

Abstract
Abdominal aortic aneurysm (AAA) is a common degenerative vascular disease whose pathogenesis is associated with activation of multiple signaling pathways including Jun NH2-terminal kinases (JNK) and NF-κB. It is now well recognized that these pathways are chaperoned by the heat shock protein 90 (Hsp90), suggesting that inhibition of Hsp90 may be a novel strategy for inhibiting AAAs. The aim of this study is to investigate whether inhibition of Hsp90 by 17-DMAG (17-dimethyl-aminothylamino-17-demethoxy-geldanamycin) attenuates ANG II-induced AAA formation in mice, and, if so, to elucidate the mechanisms involved. Apolipoprotein E-null mice were infused with ANG II to induce AAA formation and simultaneously treated by intraperitoneal injection with either vehicle or 17-DMAG for 4 wk. ANG II infusion induced AAA formation in 80% of mice, which was accompanied by increased matrix metalloproteinase (MMP) activity, enhanced tissue inflammation, oxidative stress, and neovascularization. Importantly, these effects were inhibited by 17-DMAG treatment. Mechanistically, we showed that 17-DMAG prevented the formation and progression of AAA through its inhibitory effects on diverse biological pathways including 1) by blocking ANG II-induced phosphorylation of ERK1/2 and JNK that are critically involved in the regulation of MMPs in vascular smooth muscle cells, 2) by inhibiting IκB kinase expression and expression of MCP-1, and 3) by attenuating ANG II-stimulated angiogenic processes critical to AAA formation. Our results demonstrate that inhibition of Hsp90 by 17-DMAG effectively attenuates ANG II-induced AAA formation by simultaneously inhibiting vascular inflammation, extracellular matrix degradation, and angiogenesis, which are critical in the formation and progression of AAAs.
Keywords: aneurysms, angiotensin II, apolipoprotein E-null mouse, heat shock protein 90
abdominal aortic aneurysms (AAAs) occur in ∼9% of older men and account for more than 15,000 deaths annually in the United States (9, 51). Pathological processes of AAA are complex, but mainly characterized by significant degradation of extracellular matrix including elastin and collagen, increased activity of matrix metalloproteinases (MMPs), excessive local inflammation, and neovascularization of the media and adventitia (7, 16, 32). Currently there is no effective pharmacological therapy available to prevent the development and progression of AAA, and surgical repair of late-stage disease remains the only effective method of reducing aneurysm-related mortality. Therefore, identification of novel molecular targets is of considerable scientific and therapeutic interest (32).
Infusion of ANG II with subcutaneous osmotic minipumps in apolipoprotein E-null (apoE−/−) mice is a widely used mouse model of AAA, which exhibits multiple characteristics of human AAAs, including disruption of media, secretion of MMPs, rupture of elastic layer, macrophage infiltration, and neovascularization (2, 10). Moreover, ANG II has been shown to activate multiple signaling pathways, such as c-Jun NH2-terminal kinase (JNK) and NF-κB pathways in vascular smooth muscle cells (VSMCs), which are critically involved in AAA formation (11, 38). Accordingly, targeted inhibition of JNK pathway, by either genetic or pharmacological approaches, has been shown to prevent experimental AAA formation (53). Similarly, inhibition of NF-κB signaling pathway has been reported to inhibit macrophage infiltration and inflammation in the adventitia and media of AAA model (33). Nevertheless, these studies highlight the critical importance of both JNK and NF-κB pathways in the development of AAA.
Heat shock protein 90 (Hsp90) is an evolutionarily conserved and abundant molecular chaperone that participates in stabilizing and activating more than 200 proteins including kinases, signaling molecules, and transcriptional factors, such as v-Src, Raf-1, ErbB2, HIF-1, HSF1, and some growth factor receptors (41, 45). Because many Hsp90 client proteins are classified as oncogenic proteins, which promote cancer cell growth or survival or both (37), a number of highly potent and pharmaceutically improved Hsp90 inhibitors have been developed and entered into clinical trials for cancer treatment (45). 17-Dimethyl-aminothylamino-17-demethoxy-geldanamycin (17-DMAG) is a selective Hsp90 inhibitor, which blocks the ATP binding site of Hsp90 to induce the degradation of some client proteins (54). Recently, inhibition of Hsp90 by 17-DMAG has been shown to attenuate inflammatory responses and oxidative stress in experimental atherosclerosis, indicating that Hsp90 inhibitors may have potentials for treatment of certain cardiovascular diseases (27, 28). Because many important signaling proteins, including extracellular signal-regulated kinase (ERK), JNK, and NF-κB, which are critically involved in formation and progression of AAA, are the Hsp90 client proteins (8, 18), we speculated that inhibition of Hsp90 could be an effective therapeutic strategy for the treatment of AAA. Thus, in the present study, we investigated whether inhibition of Hsp90 by 17-DMAG attenuates AAA formation in mice, and if so, to determine the mechanism(s) involved.
MATERIALS AND METHODS
Mice.
Male, apoE−/− mice at 10 wk of age on a C57BL/6 background were obtained from the Model Animal Research Center of Nanjing University, China and bred in pathogen-free environment. The research conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996), and the protocol was approved by the Institutional Animal Care Committee at Shanghai Jiaotong University School of Medicine. Mice were euthanized by using a gradually filling the chamber with CO2.
Drug treatment.
Osmotic pumps (model 2004; Alzet) containing either ANG II (1,000 ng·min−1·kg−1; Sigma-Aldrich; n = 40) or saline (n = 10) were subcutaneously implanted into 10-wk-old male apoE−/− mice as described previously (15, 40). ANG II-treated mice were intraperitoneally injected with 5 mg/kg of 17-DMAG (Lclab), or vehicle every other day (3 times per wk) during 4 wk. This treatment regime is based on the previous notion indicating that low-dose 17-DMAG therapy (5 mg/kg ip, 3 times per wk) could efficiently inhibit Hsp90 activity without obvious toxic effects in mice (23, 43). The inhibitory effect of 17-DMAG on Hsp90 was determined by the induction of Hsp70 expression (42).
Blood pressure measurements.
Blood pressure was measured in conscious mice by a tail-cuff system (Bp98A, softron, China).
Analysis and quantification of AAA.
After perfusion with 4% paraformaldehyde, the abdominal aortas were harvested and immediately placed in PBS and cleaned of adventitial fat. The outer diameter of the suprarenal aorta was measured with a caliper under a dissecting microscope while the aortas were in PBS without physical stretching. To quantify aneurysm incidence, an aneurysm was defined as >50% increase in external diameter of suprarenal aorta compared with aortas from saline-infused mice, which is consistent with the clinical standard to diagnose abdominal aortic aneurysm (47). AAA severity was determined with a classification scheme described previously (14), where Type 1 represents a simple dilation of the abdominal aorta with an external diameter of 1.5–2 mm, Type 2 represents a AAA with the external diameter of 2 to 3 mm, and Type 3 represents a pronounced bulbous containing a thrombus and an external aortic diameter of >3 mm. Mice in the Type 4 AAA category were those that died of aneurysmal rupture and resultant bleeding in the peritoneal cavity. AAA severity was also evaluated by measuring the wet weights of the abdominal aortas.
Histology and immunohistochemistry.
Anesthetized mice were perfused with normal saline and fixed with 10% PBS and formalin for 5 min. Whole aortas were harvested, fixed for 24 h, and embedded in paraffin, and cross-sections (5 μm) were prepared. Paraffin sections were stained with hematoxylin and eosin and Vehoeff-van Geisen for elastin, MAC3 for macrophages, and CD31 for endothelial cells. Antibody binding was detected using the Vectastain Elite ABC kit and di-amino benzidine (DAB) staining using manufacturer's instructions (Vector, Burlingame, CA). Quantitation of immuno-positive cells was performed by determining the ratio of the number of positive cells to the total number of hematoxylin-positive cells in a defined field on more than 10 slides per mouse.
Cell culture.
Mouse VSMCs were cultured in DMEM supplemented with 10% FBS, 100 U/l penicillin, and 100 μg/ml streptomycin. Human umbilical cord vein endothelial cells (HUVECs) were isolated and cultured as previously described (13). In all experiments, the cells were used between passages 3 and 8.
Measurement of MMPs activity and MCP-1 secretion.
The evaluation of MMP-2 and MMP-9 activities in conditioned media form cells cultures or homogenates of aortic tissue was performed by zymography as described previously (50). MCP-1 secretion was measured by ELISA (Pierce, Rockford, IL).
Quantitation of mRNA expression.
Total RNA was extracted from cells or aortic tissue using TRIzol (Invitrogen). Total RNA (1 μg) was used to perform the reverse transcription with High Capacity cDNA Archive Kit (Applied Biosystem). Real-time quantitative PCR analysis for MCP-1, MMP-2, and MMP-9 was performed using TaqMan gene expression assays and the ΔΔCt method with housekeeping gene 18S as the endogenous control. The primers used for the quantitative RT-PCR are listed in Table 1.
Table 1.
Primers for PCR
| Mouse MMP-2 promoter | 5′-AGCTAGTGGCTGCCATATGGAAACTGG-3′ |
| 5′-GGTACCTGGTGGGAGCAGAACACACAT-3′ | |
| Mouse MMP-9 promoter | 5′-GTA GTG TAA ACA CAC ACA CACA-3′ and 5′-AGT AAA ACG GAA TCA GTG ACC C-3′ for the distal AP-1 site; |
| 5′-CCC CAC ACT GTA GGT TCT ATC C-3 and 5-ATC CTG CCT CAA AGA GCCT-3′ for the proximal AP-1 site | |
| Mouse MMP-2 | 5′-ACCAAGAACTTCCGATTATCCC-3′ |
| 5′-CAGTACCAGTGTCAGTATCAGC-3′ | |
| Mouse MMP-9 | 5′-GATCCCCAGAGCGTCATTC-3′ |
| 5′-CCACCTTGTTCACCTCATTTTG-3′ | |
| 18S rRNA | 5′-TCAAGAACGAAAGTCGGAGG-3′ |
| 5′-GGACATCTAAGGGCATCAC-3′ | |
| Mouse MCP-1 | 5′-GTCCCTGTCATGCTTCTGG-3′ |
| 5′-GCTCTCCAGCCTACTCATTG-3′ | |
| Human VEGF | 5′-ATCATGCGGATCAAACCTCACC-3′ |
| 5′-GGTCTGCATTCACATCTGCTATGC-3′ |
Measurement of malondialdehyde.
The protein concentration was measured by using the BCA protein assay kit (Pierce). Malondialdehyde (MDA) levels were measured by an ELISA kit (Mybiosource, San Diego, CA), which applies the competitive enzyme immunoassay technique using a monoclonal anti-MDA antibody and an MDA-HRP conjugate. Briefly, the assay sample and buffer were incubated together with MDA-HRP conjugate in precoated plate for 1 h. After the incubation period, the wells were washed five times and then incubated with a substrate for HRP enzyme. A stop solution was added, and the intensity of color was measured spectrophotometrically at 450 nm in a microplate reader. The intensity of the color is inversely proportional to the MDA concentration since MDA from samples and MDA-HRP conjugate compete for the anti-MDA antibody binding site.
Western blot.
Total proteins were extracted from corresponding area of aneurysm tissue or cells. Equal amounts of protein lysates were separated by 10% SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membrane, and then immunoblotted with antibodies against ERK1/2 (1:1,000 dilution; Cell Signaling Technology), phosphor-ERK1/2 (1:1,000 dilution; Cell Signaling Technology), JNK (1:1,000 dilution; Cell Signaling Technology), and phosphor-JNK (1:500 dilution; Cell Signaling Technology). Protein bands were visualized by Odyssey imagining system (LICOR, Lincoln, NE).
Electrophoretic mobility shift assay.
Electrophoretic mobility shift assay (EMSA) were performed with Odyssey IRDye 700 infrared dye labeled double-stranded oligonucleotides coupled with the EMSA buffer kit (LI-COR Bioscience) according to manufacturer's instructions.
Chromatin immunoprecipitation assay for AP-1 and MMPs promoters binding.
Chromatin immunoprecipitation (ChIP) assays of VSMCS were performed with a chip kit (Millipore, Billerica, MA) according to manufacturer's instructions. Briefly, chromatin was cross-linked by adding formaldehyde to cell culture medium for 10 min at room temperature. Crosslinking reaction was ceased by glycine solution. Cross-linked chromatin was sheared by supersonic at 4°C. Debris was removed by centrifugation, and supernatants were collected. The purified chromatin was immunoprecipitated with c-Jun and c-Fos antibodies from Santa Cruz Biotechnology and normal rabbit IgG from Millipore. The DNA/Protein complexes were collected by the use of protein G-argrose beads. Beads were washed and bound DNA was eluted. After reverse cross-linking reaction and proteinase K digestion, the eluted DNA was used in 35 cycles of PCR amplification with the AP-1 binding site specific primers, which are listed in Table 1.
Statistical analysis.
Quantitative data are presented as means ± SE. Parameters between two groups were compared by two-tailed Student's t-test. Comparisons of parameters among multiple groups were made by one-way ANOVA, and comparisons of different parameters between each group were made by Bonferroni's post hoc test. Exact χ2 test was used to assess the statistical significance of aneurysm subtype variables. A χ2 test was used to compare AAA incidence and survival rate. A P value <0.05 was considered to be statistically significant.
RESULTS
Hsp90 inhibitor 17-DMAG inhibits the incidence and severity of ANG II-induced AAAs.
To elucidate whether Hsp90 participates in the development of AAA, we induced the AAA formation by the infusion of ANG II to apoE−/− mice for 4 wk and the expression of Hsp90 was determined by Western blot. As shown in Fig. 1A, Hsp90 expression was significantly increased in the mouse AAA lesions. This observation prompted us to investigate whether inhibition of Hsp90 could attenuate the AAA formation in mice. To this end, we induced the AAA by the infusion of ANG II to apoE−/− mice and treated the mice simultaneously with intraperitoneal injection of either vehicle or 17-DMAG (5 mg/kg, every 2 days) for 4 wk. The dose of the drug was chosen based on studies showing effective treatment and minimal toxicity at dosages under 15 mg/kg administered 3 days per wk for 6 wk (23) (43). Hsp90 inhibitors have been shown to disassociate the interaction of Hsp90 with HSF1, which in turn induces the expression of Hsp70 (6). Therefore, assessment of HSP70 induction has been used as a useful pharmacodynamic marker of HSP90 inhibition. Indeed, treatment of mice with 5 mg/kg 17-DMAG markedly increased Hsp70 expression in abdominal aorta by approximately twofold (Fig. 1B), but barely affected the body weight of the mice (Fig. 1C), suggesting that the current treatment regimen is indeed effective for inhibiting Hsp90 in abdominal aorta. Furthermore, ANG II infusion for 4 wk induced an 80% incidence (16 of 20 mice) of AAA in apoE−/− mice, whereas the incidence was significantly reduced to 10% (2 of 20 mice) in 17-DMAG-treated group (Fig. 2, A and B). 17-DMAG also significantly decreased the severity of ANG II-induced AAAs, as determined by a classification scheme based on the external diameter of the abdominal aortas (Fig. 2C) as well as by measuring the wet weights of the abdominal aortas (Fig. 2D).
Fig. 1.
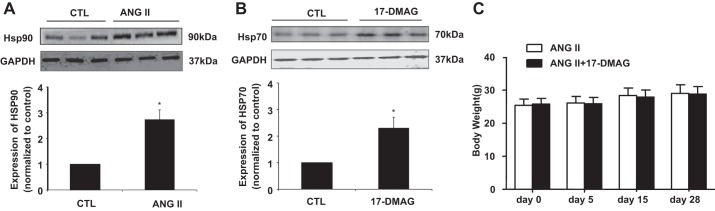
Expression of heat shock protein 90 (Hsp90) and heat shock protein 70 (Hsp70) in mouse abdominal aorta. A: saline or ANG II (1,000 ng·min−1·kg−1) was administrated to mice for 4 wk; the expression of Hsp90 was determined in mouse abdominal aorta by Western blot. *P < 0.05 compared with control group; n = 3 in each group. B: apoE−/− mice received either vehicle or 17-dimethyl-aminothylamino-17-demethoxy-geldanamycin (17-DMAG; 5 mg/kg ip). Fourteen hours after injection of 17-DMAG, expression of Hsp70 in abdominal aorta was determined by Western blot. Hsp70 expression was then quantitated by densitometric analysis. *P < 0.05 compared with control (CTL) group; n = 3 in each group. C: saline or ANG II (1,000 ng·min−1·kg−1) was administrated to mice for 4 wk with or without 17-DMAG administration (5 mg/kg per 2 days, 3 times per wk). The body weight was then measured.
Fig. 2.

17-DMAG reduces the incidence and severity of ANG II-induced abdominal aortic aneurysm (AAA) in apoE−/− mice. Saline or ANG II (1,000 ng·min−1·kg−1) was administrated to mice for 4 wk with or without 17-DMAG administration (5 mg/kg per 2 days, 3 times per wk). The severity of AAA was determined by the aorta wet weights and maximal abdominal aortic diameter. A: representative photographs showing the macroscopic features of aneurysms from indicated groups. B: effects of 17-DMAG on the incidence of ANG II-induced AAA. C: AAAs were scored from type 1 to type 4 pathology on the basis of the external diameter. Exact χ2-test indicated the significance (P < 0.01) between ANG II and ANG II + 17-DMAG groups. D: effects of 17-DMAG on abdominal aortic wet weights. *P < 0.05 compared with control group; #P < 0.05 compared with ANG II infusion group; n = 20 in each group. The P values were obtained by a χ2-test in B and 1-way ANOVA plus a post hoc analysis using a Bonferronia test in D.
17-DMAG attenuates remodeling of the aortic wall, inflammatory responses, and neovascularization in ANG II-induced AAA.
HE staining of cross sections of the abdominal aortas showed that ANG II treatment induced severe thickening and considerable destruction of the abdominal aortic walls (Fig. 3A). In addition, elastin staining demonstrated that ANG II treatment disrupted the elastin fibers in the suprarenal region of the aorta (Fig. 3, B and E) and increased infiltration of Mac-3 positive macrophages (Fig. 3, C and F) and formation of capillary vessels, as determined by CD31 immunochemical studies (Fig. 3, D and G). Remarkably, all these parameters associated with AAA formation observed in ANG II-infused mice were substantially inhibited by treatment of mice with Hsp90 inhibitor 17-DMAG (Fig. 3, A–G).
Fig. 3.

17-DMAG decreases remodeling of aortic wall, angiogenesis, and inflammatory responses in ANG II-induced AAA. Saline or ANG II (1,000 ng·min−1·kg−1) was administrated to mice for 4 wk with or without 17-DMAG administration (5 mg/kg, 3 days). The histological features of abdominal aortas from indicated groups were analyzed. Representative photomicrographs of aortic cross sections of hematoxylin and eosin (HE) staining (A), Vehoeff-van Geisen (VVG) staining (B), and macrophage staining (C) in the abdominal aortas of indicated groups are shown. D: CD31+ staining. E: quantification of elastin degradation. F: quantification of MAC3 immuno-positive cells. G: quantification of CD31 immuno-positive cells. *P < 0.05 compared with control group; #P < 0.05 compared with ANG II infusion group; n = 5 in each group.
17-DMAG attenuates MMP-2 and MMP-9 expression, MDA levels, and MCP-1 secretion in ANG II-induced AAA.
Given important roles of MMPs, reactive oxygen species (ROS), and MCP-1 in ANG II-induced AAA formation (32), we sought to investigate the effect of Hsp90 inhibition on expression of MMP-2 and MMP-9, ROS production, and MCP-1 secretion in homogenates of AAAs. As expected, MMP-2 and MMP-9 activity in homogenates of abdominal aortas were both increased significantly as compared with the abdominal aortas from the control group (Fig. 4A). Both MMP-2 and MMP-9 activities, however, were significantly lower in ANG II plus 17-DMAG-treated group as compared with ANG II-treated alone (P < 0.05) (Fig. 4A). Likewise, both MCP-1 and lipid peroxides, as determined by MDA levels in homogenates of abdominal aortas, were significantly increased by ANG II infusion as compared with the control group, and those increases were markedly attenuated by 17-DMAG treatment (Fig. 4, B and C). Furthermore, ANG II infusion markedly increased the phosphorylation of ERK1/2 and JNK MAP kinases in abdominal aortas, which was blunted by cotreatment of mice with 17-DMAG (Fig. 4D).
Fig. 4.

17-DMAG attenuates matrix metalloproteinase (MMP)-2 and MMP-9 expression, malondialdehyde (MDA) formation, and MCP-1 secretion in ANG II-induced AAA. Saline or ANG II (1,000 ng·min−1·kg−1) was administrated to mice for 28 days with or without 17-DMAG administration (5 mg/kg per 2 days, 3 times per wk). A: representative zymogram of aortic tissue homogenates from indicated group mice (top) and the densitometric quantification of gelatinolytic activity (bottom). B: effects of 17-DMAG on concentration of MCP-1 in aortic tissues assessed by ELISA. C: effects of 17-DMAG on MDA levels in aortic tissues assessed by ELISA. *P < 0.05 compared with control group; #P < 0.05 compared with ANG II infusion group; n = 5 in each group. D: effects of 17-DMAG on the activation of MAP kinases in aortic tissues assessed by Western blot. *P < 0.05 compared with control group; #P < 0.05 compared with ANG II infusion group; n = 4 in each group.
17-DMAG decreases MMP-2 and MMP-9 expression in VSMCS via inhibition of AP-1 pathway.
Because AP-1 transcription factor is essentially involved in regulating MMP-2 and MMP-9 expression in VSMCs (55), we investigated whether 17-DMAG inhibits expression of MMPs via inhibition of AP-1 transcriptional activity. Indeed, treatment of mouse VSMCs with ANG II markedly increased the activity and mRNA levels of MMP-2 and MMP-9, which were significantly inhibited by treatment of cells with 17-DMAG (Fig. 5, A and B). The concentration of the drug used in these experiments was based on the literature and our own data showing minimal toxicity after 24-h treatments of mouse VSMCs with concentrations of 17-DMAG below 1 μM (16, 38).
Fig. 5.

17-DMAG decreases MMP-2 and MMP-9 expression in vascular smooth muscle cells (VSMCs) via inhibiting AP-1 pathway. Cultured mouse VSMCs pretreated with or without 17-DMAG (400 nmol/l) for 6 h were treated with either vehicle or ANG II for 24 h. A: MMPs activity in culture medium were detected by zymography (top) and then quantitated by densitometric analysis (bottom). B: effects of 17-DMAG on the mRNA expression of MMPs were analyzed by quantitative PCR. C: cultured rat VSMCs pretreated with or without 17-DMAG (400 nmol/l) for 6 h were treated with either vehicle or ANG II for 2 h. The effects of 17-DMAG on the binding of AP-1 to MMP-2 and MMP-9 gene promoters were detected by chromatin immunoprecipitation (ChIP) analysis. *P < 0.05 compared with cells with vehicle treatment alone; #P < 0.05 compared with cells treated with ANG II alone; n = 5 independent experiments for all quantitative data. D: cultured VSMCs were incubated with ANG II (1 μmol/l) for 2 h with or without the pretreatment of 17-DMAG (400 nmol/l) for 6 h. AP-1 DNA binding activity was measured by electrophoretic mobility shift assay (EMSA).
The promoter region of MMP-9 has a proximal AP-1 binding site and MMP-2 has a noncanonical AP-1 site that is functionally critical for the augmented production of MMP-2 (3, 24, 52). We then investigated the effect of 17-DMAG on AP-1 binding to the MMP-2 and MMP-9 promoters in mouse VSMCs by ChIP assays. In mouse VSMCs, ANG II treatment markedly increased the binding of AP-1 to the MMP-2 and MMP-9 promoters, as shown in ANG II-treated cells, after immunoprecipitation with primary antibody to AP-1 (c-fos and c-Jun), but not with control immunoglobulin G (IgG). Importantly, 17-DMAG markedly attenuated the AP-1 binding to both MMP-2 and MMP-9 promoters (Fig. 5C). Moreover, we examined the effect of 17-DMAG on AP-1 activity by an electrophoretic mobility shift assay (EMSA). Stimulation of VSMCs with ANG II increased the binding AP-1 to its conserved sites, which was markedly decreased in the presence of 17-DMAG (Fig. 5D). Taken together, these results indicate that 17-DMAG inhibits ANG II-induced expression of MMP-2 and MMP-9, at least in part, via inhibition of AP-1 transcriptional activity.
17-DMAG inhibits AP-1 activity via disruption of MAPK pathways.
Because the activation of MAPK pathway, such as ERK1/2 and JNK, is involved in the activation of AP-1 pathway and MMP expression (19), we attempted to investigate the effect of 17-DMAG on the activation of ERK1/2 and JNK in mouse VSMCs. Indeed, ANG II stimulation caused a rapid and transient activation of ERK1/2 at 5 min, which was dramatically inhibited by 17-DMAG, at all tested time points (Fig. 6A). Similarly, JNK was activated at 30 min after stimulation with ANG II, which was also markedly attenuated in the presence of 17-DMAG (Fig. 6B).
Fig. 6.

17-DMAG inhibits phosphorylation and nucleus translocation of ERK1/2 and JNK in VSMCs. A: cultured mouse VSMCs pretreated with either vehicle or 17-DMAG were treated with ANG II for indicated time points. Effect of 17-DMAG on ERK1/2 phosphorylation induced by ANG II (top) was determined by Western blot analysis and then quantitated by densitometric analysis (bottom). B: effect of 17-DMAG on JNK phosphorylation induced by ANG II was determined by Western blot (top) and then quantitated by densitometric analysis (bottom). C: cultured mouse VSMCs pretreated with or without 17-DMAG were treated with ANG II for 2 h. Western blot was performed to determine the subcellular localization of ERK and JNK. D: Western blot analysis showing the effect of 17-DMAG on c-Jun phosphorylation in mouse VSMCs at different time points in response to ANG II stimulation. *P < 0.05, compared with cells treated with only vehicle; #P < 0.05, compared with cells without 17-DMAG treatment at same time point; n = 5 independent experiments for all quantitative data.
Inhibition of ANG II-induced ERK1/2 and JNK activity by 17-DMAG might result in a prevention of nuclear translocation of JNK and ERK1/2. Therefore, we investigated whether 17-DMAG inhibits ANG II-induced ERK1/2 and JNK nuclear translocation in VSMCs. Indeed, as determined by Western blot analysis, ANG II stimulation of VSMCs for 2 h induced a significant nuclear translocation of ERK1/2 and JNK, which was markedly inhibited by 17-DMAG treatment (Fig. 6C). Accordingly, ANG II-induced phosphorylation of c-Jun, a key component in AP-1 complex (1), was also substantially inhibited by 17-DMAG (Fig. 6D). Taken together, these results show that inhibition of Hsp90 by 17-DMAG attenuates the AP-1 transcriptional activity via inhibition of the MAPK signaling pathway.
17-DMAG modulates inflammatory signaling pathways in VSMCs.
AAA is also considered an immune and inflammatory disease, and macrophages, lymphocytes, and mast cells participate in its development (40). In the animal models, we found that 17-DMAG markedly inhibited the infiltration of macrophage and MCP-1 production, which is a key chemokine for the migration and infiltration of macrophage in AAA formation. To determine the molecular mechanism involved, we examined the effect of 17-DMAG on the activation of NF-κB, which is critically involved in the regulation of MCP-1 expression in mouse VSMCs. Because Hsp90 has been shown to be associated with IKKs implicated in the NF-κB activation, we examined the effect of 17-DMAG on the protein levels of IKK-α and IKK-β in VSMCs. As shown in Fig. 7A, treatment of mouse VSMCs with 17-DMAG markedly attenuated the protein levels of both IKK-α and IKK-β. Similarly, ANG II-induced p65 nuclear translocation, as determined by Western blot, was significantly inhibited (Fig. 7, B and C). Accordingly, ANG II-induced MCP-1 expression was inhibited in mouse VSMCs, as determined by quantitative RT-PCR (Fig. 7D). Together, these results suggest that 17-DMAG inhibits inflammatory responses in VSMCs through attenuating the IKK/NF-κB pathway.
Fig. 7.

17-DMAG modulates inflammatory signaling pathways in VSMCs. Cultured mouse VSMCs pretreated with or without 17-DMAG (400 nmol/l) for 6 h were then treated with either vehicle or ANG II for 2 h. A: Western blot showing the effect of 17-DMAG on IKK expression. B: Western blot showing the effect of 17-DMAG on the subcellular localization of p65 in response to ANG II stimulation. Histone H3 was used as a nuclear marker. C: p65 levels in the nucleus of VSMCs were quantitated by densitometric analysis. D: effect of 17-DMAG on mRNA expression of MCP-1 was analyzed by quantitative PCR. *P < 0.05 compared with cells with vehicle treatment alone; #P < 0.05 compared with cells treated with ANG II alone; n = 5 independent experiments for all quantitative data.
17-DMAG inhibits ANG II-induced tube formation and MMP-2 release by HUVECs.
Accumulating evidence suggests that neovascularization plays an essential role in the pathogenesis of ANG II-induced AAA (7, 17). Therefore, we investigated the effect of 17-DMAG on ANG II-stimulated tube formation in HUVECs on Matrigel. Tube formation was significantly increased in ANG II-treated cells, as compared with the control. However, ANG II-induced tube formation was significantly inhibited by 17-DMAG (Fig. 8A). Because ANG II has been shown to induce neovascularization via VEGF pathway, we then investigated the effect of 17-DMAG on VEGF expression in HUVECs. As shown in Fig. 8B, 17-DMAG treatment markedly inhibited the ANG II-induced increased VEGF expression (Fig. 8B). Because endothelial MMP-2 is essentially implicated in the angiogenic process, the effect of 17-DMAG on MMP-2 activity in HUVECs in the presence and absence of ANG II stimulation was determined. As shown in Fig. 8C, 17-DMAG significantly attenuated both basal and ANG II- induced MMP-2 release from HUVECs. Taken together, our results suggest that 17-DMAG may inhibit ANG II-induced angiogenesis through suppressing ANG II-induced both VEGF expression and MMP-2 activation in vascular HUVECs.
Fig. 8.
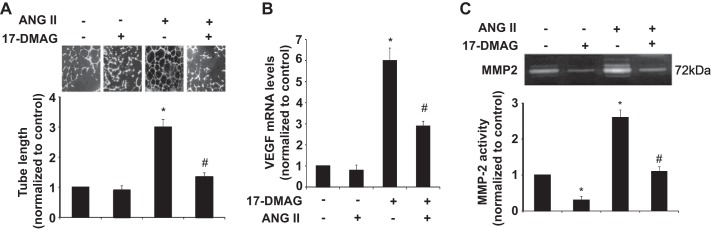
17-DMAG attenuates ANG II-induced tube formation and MMP-2 secretion by human umbilical cord vein endothelial cells (HUVECs). HUVECs were cultured with vehicle, 400 nmol/l ANG II, or 400 nmol/l ANG II plus 17-DMAG (top) for 24 h. A: photomicrographs showing the effects of 17-DMAG on ANG II-induced tube formation on the Matrigel. B: effect of 17-DMAG on VEGF mRNA expression was determined by quantitative RT-PCR. C: effect of 17-DMAG on MMP-2 activity in the medium of HUVECs (top) was determined by zymography and then quantitated by densitometric analysis (bottom). *P < 0.05 compared with cells with vehicle treatment alone; #P < 0.05 compared with cells treated with ANG II alone; n = 5 independent experiments for all quantitative data.
DISCUSSION
Here we show that both the incidence and severity of ANG II-induced AAA in apoE−/− mice were markedly reduced by treatment of the mice with 17-DMAG, a specific HSP90 inhibitor. Our data further demonstrate that 17-DMAG inhibits ANG II-induced MAPK (mainly ERK1/2 and JNK) phosphorylation and the subsequent nuclear translocation, which results in an inhibition of AP-1 activation and a decreased expression of MMPs, under both in vivo and in vitro conditions. In addition, both inflammatory responses and revascularization, as induced by ANG II in AAA formation, were significantly attenuated by inhibition of Hsp90. In this regard, our results demonstrate, for the first time, an effective treatment that prevents the development of AAA by targeting Hsp90, a key chaperone protein involved in controlling both AP-1 and NF-κB pathways critical to the pathogenesis of AAA.
Aberrant vascular remodeling and degradation of extracellular matrix is a well-recognized feature of human AAAs. MMPs are the predominant proteinases participating in the process of the destruction of the human aortic wall. Importantly, MMP-2 and MMP-9 protein levels have been shown to be significantly elevated in human AAA, and their expressions are correlated with aneurysm diameter (16, 31, 35). VSMCs are the major sources of proteolytic enzymes, which can degrade components of the extracellular matrix and impair the structural integrity of the vascular wall, thus leading to AAA formation (22). Indeed, inhibition of MMPs by the broad-spectrum MMP inhibitor doxycycline has been shown to inhibit AAA formation in several experimental AAA models (30, 36). In agreement with previous studies, our data demonstrated that ANG II significantly increased the activities of both MMP-2 and MMP-9 in the mouse aortic wall as well as in cultured VSMCs, which was substantially inhibited by Hsp90 inhibitor 17-DMAG. At this point, the molecular mechanism(s) underling the regulation of expression of MMPs by Hsp90 inhibitor 17-DMAG remains elusive, but it is mainly involved in inhibiting MAP-mediated activation of AP-1 pathway in VSMCs (4). Indeed, our results demonstrated that 17-DMAG markedly inhibited the ANG II-induced binding of AP-1 to the promoter regions of MMP-9 and MMP-2. In addition to its roles in regulation of MMP expression, Hsp90 has been shown to participate in the activation of MMPs, through its chaperone activity, in some cancer cells (46). Thus it is highly likely that inhibition of Hsp90 may limit the activity of MMPs through inhibiting both expression and activation of MMPs in ANG II-stimulated VSMCs, thus exerting potent inhibitory effects on AAA formation in ANG II-infused apoE−/− mice, as observed in the present study.
Accumulating evidence suggests that ROS plays essential roles in the development of AAA. The production of ROS is significantly increased in human AAA and experimental AAA lesions. Indeed, ANG II has been shown to induce the production of ROS in VSMCs, which leads to the phosphorylation of ERK1/2 and JNK, and a subsequent activation of AP-1 transcriptional activity, which contributes to the increased expression of MMPs in AAA lesions. In addition, ROS has been shown to induce NF-κB activation, hence promoting AAA formation by locally enhancing inflammatory cell infiltration and cytokine production in the abdominal aorta. Accordingly, genetic and pharmacological or genetic inhibition of ROS production has been shown to markedly suppress aneurysm formation in ANG II-infused apoE−/− mice through inhibiting macrophage infiltration and MMP2 activity in the abdominal aorta. Importantly, accumulating evidence suggests that Hsp90 plays a key regulatory role in ROS production through its interaction with the nicotinamide adenine dinucleotide phosphate oxidase (Nox) family of enzymes that are critically involved in ROS production (6). In fact, inhibition of Hsp90 has been shown to induce Nox degradation, thus inhibiting the ROS production. In contrast with the role of Hsp90 in stabilizing Nox, Hsp70 has been shown to promote Nox degradation through the Hsp70-regulated ubiquitin ligase (CHIP) (27) (6); therefore, the inhibition of Hsp90 can indirectly facilitate the Nox degradation through upregulating Hsp70 expression. In this regard, Hsp90 inhibitors may represent the potent therapeutic agents for anti-oxidative therapy (46, 53).
Inflammation has been well documented as a hallmark of AAA pathology, which is an early event in clinical aneurysm formation and ANG II-infusion AAA formation in animal models (40). Recently, it has been increasingly recognized that activation of NF-κB pathway plays an important role in promoting expression of both MMPs and MCP-1, which contributes significantly to the macrophage infiltration during AAA formation. Indeed, both genetic and pharmacological inhibition of NF-κB pathway has been reported to inhibit the development of experimental AAA (44). Of particular, ANG II has been reported to stimulate phosphorylation of IKK, resulting in increased phosphorylation of p65 and expression of its target genes in VSMCs (56). Because IKK exists in complexes with Hsp90, disruption of these complexes by Hsp90 inhibitors may block IKK function and subsequent NF-κB activation. In accordance with this hypothesis, we found that inhibition of Hsp90 by 17-DMAG markedly attenuated the ANG II-induced increase of macrophage infiltration and MCP-1 secretion in ANG II-induced AAA model. Likewise, we further demonstrated that 17-DMAG potently inhibited the protein expression of IKK, which leads to an inhibition of ANG II-induced NF-κB activation and MCP-1 expression in VSMCs. In these regard, our findings are consistent with recent studies showing that 17-DMAG inhibits INF-γ and IL-6-induced NF-κB activation and MCP-1 expression in macrophages (28). In addition to its critical roles in inflammatory response, NF-κB has been shown to be involved in regulating expression of MMP-2 and MMP-9 (5, 20). Therefore, it is highly likely that 17-DMAG may inhibit the activity of MMPs, through limiting the activation of both NF-κB and AP-1 pathways in VSMCs.
It has been reported that ANG II-induced rupture of AAA is associated with increased medial neovascularization (7). Indeed, ANG II has been demonstrated to stimulate neovascularization via the VEGF/eNOS pathway, under both in vivo and in vitro circumstances (12, 39, 49, 55). Our previous study also demonstrated that inhibition of Hsp90 decreases VEGF-induced angiogenesis via inhibition of eNOS gene transcription and Akt-mediated phosphorylation of eNOS in HUVECs, further suggesting an important role of Hsp90 in neovascularization (48). The molecular mechanism underlying inhibition of VEGF expression by 17-DMAG in HUVECs remains elusive, but may be involved in suppressing both HIF-1 dependent transcriptional activity and ROS production, as HIF-1 has been shown to be associated with Hsp90 in various types of cells, including HUVECs (26, 27). As MMPs have been shown to induce inflammation via proteolytic activation of growth factors and other proteins, MMPs released by both vascular VSMCs and HUVECs induced by ANG II may play a critical role in both inflammatory and angiogenic responses in the wall of the aorta and the disruption of aortic tissue (34). In this regard, our data clearly demonstrated that in addition to inhibiting the activity of MMP-9 and MMP-2 in VSMCs, 17-DMAG markedly inhibited ANG II-induced MMP-2 activation in HUVECs, thus exerting powerful synergistic effects in terms of inhibiting both inflammatory and angiogenic responses during AAA development.
In summary, in agreement with human AAA tissue, ANG II-induced AAA formation in apoE−/− mice is associated with a significant increase in inflammation, protease activation, and ROS production in the aortic wall. Inhibition of Hsp90 by 17-DMAG markedly attenuated ANG II-induced AAA formation through simultaneously inhibiting several key signaling and transcriptional pathways implicated in ROS production, MMP expression, vascular inflammation, and angiogenesis during the AAA formation and development (Fig. 9). In this regard, the pleiotropic effects of Hsp90 inhibitors could provide more potent or synergistic therapeutic effects for the treatment of AAA than MMP inhibitors or angiotensin-converting enzyme inhibitors, which have been previously shown to attenuate AAA development (25, 29, 30). Currently, 17-DMAG has been withdrawn from the clinical trials for the treatment of cancer patients due to potential side effects and safety issues. Today, more than 10 Hsp90 inhibitors representing multiple drug classes, with different mechanisms of action, are undergoing clinical evaluations (21). These novel Hsp90 inhibitors with greater activity and lower toxicity than 17-DMAG will offer further promising opportunities for the pharmacological intervention of AAA.
Fig. 9.
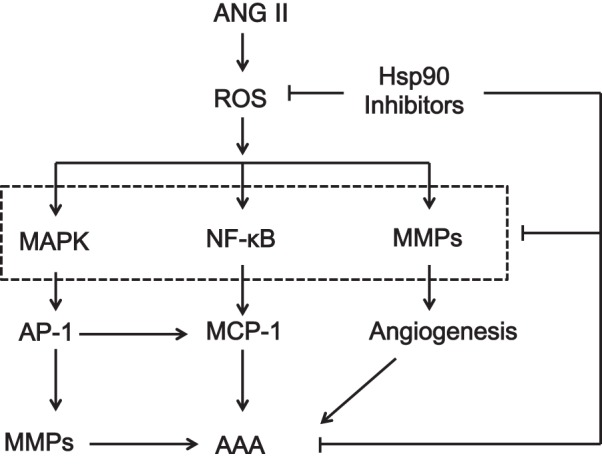
Proposed mechanisms of Hsp90 inhibition on AAA formation. Inhibition of Hsp90 inhibits simultaneously the reactive oxygen species (ROS) production, MMP expression, vascular inflammation, and angiogenesis, which collectively contribute to the attenuation of AAA formation by Hsp90 inhibitor 17-DMAG.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant R01 HL-103869, Chinese Natural Science Foundation Grants No. 381170114 (to J. Sun) and No. 81302768 (to J. Qi), Excellent Young Teachers Program of Shanghai Educational Commission Grant jdy11077 (to J. Qi), and Science and Technology Commission of Shanghai Municipality Grants No. 11DZ1972500 and No. 12DZ1930404 (to J. Zhang).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.Q., P.Y., J.Z., and J.S. conception and design of research; J.Q., P.Y., B.Y., Y.H., M.C., and J.S. performed experiments; J.Q., P.Y., B.Y., Y.H., M.C., J.Z., and J.S. analyzed data; J.Q., P.Y., B.Y., and J.S. interpreted results of experiments; J.Q., P.Y., and J.S. prepared figures; J.Q., P.Y., J.Z., and J.S. drafted manuscript; J.Z. and J.S. approved final version of manuscript; J.S. edited and revised manuscript.
ACKNOWLEDGMENTS
We thank Dr. Ross Summer for critical reading of the manuscript and fruitful discussion and Dr. Benjamin E. Leiby for the help with statistical analysis.
REFERENCES
- 1.Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta 1072: 129–157, 1991. [DOI] [PubMed] [Google Scholar]
- 2.Ayabe N, Babaev VR, Tang Y, Tanizawa T, Fogo AB, Linton MF, Ichikawa I, Fazio S, Kon V. Transiently heightened angiotensin II has distinct effects on atherosclerosis and aneurysm formation in hyperlipidemic mice. Atherosclerosis 184: 312–321, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Bergman MR, Cheng S, Honbo N, Piacentini L, Karliner JS, Lovett DH. A functional activating protein 1 (AP-1) site regulates matrix metalloproteinase 2 (MMP-2) transcription by cardiac cells through interactions with JunB-Fra1 and JunB-FosB heterodimers. Biochem J 369: 485–496, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Browatzki M, Larsen D, Pfeiffer CA, Gehrke SG, Schmidt J, Kranzhofer A, Katus HA, Kranzhofer R. Angiotensin II stimulates matrix metalloproteinase secretion in human vascular smooth muscle cells via nuclear factor-kappaB and activator protein 1 in a redox-sensitive manner. J Vasc Res 42: 415–423, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Chase AJ, Bond M, Crook MF, Newby AC. Role of nuclear factor-kappa B activation in metalloproteinase-1, -3, and -9 secretion by human macrophages in vitro and rabbit foam cells produced in vivo. Arterioscler Thromb Vasc Biol 22: 765–771, 2002. [DOI] [PubMed] [Google Scholar]
- 6.Chen F, Yu Y, Qian J, Wang Y, Cheng B, Dimitropoulou C, Patel V, Chadli A, Rudic RD, Stepp DW, Catravas JD, Fulton DJ. Opposing actions of heat shock protein 90 and 70 regulate nicotinamide adenine dinucleotide phosphate oxidase stability and reactive oxygen species production. Arterioscler Thromb Vasc Biol 32: 2989–2999, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choke E, Thompson MM, Dawson J, Wilson WR, Sayed S, Loftus IM, Cockerill GW. Abdominal aortic aneurysm rupture is associated with increased medial neovascularization and overexpression of proangiogenic cytokines. Arterioscler Thromb Vasc Biol 26: 2077–2082, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Citri A, Harari D, Shohat G, Ramakrishnan P, Gan J, Lavi S, Eisenstein M, Kimchi A, Wallach D, Pietrokovski S, Yarden Y. Hsp90 recognizes a common surface on client kinases. J Biol Chem 281: 14361–14369, 2006. [DOI] [PubMed] [Google Scholar]
- 9.Creager MA, Jones DW, Easton JD, Halperin JL, Hirsch AT, Matsumoto AH, O′Gara PT, Safian RD, Schwartz GL, Spittell JA, American Heart Association. Atherosclerotic Vascular Disease Conference: Writing Group V: medical decision making and therapy. Circulation 109: 2634–2642, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Daugherty A, Cassis LA. Mouse models of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol 24: 429–434, 2004. [DOI] [PubMed] [Google Scholar]
- 11.Eguchi S, Dempsey PJ, Frank GD, Motley ED, Inagami T. Activation of MAPKs by angiotensin II in vascular smooth muscle cells. Metalloprotease-dependent EGF receptor activation is required for activation of ERK and p38 MAPK but not for JNK. J Biol Chem 276: 7957–7962, 2001. [DOI] [PubMed] [Google Scholar]
- 12.Feliers D, Gorin Y, Ghosh-Choudhury G, Abboud HE, Kasinath BS. Angiotensin II stimulation of VEGF mRNA translation requires production of reactive oxygen species. Am J Physiol Renal Physiol 290: F927–F936, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Gimbrone MA., Jr Culture of vascular endothelium. Prog Hemost Thromb 3: 1–28, 1976. [PubMed] [Google Scholar]
- 14.Gitlin JM, Trivedi DB, Langenbach R, Loftin CD. Genetic deficiency of cyclooxygenase-2 attenuates abdominal aortic aneurysm formation in mice. Cardiovasc Res 73: 227–236, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Goergen CJ, Azuma J, Barr KN, Magdefessel L, Kallop DY, Gogineni A, Grewall A, Weimer RM, Connolly AJ, Dalman RL, Taylor CA, Tsao PS, Greve JM. Influences of aortic motion and curvature on vessel expansion in murine experimental aneurysms. Arterioscler Thromb Vasc Biol 31: 270–279, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodall S, Crowther M, Hemingway DM, Bell PR, Thompson MM. Ubiquitous elevation of matrix metalloproteinase-2 expression in the vasculature of patients with abdominal aneurysms. Circulation 104: 304–309, 2001. [DOI] [PubMed] [Google Scholar]
- 17.Herron GS, Unemori E, Wong M, Rapp JH, Hibbs MH, Stoney RJ. Connective tissue proteinases and inhibitors in abdominal aortic aneurysms. Involvement of the vasa vasorum in the pathogenesis of aortic aneurysms. Arterioscler Thromb 11: 1667–1677, 1991. [DOI] [PubMed] [Google Scholar]
- 18.Hostein I, Robertson D, DiStefano F, Workman P, Clarke PA. Inhibition of signal transduction by the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin results in cytostasis and apoptosis. Cancer Res 61: 4003–4009, 2001. [PubMed] [Google Scholar]
- 19.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem 270: 16483–16486, 1995. [DOI] [PubMed] [Google Scholar]
- 20.Kim H, Koh G. Lipopolysaccharide activates matrix metalloproteinase-2 in endothelial cells through an NF-kappaB-dependent pathway. Biochem Biophys Res Commun 269: 401–405, 2000. [DOI] [PubMed] [Google Scholar]
- 21.Kim T, Keum G, Pae AN. Discovery and development of heat shock protein 90 inhibitors as anticancer agents: a review of patented potent geldanamycin derivatives. Expert Opin Ther Pat 23: 919–943, 2013. [DOI] [PubMed] [Google Scholar]
- 22.Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res 95: 194–204, 2012. [DOI] [PubMed] [Google Scholar]
- 23.Lang SA, Klein D, Moser C, Gaumann A, Glockzin G, Dahlke MH, Dietmaier W, Bolder U, Schlitt HJ, Geissler EK, Stoeltzing O. Inhibition of heat shock protein 90 impairs epidermal growth factor-mediated signaling in gastric cancer cells and reduces tumor growth and vascularization in vivo. Mol Cancer Ther 6: 1123–1132, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Lee JG, Dahi S, Mahimkar R, Tulloch NL, Alfonso-Jaume MA, Lovett DH, Sarkar R. Intronic regulation of matrix metalloproteinase-2 revealed by in vivo transcriptional analysis in ischemia. Proc Natl Acad Sci USA 102: 16345–16350, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liao S, Miralles M, Kelley BJ, Curci JA, Borhani M, Thompson RW. Suppression of experimental abdominal aortic aneurysms in the rat by treatment with angiotensin-converting enzyme inhibitors. J Vasc Surg 33: 1057–1064, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Mabjeesh NJ, Post DE, Willard MT, Kaur B, Van Meir EG, Simons JW, Zhong H. Geldanamycin induces degradation of hypoxia-inducible factor 1alpha protein via the proteosome pathway in prostate cancer cells. Cancer Res 62: 2478–2482, 2002. [PubMed] [Google Scholar]
- 27.Madrigal-Matute J, Fernandez-Garcia CE, Gomez-Guerrero C, Lopez-Franco O, Munoz-Garcia B, Egido J, Blanco-Colio LM, Martin-Ventura JL. HSP90 inhibition by 17-DMAG attenuates oxidative stress in experimental atherosclerosis. Cardiovasc Res 95: 116–123, 2012. [DOI] [PubMed] [Google Scholar]
- 28.Madrigal-Matute J, Lopez-Franco O, Blanco-Colio LM, Munoz-Garcia B, Ramos-Mozo P, Ortega L, Egido J, Martin-Ventura JL. Heat shock protein 90 inhibitors attenuate inflammatory responses in atherosclerosis. Cardiovasc Res 86: 330–337, 2010. [DOI] [PubMed] [Google Scholar]
- 29.Malekzadeh S, Fraga-Silva RA, Trachet B, Montecucco F, Mach F, Stergiopulos N. Role of the renin-angiotensin system on abdominal aortic aneurysms. Eur J Clin Invest 43: 1328–1338, 2013. [DOI] [PubMed] [Google Scholar]
- 30.Manning MW, Cassis LA, Daugherty A. Differential effects of doxycycline, a broad-spectrum matrix metalloproteinase inhibitor, on angiotensin II-induced atherosclerosis and abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol 23: 483–488, 2003. [DOI] [PubMed] [Google Scholar]
- 31.McMillan WD, Tamarina NA, Cipollone M, Johnson DA, Parker MA, Pearce WH. Size matters: the relationship between MMP-9 expression and aortic diameter. Circulation 96: 2228–2232, 1997. [DOI] [PubMed] [Google Scholar]
- 32.Miyake T, Morishita R. Pharmacological treatment of abdominal aortic aneurysm. Cardiovasc Res 83: 436–443, 2009. [DOI] [PubMed] [Google Scholar]
- 33.Nakashima H, Aoki M, Miyake T, Kawasaki T, Iwai M, Jo N, Oishi M, Kataoka K, Ohgi S, Ogihara T, Kaneda Y, Morishita R. Inhibition of experimental abdominal aortic aneurysm in the rat by use of decoy oligodeoxynucleotides suppressing activity of nuclear factor kappaB and ets transcription factors. Circulation 109: 132–138, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Pearce WH, Shively VP. Abdominal aortic aneurysm as a complex multifactorial disease: interactions of polymorphisms of inflammatory genes, features of autoimmunity, and current status of MMPs. Ann N Y Acad Sci 1085: 117–132, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Petersen E, Gineitis A, Wagberg F, Angquist KA. Activity of matrix metalloproteinase-2 and -9 in abdominal aortic aneurysms. Relation to size and rupture. Eur J Vasc Endovasc Surg 20: 457–461, 2000. [DOI] [PubMed] [Google Scholar]
- 36.Petrinec D, Liao S, Holmes DR, Reilly JM, Parks WC, Thompson RW. Doxycycline inhibition of aneurysmal degeneration in an elastase-induced rat model of abdominal aortic aneurysm: preservation of aortic elastin associated with suppressed production of 92 kD gelatinase. J Vasc Surg 23: 336–346, 1996. [DOI] [PubMed] [Google Scholar]
- 37.Porter JR, Fritz CC, Depew KM. Discovery and development of Hsp90 inhibitors: a promising pathway for cancer therapy. Curr Opin Chem Biol 14: 412–420, 2010. [DOI] [PubMed] [Google Scholar]
- 38.Ruiz-Ortega M, Lorenzo O, Ruperez M, Konig S, Wittig B, Egido J. Angiotensin II activates nuclear transcription factor kappaB through AT1 and AT2 in vascular smooth muscle cells: molecular mechanisms. Circ Res 86: 1266–1272, 2000. [DOI] [PubMed] [Google Scholar]
- 39.Sanchez-Lopez E, Lopez AF, Esteban V, Yague S, Egido J, Ruiz-Ortega M, Alvarez-Arroyo MV. Angiotensin II regulates vascular endothelial growth factor via hypoxia-inducible factor-1alpha induction and redox mechanisms in the kidney. Antioxid Redox Signal 7: 1275–1284, 2005. [DOI] [PubMed] [Google Scholar]
- 40.Saraff K, Babamusta F, Cassis LA, Daugherty A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 23: 1621–1626, 2003. [DOI] [PubMed] [Google Scholar]
- 41.Scaltriti M, Dawood S, Cortes J. Molecular pathways: targeting hsp90—who benefits and who does not. Clin Cancer Res 18: 4508–4513, 2012. [DOI] [PubMed] [Google Scholar]
- 42.Schmitt E, Gehrmann M, Brunet M, Multhoff G, Garrido C. Intracellular and extracellular functions of heat shock proteins: repercussions in cancer therapy. J Leukoc Biol 81: 15–27, 2007. [DOI] [PubMed] [Google Scholar]
- 43.Shimp SK 3rd, Chafin CB, Regna NL, Hammond SE, Read MA, Caudell DL, Rylander M, Reilly CM. Heat shock protein 90 inhibition by 17-DMAG lessens disease in the MRL/lpr mouse model of systemic lupus erythematosus. Cell Mol Immunol 9: 255–266, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shiraya S, Miwa K, Aoki M, Miyake T, Oishi M, Kataoka K, Ohgi S, Ogihara T, Kaneda Y, Morishita R. Hypertension accelerated experimental abdominal aortic aneurysm through upregulation of nuclear factor kappaB and Ets. Hypertension 48: 628–636, 2006. [DOI] [PubMed] [Google Scholar]
- 45.Soga S, Akinaga S, Shiotsu Y. Hsp90 inhibitors as anti-cancer agents, from basic discoveries to clinical development. Curr Pharm Des 19: 366–376, 2013. [DOI] [PubMed] [Google Scholar]
- 46.Song X, Wang X, Zhuo W, Shi H, Feng D, Sun Y, Liang Y, Fu Y, Zhou D, Luo Y. The regulatory mechanism of extracellular Hsp90α on matrix metalloproteinase-2 processing and tumor angiogenesis. J Biol Chem 285: 40039–40049, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sparks AR, Johnson PL, Meyer MC. Imaging of abdominal aortic aneurysms. Am Fam Physician 65: 1565–1570, 2002. [PubMed] [Google Scholar]
- 48.Sun J, Liao JK. Induction of angiogenesis by heat shock protein 90 mediated by protein kinase Akt and endothelial nitric oxide synthase. Arterioscler Thromb Vasc Biol 24: 2238–2244, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tamarat R, Silvestre JS, Durie M, Levy BI. Angiotensin II angiogenic effect in vivo involves vascular endothelial growth factor- and inflammation-related pathways. Lab Invest 82: 747–756, 2002. [DOI] [PubMed] [Google Scholar]
- 50.Wang YX, Martin-McNulty B, da Cunha V, Vincelette J, Lu X, Feng Q, Halks-Miller M, Mahmoudi M, Schroeder M, Subramanyam B, Tseng JL, Deng GD, Schirm S, Johns A, Kauser K, Dole WP, Light DR. Fasudil, a Rho-kinase inhibitor, attenuates angiotensin II-induced abdominal aortic aneurysm in apolipoprotein E-deficient mice by inhibiting apoptosis and proteolysis. Circulation 111: 2219–2226, 2005. [DOI] [PubMed] [Google Scholar]
- 51.Weintraub NL. Understanding abdominal aortic aneurysm. N Engl J Med 361: 1114–1116, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Westermarck J, Kahari VM. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB 13: 781–792, 1999. [PubMed] [Google Scholar]
- 53.Yoshimura K, Aoki H, Ikeda Y, Fujii K, Akiyama N, Furutani A, Hoshii Y, Tanaka N, Ricci R, Ishihara T, Esato K, Hamano K, Matsuzaki M. Regression of abdominal aortic aneurysm by inhibition of c-Jun N-terminal kinase. Nat Med 11: 1330–1338, 2005. [DOI] [PubMed] [Google Scholar]
- 54.Zhang H, Burrows F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med (Berl) 82: 488–499, 2004. [DOI] [PubMed] [Google Scholar]
- 55.Zhang HW, Wang X, Zong ZH, Huo X, Zhang Q. AP-1 inhibits expression of MMP-2/9 and its effects on rat smooth muscle cells. J Surg Res 157: e31–e37, 2009. [DOI] [PubMed] [Google Scholar]
- 56.Zhang L, Cheng J, Ma Y, Thomas W, Zhang J, Du J. Dual pathways for nuclear factor kappaB activation by angiotensin II in vascular smooth muscle: phosphorylation of p65 by IkappaB kinase and ribosomal kinase. Circ Res 97: 975–982, 2005. [DOI] [PubMed] [Google Scholar]