

Abstract
The nucleus of the solitary tract (NTS) is a medullary integrative center with critical roles in the coordinated control of energy homeostasis. Here, we used whole cell current-clamp recordings on rat NTS neurons in slice preparation to identify the presence of physiologically relevant glucose-sensing neurons. The majority of NTS neurons (n = 81) were found to be glucose-responsive, with 35% exhibiting a glucose-excited (GE) phenotype (mean absolute change in membrane potential: 9.5 ± 1.1 mV), and 21% exhibiting a glucose-inhibited (GI) response (mean: 6.3 ± 0.7 mV). Furthermore, we found glucose-responsive cells are preferentially influenced by the anorexigenic peptide α-melanocyte-stimulating hormone (α-MSH), but not nesfatin-1. Accordingly, alterations in glycemic state have profound effects on the responsiveness of NTS neurons to α-MSH, but not to nesfatin-1. Indeed, NTS neurons showed increasing responsiveness to α-MSH as extracellular glucose concentrations were decreased, and in hypoglycemic conditions, all NTS neurons were depolarized by α-MSH (mean 10.6 ± 3.2 mV; n = 8). Finally, decreasing levels of extracellular glucose correlated with a significant hyperpolarization of the baseline membrane potential of NTS neurons, highlighting the modulatory effect of glucose on the baseline excitability of cells in this region. Our findings reveal individual NTS cells are capable of integrating multiple sources of metabolically relevant inputs, highlight the rapid capacity for plasticity in medullary melanocortin circuits, and emphasize the critical importance of physiological recording conditions for electrophysiological studies pertaining to the central control of energy homeostasis.
Keywords: nucleus of the solitary tract, glucose-sensing neuron, nesfatin-1, α-melanocyte stimulating hormone, electrophysiology
the medullary nucleus of the solitary tract (NTS) is a complex integrative autonomic center with critical roles in the coordinated control of ingestive behaviors. The NTS receives vagal inputs arising from the gastrointestinal tract, which transmit information regarding the mechanical status of, and presence of nutrients in, the gut, as well as descending inputs from higher brain areas involved in the control of feeding behaviors, such as the hypothalamus (3, 17). In addition to its role as a relay center for multiple sources of inputs, the NTS also produces and senses numerous neuropeptides involved in the control of energy balance. Among these peptides are nesfatin-1 (15, 30) and α-melanocyte-stimulating hormone (α-MSH) (7), as well as the α-MSH receptor, the melanocortin 4 receptor (MC4R) (21). Fourth-ventricle injections of nesfatin-1 (39) and the melanocortin 3 and 4 receptor (MC3/4R) agonist melanotan II (16, 49) lead to profound decreases in food intake, as do more localized injections of both peptides into the dorsal vagal complex [including NTS and the dorsal motor nucleus of the vagus (DMV)] (12, 45). Furthermore, the receptors mediating the medullary anorexigenic effects of melanocortins appear to be located in the NTS, as antagonism of MC3/4Rs specifically in the NTS abolishes α-MSH-induced decreases in feeding (50).
In addition to its ability to sense peptides involved in the control of feeding, NTS neurons can also detect fluctuations in extracellular glucose levels, leading to alterations in gastric motility (14), glucagon secretion (22), and to the stimulation of feeding in hypoglycemic conditions (34). Indeed, both glucose-excited (GE) and glucose-inhibited (GI) neurons have been described in vitro in this region (2, 10, 28, 40), although these studies have used of supraphysiological concentrations of glucose, or very large changes in glucose concentrations not likely to be encountered by central nervous system (CNS) neurons in vivo. Furthermore, although it is clear that individual NTS neurons process multiple sources and types of converging inputs regarding the metabolic status of the organism, many in vitro studies in this region evaluate the effects of only one individual input at a time, rather than assessing the integrative properties of these cells. In addition, many such studies are conducted at only one extracellular glucose level, thereby mimicking only one feeding state of the animal. This approach evidently does not permit a full appreciation of the range of actions of peptides across the spectrum of hunger to satiety.
The present study was thus undertaken to provide evidence of physiologically relevant glucose-sensing neurons in the NTS, defined as such on the basis of their ability to respond to glucose concentrations similar to those observed in vivo. Furthermore, we evaluated the ability of these cells to integrate additional metabolically relevant signals and assessed whether the responsiveness of NTS neurons to anorexigenic peptides is altered as extracellular glucose concentrations are modified in vitro.
MATERIALS AND METHODS
Preparation of NTS slices for electrophysiology.
Coronal slices at the level of the NTS were prepared daily from 3- to 4-wk-old male Sprague-Dawley rats fed ad libitum (Charles River, Quebec, Canada). Unanesthetized rats were decapitated, and their brains were submerged in ice-cold slicing solution made of (in mM) 87 NaCl, 2.5 KCl, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 glucose, 75 sucrose bubbled with 95% O2-5% CO2. A region of the brain stem containing the NTS was isolated, and 300-μm coronal sections were cut with a vibratome (Leica, Nussloch, Germany). Slices were then incubated at 32°C in artificial cerebrospinal fluid (aCSF) made of (in mM) 124 NaCl, 2.5 KCl, 20 NaHCO3, 2 CaCl2, 1.3 MgSO4, 1.24 KH2PO4 bubbled with 95% O2-5% CO2. The glucose concentration of the aCSF varied and was either 5, 3, or 0.2 mM. Slices were only incubated at one glucose concentration (i.e., they were not switched from one concentration to another during the incubation period). At each concentration, the osmolarity of the aCSF was adjusted to that of standard aCSF containing 10 mM glucose by equimolar substitution with sucrose. Slices were incubated at 32°C for a minimum of 1 h before electrophysiological recordings were performed. All procedures were in accordance with the ethical criteria established by the Canadian Council on Animal Care and were approved by the Queen's University Animal Care Committee.
Electrophysiology.
Brain slices were moved to a recording chamber perfused with carbogenated aCSF warmed to ∼32°C at a flow rate of 1.5–2 ml/min. The baseline glucose concentration of the aCSF in the recording chamber was the same as that used during the incubation period following slice preparation. An upright differential interference contrast microscope at ×40 was used to visualize neurons (Scientifica, East Sussex, United Kingdom). Borosilicate glass electrodes (World Precision Instruments, Sarasota, FL) were pulled on a Sutter Instruments P97 flaming micropipette puller and filled with an intracellular solution made of (in mM) 125 potassium gluconate, 10 KCl, 2 MgCl2, 0.1 CaCl2, 5.5 EGTA, 10 HEPES, 2 NaATP (pH 7.2 with KOH). Electrodes had a resistance of 3–5.5 MΩ when filled with the intracellular solution. After obtaining a high-resistance seal (minimum 1 GΩ), brief suction was used to rupture the membrane and obtain whole cell access. Whole cell recordings were made with a MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA) sampled at 10 kHz, filtered at 2.4 kHz using a Micro 1401 interface. Data were collected with Spike 2 software for offline analysis (Cambridge Electronic Devices, Cambridge, UK). At all glucose concentrations used, neurons selected for experimentation had action potentials with an amplitude of at least 60 mV and a stable baseline membrane potential. Of note, while it was more challenging to obtain recordings as extracellular glucose concentrations were decreased, once cells were obtained, there was no appreciable difference in the quality or duration of recordings at low glucose concentrations. Furthermore, we note that we have chosen the whole cell configuration 1) because previous studies have demonstrated the validity of this recording configuration in assessing the responsiveness of NTS neurons to changes in extracellular glucose concentration (2), and 2) we wished to maintain consistency and to allow for comparisons with previous work from our laboratory.
All solutions were applied to slices via bath perfusion. Responses to changes in glucose concentration, to α-MSH, or to nesfatin-1 were assessed by comparing the membrane potential of the neuron before and after glucose or peptide application. A response was deemed significant when the change in membrane potential after glucose or peptide application was at least twice the standard deviation of the baseline membrane potential obtained during a 100-s period immediately before application of the test solution. Postapplication membrane potential was the peak membrane potential averaged over 100 s of the recording showing a maximal effect. A calculated liquid junction potential of 15 mV has been subtracted from all membrane potentials.
Glucose concentrations and characterization of glucose responsiveness.
We selected 5 mM glucose to mimic central hyperglycemia, 3 mM glucose to mimic central normoglycemia, and 0.2 mM glucose to mimic central hypoglycemia. These concentrations were chosen on the basis of measurements obtained in vivo from the rat brain (20, 31, 36, 37), as well as previous in vitro electrophysiological experiments in rat brain slices (8, 9, 22, 25, 38, 43, 46). To further mimic physiological transitions in glucose concentrations, steps were only made from normoglycemic conditions to hypoglycemic or hyperglycemic conditions, or vice versa (i.e., no steps from hypoglycemic directly to hyperglycemic conditions, or vice versa, were performed). Neurons were considered GE if they depolarized in response to an increase in glucose concentration or hyperpolarized in response to a decrease in glucose concentration. Cells were classified as GI if they depolarized in response to a decrease in glucose concentration or hyperpolarized in response to an increase in glucose concentration (24, 38, 43). The mean responses to alterations in glucose concentration in GE and GI neurons are reported as absolute changes in membrane potential (i.e., the sign removed from hyperpolarizations and pooled with depolarizations).
Chemicals and drugs.
Slicing solution, aCSF, and intracellular recording solution were made with salts obtained from Sigma Pharmaceuticals (Oakville, Ontario, Canada). Nesfatin-1 and α-MSH were obtained from Phoenix Pharmaceuticals (Belmont, CA).
RESULTS
NTS contains physiologically relevant glucose-sensing neurons.
We first assessed whether NTS neurons respond to changes in glucose concentrations that fall within the physiological range measured in the CNS. To this end, we bath-perfused varying concentrations of extracellular glucose (5, 3, and 0.2 mM, see Experimental procedures for rationale) on a total of 80 medial NTS neurons during current-clamp recordings. The majority (56%) of the cells examined were found to be glucose-responsive. Overall, 28 cells were GE neurons (35%, mean absolute change in membrane potential: 9.5 ± 1.1 mV, Fig. 1, A, C, and D), and 17 cells were GI neurons (21%, mean absolute change in membrane potential: 6.3 ± 0.7 mV, Fig. 1, B, C, and D). The remaining 35 neurons (44%) did not respond to changes in glucose concentrations. In the 23 GE cells in which the extracellular glucose concentration was returned to initial baseline levels, 17 cells showed a recovery toward baseline membrane potential. Furthermore, in the 13 GI cells in which the extracellular glucose concentration was returned to baseline levels, 9 showed a recovery toward baseline membrane potential. Finally, there was no correlation between baseline membrane potential and the cellular response to changes in glucose concentration, as the baseline membrane potential of GE, GI, and glucose nonresponsive (NR) cells were not different from one another (mean baseline membrane potential GE neurons: −69.5 ± 1.4 mV, GI neurons: −71.6 ± 1.2 mV, and NR neurons: −69.1 ± 0.9 mV, one-way ANOVA with Tukey's multiple-comparison test, P = 0.34).
Fig. 1.

The nucleus of the solitary tract (NTS) contains physiologically relevant glucose-sensing neurons. A and B: representative current-clamp recordings from four separate NTS neurons in slice preparation. An increase in glucose concentration from a baseline of 3 mM (horizontal medium gray bar) to 5 mM (horizontal dark gray bar) (A, i) caused a reversible depolarization in this glucose excited (GE) cell, while a decrease in glucose concentration from 3 to 0.2 mM (horizontal pale gray bar) elicited a hyperpolarization in the GE neuron (A, ii). An increase from 3 to 5 mM glucose caused a reversible hyperpolarization in the glucose-inhibited (GI) cell (B, i), while a decrease from 3 to 0.2 mM glucose elicited a reversible depolarization in the GI neuron (B, ii). C: scatterplot depicting the range of responses elicited by changes in glucose concentrations in NTS neurons. Black bars represent the absolute mean change (± SE) in membrane potential induced by altered glucose concentrations in GE and GI neurons, while each single point represents the response of a single neuron. Mean GE response: was 9.5 ± 1.1 mV (n = 28), and mean GI response was 6.3 ± 0.7 mV (n = 17). D: bar graph showing the percentage of NTS neurons that displayed a GE, GI, or nonresponsive (NR) phenotype in response to changes in glucose concentration. E: bar graph illustrating the percentage of NTS neurons that exhibit a GE, GI, or NR phenotype at each step in glucose concentration employed.
We also examined the distribution of GE, GI, and NR phenotypes at each glucose step performed, as summarized in Fig. 1E. When extracellular glucose levels were changed from 0.2 to 3 mM (n = 15), we found 73% of neurons were GE, 20% were GI, and 7% were nonresponsive to changes in glucose (NR). Conversely, when glucose concentrations were decreased from 3 to 0.2 mM (n = 24), 17% of cells were GE, 25% were GI, and 58% were NR. Furthermore, when stepping from 3 to 5 mM glucose (n = 20), 50% of neurons were GE, 20% were GI, and 30% were NR. Conversely, when glucose levels were decreased from 5 to 3 mM (n = 21), 14% of cells were GE, 19% were GI, and 67% were NR. Thus, these findings suggest NTS neurons are more responsive to increases in extracellular glucose concentrations than to decreases in glucose levels (χ2, P < 0.0003 for all comparisons of increases vs. decreases in glucose concentration, P > 0.1 for all comparisons of changes in glucose concentration in the same direction).
Nesfatin-1 exerts heterogeneous effects on both glucose-responsive and glucose-NR NTS neurons.
Since nesfatin-1-expressing NTS neurons exhibit c-Fos immunoreactivity in response to systemic hypoglycemia in rats (4), and since we have previously demonstrated effects of nesfatin-1 on the excitability of NTS neurons at the traditional 10-mM extracellular glucose concentration used in electrophysiological studies (27), we next investigated whether nesfatin-1 may exert preferential effects on a particular subtype of glucose-sensing cell. To this end, 40 of the neurons tested for glucose responsiveness were also evaluated for nesfatin-1 responsiveness. Nesfatin-1 (10 nM) exerted heterogeneous effects on GE (n = 13), GI (n = 8), and NR (n = 19) cells in this region, as summarized in Fig. 2, C and D. More specifically, nesfatin-1 depolarized 38% of GE and GI neurons (mean depolarization: 6.4 ± 3.2 mV and 6.6 ± 3.0 mV, respectively) hyperpolarized 24% of GE and GI neurons (mean hyperpolarization: −6.7 ± 1.5 mV and −5.6 ± 1.0 mV, respectively; Fig. 2A) and exerted no effect on the remaining 38% of GE and GI neurons. Furthermore, 42% of NR cells were depolarized by nesfatin-1 (mean depolarization: 5.2 ± 1.0 mV; Fig. 2B), 11% of NR neurons hyperpolarized in response to nesfatin-1 (mean hyperpolarization: −6.3 ± 0.6 mV), and the remaining 47% did not respond to nesfatin-1. Thus, both the amplitude (one-way ANOVA with Tukey's multiple-comparison test, P > 0.8; Fig. 2C) and the distribution (χ2, P > 0.3; Fig. 2D) of the effects of nesfatin-1 on the membrane potential of NTS neurons were consistent between GE, GI, and NR cells.
Fig. 2.
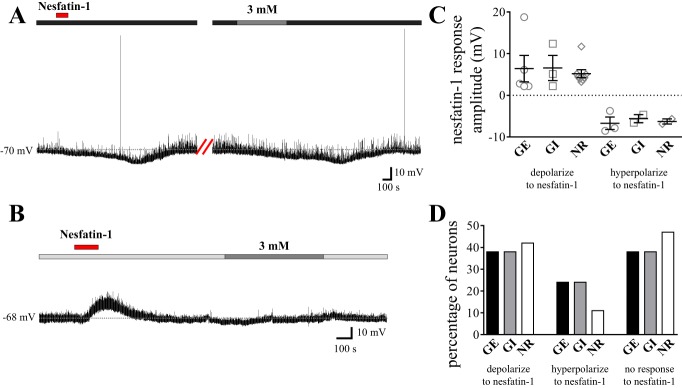
Nesfatin-1 exerts heterogeneous effects on both glucose-responsive and glucose-nonresponsive NTS neurons. A and B: representative current-clamp recordings from two separate NTS neurons. A: nesfatin-1 (10 nM; horizontal red bar) elicited a reversible hyperpolarization in this neuron subsequently identified as GE following a decrease in glucose concentration from a baseline of 5 mM (horizontal dark gray bar) to 3 mM (horizontal medium gray bar). B: nesfatin-1 elicited a reversible depolarization in this NR NTS neuron bathed in artificial cerebrospinal fluid (aCSF) containing 0.2 mM glucose at baseline [horizontal pale gray bar; there was no response upon increasing to 3 mM extracellular glucose (horizontal medium gray bar)]. C: scatterplot depicting the range of responses elicited by 10 nM nesfatin-1 in GE, GI, and NR NTS neurons. Mean nesfatin-1-induced depolarization GE: 6.4 ± 3.2 mV, GI: 6.6 ± 3.0 mV, and NR: 5.2 ± 1.0 mV. Mean nesfatin-1-induced hyperpolarization GE: −6.7 ± 1.5 mV, GI: −5.6 ± 1.0 mV, and NR: −6.3 ± 0.6 mV. D: bar graph showing the percentage of GE (n = 13), GI (n = 8), and NR (n = 19) NTS neurons that also responded to nesfatin-1. There is no difference in the amplitude (ANOVA with Tukey's multiple-comparison test, P > 0.8) or distribution (χ2, P > 0.3) of nesfatin-1 responses across subtypes of glucose-sensing NTS neurons.
Effects of nesfatin-1 on NTS neurons are consistent across glucose concentrations.
We next evaluated whether the effects of nesfatin-1 on the excitability of NTS neurons may be altered as extracellular glucose concentrations are modified. Therefore, we maintained rat brain slices containing the NTS at either 5, 3, or 0.2 mM glucose and subsequently bath-perfused 10 nM nesfatin-1 to evaluate the effects of this peptide on the membrane potential of NTS cells. Our findings are summarized in Fig. 3. Previous studies from our laboratory using slices maintained at 10 mM glucose showed effects of nesfatin-1 (10 nM) on 63% of NTS neurons, 42% of which depolarized (n = 39; mean 7.8 ± 0.8 mV), 21% of which hyperpolarized (n = 20; −8.2 ± 1.0 mV), and the remaining 37% of which did not respond to nesfatin-1 (n = 34) (27). At 5 mM extracellular glucose, 64% of NTS neurons (n = 14) responded to nesfatin-1, 36% with a reversible depolarization (n = 5; mean 5.6 ± 0.8 mV) and 28% with a hyperpolarization (n = 4; mean: −6.1 ± 1.0 mV, 2 reversible). When extracellular glucose concentrations were decreased to 3 mM, 59% of NTS neurons (n = 32) responded to nesfatin-1, 41% with a depolarization (n = 13; mean 6.7 ± 1.2 mV, 11 at least partially reversible, Fig. 3A, i) and 18% with a hyperpolarization (n = 6; mean: −7.5 ± 1.0 mV, 3 reversible, Fig. 3B, i). Finally, at 0.2 mM extracellular glucose, 71% of NTS neurons (n = 17) responded to nesfatin-1, 47% with a depolarization (n = 8; mean: 5.8 ± 1.4 mV, six at least partially reversible; Fig. 3A, ii), and 24% with a reversible hyperpolarization (n = 4; mean: −6.6 ± 1.0 mV, Fig. 3B, ii). Thus, there was no difference in the amplitude of the depolarizations or hyperpolarizations observed at all extracellular glucose concentrations employed (one-way ANOVA with Tukey's multiple-comparison test; P = 0.58 and 0.70 for depolarizations and hyperpolarizations, respectively; Fig. 3C). Secondly, there was no change in the proportion of NTS neurons depolarized, hyperpolarized, or unaffected by nesfatin-1 across the range of glucose concentrations used (χ2, P > 0.5 for all possible pairs, Fig. 3D). These findings, therefore, indicate the effects of nesfatin-1 on the membrane potential of NTS neurons are unchanged as extracellular glucose concentrations are altered.
Fig. 3.

The effects of nesfatin-1 on NTS neurons are consistent across glucose concentrations. A and B: representative current-clamp recordings from four separate NTS neurons. A, i and B, i: neurons are bathed in aCSF containing 3 mM glucose (horizontal medium gray bar). A, ii and B, ii: neurons are bathed in aCSF with 0.2 mM glucose (horizontal pale gray bar). A, i and A, ii: bath-applied nesfatin-1 (10 nM, horizontal red bar) caused a depolarizing effect of similar amplitude in both these cells. B, i and ii: nesfatin-1 elicited a hyperpolarization of similar amplitude in these neurons. C: scatterplot depicting the range of responses elicited by 10 nM nesfatin-1 at varying extracellular glucose concentrations. Mean depolarization at 10 mM was 7.8 ± 0.8 mV (n = 39), 5 mM, 5.6 ± 0.8 mV (n = 5), 3 mM, 6.7 ± 1.2 mV (n = 13), and 0.2 mM, 5.8 ± 1.4 mV (n = 8). Mean hyperpolarization at 10 mM was −8.2 ± 1.0 mV (n = 20), 5 mM, −6.1 ± 1.0 mV (n = 4), 3 mM, −7.5 ± 1.0 mV (n = 6), and 0.2 mM, −6.6 ± 1.0 mV (n = 4). D: bar graph showing the percentage of NTS neurons that respond to nesfatin-1 with a depolarization, hyperpolarization, or no response across all glucose concentrations used. There is no difference in the amplitude (ANOVA with Tukey's multiple-comparison test, P > 0.5) or distribution of nesfatin-1 responses across groups (χ2, P > 0.5). Data at 10 mM glucose are obtained from Ref. 27.
GE NTS neurons are more responsive to α-MSH than NR cells, and are selectively depolarized by α-MSH.
Because the anorexigenic peptide α-MSH and the MC4R have been shown to be involved in the autonomic processes regulating glucose homeostasis (13, 29, 35) and because we have previously demonstrated effects of α-MSH on the membrane potential of NTS cells at 10 mM glucose (26), we conducted parallel experiments evaluating whether α-MSH exerts selective effects on the excitability of a particular subtype of glucose-sensing NTS neuron. Our results are summarized in Fig. 4. We found 75% of GE neurons were depolarized by α-MSH (n = 6; mean depolarization: 9.4 ± 4.2 mV, Fig. 4A), while the remaining 25% of GE cells were unaffected by the peptide (n = 2). Thus, we did not observe hyperpolarizing effects of α-MSH on GE neurons. In addition, 57% of GI neurons depolarized (n = 4; mean depolarization: 9.6 ± 2.6 mV; Fig. 4B), 29% hyperpolarized (n = 2; mean hyperpolarization: −8.6 ± 1.3 mV), and 14% did not respond (n = 1) to α-MSH. Finally, 33% of NR neurons depolarized (n = 4; mean depolarization: 4.5 ± 1.3 mV), 17% hyperpolarized (n = 2; mean hyperpolarization: −6.9 ± 0.02 mV), and 50% did not respond (n = 6) to α-MSH. Although there was no difference in the amplitude of depolarizations (one-way ANOVA with Tukey's multiple-comparison test; P = 0.56) or hyperpolarizations (unpaired t-test; P = 0.34) between groups, our findings do indicate GE NTS neurons show significantly more depolarizing responses to α-MSH than NR cells (χ2, P = 0.04). Furthermore, our results show glucose-responsive cells collectively (i.e., GE and GI combined) are significantly more responsive to α-MSH than NR neurons (χ2, P = 0.02).
Fig. 4.

GE NTS neurons are more responsive to α-MSH than NR cells and are selectively depolarized by α-MSH. A and B: current-clamp recordings from two separate NTS neurons. A: α-MSH (500 nM; horizontal blue bar) elicited a reversible depolarization in this neuron subsequently identified as GE following an increase in glucose concentration from a baseline of 0.2 mM (horizontal pale gray bar) to 3 mM (horizontal medium gray bar). We note the amplitudes of these effects are above average for the population sampled. B: α-MSH elicited a reversible depolarization in this cell subsequently identified as GI following a decrease in glucose concentration from a baseline of 5 mM (horizontal dark gray bar) to 3 mM (horizontal medium gray bar). C: scatterplot depicting the range of responses elicited by 500 nM α-MSH in GE, GI, and NR NTS neurons. Mean α-MSH-induced depolarization: GE: 9.4 ± 4.2 mV, GI: 9.6 ± 2.6 mV, and NR: 4.5 ± 1.3 mV. Mean α-MSH-induced hyperpolarization: GI: −8.6 ± 1.3 mV, NR: −6.9 ± 0.02 mV. D: bar graph showing the percentage of GE (n = 8), GI (n = 7), and NR (n = 12) NTS neurons which also responded to α-MSH. More GE neurons exhibit depolarizing responses to α-MSH than NR NTS neurons (χ2; P = 0.04). There was no difference in the amplitude of α-MSH-induced effects across groups (ANOVA with Tukey's multiple-comparison test; P = 0.52 for depolarizations, unpaired t-test; P = 0.34 for hyperpolarizations).
NTS neurons show increased responsiveness to α-MSH as extracellular glucose concentrations decrease.
We next investigated whether the effects of α-MSH on the membrane potential of NTS neurons would be modified by changes in extracellular glucose concentrations. Again, we maintained rat brain slices containing the NTS at either 5, 3, or 0.2 mM glucose and subsequently bath-perfused 500 nM α-MSH to evaluate the effects of this peptide on the excitability of NTS cells. Our findings are summarized in Fig. 5. Previous studies from our laboratory using slices maintained at 10 mM glucose revealed effects of α-MSH on 61% of NTS neurons, with 39% of cells showing a depolarization (n = 16; mean 6.1 ± 0.5 mV), 22% of cells showing a hyperpolarization (n = 9; −6.8 ± 1.0 mV), and the remaining 39% of cells showing no response (n = 16) (26). After decreasing the extracellular glucose concentration to 5 mM, 64% of NTS neurons responded to α-MSH. More specifically, 53% of neurons showed a depolarization (n = 10; mean: 6.7 ± 1.4 mV), 11% showed a hyperpolarization (n = 2, −9.7 ± 0.3 mV), and 36% showed no response (n = 7) to α-MSH. All but one of the responses to α-MSH at 5 mM glucose were at least partially reversible upon washout. At 3 mM extracellular glucose, 71% of neurons responded to α-MSH, with 50% of cells showing a depolarization (n = 12; mean: 7.3 ± 0.8 mV, 9 at least partially reversible), 21% showing a reversible hyperpolarization (n = 5; −7.7 ± 1.8 mV), and 29% showing no response (n = 7) to α-MSH. Finally, at 0.2 mM extracellular glucose, 100% of neurons depolarized to α-MSH (n = 8; mean: 10.6 ± 3.2 mV, 5 at least partially reversible). The amplitudes of the depolarizations and hyperpolarizations to α-MSH across all four glucose concentrations were not different from one another (one-way ANOVA with Tukey's multiple-comparison test, P = 0.18 and P = 0.55 for depolarizations and hyperpolarizations, respectively). The distribution of responses to α-MSH was, however, significantly different between 0.2 and 10 mM, 0.2 and 5 mM, and 0.2 and 3 mM extracellular glucose (χ2, P = 0.0019, P = 0.03, and P = 0.02, respectively). Thus, these findings suggest an increased responsiveness of NTS neurons to α-MSH with decreasing extracellular glucose concentrations, specifically with reference to depolarizing effects of this peptide.
Fig. 5.

NTS neurons show increased responsiveness to α-MSH as extracellular glucose concentrations decrease. A and B: representative current-clamp recordings from five separate NTS neurons. A: bath-applied α-MSH (500 nM, horizontal blue bar) induced reversible depolarizing effects on a neuron bathed in aCSF containing 5 mM extracellular glucose (horizontal dark gray bar) (i), one bathed in 3 mM extracellular glucose (horizontal medium gray bar) (ii), and one bathed in 0.2 mM extracellular glucose (horizontal pale gray bar) (iii). B: α-MSH induced reversible hyperpolarizing effects in NTS cells at both 5 mM glucose (horizontal dark gray bar) (i) and 3 mM glucose (horizontal medium gray bar) (ii). C: scatterplot depicting the range of responses elicited by 500 nM α-MSH at varying extracellular glucose concentrations. Mean depolarization: 10 mM: 6.1 ± 0.5 mV (n = 16), 5 mM: 6.7 ± 1.4 mV (n = 10), 3 mM: 7.3 ± 0.8 mV (n = 12), and 0.2 mM: 10.6 ± 3.2 mV (n = 8). Mean hyperpolarization: 10 mM −6.8 ± 1.0 mV (n = 9), 5 mM: −9.7 ± 0.3 mV (n = 2), and 3 mM: −7.7 ± 1.8 mV (n = 5). D: bar graph showing the percentage of NTS neurons that respond to α-MSH with a depolarization, hyperpolarization, or no response across all glucose concentrations used. Neurons at 0.2 mM glucose are significantly more responsive to α-MSH than cells at 3, 5, and 10 mM glucose. There is no difference in the amplitude of α-MSH responses across groups (ANOVA with Tukey's multiple comparisons test, P > 0.15). Data at 10 mM glucose obtained from Ref. 26.
The resting membrane potential of NTS neurons is more hyperpolarized as extracellular glucose concentrations decrease.
In performing the series of experiments at 5, 3, and 0.2 mM glucose, we noted the baseline membrane potential of NTS neurons was more hyperpolarized as extracellular glucose concentrations were decreased. The mean baseline membrane potential of neurons without holding current at 10 mM was −64.2 ± 0.9 mV (n = 82; from Refs. 27 and 26), at 5 mM was −69.1 ± 0.9 mV (n = 21), at 3 mM was −70.0 ± 0.8 mV (n = 40), and at 0.2 mM was −74.7 ± 1.7 mV (n = 21). Indeed, the baseline membrane potential of neurons at 0.2 mM extracellular glucose was significantly more hyperpolarized than that at both 5 mM and 10 mM glucose, as was the baseline membrane potential of cells at 3 and 5 mM glucose vs. 10 mM (one-way ANOVA with Tukey's multiple-comparison test; P < 0.0001). These observations suggest that altering extracellular glucose concentrations has functionally relevant effects on the baseline excitability of NTS neurons.
DISCUSSION
The present study provides conclusive evidence of the integrative capacity of single NTS neurons. Furthermore, we have shown that the responses of these neurons to α-MSH are dynamically altered in accordance with the energy status of the region and, thus, the feeding status of the organism. This is, to the best of our knowledge, the first documentation of changes in the responsiveness of individual NTS neurons to peptide signals as a consequence of altered extracellular glucose concentrations, as well as of the potential rapid plasticity in melanocortin circuits in this medullary center.
We first sought to establish whether NTS neurons respond to physiological changes in extracellular glucose concentration in vitro. Studies simultaneously measuring circulating blood glucose levels in relation to central extracellular glucose levels in rats have found the latter range from 0.16 ± 0.03 in hypoglycemia to 4.5 ± 0.4 in hyperglycemia (37). Our chosen concentrations of 0.2, 3, and 5 mM extracellular glucose, therefore, provide a closer mimic of the range of concentrations that may be encountered by NTS neurons in vivo than previous whole cell patch-clamp studies in the region, where large decreases from 10 to 3 or 0 mM, or large increases from elevated baselines of 10 mM to 20 or 30 mM glucose have been used (2, 40). These studies, as well as others using extracellular recordings, have found between 20 and 81% of NTS neurons respond to changes in extracellular glucose concentrations, with contradicting reports of the predominance of GE vs. GI phenotypes (2, 10, 28, 40, 41, 47). Our observation that 56% of NTS neurons are glucose-responsive is similar to those of the most physiological previous studies, where 51–56% of NTS neurons responded to 2 mM changes in glucose concentrations. Indeed, as we have shown, these studies also found more GE vs. GI neurons (10, 11).
We also noted a gradual hyperpolarization of the membrane potential of NTS cells with decreasing glucose concentrations. Because the frequency of excitatory postsynaptic potentials increases as extracellular glucose concentrations rise (40), it can be assumed the opposite would be true as glucose levels decrease. This expected decline in excitatory inputs to NTS neurons at low extracellular glucose concentrations may explain the hyperpolarizing trend of the membrane potential that we observed. In future studies, it will be interesting to note whether there is also a correlation between extracellular glucose levels and inhibitory postsynaptic potentials in the NTS.
Unexpectedly, we found no cellular evidence to indicate nesfatin-1 preferentially affects glucose sensing over glucose-unresponsive cells in the NTS. Our findings contrast those of a recent study by Dong et al. (12), who noted predominantly excitatory effects of nesfatin-1 on GE neurons, and inhibitory effects of the peptide on GI cells. These authors, however, recorded from both the NTS and neighboring DMV, and it is, therefore, possible that their findings are more reflective of actions of nesfatin-1 in the DMV. Furthermore, glucose-sensing neurons were characterized on the basis of a step from 25 to 5 mM extracellular glucose, vs. the more physiological changes in glucose concentration used herein. While hypoglycemia has been shown to activate nesfatin-1-expressing neurons in the NTS, this in vivo manipulation also elicits activation of several hypothalamic areas with projections to the NTS, thereby making it difficult to determine whether hypoglycemia-induced medullary activation is a direct or a downstream effect of hypothalamic excitation. Moreover, nesfatin-1-immunoreactive NTS neurons express numerous other neuropeptides (5, 15), which may, indeed, be the relevant mediators of glucose regulation in the NTS, rather than nesfatin-1 itself. Thus, on the basis of these considerations and our current findings, we reason that actions of nesfatin-1 in the NTS may not, in fact, be involved in the coordination of physiological responses to alterations in central glucose levels. Furthermore, since we noted the effects of nesfatin-1 are entirely consistent across the range of glucose concentrations that we employed, which parallel states of hunger to satiety in the animal, we conclude that nesfatin-1 may not act as a functionally relevant regulator of energy status in the NTS. Instead, nesfatin-1 may act in the NTS to affect food intake not through its own intrinsic, direct actions on neurons in this region, but rather by modulating the effects of other feeding-related neuropeptides produced either locally or released from descending hypothalamic inputs (23). Indeed, nesfatin-1 upregulates POMC mRNA expression in the NTS, which may lead to altered α-MSH release and subsequent effects on glycemia or food intake (44). Thus, although it is evident further studies are required to accurately assess the precise role of the NTS in mediating the anorexigenic effects of nesfatin-1, our findings suggest this peptide may act predominantly as a local autocrine and/or paracrine modulator of neuronal function in the NTS rather than a metabolically relevant signaling molecule.
Our studies have revealed preferential actions of α-MSH on glucose- responsive NTS neurons, with a striking 80% of these cells being affected by this melanocortin. Moreover, GE neurons were universally depolarized by α-MSH, and GI neurons were predominantly (57%) depolarized by this peptide in the NTS. Our findings are, thus, the first to reveal a clear overlap in glucose sensing and melanocortin signaling in individual NTS neurons, highlighting the important integrative properties of these cells. In light of our previous observations of excitatory actions of α-MSH on GABAergic NTS cells (26), it is interesting to note that others have hypothesized GE NTS neurons are, in fact, GABAergic (14, 40). Furthermore, activation of NTS GE neurons leads to decreases in gastric motility and tone via modulation of cholinergic parasympathetic signaling (40). Moreover, activation of at least a subset of GI NTS neurons results in glucagon secretion (22). As we noted both depolarizing and hyperpolarizing effects of α-MSH on NTS GI neurons, we can speculate that cellular actions of α-MSH in this region may modulate circulating glucagon levels to contribute to the maintenance of normoglycemia in the animal. Taken together, our findings suggest the convergence of metabolically relevant signals on single glucose-sensing NTS cells may provide critical feedback in the coordination of behaviors pertaining to gastrointestinal function, food intake, and glucose homeostasis in the organism.
Finally, we have shown a gradual increase in the responsiveness of NTS neurons to α-MSH with decreasing concentrations of extracellular glucose. Most profoundly, all cells that were recorded in hypoglycemic conditions showed depolarizing responses to α-MSH, in stark contrast to the heterogeneous responses to the peptide in normoglycemic and hyperglycemic conditions. We emphasize the remarkably rapid time course, on the order of hours, of this shift in responsiveness of NTS neurons, an observation that suggests potential plasticity in the neurocircuits of this medullary structure. As it has been observed that POMC neurons in the NTS play important roles in acute, but not chronic, control of feeding (48), our current findings add to the possibility that NTS melanocortin signaling may preferentially contribute to the rapid regulation of ingestive behaviors and energy homeostasis. It is also interesting to note fasting leads to increased levels of α-MSH in the NTS (32), and to increased availability of MC4R binding sites in several hypothalamic nuclei (18), although similar studies have not yet been conducted in the NTS. Taken together, it is tempting to speculate that these potential changes in α-MSH expression and MC4R activity may explain the shift toward more depolarizing actions of this peptide in hypoglycemic conditions. Furthermore, as our previous studies have demonstrated the necessity for presynaptic, GABAergic signaling in mediating the hyperpolarizing actions of α-MSH (26), the elimination of these inhibitory effects in hypoglycemia may be a consequence of the hyperpolarized membrane potential of NTS neurons maintained in low glucose. At these resting membrane potentials, the passage of Cl− through ionotropic GABAA receptors may depolarize neurons, thereby abolishing the hyperpolarizing effects of α-MSH.
Perspectives and Significance
In conclusion, our findings demonstrate the presence of physiologically relevant glucose-sensing neurons in the NTS, which are preferentially influenced by α-MSH, but not nesfatin-1. Accordingly, alterations in glycemic state have profound effects on the responsiveness of NTS neurons to α-MSH, but not to nesfatin-1. Furthermore, the progressive shift toward more depolarizing responses to α-MSH, and a complete lack of hyperpolarizing responses, with decreasing extracellular glucose concentrations provides indisputable evidence of the ability of alterations in local glucose levels to profoundly modify the responsiveness of NTS neurons to, at the very least, metabolically relevant signals. In light of the fact that many electrophysiological studies in the region are conducted at elevated levels of glucose (5 and 10 mM; see Refs. 1, 6, 19, 26, 33, and 42, for example), these findings are of particular relevance and highlight a pressing need to adopt the usage of more physiological recording conditions to obtain the most relevant, accurate, and translational in vitro results. Indeed, we have demonstrated that elevated levels of glucose are not necessary to maintain neuronal integrity, as cells bathed at 0.2 mM glucose are fully capable of firing action potentials of high amplitude, maintaining a stable baseline membrane potential, being held for long-duration electrophysiological recordings, and surviving for hours in vitro. Overall, our observations have important implications for future electrophysiological studies on the effects of feeding-related peptides not only in the NTS, but in other central regions regulating the control of ingestive behaviors.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: A.M. and A.V.F. conception and design of research; A.M. performed experiments; A.M. analyzed data; A.M. and A.V.F. interpreted results of experiments; A.M. prepared figures; A.M. drafted manuscript; A.M. and A.V.F. edited and revised manuscript; A.M. and A.V.F. approved final version of manuscript.
ACKNOWLEDGMENTS
This work was supported by funding from the Natural Sciences and Engineering Research Council of Canada, Le Fonds québécois de la recherche sur la nature et les technologies, and the Heart and Stroke Foundation of Ontario.
REFERENCES
- 1.Appleyard SM, Bailey TW, Doyle MW, Jin YH, Smart JL, Low MJ, Andresen MC. Proopiomelanocortin neurons in nucleus tractus solitarius are activated by visceral afferents: regulation by cholecystokinin and opioids. J Neurosci 25: 3578–3585, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balfour RH, Hansen AM, Trapp S. Neuronal responses to transient hypoglycaemia in the dorsal vagal complex of the rat brainstem. J Physiol 570: 469–484, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berthoud HR. The vagus nerve, food intake and obesity. Regul Pept 149: 15–25, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonnet MS, Djelloul M, Tillement V, Tardivel C, Mounien L, Trouslard J, Troadec JD, Dallaporta M. Central NUCB2/Nesfatin-1-expressing neurones belong to the hypothalamic-brainstem circuitry activated by hypoglycaemia. J Neuroendocrinol 25: 1–13, 2013. [DOI] [PubMed] [Google Scholar]
- 5.Brailoiu GC, Dun SL, Brailoiu E, Inan S, Yang J, Chang JK, Dun NJ. Nesfatin-1: distribution and interaction with a G protein-coupled receptor in the rat brain. Endocrinology 148: 5088–5094, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Branchereau P, Champagnat J, Denavit-Saubie M. Cholecystokinin-gated currents in neurons of the rat solitary complex in vitro. J Neurophysiol 70: 2584–2595, 1993. [DOI] [PubMed] [Google Scholar]
- 7.Bronstein DM, Schafer MK, Watson SJ, Akil H. Evidence that β-endorphin is synthesized in cells in the nucleus tractus solitarius: detection of POMC mRNA. Brain Res 587: 269–275, 1992. [DOI] [PubMed] [Google Scholar]
- 8.Burdakov D, Gerasimenko O, Verkhratsky A. Physiological changes in glucose differentially modulate the excitability of hypothalamic melanin-concentrating hormone and orexin neurons in situ. J Neurosci 25: 2429–2433, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burdakov D, Jensen LT, Alexopoulos H, Williams RH, Fearon IM, O'Kelly I, Gerasimenko O, Fugger L, Verkhratsky A. Tandem-pore K+ channels mediate inhibition of orexin neurons by glucose. Neuron 50: 711–722, 2006. [DOI] [PubMed] [Google Scholar]
- 10.Dallaporta M, Himmi T, Perrin J, Orsini JC. Solitary tract nucleus sensitivity to moderate changes in glucose level. Neuroreport 10: 2657–2660, 1999. [DOI] [PubMed] [Google Scholar]
- 11.Dallaporta M, Perrin J, Orsini JC. Involvement of adenosine triphosphate-sensitive K+ channels in glucose-sensing in the rat solitary tract nucleus. Neurosci Lett 278: 77–80, 2000. [DOI] [PubMed] [Google Scholar]
- 12.Dong J, Guan HZ, Jiang ZY, Chen X. Nesfatin-1 influences the excitability of glucosensing neurons in the dorsal vagal complex and inhibits food intake. PLoS One 9: e98967, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan W, Dinulescu DM, Butler AA, Zhou J, Marks DL, Cone RD. The central melanocortin system can directly regulate serum insulin levels. Endocrinology 141: 3072–3079, 2000. [DOI] [PubMed] [Google Scholar]
- 14.Ferreira M Jr, Browning KN, Sahibzada N, Verbalis JG, Gillis RA, Travagli RA. Glucose effects on gastric motility and tone evoked from the rat dorsal vagal complex. J Physiol 536: 141–152, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foo KS, Brismar H, Broberger C. Distribution and neuropeptide coexistence of nucleobindin-2 mRNA/nesfatin-like immunoreactivity in the rat CNS. Neuroscience 156: 563–579, 2008. [DOI] [PubMed] [Google Scholar]
- 16.Grill HJ, Ginsberg AB, Seeley RJ, Kaplan JM. Brainstem application of melanocortin receptor ligands produces long-lasting effects on feeding and body weight. J Neurosci 18: 10128–10135, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grill HJ, Hayes MR. Hindbrain neurons as an essential hub in the neuroanatomically distributed control of energy balance. Cell Metab 16: 296–309, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harrold JA, Widdowson PS, Williams G. Altered energy balance causes selective changes in melanocortin-4(MC4-R), but not melanocortin-3 (MC3-R), receptors in specific hypothalamic regions: further evidence that activation of MC4-R is a physiological inhibitor of feeding. Diabetes 48: 267–271, 1999. [DOI] [PubMed] [Google Scholar]
- 19.Hisadome K, Reimann F, Gribble FM, Trapp S. Leptin directly depolarizes preproglucagon neurons in the nucleus tractus solitarius: electrical properties of glucagon-like Peptide 1 neurons. Diabetes 59: 1890–1898, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu Y, Wilson GS. Rapid changes in local extracellular rat brain glucose observed with an in vivo glucose sensor. J Neurochem 68: 1745–1752, 1997. [DOI] [PubMed] [Google Scholar]
- 21.Kishi T, Aschkenasi CJ, Lee CE, Mountjoy KG, Saper CB, Elmquist JK. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J Comp Neurol 457: 213–235, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Lamy CM, Sanno H, Labouebe G, Picard A, Magnan C, Chatton JY, Thorens B. Hypoglycemia-activated GLUT2 neurons of the nucleus tractus solitarius stimulate vagal activity and glucagon secretion. Cell Metab 19: 527–538, 2014. [DOI] [PubMed] [Google Scholar]
- 23.Maejima Y, Sedbazar U, Suyama S, Kohno D, Onaka T, Takano E, Yoshida N, Koike M, Uchiyama Y, Fujiwara K, Yashiro T, Horvath TL, Dietrich MO, Tanaka S, Dezaki K, Oh I, Hashimoto K, Shimizu H, Nakata M, Mori M, Yada T. Nesfatin-1-regulated oxytocinergic signaling in the paraventricular nucleus causes anorexia through a leptin-independent melanocortin pathway. Cell Metab 10: 355–365, 2009. [DOI] [PubMed] [Google Scholar]
- 24.Medeiros N, Dai L, Ferguson AV. Glucose-responsive neurons in the subfornical organ of the rat—a novel site for direct CNS monitoring of circulating glucose. Neuroscience 201: 157–165, 2012. [DOI] [PubMed] [Google Scholar]
- 25.Melnick IV, Price CJ, Colmers WF. Glucosensing in parvocellular neurons of the rat hypothalamic paraventricular nucleus. Eur J Neurosci 34: 272–282, 2011. [DOI] [PubMed] [Google Scholar]
- 26.Mimee A, Kuksis M, Ferguson AV. α-MSH exerts direct postsynaptic excitatory effects on NTS neurons and enhances GABAergic signaling in the NTS. Neuroscience 262: 70–82, 2014. [DOI] [PubMed] [Google Scholar]
- 27.Mimee A, Smith PM, Ferguson AV. Nesfatin-1 influences the excitability of neurons in the nucleus of the solitary tract and regulates cardiovascular function. Am J Physiol Regul Integr Comp Physiol 302: R1297–R1304, 2012. [DOI] [PubMed] [Google Scholar]
- 28.Mizuno Y, Oomura Y. Glucose responding neurons in the nucleus tractus solitarius of the rat: in vitro study. Brain Res 307: 109–116, 1984. [DOI] [PubMed] [Google Scholar]
- 29.Obici S, Feng Z, Tan J, Liu L, Karkanias G, Rossetti L. Central melanocortin receptors regulate insulin action. J Clin Invest 108: 1079–1085, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oh-IS, Shimizu H, Satoh T, Okada S, Adachi S, Inoue K, Eguchi H, Yamamoto M, Imaki T, Hashimoto K, Tsuchiya T, Monden T, Horiguchi K, Yamada M, Mori M. Identification of nesfatin-1 as a satiety molecule in the hypothalamus. Nature 443: 709–712, 2006. [DOI] [PubMed] [Google Scholar]
- 31.Ono T, Steffens AB, Sasaki K. Influence of peripheral and intracerebroventricular glucose and insulin infusions on peripheral and cerebrospinal fluid glucose and insulin levels. Physiol Behav 30: 301–306, 1983. [DOI] [PubMed] [Google Scholar]
- 32.Perello M, Stuart RC, Nillni EA. Differential effects of fasting and leptin on proopiomelanocortin peptides in the arcuate nucleus and in the nucleus of the solitary tract. Am J Physiol Endocrinol Metab 292: E1348–E1357, 2007. [DOI] [PubMed] [Google Scholar]
- 33.Peters JH, McDougall SJ, Kellett DO, Jordan D, Llewellyn-Smith IJ, Andresen MC. Oxytocin enhances cranial visceral afferent synaptic transmission to the solitary tract nucleus. J Neurosci 28: 11731–11740, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ritter S, Dinh TT, Zhang Y. Localization of hindbrain glucoreceptive sites controlling food intake and blood glucose. Brain Res 856: 37–47, 2000. [DOI] [PubMed] [Google Scholar]
- 35.Rossi J, Balthasar N, Olson D, Scott M, Berglund E, Lee CE, Choi MJ, Lauzon D, Lowell BB, Elmquist JK. Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab 13: 195–204, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Routh VH. Glucose-sensing neurons: are they physiologically relevant? Physiol Behav 76: 403–413, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Silver IA, Erecinska M. Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J Neurosci 14: 5068–5076, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song Z, Levin BE, McArdle JJ, Bakhos N, Routh VH. Convergence of pre- and postsynaptic influences on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 50: 2673–2681, 2001. [DOI] [PubMed] [Google Scholar]
- 39.Stengel A, Goebel M, Wang L, Rivier J, Kobelt P, Monnikes H, Lambrecht NW, Tache Y. Central nesfatin-1 reduces dark-phase food intake and gastric emptying in rats: differential role of corticotropin-releasing factor2 receptor. Endocrinology 150: 4911–4919, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wan S, Browning KN. d-glucose modulates synaptic transmission from the central terminals of vagal afferent fibers. Am J Physiol Gastrointest Liver Physiol 294: G757–G763, 2008. [DOI] [PubMed] [Google Scholar]
- 41.Wan S, Browning KN. Glucose increases synaptic transmission from vagal afferent central nerve terminals via modulation of 5-HT3 receptors. Am J Physiol Gastrointest Liver Physiol 295: G1050–G1057, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wan S, Browning KN, Coleman FH, Sutton G, Zheng H, Butler A, Berthoud HR, Travagli RA. Presynaptic melanocortin-4 receptors on vagal afferent fibers modulate the excitability of rat nucleus tractus solitarius neurons. J Neurosci 28: 4957–4966, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang R, Liu X, Hentges ST, Dunn-Meynell AA, Levin BE, Wang W, Routh VH. The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes 53: 1959–1965, 2004. [DOI] [PubMed] [Google Scholar]
- 44.Wernecke K, Lamprecht I, Johren O, Lehnert H, Schulz C. Nesfatin-1 increases energy expenditure and reduces food intake in rats. Obesity (Silver Spring) 22: 1662–1668, 2014. [DOI] [PubMed] [Google Scholar]
- 45.Williams DL, Kaplan JM, Grill HJ. The role of the dorsal vagal complex and the vagus nerve in feeding effects of melanocortin-3/4 receptor stimulation. Endocrinology 141: 1332–1337, 2000. [DOI] [PubMed] [Google Scholar]
- 46.Williams RH, Alexopoulos H, Jensen LT, Fugger L, Burdakov D. Adaptive sugar sensors in hypothalamic feeding circuits. Proc Natl Acad Sci USA 105: 11975–11980, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yettefti K, Orsini JC, Perrin J. Characteristics of glycemia-sensitive neurons in the nucleus tractus solitarii: possible involvement in nutritional regulation. Physiol Behav 61: 93–100, 1997. [DOI] [PubMed] [Google Scholar]
- 48.Zhan C, Zhou J, Feng Q, Zhang JE, Lin S, Bao J, Wu P, Luo M. Acute and long-term suppression of feeding behavior by POMC neurons in the brainstem and hypothalamus, respectively. J Neurosci 33: 3624–3632, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng H, Patterson LM, Phifer CB, Berthoud HR. Brain stem melanocortinergic modulation of meal size and identification of hypothalamic POMC projections. Am J Physiol Regul Integr Comp Physiol 289: R247–R258, 2005. [DOI] [PubMed] [Google Scholar]
- 50.Zheng H, Patterson LM, Rhodes CJ, Louis GW, Skibicka KP, Grill HJ, Myers MG Jr., Berthoud HR. A potential role for hypothalamomedullary POMC projections in leptin-induced suppression of food intake. Am J Physiol Regul Integr Comp Physiol 298: R720–R728, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]