

Abstract
In a rat model, downregulation of homeostatic mesenchymal peroxisome proliferator-activated receptor-γ (PPAR-γ) signaling following perinatal nicotine exposure contributes to offspring asthma, which can be effectively prevented by concomitant administration of PPAR-γ agonist rosiglitazone (RGZ). However, whether perinatal nicotine exposure-induced asthma can be reversed is not known. We hypothesized that perinatal nicotine exposure-induced asthma would be reversed by PPAR-γ agonist RGZ. Pregnant rat dams received either placebo or nicotine from embryonic day 6 until term. Following spontaneous delivery at term, dams were continued on the assigned treatments, up to postnatal day 21 (PND21). However, at delivery, pups were divided into two groups; one group received placebo, and the other group received RGZ from PND1 to PND21. At PND21, pulmonary function and the expression of mesenchymal markers of airway contractility (α-smooth muscle actin, calponin, fibronectin, collagen I, and collagen III) were determined by immunoblotting and immunostaining for the evidence of reversibility of perinatal nicotine exposure-induced lung effects. Compared with controls, perinatal nicotine exposure caused 1) a significant increase in airway resistance and a decrease in airway compliance following methacholine challenge, 2) a significant increase in acetylcholine-induced tracheal constriction, and 3) increased pulmonary and tracheal expression of the mesenchymal markers of contractility. Treatment with RGZ, starting on PND1, reversed all of the nicotine-induced molecular and functional pulmonary effects, virtually normalizing the pulmonary phenotype of the treated animals. We conclude that perinatal nicotine exposure-induced functional and molecular alterations in upper and lower airways can be reversed by PPAR-γ agonist RGZ, allowing an effective intervention even when started postnatally.
Keywords: nicotine, peroxisome proliferator-activated receptor, pregnancy, smoking, asthma
strong epidemiological and experimental evidence supports the notion that exposure of the developing lung to maternal smoking results in detrimental long-term effects on lung growth and function including risk of asthma (8, 20). Given that there are more than 4,000 known chemicals in the combustible tobacco smoke, it is difficult to discern which of these chemicals are responsible for the perinatal smoke-induced hyperreactive pulmonary phenotype. However, there is clear experimental evidence to show that nicotine directly affects fetal lung development, resulting in asthma phenotype in the exposed offspring (13, 15, 20, 24, 29). Moreover, with the increasing use of nicotine-replacement therapy by pregnant women (7) and the increasing popularity of electronic cigarettes among reproductive-age women (1, 16), a better understanding of the specific effects of nicotine alone on the developing offspring has acquired increasing public health significance.
Experimentally, perinatal nicotine exposure is associated with offspring asthma, mediated by decreased mesenchymal peroxisome proliferator-activated receptor-γ (PPAR-γ) signaling (11, 12, 23). Importantly, the concomitant administration of PPAR-γ agonist rosiglitazone (RGZ) has been shown to effectively block the perinatal nicotine exposure-induced asthma (11, 12, 17, 18). However, whether perinatal nicotine exposure-induced asthma can be reversed is not known. Therefore, the objective of this study was to determine whether PPAR-γ agonist RGZ, administered postnatally, could reverse the perinatal nicotine exposure-induced asthma. We hypothesized that the effects of perinatal nicotine exposure on offspring pulmonary function and mesenchymal markers of airway contractility could be reversed by PPAR-γ agonist RGZ. Using a well-established rat model of perinatal nicotine exposure-induced offspring asthma (10, 11, 12, 17, 18), we determined that treatment with RGZ, starting on postnatal day 1 (PND1), reversed all of the perinatal nicotine-induced molecular and functional pulmonary effects on the exposed offspring, virtually normalizing the pulmonary phenotype of the treated animals. These data suggest that perinatal smoke exposure-induced asthma can be, not only prevented, but also reversed. Therefore, we conclude that the functional and molecular alterations in upper and lower airways in a rodent model of perinatal nicotine exposure were reversed by PPAR-γ agonist RGZ, allowing us to speculate on the effectiveness of PPAR-γ treatment even when it is started postnatally following the exposure to maternal smoke throughout gestation.
MATERIALS AND METHODS
Animals.
In line with the previous description of this model (10–12, 17, 18), pair-fed, time-mated, first-time pregnant Sprague Dawley rat dams received either placebo (saline) or nicotine (1 mg/kg sc) in 100-μl volumes once daily from embryonic day 6 until term. The animals were maintained in a 12-h:12-h light/dark cycle, pair fed in accordance with the consumption of the previous day by the nicotine-treated group, and allowed free access to water. Pups delivered spontaneously at term and breast fed ad libitum. Dams continued to get treatment according to their assigned groups up to PND21. However, at delivery, pups were divided into two groups; one group received placebo, and the other group received RGZ (3 mg/kg ip) in 50-μl volumes once daily from PND1 to PND21. At PND21, pups were either subjected to tracheal tension studies or killed to obtain their lungs and tracheas to determine the relevant protein markers of airway contractility. In a subset of animals at 2–3 wk of age, pulmonary function was evaluated, as described previously (12). All animal procedures were performed following the National Institutes of Health guidelines for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committees at the Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center and the University of Southern California.
Pulmonary function testing.
Pulmonary function was measured using a plethysmograph for restrained animals (Buxco, Troy, NY), with rat pups in the supine position. Dynamic compliance and resistance of the respiratory system were simultaneously measured on the anesthetized, tracheotomized, and ventilated rats at baseline and immediately following serial 3-min nebulizations of saline and methacholine (MCh, 0 to 20 mg/ml), with 3-min recovery period after each exposure to MCh, as previously described (11). The pups were deeply anesthetized and sedated with ketamine (70 mg/kg; Bioniche Teoranta Inverin, Galway, Ireland) and xylazine (7 mg/kg; Akorn, Decatur, IL) and then ventilated at 7–8 ml/kg with a small-animal ventilator (MiniVent; Harvard Apparatus, Cambridge, MA) for the duration of the procedure.
Tracheal tension studies.
The whole trachea was excised immediately after death and dissected free of connective tissue in ice-cold modified Krebs-Ringer bicarbonate buffer (in mM: 118.3 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25.0 NaHCO3, and 11.1 glucose). Subsequently, a 6-mm tracheal ring was resected from the middle of the trachea and used for tracheal tension studies. The tracheal ring was suspended in an organ chamber filled with 10 ml of modified Krebs-Ringer bicarbonate solution maintained at 37 ± 0.5°C and aerated with 95% O2-5% CO2 (pH 7.4). Each ring was suspended via two stirrups that were passed through the lumen; one stirrup was anchored to the bottom of the organ chamber, and the other stirrup was connected to a strain gauge (model FT03C; Grass Instruments, Quincy, MA) for the measurement of isometric force, as described previously (12). At the beginning of the experiment, each tracheal ring was stretched to its optimal resting tension, which was achieved by step-wise stretching in 0.1-g increments, until the contractile response to 100 mM KCl reached a plateau. The optimal resting tension was measured, and then each tracheal ring was allowed to equilibrate for 1 h after it was brought to its optimal resting tension. The effects of acetylcholine (ACh) were determined at least 30 min after the administration of nitro-l-arginine (1 × 10−4 M, an inhibitor of nitric oxide synthase). In all experiments, indomethacin (1 × 10−5 M) was added to the bath to prevent the possible interference by prostanoids.
In vitro confirmation that effects of PPAR-γ agonists on nicotine-mediated alterations in alveolar fibroblast differentiation are specifically PPAR-γ mediated.
Embryonic day 19 fetal rat lung fibroblasts were isolated and cultured using our previously described methods (26). At 80–90% confluence, cells were treated with nicotine (1 × 10−6 M) for 24 h with or without pioglitazone (1 × 10−5 M, catalog no. E6910; Sigma-Aldrich, St. Louis, MO) or prostaglandin J2 (PGJ2, 5 × 10−6 M, catalog no. D8440; Sigma-Aldrich) pretreatment for 1 h. Experiments were also conducted using pretreatment with a specific PPAR-γ antagonist GW9662 (1 × 10−5 M, catalog no. M6191; Sigma-Aldrich) before nicotine and pioglitazone or PGJ2 treatments. Subsequently, immunoblotting was performed for PPAR-γ, lymphoid enhancer-binding factor 1 (LEF-1), fibronectin, α-smooth muscle actin (α-SMA), and calponin, as outlined next.
Immunoblot analysis.
Following experimental conditions, the isolated tracheas, lungs, and alveolar fibroblasts were flash frozen in liquid nitrogen and then homogenized and sonicated in ice-cold lysis buffer containing 50 mM glycerophosphate (pH 7.4), 150 mM NaCl, 1.5 mM EGTA, 1 mM EDTA, 1% Triton X-100, 100 mM NaF, 2 mM Na3VO4, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 2 μg/ml pepstatin A. After centrifugation at 13,200 g for 15 min at 4°C, the supernatant was used for Western blot analysis for fibronectin, α-SMA, calponin, collagen I, collagen III, cholinergic ACh receptor-α3 and -α7 (AChRα3 and AChRα7), β-catenin, LEF-1, PPAR-γ, and phosphorylated PPAR-γ (phospho-PPAR-γ). The protein concentration of the supernatant was measured by the Bradford method, with bovine serum albumin as the standard. Aliquots of the supernatant, each containing 30 μg of protein, were separated by SDS-PAGE and electrically transferred to nitrocellulose membranes. Nonspecific binding sites were blocked with Tris-buffered saline (TBS) containing 5% nonfat dry powdered milk (wt/vol) for 1 h at room temperature. After a brief rinse with TBS containing 0.1% Tween 20 (TBST), the protein blots were incubated in 1:250 diluted anti-fibronectin monoclonal antibody (cat. no. 610078; BD Biosciences, San Jose, CA), 1:10,000 diluted anti-α-SMA monoclonal antibody (cat. no. A2547, Sigma), 1:6,000 diluted anti-calponin monoclonal antibody (cat. no. C-2687, Sigma), 1:500 diluted anti-collagen I polyclonal antibody (cat. no. RDI-MCOII1abr; Fitzgerald Industries, Concord, MA), 1:1,000 diluted anti-collagen III monoclonal antibody (cat. no. C7805, Sigma), 1:400 diluted anti-nicotinic AChRα3 (cat. no. sc-5590; Santa Cruz Biotechnology, Santa Cruz, CA), 1:20,000 diluted anti-nicotinic AChRα7 (cat. no. N8158, Sigma), 1:1,000 diluted anti-β-catenin (cat. no. sc-7963, Santa Cruz Biotechnology), 1:500 diluted anti-LEF-1 (cat. no. sc-28687, Santa Cruz Biotechnology), 1:1,000 diluted anti-PPAR-γ (cat. no. sc-7196, Santa Cruz Biotechnology), 1:1,000 diluted anti-phospho-PPAR-γ (cat. no. ab60953; Abcam, Cambridge, MA), and 1:4,000 diluted anti-GAPDH monoclonal antibody (cat. no. MAB374; Millipore, Billerica, MA) overnight at 4°C. After they were washed three times with TBST, the blots were incubated in 1:1,000 (fibronectin), 1:10,000 (α-SMA), 1:6,000 (calponin), 1:2,500 (collagen I), 1:3,000 (AChRα3), 1:20,000 (AChRα7), 1:2,500 (β-catenin, LEF-1, PPAR-γ, and phospho-PPAR-γ), 1:4,000 (GAPDH) diluted horseradish peroxidase-conjugated anti-mouse, or anti-rabbit secondary antibody for 1 h at room temperature. After three more washes in TBST, the blots were exposed to X-ray film using SuperSignal West Pico Chemiluminescent Substrate (Pierce Biotechnology, Rockford, IL) and developed. The relative densities of the protein bands were determined with UN-SCAN-IT software (Silk Scientific, Orem, UT) and normalized to the density of GAPDH.
Immunofluorescence staining.
Immunofluorescence staining of α-SMA, calponin, PPAR-γ, β-catenin, and LEF-1 was performed as previously described (12) and was merged using ImageJ software (National Institutes of Health, Bethesda, MD).
Statistical analysis.
All values shown are expressed as means ± SE. Comparisons between the different groups were performed using either Student's t-test or a mixed-model repeated-measures ANOVA with unstructured covariance. P values of <0.05 were considered statistically significant. Male and female data were combined for each experimental condition and analyzed as mixed-sex data.
RESULTS
We first determined the effects of perinatal nicotine exposure on total airway resistance and total airway compliance and how RGZ started on PND1 affects these parameters (Fig. 1). Compared with controls, perinatal nicotine exposure alone increased total airway resistance and decreased total airway compliance, which were reversed by RGZ administration; of note, RGZ administration alone did not affect these parameters. We next determined the ACh-induced tracheal constriction response following perinatal nicotine exposure and how RGZ affects it (Fig. 2). There was significant increase in tracheal constriction with perinatal nicotine exposure alone, which was also reversed by RGZ administration. As noted for the total airway resistance and total airway compliance, RGZ administration alone did not affect tracheal contractility.
Fig. 1.
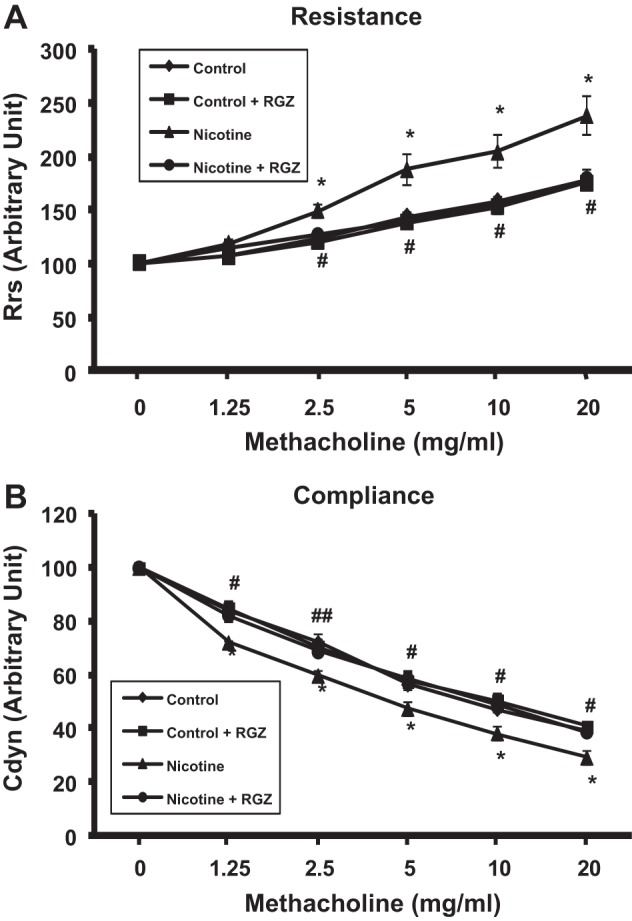
Effect of postnatally administered rosiglitazone (RGZ) on perinatal nicotine exposure-induced alterations in total airway resistance and compliance at baseline and after methacholine (MCh) challenge. Compared with the control group, with nicotine administration there was a significant increase in total airway resistance, both at baseline and following MCh challenge, which was reversed by postnatal RGZ administration. Values are means ± SE. n = 5–6 for each group. *P < 0.05 vs. control; #P < 0.05, ##P < 0.01 vs. nicotine. Rrs, respiratory system resistance; Cdyn, dynamic compliance.
Fig. 2.
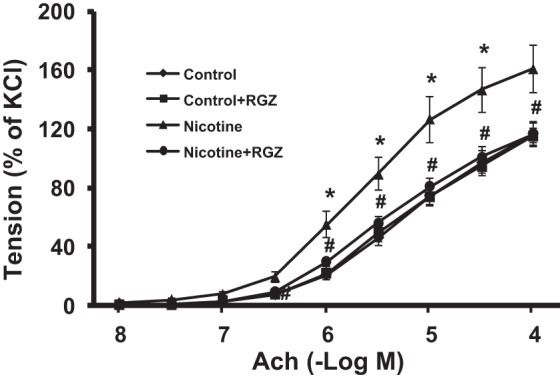
Effect of postnatally administered RGZ on perinatal nicotine exposure-induced alterations in tracheal constriction response to acetylcholine (ACh). Compared with the control group, with nicotine administration, there was a significant increase in tracheal constriction response to ACh, which was almost completely reversed in the RGZ-treated group. Values are means ± SE; n = 5–6 for each group. *P < 0.05 vs. control; #P < 0.05 vs. nicotine.
Having determined the effect of postnatally administered RGZ on perinatal nicotine-induced pulmonary hyperreactivity, we then examined the effect of RGZ on perinatal nicotine exposure-induced alterations in mesenchymal markers of airway contractility in the lung (Fig. 3) and trachea (Fig. 4). Protein levels of fibronectin, α-SMA, calponin, and collagen I and III, as determined by the Western analysis of lung lysates, increased significantly with perinatal nicotine exposure, with reversal of all of these protein levels to control levels in the RGZ-administered group. Again, RGZ administration alone did not affect any of these parameters (Fig. 3). Similarly, the tracheal protein levels of fibronectin, α-SMA, calponin, and collagen I increased significantly with perinatal nicotine exposure, an effect that was blocked by postnatal RGZ administration (Fig. 4).
Fig. 3.

Effect of postnatally administered RGZ on perinatal nicotine exposure-induced alterations in mesenchymal markers of airway contractility in rat lung. Compared with the control group, with nicotine administration, the protein levels of fibronectin, α-smooth muscle actin (α-SMA), calponin, and collagens I or III increased significantly, and this effect was reversed in the RGZ-treated group. Top: representative Western blots for the markers and GAPDH. Bottom: densitometric values of the markers normalized to GAPDH. Values are means ± SE; n = 4 for each group. **P < 0.01 vs. control; #P < 0.05, ##P < 0.01 vs. nicotine.
Fig. 4.

Effect of postnatally administered RGZ on perinatal nicotine exposure-induced alterations in mesenchymal markers of airway contractility in rat trachea. Compared with the control group, with nicotine administration, the protein levels of fibronectin, α-SMA, calponin, and collagen I increased significantly, and this effect was reversed in the RGZ-treated group. Top: representative Western blots for the markers and GAPDH. Bottom: densitometric values of the markers normalized to GAPDH. Values are means ± SE; n = 4 for each group. *P < 0.05, **P < 0.01 vs. control; #P < 0.05, ##P < 0.01 vs. nicotine.
Because we have previously determined that the nicotine-induced increase in the alveolar myogenic phenotype is mediated by nicotinic AChRα3 and AChRα7, we also determined the levels of these receptors in the lung and tracheal protein lysates. The levels of both of these receptors increased in the lungs of the nicotine-treated group, with their reversal in the RGZ-treated group (Fig. 5). Because the perinatal nicotine exposure-induced pulmonary hyperreactivity, mechanistically, has been shown to be mediated via the downregulation of PPAR-γ and upregulation of Wnt signaling proteins, the levels of PPAR-γ, β-catenin, and LEF-1 were determined at both lung and tracheal levels. In line with the rest of the data outlined above, perinatal nicotine exposure decreased the levels of PPAR-γ and increased levels of β-catenin and LEF-1 in both lung and trachea, with reversal of these changes in the RGZ-administered group (Fig. 6). Immunostaining for α-SMA, calponin, PPAR-γ, LEF-1, and β-catenin on lung sections from different experimental conditions corroborated the immunoblotting data (Fig. 7).
Fig. 5.

Effect of RGZ on perinatal nicotine exposure-induced alterations in nicotinic acetylcholine receptors-α3 and -α7 (AChRα3 and AChRα7) in rat lung (A) and trachea (B). Compared with the control group, with nicotine administration, the levels of both receptors in the lungs and tracheas of the nicotine-treated animals increased significantly, and this effect was reversed in the RGZ-treated group. Representative Western blots for the receptors and GAPDH and density histograms are shown. Values are means ± SE; n = 4 for each group. *P < 0.05, **P < 0.01 vs. control; #P < 0.05, ##P < 0.01 vs. nicotine.
Fig. 6.

Effect of RGZ on perinatal nicotine exposure-induced alterations in total and phosphorylated peroxisome proliferator-activated receptor-γ (p-PPAR-γ), β-catenin, and lymphoid enhancer-binding factor 1 (LEF-1) in rat lung and trachea. Compared with the control group, with nicotine administration, the levels of PPAR-γ and p-PPAR-γ decreased, whereas levels of β-catenin and LEF-1 increased significantly in the lungs and tracheas of the nicotine-treated animals, which were reversed in the RGZ-treated group. Representative Western blots for the receptors and GAPDH and density histograms are shown. Values are means ± SE; n = 4 for each group. *P < 0.05, **P < 0.01 vs. control; #P < 0.05, ##P < 0.01 vs. nicotine.
Fig. 7.

Effect of RGZ on perinatal nicotine exposure-induced alterations in mesenchymal markers of airway contractility (α-SMA and calponin) and the mechanistic markers (PPAR-γ, β-catenin, and LEF-1) mediating this effect in the rat lung, demonstrated by immunofluorescence staining. Corroborating the Western data, immunofluorescence staining demonstrated increased α-SMA, calponin, β-catenin, and LEF-1 staining but decreased PPAR-γ staining in lung sections from the perinatally nicotine-exposed animals, with reversal of these findings in the lung sections from the postnatally RGZ-treated group. Representative immunofluorescence stained sections are shown.
Lastly, because RGZ has both PPAR-γ-dependent and PPAR-γ-independent effects, to confirm that the effects of RGZ on nicotine-induced pulmonary phenotype are PPAR-γ dependent, in vitro experiments were performed using cultured embryonic day 19 alveolar interstitial fibroblasts. We determined that, in addition to RGZ, as demonstrated by us previously (21, 22), PPAR-γ agonists pioglitazone and PGJ2 also blocked the nicotine-induced decrease in PPAR-γ and increase in LEF-1 protein levels, as well as the levels of other myogenic marker proteins such as fibronectin, α-SMA, and calponin. More importantly, pretreatment with a specific PPAR-γ antagonist GW9662 blocked the protection provided by both pioglitazone and PGJ2 against all nicotine-induced molecular changes in alveolar fibroblast differentiation (Fig. 8), suggesting that the protection provided by PPAR-γ agonists against the nicotine-induced alteration in alveolar fibroblast differentiation is specifically mediated via PPAR-γ.
Fig. 8.

Pretreatment with a specific PPAR-γ antagonist, GW9662 (1 × 10−5 M), completely blocked the molecular protection (increase in PPAR-γ and a decrease in LEF-1, fibronectin, α-SMA, and calponin protein levels) by pioglitazone and prostaglandin J2 (PGJ2) following concomitant treatment with nicotine (1 × 10−6 M) for 24 h. Values are means ± SE; n = 4 for each group. **P < 0.01 vs. control; ##P < 0.01 vs. nicotine; ^^P < 0.01 vs. nicotine + pioglitazone; $$P < 0.01 vs. nicotine + PGJ2.
DISCUSSION
In a well-published rat model of perinatal nicotine exposure-induced offspring airway hyperreactivity (10, 11, 12, 17, 18), in line with our previous findings, compared with the control group, perinatal nicotine exposure caused a significant increase in airway resistance and a decrease in airway compliance following MCh challenge, a significant increase in ACh-induced tracheal constriction, and increased pulmonary and tracheal expression of the mesenchymal markers of contractility, all features consistent with an asthma phenotype. Administration of PPAR-γ agonist RGZ, initiated postnatally, i.e., well after the nicotine exposure was initiated in utero, reversed all of the nicotine-induced molecular and functional pulmonary effects, virtually normalizing the pulmonary phenotype of the treated animals. From these data, we conclude that perinatal nicotine exposure-induced functional and molecular alterations in upper and lower airways can be reversed by PPAR-γ agonist RGZ, allowing a possible intervention against perinatal smoke-induced asthma, even when this intervention is started postnatally.
It is well established that maternal smoking is a major modifiable health risk factor, the elimination of which could significantly reduce the prevalence of childhood asthma; however, because the tobacco industry continues to spend billions of dollars on advertising to attract young smokers (4), it is unlikely that the problem of smoking during pregnancy will go away in the near future. Approximately 250,000,000 women smoke daily worldwide (28). Twelve percent of US women smoke during pregnancy, resulting in the births of at least 400,000 smoke-exposed infants per year in the US alone (2, 14). Additionally, it is important to point out that, in addition to exposure to smoke, pregnant women are also commonly exposed to nicotine as part of nicotine-replacement therapy (3) and/or via the use of electronic cigarettes because the popularity of electronic cigarettes has risen exponentially in recent years (1), rendering perinatal nicotine exposure, a highly relevant issue on its own. Given this, any intervention that can safely prevent, or even better, reverse the smoke/nicotine-induced pulmonary effects could be an effective and probably a more practical strategy to prevent perinatal smoke/nicotine exposure-induced asthma.
We have previously demonstrated that PPAR-γ agonists, by blocking the nicotine-induced mesenchymal PPAR-γ inhibition and Wnt activation, prevent in vitro (22, 23) and in vivo (10, 11, 12, 17, 18) nicotine-induced lung injury. Furthermore, this approach was also shown to reverse the nicotine-induced lipofibroblast-to-myofibroblast differentiation in an embryonic lung fibroblast culture model (21); however, up until now, whether the same approach would be effective under in vivo conditions remained unknown. The data included here show the validity of PPAR-γ intervention in reversing the perinatal nicotine-induced asthma, providing an effective and logistically more practical approach in tackling the hazards of smoke-related damage to the developing lung. It is important to point out that, on PND1, i.e., at the beginning of RGZ administration, the lungs and alveolar interstitial fibroblasts, examined following the same intrauterine nicotine exposure regimen as used in this study, have been shown to exhibit an asthmatic phenotype, as evidenced by the increased levels of myogenic markers such as α-SMA and fibronectin and decreased levels of homeostatic markers such as parathyroid hormone-related protein receptor and PPAR-γ (10). Therefore, these data suggest a reversal rather than simply a blockage of the perinatal nicotine-induced asthmatic phenotype. However, it is also important to point out that, although the perinatal nicotine exposure-induced lung phenotype in the rat model utilized here clearly demonstrates airway hyperresponsiveness, this might not exactly mimic childhood asthma seen in human infants because evidence of enhanced allergic response has not been reported in this model so far.
There are few other important issues to note. Because the PPAR-γ agonist therapy was started very early in the postnatal period, i.e., on PND1, up to how late in the postnatal period this approach is effective remains to be seen. Additionally, although it is reassuring to note that the effects against the nicotine-induced lung phenotype observed by us seem to be PPAR-γ specific and the limited human and animal data suggest safety of PPAR-γ agonists when used during the developing period (5, 6, 9, 25, 27, 30), because of significant side effects with their use in adults, detailed safety, pharmacokinetic, and pharmacodynamic studies and a search for novel safer and selective PPAR-γ agonists are needed before the use of this class of agents in human infants can be recommended clinically.
GRANTS
This work was supported by NIH (HD51857, HD058948, HL107118, and HD071731) and TRDRP (15IT-0250, 17RT-0170, and 23RT-0018).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: J.L. and R.S. performed experiments; J.L. and V.K.R. analyzed data; J.L. and R.S. prepared figures; J.L. and V.K.R. edited and revised manuscript; J.L., R.S., and V.K.R. approved final version of manuscript; V.K.R. conception and design of research; V.K.R. interpreted results of experiments; V.K.R. drafted manuscript.
REFERENCES
- 1.Arrazola RA, Kuiper NM, Dube SR. Patterns of current use of tobacco products among US high school students for 2000–2012-findings from the National Youth Tobacco Survey. J Adolesc Health 54: 54–60, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control Cigarette Smoking in the United States: Current Estimates (Online). http://www.cdc.gov/tobacco/data_statistics/fact_sheets/adult_data/cig_smoking/[20 August 2014].
- 3.Clark SM, Nakad R. Pharmacotherapeutic management of nicotine dependence in pregnancy. Obstet Gynecol Clin North Am 38: 297–311, 2011. [DOI] [PubMed] [Google Scholar]
- 4.Federal Trade Commission. Federal Trade Commission Cigarette Report for 2002 (Online) http://www.ftc.gov/reports/cigarette/041022cigaretterpt.pdf[20 August 2014].
- 5.Feig DS, Briggs GG, Koren G. Oral antidiabetic agents in pregnancy and lactation: a paradigm shift? Ann Pharmacother 41: 1174–1180, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Haddad GF, Jodicke C, Thomas MA, Williams DB, Aubuchon M. Case series of rosiglitazone used during the first trimester of pregnancy. Reprod Toxicol 26: 183–184, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Hartmann-Boyce J, Stead LF, Cahill K, Lancaster T. Efficacy of interventions to combat tobacco addiction: Cochrane update of 2013 reviews. Addiction 109: 1414–1425, 2014. [DOI] [PubMed] [Google Scholar]
- 8.Hollams EM, de Klerk NH, Holt PG, Sly PD. Persistent effects of maternal smoking during pregnancy on lung function and asthma in adolescents. Am J Respir Crit Care Med 189: 401–407, 2014. [DOI] [PubMed] [Google Scholar]
- 9.Kalyoncu NI, Yaris F, Ulku C, Kadioglu M, Kesim M, Unsal M, Dikici M, Yaris E. A case of rosiglitazone exposure in the second trimester of pregnancy. Reprod Toxicol 19: 563–564, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Krebs M, Sakurai R, Torday JS, Rehan VK. Evidence for in vivo nicotine-induced alveolar interstitial fibroblast-to-myofibroblast transdifferentiation. Exp Lung Res 36: 390–398, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J, Naeem E, Tian J, Lombardi V, Kwong K, Akbari O, Torday JS, Rehan VK. Gender-specific perinatal nicotine-induced asthma in rat offspring. Am J Respir Cell Mol Biol 48: 53–62, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J, Sakurai R, O'Roark EM, Kenyon NJ, Torday JS, Rehan VK. PPAR-γ agonist rosiglitazone prevents perinatal nicotine exposure-induced asthma in rat offspring. Am J Physiol Lung Cell Mol Physiol 300: L710–L717, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maritz GS, Harding R. Life-long programming implications of exposure to tobacco smoking and nicotine before and soon after birth: evidence for altered lung development. Int J Environ Res Public Health 8: 875–898, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin JA, Hamilton BE, Sutton PD, Ventura SJ, Menacker F, Munson ML. Births: final data for 2002. Natl Vital Stat Rep 52: 1–113, 2003. [PubMed] [Google Scholar]
- 15.Pierce RA, Nguyen NM. Prenatal nicotine exposure and abnormal lung function. Am J Respir Cell Mol Biol 26: 10–13, 2002. [DOI] [PubMed] [Google Scholar]
- 16.Regan AK, Promoff G, Dube SR, Arrazola R. Electronic nicotine delivery systems: adult use and awareness of the ‘e-cigarette’ in the USA. Tob Control 22: 19–23, 2013. [DOI] [PubMed] [Google Scholar]
- 17.Rehan VK, Liu J, Sakurai R, Torday JS. Perinatal nicotine-induced transgenerational asthma. Am J Physiol Lung Cell Mol Physiol 305: L501–L507, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rehan VK, Liu J, Naeem E, Tian J, Lombardi V, Kwong K, Akbari O, Torday JS. Perinatal nicotine exposure induces asthma in second generation offspring. BMC Med 10: 129, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rehan VK, Sakurai R, Corral J, Krebs M, Ibe B, Ihida-Stansbury K, Torday JS. Antenatally administered PPAR-γ agonist rosiglitazone prevents hyperoxia-induced neonatal rat lung injury. Am J Physiol Lung Cell Mol Physiol 299: L672–L680, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rehan VK, Asotra K, Torday JS. The effects of smoking on the developing lung: insights from a biologic model for lung development, homeostasis and repair. Lung 187: 281–289, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rehan VK, Sakurai R, Wang Y, Santos J, Huynh K, Torday JS. Reversal of nicotine-induced alveolar lipofibroblast-to-myofibroblast transdifferentiation by stimulants of parathyroid hormone-related protein signaling. Lung 185: 151–159, 2007. [DOI] [PubMed] [Google Scholar]
- 22.Rehan VK, Wang Y, Sugano S, Romero S, Chen X, Santos J, Khazanchi A, Torday JS. Mechanism of nicotine-induced pulmonary fibroblast transdifferentiation. Am J Physiol Lung Cell Mol Physiol 289: L667–L676, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Sakurai R, Cerny LM, Torday JS, Rehan VK. Mechanism for nicotine-induced up-regulation of Wnt signaling in human alveolar interstitial fibroblasts. Exp Lung Res 37: 144–154, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sekhon HS, Keller JA, Benowitz NL, Spindel ER. Prenatal nicotine exposure alters pulmonary function in newborn rhesus monkeys. Am J Respir Crit Care Med 164: 989–994, 2001. [DOI] [PubMed] [Google Scholar]
- 25.Sierra H, Sakurai R, Lee WN, Truong NC, Torday JS, Rehan VK. Prenatal rosiglitazone administration to neonatal rat pups does not alter the adult metabolic phenotype. PPAR Res 2012: 604216, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Torday JS, Rehan VK. Stretch-stimulated surfactant synthesis is coordinated by the paracrine actions of PTHrP and leptin. Am J Physiol Lung Cell Mol Physiol 283: L130–L135, 2002. [DOI] [PubMed] [Google Scholar]
- 27.Truong NC, Abbasi A, Sakurai R, Lee WN, Torday JS, Rehan VK. Postnatal rosiglitazone administration to neonatal rat pups does not alter the young adult metabolic phenotype. Neonatology 101: 217–224, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.WHO. The tobacco atlas. Tobacco Free Initiative, June 2003 (Online). http://www.who.int/tobacco/resources/publications/tfi_final_26Jan.pdf[20 August 2014].
- 29.Wongtrakool C, Wang N, Hyde DM, Roman J, Spindel ER. Prenatal nicotine exposure alters lung function and airway geometry through alpha7 nicotinic receptors. Am J Respir Cell Mol Biol 46: 695–702, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yaris F, Yaris E, Kadioglu M, Ulku C, Kesim M, Kalyoncu NI. Normal pregnancy outcome following inadvertent exposure to rosiglitazone, gliclazide, and atorvastatin in a diabetic and hypertensive woman. Reprod Toxicol 18: 619–621, 2004. [DOI] [PubMed] [Google Scholar]