuly R, Stenmark KR, Morrell NW. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol 62: D4–D12, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wan J, Yamamura A, Zimnicka AM, Voiriot G, Smith KA, Tang H, Ayon RJ, Choudhury MS, Ko EA, Wang J, Wang C, Makino A, Yuan JX. Chronic hypoxia selectively enhances L- and T-type voltage-dependent Ca2+ channel activity in pulmonary artery by upregulating CaV1.2 and CaV3.2. Am J Physiol Lung Cell Mol Physiol 305: L154–L164, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 98: 1528–1537, 2006. [DOI] [PubMed] [Google Scholar]
- 43.Xu M, Platoshyn O, Makino A, Dillmann WH, Akassoglou K, Remillard CV, Yuan JX. Characterization of agonist-induced vasoconstriction in mouse pulmonary artery. Am J Physiol Heart Circ Physiol 294: H220–H228, 2008. [DOI] [PubMed] [Google Scholar]
- 44.Yoo HY, Zeifman A, Ko EA, Smith KA, Chen J, Machado RF, Zhao YY, Minshall RD, Yuan JX. Optimization of isolated perfused/ventilated mouse lung to study hypoxic pulmonary vasoconstriction. Pulm Circ 3: 396–405, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JX. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci USA 101: 13861–13866, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu Y, Keller SH, Remillard CV, Safrina O, Nicholson A, Zhang SL, Jiang W, Vangala N, Landsberg JW, Wang JY, Thistlethwaite PA, Channick RN, Robbins IM, Loyd JE, Ghofrani HA, Grimminger F, Schermuly RT, Cahalan MD, Rubin LJ, Yuan JX. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 119: 2313–2322, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu Y, Sweeney M, Zhang S, Platoshyn O, Landsberg J, Rothman A, Yuan JX. PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol Cell Physiol 284: C316–C330, 2003. [DOI] [PubMed] [Google Scholar]
- 48.Zhang S, Dong H, Rubin LJ, Yuan JX. Upregulation of Na+/Ca2+ exchanger contributes to the enhanced Ca2+ entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol 292: C2297–C2305, 2007. [DOI] [PubMed] [Google Scholar]