

Abstract
Neurohormonal excitation and dyspnea are the hallmarks of heart failure (HF) and have long been associated with poor prognosis in HF patients. Sympathetic nerve activity (SNA) and ventilatory equivalent of carbon dioxide (VE/VO2) are elevated in moderate HF patients and increased even further in severe HF patients. The increase in SNA in HF patients is present regardless of age, sex, and etiology of systolic dysfunction. Neurohormonal activation is the major mediator of the peripheral vasoconstriction characteristic of HF patients. In addition, reduction in peripheral blood flow increases muscle inflammation, oxidative stress, and protein degradation, which is the essence of the skeletal myopathy and exercise intolerance in HF. Here we discuss the beneficial effects of exercise training on resting SNA in patients with systolic HF and its central and peripheral mechanisms of control. Furthermore, we discuss the exercise-mediated improvement in peripheral vasoconstriction in patients with HF. We will also focus on the effects of exercise training on ventilatory responses. Finally, we review the effects of exercise training on features of the skeletal myopathy in HF. In summary, exercise training plays an important role in HF, working synergistically with pharmacological therapies to ameliorate these abnormalities in clinical practice.
Keywords: heart failure, exercise training, sympathetic nerve activity, vasoconstriction, skeletal myopathy
this article is part of a collection on Exercise Training in Cardiovascular Disease: Cell, Molecular, and Integrative Perspectives. Other articles appearing in this collection, as well as a full archive of all collections, can be found online at http://ajpheart.physiology.org/.
Chronic heart failure (HF) afflicts almost 6 million of Americans, is responsible for more than 1 million primary hospitalizations yearly, and is directly associated with 1 in 9 deaths in the United States. Nearly half of these HF patients suffer from systolic dysfunction, and the progression of their HF is attributable to neurohormonal excitation leading to adverse cardiac remodeling and multi-organ dysfunction (38). Pharmacological therapies that target and counteract this neurohormonal activation lead to decreased morbidity and improved survival in HF patients. Exercise training also targets neurohormonal excitation and is now recommended as part of the standard therapeutic armamentarium for stable HF patients (111). Exercise training leads to improved neurovascular control and also has been shown to reverse features of the skeletal myopathy and abnormal ventilatory responses in HF, leading to improved functional status (66).
Neurohormonal excitation, specifically, increased resting sympathetic nerve activity (SNA), first measured by plasma norepinephrine levels, has long been recognized as a marker of poor prognosis in untrained HF patients (23, 32). Muscle SNA (MSNA) can be measured directly and quantitatively with microneurography, and increased MSNA is directly correlated with the clinical severity of HF. Resting MSNA is elevated in moderate HF patients and is increased even further in severe HF patients (55, 78). This increase in MSNA in untrained HF patients is present regardless of age, sex, and etiology of systolic dysfunction (4, 5, 79). The mechanisms involved in the sustained increase in SNA in HF remain poorly understood, but investigations in animals and humans have shown that abnormalities of peripheral reflex control and central neural integration play important roles (33, 77, 115).
Neurohormonal activation is the principle mediator of the peripheral vasoconstriction characteristic of untrained HF patients. Intra-arterial infusion of phentolamine (α-adreno-receptor antagonist) significantly reverses this vasoconstriction and increases muscle blood flow in resting HF patients (75). Moreover, phentolamine infusion significantly increases muscle vasodilatory responses during mental stress and exercise, and reverses the paradoxical muscle vasoconstriction during hypoxia in HF patients (2, 75, 92). Non-neuronal factors, including blunted nitric oxide-mediated endothelial reactivity, also contribute to the vasoconstriction in untrained HF patients (76). In addition to muscle vasoconstriction, patients with systolic HF have diminished resting renal cortical blood flow when compared with age-matched healthy individuals (69). Reduced renal blood flow likely further exacerbates neurohormonal activation, resulting in activation of the renin angiotensin system, and contributing to the positive feedback of progressive HF (18).
In addition to neurohormonal activation, HF causes dyspnea and alterations in the respiratory pattern. Patients with HF have lower ventilatory efficiency when compared with healthy individuals, and periodic breathing is often observed. Ventilatory equivalent of carbon dioxide (VE/VCO2 ratio) is increased and associated with exercise intolerance in HF patients (63). Heightened arterial chemoreflex sensitivity likely underlies the altered pattern of ventilation characteristic of HF (26). Finally, HF causes skeletal myopathy, which is thought to be the major mediator of exercise intolerance in patients with HF (Table 1) [for review, Middlekauff (66)]. Peripheral vasoconstriction slows oxygen kinetics, which shifts the energy production from oxidative metabolism to glycolytic metabolism in skeletal muscle (84, 93, 94). Elevated ANG II increases oxidative stress and muscle protein degradation (discussed below), contributing to the reduced muscle mass (28, 96, 113). HF also is associated with a shift from oxidative muscle fibers to glycolytic muscle fibers (93, 94). These fiber changes result in increased glycolysis and premature acidosis independently of changes in blood flow (59). Mancini et al. (60) reported a significant relationship between fiber type and peak oxygen consumption (VO2), suggesting that the change in fiber type is one more factor contributing to the exercise intolerance in HF. A longstanding controversy is whether the myopathy of HF is attributable to disuse and deconditioning, or if it is an active process driven by the neurohormonal and inflammatory mediators characteristic of the HF milieu (85).
Table 1.
Features of the skeletal myopathy of heart failure
| Features | Correlated to Exercise Capacity? | References | Exercise-training Effect? | References |
|---|---|---|---|---|
| Decreased muscle blood flow | Yes* | 3 | Improved | 4, 5, 30 |
| Decreased endothelial function | Yes | 44, 74 | Improved | 44, 58 |
| Capillary rarefaction | Yes | 11, 93 | Improved | 11, 30 |
| Fiber shift from type I to type II fibers | Yes | 93, 94 | Improved | 11, 45 |
| Abnormal Ca2+ cycling | Unknown | 16, 71 | Improved | 16, 110 |
| Decreased mitochondrial volume | Yes | 27 | Improved | 30, 91 |
| Decreased mitochondrial enzyme content | Yes** | 64 | Improved | 11, 30, 52 |
| Reduced metabolic capacity | Yes | 17, 53, 80, 101 | Improved | 1, 72 |
| Increased inflammation | Yes | 105 | Improved | 6, 36 |
| Reduced cross-sectional area | Yes | 19, 46 | Improved | 24, 30, 37, 47 |
Only related with exercise capacity in the presence of cachexia;
not found in heart failure patients on optimal medical and device therapy (Ref. 70).
In this review, we will discuss the beneficial effects of exercise training on resting SNA in patients with systolic HF. Furthermore, we will review recent work on potential mechanisms underlying this sympathetic excitation, focusing particularly on the interstitial muscle afferent neurons and the central neural integration, and its implications in exercise capacity. We will also focus on the effects of exercise training on ventilatory responses and the underlying augmented arterial chemoreflex sensitivity. Finally, we will review the effects of exercise training on sympathetically mediated skeletal muscle vasoconstriction and on features of the skeletal myopathy of HF. We argue that exercise training should assume a major role in the treatment of HF, working synergistically with pharmacological therapies, to ameliorate these abnormalities in clinical practice.
Effects of Exercise Training on SNA
According to the American Heart Association/American College of Cardiology guideline for the management of HF, exercise training is now a Class I recommendation for stable HF patients (111). One of the most remarkable effects of exercise training in HF patients is the reduction in resting sympathetic activation. First reported as a reduction in plasma norepinephrine levels in HF patients following training (21), the decrease in SNA has been precisely and repeatedly demonstrated with direct microneurographic recordings of MSNA (Fig. 1). Our group has consistently shown that 4 months of supervised moderate exercise training significantly decreases MSNA in patients with chronic systolic HF (4, 5, 31, 88). Before β-blocker therapy became standard in the treatment of HF patients, we compared the impact of exercise training in the MSNA levels in HF patients and found that training significantly decreased resting MSNA compared with that of untrained HF patients, and in fact MSNA was no longer elevated above normal (31). In a follow-up study, we found that the training benefits were similarly observed in HF patients on β-blocker therapy (31) (Fig. 2). Furthermore, middle-aged and older HF patients derived similar sympatholytic benefits from exercise training (4). Likewise, the reduction in MSNA with exercise training was not different between women and men with HF (5). Finally, Ueno and colleagues (106) demonstrated that exercise training decreased the severity of obstructive sleep apnea and decreased resting MSNA in HF patients with sleep apnea. In fact, the neurohormonal benefits were even more pronounced in this cohort.
Fig. 1.

Direct recording of muscle sympathetic nerve activity (MSNA) in patients with systolic heart failure, before (pre) and after (post) exercise training. Observe that exercise training causes a remarkable reduction in MSNA.
Fig. 2.
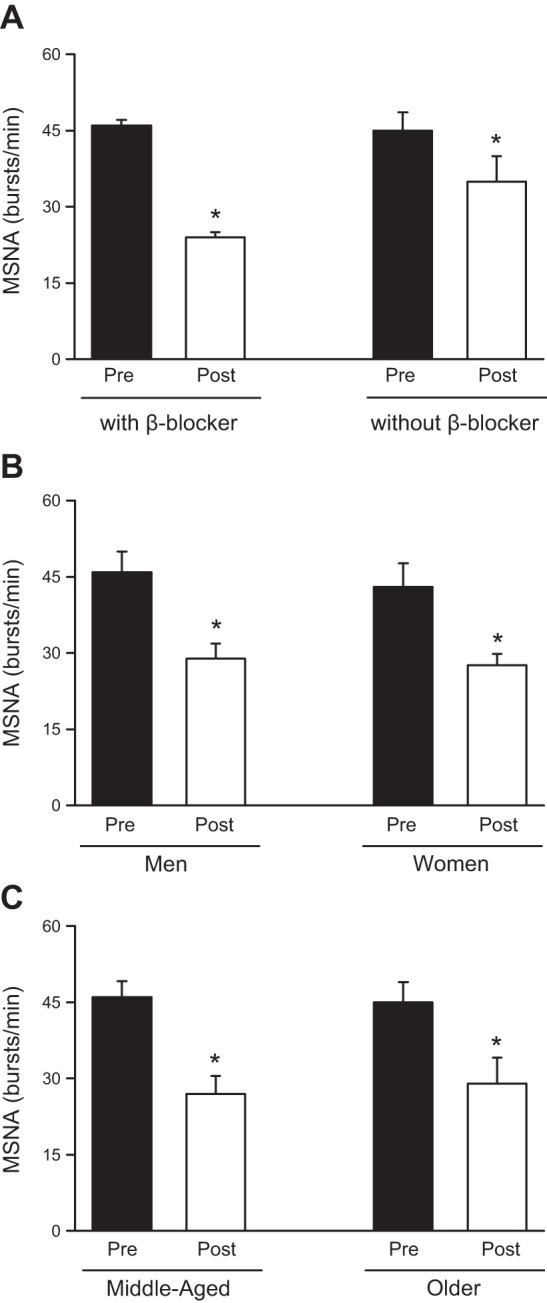
Effects of exercise training on MSNA in patients with systolic heart failure. Observe that exercise training reduces MSNA independently of pharmacological β-blocker therapy (A), sex (B), and age (C). *P < 0.05, within group difference.
The mechanisms underlying the reduction in the sympathetic modulation in exercise-trained HF patients remain uncertain, but several likely contributors will be discussed. In patients with chronic myocardial infarction but without HF, training-related reduction in MSNA is associated with improvement in baroreflex control (62). In the rat infarct model of HF, exercise training increases the inhibitory aortic afferent nerve discharge during fluctuations in blood pressure, consistent with improved baroreflex function localized to the afferent limb (87). Because baroreflex dysfunction has long been posited as an important mechanism underlying the sympatho-excitation in chronic HF (61), it is plausible that improved baroreflex function with exercise training contributes to its normalization. Exercise training has also been shown to improve arterial chemoreflex control of SNA in the rabbit pacing model of HF. Following exercise training in the HF rabbit, the exaggerated increase in renal sympathetic nerve activity (RSNA) in response to oxygen arterial partial pressure variation is attenuated (56). Moreover, these changes seem to be due to an improvement in carotid body nitric oxide production (102, 103). In summary, exercise training has beneficial effects on both arterial baroreflex and arterial chemoreflex control of sympathetic nerve activity, which likely contribute to the observed reductions in resting SNA in trained HF patients.
Most recently, our laboratory has focused on skeletal muscle afferent nerve fibers, including muscle metaboreceptors, which are unmyelinated group IV fibers sensitive to ischemic metabolites, and muscle mechanoreceptors, thinly myelinated group III fibers sensitive to mechanical stimuli. Data from animal HF models, as well as from untrained HF patients, support the concept that muscle metaboreflex control of MSNA is blunted and muscle mechanoreflex sensitivity is augmented (35, 67, 68, 100). The molecular mechanisms underlying these altered skeletal muscle reflexes in HF are not completely understood. Transient receptor potential vanilloid type-1 (TRPV1) and cannabinoid receptor type-1 (CB1) receptors are colocalized on muscle metaboreceptors. In animal models of HF, these receptors are downregulated on muscle metaboreceptors (98, 107, 109). Wang et al. (108) observed that exercise training initiated soon after myocardial infarction before the onset of HF in the rat infarct model of HF increased expression of TRPV1 receptors in dorsal root ganglia and prevented blunted metaboreflex sensitivity. Nerve growth factor (NGF) is a trophic factor for TRPV1 expression, and NGF is downregulated in HF models, suggestive of a possible mechanism to explain the decreased TRPV1 expression in HF. The impact of exercise training in HF rats on NGF has not been studied. Switching our focus to the mechanoreflex, cyclooxygenase-2 (COX) metabolites and purinergic 2X (P2X) receptors have been shown to modulate the sensitivity of muscle mechanoreceptors in animals models of HF (107). Exercise training reduces the expression of P2X receptors in skeletal muscle afferents in animals with HF (108).
We hypothesized that the reduction in resting SNA following exercise training in humans with HF would be mediated by changes in muscle mechanoreflex and metaboreflex sensitivity. To isolate the muscle metaboreceptor, we used the technique of post-exercise circulatory arrest, in which a sphygmomanometer cuff placed on the limb proximal to exercising muscle is inflated to supra-systolic levels at the conclusion of exercise, trapping ischemic metabolites in the vicinity of the muscle metaboreceptors. Moderate exercise training for 4 months significantly increased MSNA responses during metaboreceptors stimulation (6). Furthermore, this increase in muscle metaboreflex sensitivity was associated with an increase in expression of TRPV1 and CB1. Moderate exercise training also significantly decreased MSNA responses during muscle mechanoreceptor stimulation (6). The changes in mechanoreceptor sensitivity were accompanied by reduction in COX-2 expression. COX-2 is the rate limiting enzyme in the formation of prostaglandins and is induced by the inflammatory mediator NF-κB. Gielen and colleagues (36) previously reported that exercise training in chronic HF patients decreased inflammatory markers, including TNF-α, IL-1, and IL-6 levels present in skeletal muscle, but not in the circulation. These findings are consistent with an inflammatory process localized to the skeletal muscle itself, and not from spillover from a nonmuscle source. Our findings suggest that the alterations in muscle metaboreflex and mechanoreflex control of MSNA may also play a role in the reduction in resting MSNA following exercise training in HF patients.
Central command, a signal generated in the cerebral cortex proportionate to perceived effort during exercise, also increases SNA and heart rate responses during exercise. In animals with HF, Koba et al. (54) found that exercise training normalized the central command control of RSNA during electrical stimulation of the mesencephalic locomotor region, which was attributed to antioxidant effects in the medulla. In humans with end-stage renal failure in whom reflex control of the circulation is altered, central command control of HR is augmented (82); central command control of SNA has not been studied. Similarly, central command control of SNA has not been studied in humans with HF. It remains unknown, but possible, if the changes in SNA due to exercise training are mediated in part by changes in central command, and further studies are needed.
Exercise training also ameliorates abnormal central neuronal signaling, which may contribute to the observed decrease in resting SNA following exercise training [for review, Haack and Zucker (43)]. To date, data exist only in animal models to illuminate the role for the central nervous system signaling in HF. Central ANG II levels and expression of angiotensin II type 1 (AT1) receptors are increased in the rostral ventrolateral medulla (RVLM) in HF models and may contribute to the exaggerated sympathetic outflow in HF models. Exercise training reduces gene expression of ANG II receptors in the rostroventrolateral medulla in HF animals (33). Because ANG II modulates central sympathetic outflow, it is plausible that the reduction in efferent SNA following exercise training in HF is due to attenuation in ANG II activity in the sites of central neural integration. In fact, an association between the changes in central AII receptor type 1 (AT1) gene expression and the reduction in RSNA has been reported in the rabbit pacing model of HF (73). More recently, Kar et al. (50) showed that exercise training normalizes the balance between angiotensin converting enzyme (ACE) and ACE2 in the central nervous systems of animals with HF. This finding suggests an important role of ANG II/ANG 1–7 interaction in the exaggerated central sympathetic outflow in HF (50). Exercise training also increases the bioavailability of nitric oxide (NO) in the central nervous system due to an increased expression of neuronal NO synthase (nNOS) in paraventricular nucleus (114). Finally, exercise training reduces oxidative stress by reduced protein expression of NAPH oxidase subunit gp91(phox) and increased expression of CuZn superoxide dismutase in rostral ventral lateral medulla in HF rabbits (34).
In summary, exercise training decreases the elevated MSNA levels in HF toward normal and ameliorates the abnormal reflex control of MSNA during acute exercise. The finding that these beneficial effects of exercise training on sympathetic activation are accompanied by a decrease in muscle inflammation suggests an association, although it is impossible to know which factor - inflammation or SNA - is the driver. Evidence supports a role for both. To summarize (see Fig. 3): acute sympathetic excitation (likely baroreflex mediated) following a myocardial infarction or similar injury leads to an acute increase in efferent MSNA and reduced muscle and renal blood flow. This chronic muscle hypoperfusion may then generate skeletal muscle inflammation, diminished NGF levels, and altered afferent muscle neuronal receptor expression (e.g., TRPV1). Renal hypoperfusion leads to activation of the renin angiotensin system, specifically ANG II. Consequently, central reflex control of MSNA during exercise is altered, further contributing to increase in MSNA and RSNA, poor exercise tolerance, and skeletal myopathy (6). Although pharmacological therapies partially interrupt the neurohormonal excitation and improve muscle and kidney blood flow, these pharmacological strategies are insufficient. Only exercise training has been shown to restore the neurohormonal balance and reverse many key features of the skeletal myopathy in patients with HF from systolic dysfunction.
Fig. 3.

Mechanisms by which a myocardial infarction, or similar injury, provokes sympathetic excitation. Lowered cardiac output decreases the arterial baroreflex inhibition on efferent MSNA, which provokes peripheral vasoconstriction. The chronic renal hypoperfusion leads to activation of the renin angiotensin system and generates skeletal muscle inflammation and atrophy. The result of these alterations is skeletal myopathy. Abnormal chemoreflex activity and abnormal skeletal muscle reflex control lead to increased ventilation. NO, nitric oxide; RSNA, renal sympathetic nerve activity.
Effects of Exercise Training on Ventilatory Response
Dyspnea is a common symptom in patients with HF. Several mechanisms have been suggested to explain the abnormal ventilatory pattern and increased arterial chemoreflex sensitivity in HF patients. This complex issue has been attributed to pulmonary congestion, peripheral hypoxemia, and hypoperfusion in consequence of a low cardiac output [for review, Dempsey (26)]. In addition, animal data support the notion that carotid body alterations play a role in the altered respiratory pattern in HF. HF patients have enhanced chemoreflex sensitivity mediated by augmented afferent input from carotid body. These alterations are associated with upregulation of ANG II system and decreased nNOS expression in carotid body, which contribute to the enhanced carotid body sensitivity in HF (Fig. 3) (95). More recently, respiratory disturbances have been associated with abnormal afferent skeletal muscle reflex control. Olson et al. (81) demonstrated that inhibition of afferent feedback from skeletal muscle, via lumbar intrathecal injection of fentanyl, significantly decreased the ventilator responses during submaximal exercise in HF patients. Because exercise training improves muscle mechano and metaboreflex control of MSNA in patients with HF as previously reported (Antunes-Correa), it is reasonable to raise the question whether the improvement in muscle afferent reflexes following exercise training also contributes to the amelioration of the respiratory pattern in HF. This potential benefit of exercise training has not yet been investigated. Furthermore, it is likely that exercise-induced improvement in chemoreflex control also plays a role in the amelioration of respiratory pattern in HF patients. Exercise training normalizes afferent carotid body chemoreflex activity by reversing alterations in ANG II systems and nNOS expression in carotid body in animals with HF (56). The improvement in the VE/VCO2 ratio during exercise has been consistently demonstrated in exercise-trained HF patients (5, 31). This is an important response because reduction in work of breathing during exercise may increase blood flow to the skeletal muscle, thereby improving exercise capacity.
Effects of Exercise Training on Skeletal Myopathy
The skeletal myopathy of HF is recognized as an important contributor to exercise intolerance in chronic systolic HF (20, 22, 66). Exercise training has been shown to significantly improve many of the key features of the skeletal myopathy in HF patients (Table 1), including increased muscle capillarization, muscle blood flow, and flow mediated dilation (4, 11, 30, 31, 93, 99, 106). Importantly, significantly augmented circulating bone marrow-derived progenitor cells, which restore the diseased endothelium, have been detected in exercise-trained HF patients (29). All these changes in vascular architecture and function favor oxygen diffusion capacity into skeletal muscle and energy production [for review, Poole et al. (84)]. Exercise training restores aerobic metabolism in HF by improving in oxidative pathways. Exercise training increases maximal citrate synthase activity in HF (11, 72). Exercise training also increases mitochondrial volume and enzyme content, improving the metabolic capacity in HF (11, 30, 52, 91). These metabolic changes contribute, at least in part, to the improvement in functional capacity and reduction in exercise intolerance in patients with HF.
At the skeletal myocyte level, exercise training results in key changes in protein expression. Exercise training reduces skeletal muscle TNF-α gene and protein expression in patients with HF, presumably leading to a decrease in reactive oxygen species (ROS) production (28, 36). Among the many damaging effects of oxidative stress, ROS mediate the translocation of NF-κB to the nucleus, which activates catabolic genes (“an atrophy program”), leading to skeletal muscle wasting (42, 57, 90). In a mouse model of sympathetic hyperactivity-induced HF, exercise training reduces lipid hydroperoxidation and carbonylated proteins, markers of sustained ROS (24).
In humans with HF, muscle atrophy is present in some, but not all, skeletal muscle biopsies and in its most profound state is known clinically as cardiac cachexia. Exercise training ameliorates protein degradation pathways in animal models, including the mouse model of sympathetic hyperactivity-induced HF. Ubiquitin-proteasome system (UPS), the final common of proteolysis in skeletal muscle, is significantly downregulated by exercise training. Exercise training significantly reduces the components of E3 ligases; Atrogin-1, MuRF-1 and E3α gene expression are all decreased by exercise training (24, 40).
Insulin-like growth factor-1 (IGF-1) signaling pathway is a key pathway involved in skeletal muscle anabolic/catabolic balance (90). Godard et al. (39) found that IGF-1 is downregulated in skeletal muscle of HF patients, even before anatomical alterations are present. Only 4 weeks of exercise training significantly reverses the downregulation of the IGF-1 signaling pathway in humans with chronic systolic HF (37). Importantly, these changes in muscle protein degradation pathways are associated with increased quadriceps muscle cross-sectional area and mass (24). Similar findings have been reported in ischemic models of HF and in HF patients following exercise training (24, 37, 47, 72).
Pharmacological interruption of the renin angiotensin system is a mainstay of HF therapy; elevated ANG II levels despite angiotensin converting enzyme inhibitor therapy is associated with increased mortality (86). Following myocardial infarction or similar cardiac injury, elevated RSNA and decreased renal perfusion increase ANG II (97). ANG II remains elevated in the plasma, organs, and tissues contributing to the progression of HF (see Table 2) (41, 51, 86, 97). We found that ANG II levels are also elevated in the skeletal muscle in the rat infarct HF model; ANG II was increased in soleus and plantaris muscles of HF rats compared with normal controls (41). ANG II increases the ROS via NADPH oxidase and thereby produces mitochondrial dysfunction and limits exercise capacity (48, 96). In addition, ANG II reduces the all-important IGF-1 signaling pathway and energy capacity (15, 48, 104, 112). Finally, ANG II facilitates NF-κB translocation to the nucleus, thereby upregulating the UPS, with consequent proteolysis and muscle atrophy (13, 89, 96). Exercise training significantly reduces both circulating and skeletal muscle ANG II levels in HF (14, 41, 73). The ANG II reduction may be a necessary consequence of exercise training in HF to ameliorate the skeletal myopathy. We found an association between the improvement skeletal myopathy and the reduction in plasma ANG II concentrations (40). Exercise training emerges as an important strategy that works synergistically with pharmacological therapies for the comprehensive treatment of HF.
Table 2.
Adverse effects of ANG II
| Increased sympathetic nerve activity |
| Decreased renal blood flow |
| Increased reactive oxygen species |
| Decreased insulin-like growth factor |
| Increased translocation of NF-κB to nucleus |
| Activation of ubiquitin-proteasome system |
| Muscle atrophy |
Clinical Implications
The translational studies reviewed substantially extend our understanding of the role of exercise training on clinical outcomes in patients with HF. The clinical benefits accompanying the physiologic benefits of reducing SNA and the improving in respiratory pattern following exercise training in HF patients are important. SNA exacerbation precipitates cardiac arrhythmias, increases afterload, and decreases ventricular function. Increased MSNA is an independent predictor of mortality in HF patients (12). Similarly, VE/VCO2 predicts a poor prognosis in HF patients (8, 10). Reversal of the elevated MSNA increases endothelial-mediated vasodilatation in skeletal muscle (92). The increase in peripheral blood flow may improve the skeletal myopathy of HF. Finally, these exercise-induced physiological changes contribute to the improvement in exercise tolerance and quality of life in patients with HF.
Exercise Training Paradigm
What is the ideal prescription for exercise for patients with HF? During the last decades, several studies demonstrated that both supervised and nonsupervised exercise training substantially benefit patients with left systolic dysfunction, although compliance with supervised exercise is certainly better (83). In the HF-ACTION Study a large proportion of HF patients did not complete the unsupervised exercise program. Moderate aerobic exercise training has been the preferred modality of exercise training, and patients likely benefit from moderate strength training as well (4–6, 25, 31, 88, 106). Recent studies have suggested that intense exercise alternates moderate exercise during a session is safe and well tolerate in HF patients, and the benefits produced by more intense exercise seem to be superior that those achieved by moderate exercise (7, 9, 49, 110). Selection criteria for intense exercise training in HF patients are uncertain. Much work needs to be done on tailoring the exercise training program to the individual HF patient. When should exercise training start? Because inactivity accelerates the progression of the exercise intolerance and worsening of the quality of life, it is legitimate to suggest that exercise training for HF patients starts as soon as possible. An ongoing multicenter, randomized, prospective, open, controlled trial will provide more information regarding this subject in the near future (65).
Final Remarks
Exercise training improves neurovascular control and ventilatory responses in HF patients with chronic systolic dysfunction, including patients optimized on medical therapy (Fig. 4). Exercise training decreases resting MSNA in chronic HF patients, independent of age, sex, etiology of HF, and comorbidities. Exercise training reverses abnormal reflex control of MSNA and increases expression of the neuronal afferent receptors TRPV1 and CB1. Exercise training decreases muscle inflammation and ANG II levels, presumably through its sympatholytic and vasodilatory effects. Reversal of muscle inflammation and increased ANG II restores the balance of the muscle catabolic-anabolic pathways, thereby reversing muscle atrophy. Exercise training is associated with improved quality of life in patients already on optimal, guideline directed medical therapy and is playing an increasingly important therapeutic role in the treatment of HF (111).
Fig. 4.

Effects of exercise training in systolic heart failure. Note that exercise training reduces plasma and tissue renin-angiotensin system, and angiotensin II activity in the central nervous system. In addition, exercise training improves arterial baroreflex, chemoreflex, and muscle pressor reflex controls. All together, these responses lead to remarkable reduction in sympathetic nerve activity (SNA) and renal vasoconstriction and ventilatory responses (see text for more details). TRPV1, transient receptor potential vanilloid type-1.
GRANTS
This study was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; No. 2010/50048-1). C. E. Negrao was supported Conselho Nacional de Pesquisa (CNPq; 301867/2010-0). H. R. Middlekauff was supported by NIH/RO1-HL084525. I. L. Gomes-Santos and L. M. Antunes-Correa were supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; No. 2014/13690-8 and No. 2013/15651-7).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: C.E.N., H.R.M., I.L.G.-S., and L.M.A.-C. conception and design of research; C.E.N., H.R.M., I.L.G.-S., and L.M.A.-C. drafted manuscript; C.E.N. and H.R.M. edited and revised manuscript; C.E.N. approved final version of manuscript.
REFERENCES
- 1.Adamopoulos S, Coats AJ, Brunotte F, Arnolda L, Meyer T, Thompson CH, Dunn JF, Stratton J, Kemp GJ, Radda GK. Physical training improves skeletal muscle metabolism in patients with chronic heart failure. J Am Coll Cardiol 21: 1101–1106, 1993. [DOI] [PubMed] [Google Scholar]
- 2.Alves MJ, Rondon MU, Santos AC, Dias RG, Barretto AC, Krieger EM, Middlekauff HR, Negrão CE. Sympathetic nerve activity restrains reflex vasodilatation in heart failure. Clin Auton Res 17: 364–369, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Anker SD, Swan JW, Volterrani M, Chua TP, Clark AL, Poole-Wilson PA, Coats AJ. The influence of muscle mass, strength, fatigability and blood flow on exercise capacity in cachectic and non-cachectic patients with chronic heart failure. Eur Heart J 18: 259–269, 1997. [DOI] [PubMed] [Google Scholar]
- 4.Antunes-Correa LM, Kanamura BY, Melo RC, Nobre TS, Ueno LM, Franco FG, Roveda F, Braga AM, Rondon MU, Brum PC, Barretto AC, Middlekauff HR, Negrao CE. Exercise training improves neurovascular control and functional capacity in heart failure patients regardless of age. Eur J Prev Cardiol 19: 822–829, 2012. [DOI] [PubMed] [Google Scholar]
- 5.Antunes-Correa LM, Melo RC, Nobre TS, Ueno LM, Franco FG, Braga AM, Rondon MU, Brum PC, Barretto AC, Middlekauff HR, Negrao CE. Impact of gender on benefits of exercise training on sympathetic nerve activity and muscle blood flow in heart failure. Eur J Heart Fail 12: 58–65, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antunes-Correa LM, Nobre TS, Groehs RV, Alves MJ, Fernandes T, Couto GK, Rondon MU, de Oliveira PA, Lima M, Mathias W, Brum PC, Mady C, Almeida DR, Rossoni LV, Oliveira EM, Middlekauff HR, Negrao CE. Molecular basis for the improvement in muscle metaboreflex and mechanoreflex control in exercise-trained humans with chronic heart failure. Am J Physiol Heart Circ Physiol. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arena R, Cahalin LP, Borghi-Silva A, Phillips SA. Improving functional capacity in heart failure: the need for a multifaceted approach. Curr Opin Cardiol 29: 467–474, 2014. [DOI] [PubMed] [Google Scholar]
- 8.Arena R, Myers J, Aslam SS, Varughese EB, Peberdy MA. Peak VO2 and VE/VCO2 slope in patients with heart failure: a prognostic comparison. Am Heart J 147: 354–360, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Arena R, Myers J, Forman DE, Lavie CJ, Guazzi M. Should high-intensity-aerobic interval training become the clinical standard in heart failure? Heart Fail Rev 18: 95–105, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arena R, Myers J, Hsu L, Peberdy MA, Pinkstaff S, Bensimhon D, Chase P, Vicenzi M, Guazzi M. The minute ventilation/carbon dioxide production slope is prognostically superior to the oxygen uptake efficiency slope. J Card Fail 13: 462–469, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Bacurau AV, Jardim MA, Ferreira JC, Bechara LR, Bueno CR, Alba-Loureiro TC, Negrao CE, Casarini DE, Curi R, Ramires PR, Moriscot AS, Brum PC. Sympathetic hyperactivity differentially affects skeletal muscle mass in developing heart failure: role of exercise training. J Appl Physiol (1985) 106: 1631–1640, 2009. [DOI] [PubMed] [Google Scholar]
- 12.Barretto AC, Santos AC, Munhoz R, Rondon MU, Franco FG, Trombetta IC, Roveda F, de Matos LN, Braga AM, Middlekauff HR, Negrão CE. Increased muscle sympathetic nerve activity predicts mortality in heart failure patients. Int J Cardiol 135: 302–307, 2009. [DOI] [PubMed] [Google Scholar]
- 13.Bechara LR, Moreira JB, Jannig PR, Voltarelli VA, Dourado PM, Vasconcelos AR, Scavone C, Ramires PR, Brum PC. NADPH oxidase hyperactivity induces plantaris atrophy in heart failure rats. Int J Cardiol 175: 499–507, 2014. [DOI] [PubMed] [Google Scholar]
- 14.Braith RW, Welsch MA, Feigenbaum MS, Kluess HA, Pepine CJ. Neuroendocrine activation in heart failure is modified by endurance exercise training. J Am Coll Cardiol 34: 1170–1175, 1999. [DOI] [PubMed] [Google Scholar]
- 15.Brink M, Wellen J, Delafontaine P. Angiotensin II causes weight loss and decreases circulating insulin-like growth factor I in rats through a pressor-independent mechanism. J Clin Invest 97: 2509–2516, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bueno CR, Ferreira JC, Pereira MG, Bacurau AV, Brum PC. Aerobic exercise training improves skeletal muscle function and Ca2+ handling-related protein expression in sympathetic hyperactivity-induced heart failure. J Appl Physiol (1985) 109: 702–709, 2010. [DOI] [PubMed] [Google Scholar]
- 17.Chati Z, Zannad F, Robin-Lherbier B, Escanye JM, Jeandel C, Robert J, Aliot E. Contribution of specific skeletal muscle metabolic abnormalities to limitation of exercise capacity in patients with chronic heart failure: a phosphorus 31 nuclear magnetic resonance study. Am Heart J 128: 781–792, 1994. [DOI] [PubMed] [Google Scholar]
- 18.Ciccone MM, Iacoviello M, Gesualdo L, Puzzovivo A, Antoncecchi V, Doronzo A, Monitillo F, Citarelli G, Paradies V, Favale S. The renal arterial resistance index: a marker of renal function with an independent and incremental role in predicting heart failure progression. Eur J Heart Fail 16: 210–216, 2014. [DOI] [PubMed] [Google Scholar]
- 19.Cicoira M, Zanolla L, Franceschini L, Rossi A, Golia G, Zamboni M, Tosoni P, Zardini P. Skeletal muscle mass independently predicts peak oxygen consumption and ventilatory response during exercise in noncachectic patients with chronic heart failure. J Am Coll Cardiol 37: 2080–2085, 2001. [DOI] [PubMed] [Google Scholar]
- 20.Clark AL, Poole-Wilson PA, Coats AJ. Exercise limitation in chronic heart failure: central role of the periphery. J Am Coll Cardiol 28: 1092–1102, 1996. [DOI] [PubMed] [Google Scholar]
- 21.Coats AJ, Adamopoulos S, Radaelli A, McCance A, Meyer TE, Bernardi L, Solda PL, Davey P, Ormerod O, Forfar C. Controlled trial of physical training in chronic heart failure. Exercise performance, hemodynamics, ventilation, and autonomic function. Circulation 85: 2119–2131, 1992. [DOI] [PubMed] [Google Scholar]
- 22.Coats AJ, Clark AL, Piepoli M, Volterrani M, Poole-Wilson PA. Symptoms and quality of life in heart failure: the muscle hypothesis. Br Heart J 72: S36–S39, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohn JN, Levine TB, Olivari MT, Garberg V, Lura D, Francis GS, Simon AB, Rector T. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med 311: 819–823, 1984. [DOI] [PubMed] [Google Scholar]
- 24.Cunha TF, Bacurau AV, Moreira JB, Paixão NA, Campos JC, Ferreira JC, Leal ML, Negrão CE, Moriscot AS, Wisløff U, Brum PC. Exercise training prevents oxidative stress and ubiquitin-proteasome system overactivity and reverse skeletal muscle atrophy in heart failure. PLoS One 7: e41701, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Mello Franco FG, Santos AC, Rondon MU, Trombetta IC, Strunz C, Braga AM, Middlekauff H, Negrão CE, Pereira Barretto AC. Effects of home-based exercise training on neurovascular control in patients with heart failure. Eur J Heart Fail 8: 851–855, 2006. [DOI] [PubMed] [Google Scholar]
- 26.Dempsey JA. New perspectives concerning feedback influences on cardiorespiratory control during rhythmic exercise and on exercise performance. J Physiol 590: 4129–4144, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drexler H, Riede U, Münzel T, König H, Funke E, Just H. Alterations of skeletal muscle in chronic heart failure. Circulation 85: 1751–1759, 1992. [DOI] [PubMed] [Google Scholar]
- 28.Eley HL, Russell ST, Tisdale MJ. Mechanism of attenuation of muscle protein degradation induced by tumor necrosis factor-α and angiotensin II by β-hydroxy-β-methylbutyrate. Am J Physiol Endocrinol Metab 295: E1417–E1426, 2008. [DOI] [PubMed] [Google Scholar]
- 29.Erbs S, Höllriegel R, Linke A, Beck EB, Adams V, Gielen S, Möbius-Winkler S, Sandri M, Kränkel N, Hambrecht R, Schuler G. Exercise training in patients with advanced chronic heart failure (NYHA IIIb) promotes restoration of peripheral vasomotor function, induction of endogenous regeneration, and improvement of left ventricular function. Circ Heart Fail 3: 486–494, 2010. [DOI] [PubMed] [Google Scholar]
- 30.Esposito F, Reese V, Shabetai R, Wagner PD, Richardson RS. Isolated quadriceps training increases maximal exercise capacity in chronic heart failure: the role of skeletal muscle convective and diffusive oxygen transport. J Am Coll Cardiol 58: 1353–1362, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fraga R, Franco FG, Roveda F, de Matos LN, Braga AM, Rondon MU, Rotta DR, Brum PC, Barretto AC, Middlekauff HR, Negrao CE. Exercise training reduces sympathetic nerve activity in heart failure patients treated with carvedilol. Eur J Heart Fail 9: 630–636, 2007. [DOI] [PubMed] [Google Scholar]
- 32.Francis GS, Cohn JN, Johnson G, Rector TS, Goldman S, Simon A. Plasma norepinephrine, plasma renin activity, and congestive heart failure. Relations to survival and the effects of therapy in V-HeFT II The V-HeFT VA Cooperative Studies Group. Circulation 87: VI40–VI48, 1993. [PubMed] [Google Scholar]
- 33.Gao L, Wang W, Li YL, Schultz HD, Liu D, Cornish KG, Zucker IH. Sympathoexcitation by central ANG II: roles for AT1 receptor upregulation and NAD(P)H oxidase in RVLM. Am J Physiol Heart Circ Physiol 288: H2271–H2279, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Gao L, Wang W, Liu D, Zucker IH. Exercise training normalizes sympathetic outflow by central antioxidant mechanisms in rabbits with pacing-induced chronic heart failure. Circulation 115: 3095–3102, 2007. [DOI] [PubMed] [Google Scholar]
- 35.Garry MG. Abnormalities of the exercise pressor reflex in heart failure. Exerc Sport Sci Rev 39: 167–176, 2011. [DOI] [PubMed] [Google Scholar]
- 36.Gielen S, Adams V, Möbius-Winkler S, Linke A, Erbs S, Yu J, Kempf W, Schubert A, Schuler G, Hambrecht R. Anti-inflammatory effects of exercise training in the skeletal muscle of patients with chronic heart failure. J Am Coll Cardiol 42: 861–868, 2003. [DOI] [PubMed] [Google Scholar]
- 37.Gielen S, Sandri M, Kozarez I, Kratzsch J, Teupser D, Thiery J, Erbs S, Mangner N, Lenk K, Hambrecht R, Schuler G, Adams V. Exercise training attenuates MuRF-1 expression in the skeletal muscle of patients with chronic heart failure independent of age: the randomized Leipzig Exercise Intervention in Chronic Heart Failure and Aging catabolism study. Circulation 125: 2716–2727, 2012. [DOI] [PubMed] [Google Scholar]
- 38.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB, American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 129: e28–e292, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Godard MP, Whitman SA, Song YH, Delafontaine P. Skeletal muscle molecular alterations precede whole-muscle dysfunction in NYHA Class II heart failure patients. Clin Interv Aging 7: 489–497, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gomes-Santos IL. Circulating Ang II modulates the effects of exercise training on skeletal myopathy in heart failure (874.8). FASEB 28: 874.8, 2014. [Google Scholar]
- 41.Gomes-Santos IL, Fernandes T, Couto GK, Ferreira-Filho JC, Salemi VM, Fernandes FB, Casarini DE, Brum PC, Rossoni LV, de Oliveira EM, Negrao CE. Effects of exercise training on circulating and skeletal muscle renin-angiotensin system in chronic heart failure rats. PLoS One 9: e98012, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS. NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 289: 2363–2366, 2000. [DOI] [PubMed] [Google Scholar]
- 43.Haack KK, Zucker IH. Central mechanisms for exercise training-induced reduction in sympatho-excitation in chronic heart failure. Auton Neurosci 188C: 44–50, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hambrecht R, Fiehn E, Weigl C, Gielen S, Hamann C, Kaiser R, Yu J, Adams V, Niebauer J, Schuler G. Regular physical exercise corrects endothelial dysfunction and improves exercise capacity in patients with chronic heart failure. Circulation 98: 2709–2715, 1998. [DOI] [PubMed] [Google Scholar]
- 45.Hambrecht R, Fiehn E, Yu J, Niebauer J, Weigl C, Hilbrich L, Adams V, Riede U, Schuler G. Effects of endurance training on mitochondrial ultrastructure and fiber type distribution in skeletal muscle of patients with stable chronic heart failure. J Am Coll Cardiol 29: 1067–1073, 1997. [DOI] [PubMed] [Google Scholar]
- 46.Harrington D, Anker SD, Chua TP, Webb-Peploe KM, Ponikowski PP, Poole-Wilson PA, Coats AJ. Skeletal muscle function and its relation to exercise tolerance in chronic heart failure. J Am Coll Cardiol 30: 1758–1764, 1997. [DOI] [PubMed] [Google Scholar]
- 47.Höllriegel R, Beck EB, Linke A, Adams V, Möbius-Winkler S, Mangner N, Sandri M, Gielen S, Gutberlet M, Hambrecht R, Schuler G, Erbs S. Anabolic effects of exercise training in patients with advanced chronic heart failure (NYHA IIIb): impact on ubiquitin-protein ligases expression and skeletal muscle size. Int J Cardiol 167: 975–980, 2013. [DOI] [PubMed] [Google Scholar]
- 48.Inoue N, Kinugawa S, Suga T, Yokota T, Hirabayashi K, Kuroda S, Okita K, Tsutsui H. Angiotensin II-induced reduction in exercise capacity is associated with increased oxidative stress in skeletal muscle. Am J Physiol Heart Circ Physiol 302: H1202–H1210, 2012. [DOI] [PubMed] [Google Scholar]
- 49.Ismail H, McFarlane JR, Dieberg G, Smart NA. Exercise training program characteristics and magnitude of change in functional capacity of heart failure patients. Int J Cardiol 171: 62–65, 2014. [DOI] [PubMed] [Google Scholar]
- 50.Kar S, Gao L, Zucker IH. Exercise training normalizes ACE and ACE2 in the brain of rabbits with pacing-induced heart failure. J Appl Physiol (1985) 108: 923–932, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kato M, Kinugawa T, Omodani H, Osaki S, Ahmmed GU, Ogino K, Hisatome I, Miyakoda H, Thames MD. Responses of plasma norepinephrine and renin-angiotensin-aldosterone system to dynamic exercise in patients with congestive heart failure. J Card Fail 2: 103–110, 1996. [DOI] [PubMed] [Google Scholar]
- 52.Kemi OJ, Høydal MA, Haram PM, Garnier A, Fortin D, Ventura-Clapier R, Ellingsen O. Exercise training restores aerobic capacity and energy transfer systems in heart failure treated with losartan. Cardiovasc Res 76: 91–99, 2007. [DOI] [PubMed] [Google Scholar]
- 53.Kemp GJ, Thompson CH, Stratton JR, Brunotte F, Conway M, Adamopoulos S, Arnolda L, Radda GK, Rajagopalan B. Abnormalities in exercising skeletal muscle in congestive heart failure can be explained in terms of decreased mitochondrial ATP synthesis, reduced metabolic efficiency, and increased glycogenolysis. Heart 76: 35–41, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koba S, Hisatome I, Watanabe T. Central command dysfunction in rats with heart failure is mediated by brain oxidative stress and normalized by exercise training. J Physiol 592: 3917–3931, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leimbach WN Jr, Wallin BG, Victor RG, Aylward PE, Sundlof G, Mark AL. Direct evidence from intraneural recordings for increased central sympathetic outflow in patients with heart failure. Circulation 73: 913–919, 1986. [DOI] [PubMed] [Google Scholar]
- 56.Li YL, Ding Y, Agnew C, Schultz HD. Exercise training improves peripheral chemoreflex function in heart failure rabbits. J Appl Physiol 105: 782–790, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li YP, Lecker SH, Chen Y, Waddell ID, Goldberg AL, Reid MB. TNF-alpha increases ubiquitin-conjugating activity in skeletal muscle by up-regulating UbcH2/E220k. FASEB J 17: 1048–1057, 2003. [DOI] [PubMed] [Google Scholar]
- 58.Linke A, Schoene N, Gielen S, Hofer J, Erbs S, Schuler G, Hambrecht R. Endothelial dysfunction in patients with chronic heart failure: systemic effects of lower-limb exercise training. J Am Coll Cardiol 37: 392–397, 2001. [DOI] [PubMed] [Google Scholar]
- 59.Mancini DM, Coyle E, Coggan A, Beltz J, Ferraro N, Montain S, Wilson JR. Contribution of intrinsic skeletal muscle changes to 31P NMR skeletal muscle metabolic abnormalities in patients with chronic heart failure. Circulation 80: 1338–1346, 1989. [DOI] [PubMed] [Google Scholar]
- 60.Mancini DM, Walter G, Reichek N, Lenkinski R, McCully KK, Mullen JL, Wilson JR. Contribution of skeletal muscle atrophy to exercise intolerance and altered muscle metabolism in heart failure. Circulation 85: 1364–1373, 1992. [DOI] [PubMed] [Google Scholar]
- 61.Mark AL. Sympathetic dysregulation in heart failure: mechanisms and therapy. Clin Cardiol 18: I3–I8, 1995. [DOI] [PubMed] [Google Scholar]
- 62.Martinez DG, Nicolau JC, Lage RL, Toschi-Dias E, de Matos LD, Alves MJ, Trombetta IC, Dias da Silva VJ, Middlekauff HR, Negrão CE, Rondon MU. Effects of long-term exercise training on autonomic control in myocardial infarction patients. Hypertension 58: 1049–1056, 2011. [DOI] [PubMed] [Google Scholar]
- 63.Metra M, Dei Cas L, Panina G, Visioli O. Exercise hyperventilation chronic congestive heart failure, and its relation to functional capacity and hemodynamics. Am J Cardiol 70: 622–628, 1992. [DOI] [PubMed] [Google Scholar]
- 64.Mettauer B, Zoll J, Sanchez H, Lampert E, Ribera F, Veksler V, Bigard X, Mateo P, Epailly E, Lonsdorfer J, Ventura-Clapier R. Oxidative capacity of skeletal muscle in heart failure patients versus sedentary or active control subjects. J Am Coll Cardiol 38: 947–954, 2001. [DOI] [PubMed] [Google Scholar]
- 65.Mezzani A, Cacciatore F, Catanzaro R, Gualco A, Guzzetti D, Leosco D, Monelli M, Tarro Genta F, Totaro P, Traversi E, Zanelli E, Giannuzzi P. EaRly-start ExerciSe training afTer acute hemodynAmic decompensation in patients with chRonic hearT failure (RE-START). A multicenter, randomized, controlled trial on short-term feasibility and impact on functional capacity, symptoms and neurohumoral activation. Monaldi Arch Chest Dis 82: 20–22, 2014. [DOI] [PubMed] [Google Scholar]
- 66.Middlekauff HR. Making the case for skeletal myopathy as the major limitation of exercise capacity in heart failure. Circ Heart Fail 3: 537–546, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Middlekauff HR, Chiu J. Cyclooxygenase products sensitize muscle mechanoreceptors in healthy humans. Am J Physiol Heart Circ Physiol 287: H1944–H1949, 2004. [DOI] [PubMed] [Google Scholar]
- 68.Middlekauff HR, Nitzsche EU, Hoh CK, Hamilton MA, Fonarow GC, Hage A, Moriguchi JD. Exaggerated muscle mechanoreflex control of reflex renal vasoconstriction in heart failure. J Appl Physiol 90: 1714–1719, 2001. [DOI] [PubMed] [Google Scholar]
- 69.Middlekauff HR, Nitzsche EU, Hoh CK, Hamilton MA, Fonarow GC, Hage A, Moriguchi JD. Exaggerated renal vasoconstriction during exercise in heart failure patients. Circulation 101: 784–789, 2000. [DOI] [PubMed] [Google Scholar]
- 70.Middlekauff HR, Verity MA, Horwich TB, Fonarow GC, Hamilton MA, Shieh P. Intact skeletal muscle mitochondrial enzyme activity but diminished exercise capacity in advanced heart failure patients on optimal medical and device therapy. Clin Res Cardiol 102: 547–554, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Middlekauff HR, Vigna C, Verity MA, Fonarow GC, Horwich TB, Hamilton MA, Shieh P, Tupling AR. Abnormalities of calcium handling proteins in skeletal muscle mirror those of the heart in humans with heart failure: a shared mechanism? J Card Fail 18: 724–733, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moreira JB, Bechara LR, Bozi LH, Jannig PR, Monteiro AW, Dourado PM, Wisløff U, Brum PC. High- versus moderate-intensity aerobic exercise training effects on skeletal muscle of infarcted rats. J Appl Physiol (1985) 114: 1029–1041, 2013. [DOI] [PubMed] [Google Scholar]
- 73.Mousa TM, Liu D, Cornish KG, Zucker IH. Exercise training enhances baroreflex sensitivity by an angiotensin II-dependent mechanism in chronic heart failure. J Appl Physiol (1985) 104: 616–624, 2008. [DOI] [PubMed] [Google Scholar]
- 74.Nakamura M, Ishikawa M, Funakoshi T, Hashimoto K, Chiba M, Hiramori K. Attenuated endothelium-dependent peripheral vasodilation and clinical characteristics in patients with chronic heart failure. Am Heart J 128: 1164–1169, 1994. [DOI] [PubMed] [Google Scholar]
- 75.Nazaré Nunes Alves MJ, Alves MJ, dos Santos MR, Nobre TS, Martinez DG, Pereira Barretto AC, Brum PC, Rondon MU, Middlekauff HR, Negrão CE. Mechanisms of blunted muscle vasodilation during peripheral chemoreceptor stimulation in heart failure patients. Hypertension 60: 669–676, 2012. [DOI] [PubMed] [Google Scholar]
- 76.Negrão CE, Hamilton MA, Fonarow GC, Hage A, Moriguchi JD, Middlekauff HR. Impaired endothelium-mediated vasodilation is not the principal cause of vasoconstriction in heart failure. Am J Physiol Heart Circ Physiol 278: H168–H174, 2000. [DOI] [PubMed] [Google Scholar]
- 77.Negrão CE, Middlekauff HR. Adaptations in autonomic function during exercise training in heart failure. Heart Fail Rev 13: 51–60, 2008. [DOI] [PubMed] [Google Scholar]
- 78.Negrão CE, Rondon MU, Tinucci T, Alves MJ, Roveda F, Braga AM, Reis SF, Nastari L, Barretto AC, Krieger EM, Middlekauff HR. Abnormal neurovascular control during exercise is linked to heart failure severity. Am J Physiol Heart Circ Physiol 280: H1286–H1292, 2001. [DOI] [PubMed] [Google Scholar]
- 79.Negrão CE, Santos AC, Rondon MU, Franco FG, Ianni B, Rochitte CE, Braga AM, Oliveira MT, Mady C, Barretto AC, Middlekauff HR. Muscle sympathetic nerve activity in patients with Chagas' disease. Int J Cardiol 137: 252–259, 2009. [DOI] [PubMed] [Google Scholar]
- 80.Okita K, Yonezawa K, Nishijima H, Hanada A, Ohtsubo M, Kohya T, Murakami T, Kitabatake A. Skeletal muscle metabolism limits exercise capacity in patients with chronic heart failure. Circulation 98: 1886–1891, 1998. [DOI] [PubMed] [Google Scholar]
- 81.Olson TP, Joyner MJ, Eisenach JH, Curry TB, Johnson BD. Influence of locomotor muscle afferent inhibition on the ventilatory response to exercise in heart failure. Exp Physiol 99: 414–426, 2014. [DOI] [PubMed] [Google Scholar]
- 82.Park J, Middlekauff HR. Abnormal neurocirculatory control during exercise in humans with chronic renal failure. Auton Neurosci 188: 74–81, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pina IL, Apstein CS, Balady GJ, Belardinelli R, Chaitman BR, Duscha BD, Fletcher BJ, Fleg JL, Myers JN, Sullivan MJ. Exercise and heart failure: a statement from the American Heart Association Committee on exercise, rehabilitation, and prevention. Circulation 107: 1210–1225, 2003. [DOI] [PubMed] [Google Scholar]
- 84.Poole DC, Hirai DM, Copp SW, Musch TI. Muscle oxygen transport and utilization in heart failure: implications for exercise (in)tolerance. Am J Physiol Heart Circ Physiol 302: H1050–H1063, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rehn TA, Munkvik M, Lunde PK, Sjaastad I, Sejersted OM. Intrinsic skeletal muscle alterations in chronic heart failure patients: a disease-specific myopathy or a result of deconditioning? Heart Fail Rev 17: 421–436, 2012. [DOI] [PubMed] [Google Scholar]
- 86.Roig E, Perez-Villa F, Morales M, Jiménez W, Orús J, Heras M, Sanz G. Clinical implications of increased plasma angiotensin II despite ACE inhibitor therapy in patients with congestive heart failure. Eur Heart J 21: 53–57, 2000. [DOI] [PubMed] [Google Scholar]
- 87.Rondon E, Brasileiro-Santos MS, Moreira ED, Rondon MU, Mattos KC, Coelho MA, Silva GJ, Brum PC, Fiorino P, Irigoyen MC, Krieger EM, Middlekauff HR, Negrão CE. Exercise training improves aortic depressor nerve sensitivity in rats with ischemia-induced heart failure. Am J Physiol Heart Circ Physiol 291: H2801–H2806, 2006. [DOI] [PubMed] [Google Scholar]
- 88.Roveda F, Middlekauff HR, Rondon MU, Reis SF, Souza M, Nastari L, Barretto AC, Krieger EM, Negrao CE. The effects of exercise training on sympathetic neural activation in advanced heart failure: a randomized controlled trial. J Am Coll Cardiol 42: 854–860, 2003. [DOI] [PubMed] [Google Scholar]
- 89.Russell ST, Wyke SM, Tisdale MJ. Mechanism of induction of muscle protein degradation by angiotensin II. Cell Signal 18: 1087–1096, 2006. [DOI] [PubMed] [Google Scholar]
- 90.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117: 399–412, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Santoro C, Cosmas A, Forman D, Morghan A, Bairos L, Levesque S, Roubenoff R, Hennessey J, Lamont L, Manfredi T. Exercise training alters skeletal muscle mitochondrial morphometry in heart failure patients. J Cardiovasc Risk 9: 377–381, 2002. [DOI] [PubMed] [Google Scholar]
- 92.Santos AC, Alves MJ, Rondon MU, Barretto AC, Middlekauff HR, Negrão CE. Sympathetic activation restrains endothelium-mediated muscle vasodilatation in heart failure patients. Am J Physiol Heart Circ Physiol 289: H593–H599, 2005. [DOI] [PubMed] [Google Scholar]
- 93.Schaufelberger M, Eriksson BO, Grimby G, Held P, Swedberg K. Skeletal muscle fiber composition and capillarization in patients with chronic heart failure: relation to exercise capacity and central hemodynamics. J Card Fail 1: 267–272, 1995. [DOI] [PubMed] [Google Scholar]
- 94.Schaufelberger M, Eriksson BO, Held P, Swedberg K. Skeletal muscle metabolism during exercise in patients with chronic heart failure. Heart 76: 29–34, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schultz HD, Li YL. Carotid body function in heart failure. Respir Physiol Neurobiol 157: 171–185, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Semprun-Prieto LC, Sukhanov S, Yoshida T, Rezk BM, Gonzalez-Villalobos RA, Vaughn C, Michael Tabony A, Delafontaine P. Angiotensin II induced catabolic effect and muscle atrophy are redox dependent. Biochem Biophys Res Commun 409: 217–221, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sigurdsson A, Held P, Swedberg K. Short- and long-term neurohormonal activation following acute myocardial infarction. Am Heart J 126: 1068–1076, 1993. [DOI] [PubMed] [Google Scholar]
- 98.Smith SA, Williams MA, Mitchell JH, Mammen PP, Garry MG. The capsaicin-sensitive afferent neuron in skeletal muscle is abnormal in heart failure. Circulation 111: 2056–2065, 2005. [DOI] [PubMed] [Google Scholar]
- 99.Soares-Miranda L, Franco FG, Roveda F, Martinez DG, Rondon MU, Mota J, Brum PC, Antunes-Correa LM, Nobre TS, Barretto AC, Middlekauff HR, Negrao CE. Effects of exercise training on neurovascular responses during handgrip exercise in heart failure patients. Int J Cardiol 146: 122–125, 2011. [DOI] [PubMed] [Google Scholar]
- 100.Sterns DA, Ettinger SM, Gray KS, Whisler SK, Mosher TJ, Smith MB, Sinoway LI. Skeletal muscle metaboreceptor exercise responses are attenuated in heart failure. Circulation 84: 2034–2039, 1991. [DOI] [PubMed] [Google Scholar]
- 101.Sullivan MJ, Green HJ, Cobb FR. Altered skeletal muscle metabolic response to exercise in chronic heart failure. Relation to skeletal muscle aerobic enzyme activity. Circulation 84: 1597–1607, 1991. [DOI] [PubMed] [Google Scholar]
- 102.Sun SY, Wang W, Zucker IH, Schultz HD. Enhanced activity of carotid body chemoreceptors in rabbits with heart failure: role of nitric oxide. J Appl Physiol 86: 1273–1282, 1999. [DOI] [PubMed] [Google Scholar]
- 103.Sun SY, Wang W, Zucker IH, Schultz HD. Enhanced peripheral chemoreflex function in conscious rabbits with pacing-induced heart failure. J Appl Physiol (1985) 86: 1264–1272, 1999. [DOI] [PubMed] [Google Scholar]
- 104.Tabony AM, Yoshida T, Galvez S, Higashi Y, Sukhanov S, Chandrasekar B, Mitch WE, Delafontaine P. Angiotensin II upregulates protein phosphatase 2Cα and inhibits AMP-activated protein kinase signaling and energy balance leading to skeletal muscle wasting. Hypertension 58: 643–649, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Toth MJ, Ades PA, Tischler MD, Tracy RP, LeWinter MM. Immune activation is associated with reduced skeletal muscle mass and physical function in chronic heart failure. Int J Cardiol 109: 179–187, 2006. [DOI] [PubMed] [Google Scholar]
- 106.Ueno LM, Drager LF, Rodrigues AC, Rondon MU, Braga AM, Mathias W, Krieger EM, Barretto AC, Middlekauff HR, Lorenzi-Filho G, Negrão CE. Effects of exercise training in patients with chronic heart failure and sleep apnea. Sleep 32: 637–647, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang HJ, Li YL, Gao L, Zucker IH, Wang W. Alteration in skeletal muscle afferents in rats with chronic heart failure. J Physiol 588: 5033–5047, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang HJ, Li YL, Zucker IH, Wang W. Exercise training prevents skeletal muscle afferent sensitization in rats with chronic heart failure. Am J Physiol Regul Integr Comp Physiol 302: R1260–R1270, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Williams MA, Smith SA, O'Brien DE, Mitchell JH, Garry MG. The group IV afferent neuron expresses multiple receptor alterations in cardiomyopathyic rats: evidence at the cannabinoid CB1 receptor. J Physiol 586: 835–845, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wisløff U, Støylen A, Loennechen JP, Bruvold M, Rognmo Ø, Haram PM, Tjønna AE, Helgerud J, Slørdahl SA, Lee SJ, Videm V, Bye A, Smith GL, Najjar SM, Ellingsen Ø, Skjaerpe T. Superior cardiovascular effect of aerobic interval training versus moderate continuous training in heart failure patients: a randomized study. Circulation 115: 3086–3094, 2007. [DOI] [PubMed] [Google Scholar]
- 111.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL, American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 62: e240–e327, 2013. [DOI] [PubMed] [Google Scholar]
- 112.Yoshida T, Semprun-Prieto L, Sukhanov S, Delafontaine P. IGF-1 prevents ANG II-induced skeletal muscle atrophy via Akt- and Foxo-dependent inhibition of the ubiquitin ligase atrogin-1 expression. Am J Physiol Heart Circ Physiol 298: H1565–H1570, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yoshida T, Tabony AM, Galvez S, Mitch WE, Higashi Y, Sukhanov S, Delafontaine P. Molecular mechanisms and signaling pathways of angiotensin II-induced muscle wasting: potential therapeutic targets for cardiac cachexia. Int J Biochem Cell Biol 45: 2322–2332, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zheng H, Li YF, Cornish KG, Zucker IH, Patel KP. Exercise training improves endogenous nitric oxide mechanisms within the paraventricular nucleus in rats with heart failure. Am J Physiol Heart Circ Physiol 288: H2332–H2341, 2005. [DOI] [PubMed] [Google Scholar]
- 115.Zucker IH, Patel KP, Schultz HD. Neurohumoral stimulation. Heart Fail Clin 8: 87–99, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]