

Abstract
New therapies toward heart and blood vessel disorders may emerge from the development of Hsp90 inhibitors. Several independent studies suggest potent anti-inflammatory activities of those agents in human tissues. The molecular mechanisms responsible for their protective effects in the vasculature remain unclear. The present study demonstrates that the transcription factor p53, an Hsp90 client protein, is crucial for the maintenance of vascular integrity, protects again LPS-induced endothelial barrier dysfunction, and is involved in the mediation of the anti-inflammatory activity of Hsp90 inhibitors in lung tissues. p53 silencing by siRNA decreased transendothelial resistance (a measure of endothelial barrier function). A similar effect was induced by the p53 inhibitor pifithrin, which also potentiated the LPS-induced hyperpermeability in human lung microvascular endothelial cells (HLMVEC). On the other hand, p53 induction by nutlin suppressed the LPS-induced vascular barrier dysfunction. LPS decreased p53 expression in lung tissues and that effect was blocked by pretreatment with Hsp90 inhibitors both in vivo and in vitro. Furthermore, the Hsp90 inhibitor 17-allyl-amino-demethoxy-geldanamycin suppressed the LPS-induced overexpression of the p53 negative regulator MDMX as well as p53 and MDM2 (another p53 negative regulator) phosphorylation in HLMVEC. Both negative p53 regulators were downregulated by LPS in vivo. Chemically induced p53 overexpression resulted in the suppression of LPS-induced RhoA activation and MLC2 phosphorylation, whereas p53 suppression caused the opposite effects. These observations reveal new mechanisms for the anti-inflammatory actions of Hsp90 inhibitors, i.e., the induction of the transcription factor p53, which in turn can orchestrate robust vascular anti-inflammatory responses both in vivo and in vitro.
Keywords: acute lung injury, hsp90, human pulmonary endothelium, lung barrier function, p53
lipopolysaccharide (LPS) is the major component of the outer wall of gram-negative bacteria. It activates macrophages, neutrophils, dendritic, and other cells that induce inflammation, oxidative stress, and endothelial damage (6). Exposure to LPS leads to endothelial barrier dysfunction, increased endothelial permeability, acute lung injury, and acute respiratory distress syndrome (1). We have recently reported that heat shock protein 90 (Hsp90) is an important positive regulator of these effects of LPS and that inhibitors of Hsp90 prevent and repair the LPS-induced cellular damage, both in vitro and in vivo (3, 26, 59).
Hsp90 is a highly conserved cellular chaperone and one of the most abundant proteins in eukaryotic cells (27). It represents 1–2% of total cellular proteins and participates in the stabilization and activation of more than 200 “client” proteins (61). Hsp90 forms multichaperone complexes with a variety of cochaperones and proinflammatory client proteins. These complexes cycle between an open and a closed conformation of the dimeric Hsp90. Hsp90 inhibitors, such as 17-allyl-amino-demethoxy-geldanamycin (17-AAG), lock the complex in the open state, leading to client protein deactivation, destabilization, and proteasomal degradation (52).
p53 serves as an Hsp90 client protein able to regulate major metabolic pathways and cytokines, which are required for cellular growth and defense (34, 51). Hsp90 binds to a folded, nativelike conformation of p53 in vitro with micromolar affinity. The DNA-binding domain of p53 and the middle and carboxy-terminal domains of Hsp90 are responsible for this interaction, which is essential to stabilize p53 at physiological temperatures and to prevent it from irreversible thermal inactivation (44). In fact, multiple DNA binding domains interact with multiple Hsp90 sites (21).
p53 was discovered 30 years ago as a cellular partner of simian virus 40 large T antigen, an oncoprotein of this tumor virus. A decade later, it became clear that p53 is actually a tumor suppressor that is frequently mutated in most human cancers, is induced by stress, and promotes cell cycle arrest, apoptosis, and senescence (31). Additionally, p53 is involved in the induction of anti-inflammatory activities in various tissues (30, 35). Thus recent studies from several groups, including ours, have revealed a potent anti-inflammatory action of Hsp90 inhibitors in vascular tissues that involves modulation of Hsp90 activation (3, 16, 26). The molecular mechanisms that drive those effects remain unclear. We therefore tested the hypothesis that at least part of the anti-inflammatory effects of Hsp90 inhibitors may be mediated by the “guardian of the genome” and tumor suppressor protein, p53 (32).
MATERIALS AND METHODS
Reagents.
17-AAG was obtained from the National Cancer Institute (Bethesda, MD). Anti-Hsp90 antibody (624088) was purchased from BD Biosciences Transduction Laboratories (San Jose, CA). MDMX (sc-74468) antibody, p53 siRNA (sc-29435), and control siRNA (sc-37007) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). p53 (9282s), p(s15)53 (9284p), p(s392)53 (9281P), p21 Waf/Cip (2947s), pMDM2 (3521s), p-myosin light chain 2 (3674s), and RhoA (8789s) antibodies were obtained from Cell Signaling (Danvers, MA). β-Actin antibody (P8999), CelLyticM Lysis Reagent (C2978), nutlin-3a (SML0580), pifithrin a (P4359), and EZview Red Protein Affinity Gel beads (P6486) were purchased from Sigma-Aldrich (St. Louis, MO). Secondary mouse and rabbit antibodies were purchased from Licor (Lincoln, NE). Oligofectamine (12252011), Pierce BCA protein assay, and nitrocellulose membranes were obtained from Fisher Scientific (Pittsburgh, PA).
Animals.
Seven- to 8-wk-old male C57BL/6 mice from Jackson Laboratories were used in all experiments. Mice were maintained under pathogen-free conditions in a 12:12-h light-dark cycle. All animal care and experimental procedures were approved by the Old Dominion University IACUC and were in line with the principles of humane animal care adopted by the American Physiological Society.
Cell culture.
In house harvested human lung microvascular endothelial cells (HLMVEC) were maintained in M199 media supplemented with 20% FBS and antibiotics/antimycotics, as described previously (5).
Measurement of endothelial barrier function.
The barrier function of endothelial cell monolayers grown on electrode arrays (8W10E+) was estimated by the electric cell-substrate impedance sensing (ECIS) method, as previously published (3), by using an ECIS model 1600R from Applied BioPhysics. Experiments were conducted on wells that achieved at least 800-Ω baseline, steady-state resistance.
Immunoprecipitation and transfections.
Confluent cell culture dishes were placed on ice and washed three times with ice-cold PBS. PBS was removed and ice-cold lysis buffer was added. Adherent cells were scraped and the cell suspension was transferred to a cold microcentrifuge tube. Constant agitation was maintained for 30 min at 4°C and lysates were centrifuged for 30 min at 4°C. The protein concentration in supernatant was calculated by the BCA protein assay; 5 μg of appropriate antibody was added to lysates containing 500 μg protein, which were incubated overnight under agitation. The next day, 50 λ of the protein affinity gel beads were added to the lysates and the mixture was incubated at 4°C under rotary agitation. The mixture was then washed 4× with lysis buffer, and the last supernatant was removed and replaced with 1× sample buffer. The proteins were denatured and separated from the beads by 5 min boiling at 100°C. The lysates where then subjected to SDS-PAGE electrophoresis. Transfections were performed according to a standard protocol that has been previously published (3).
RhoA activity assay.
RhoA activation was detected by the Cell Signaling kit (no. 8820); the final step of the GTP-bound RhoA detection was performed by using a monoclonal rabbit RhoA antibody (Cell Signaling, no. 2117). Briefly, 500 μg of cell lysates were incubated with GST-Rhotekin-RBD fusion protein and were coupled to glutathione resin. After precipitation, the complexes were washed four times with the lysis buffer, eluted in SDS-PAGE sample buffer, immunoblotted, and probed with RhoA antibody. Aliquots were taken from supernatants prior to precipitation and were used to quantify total RhoA.
Protein isolation and Western blot analysis.
Proteins were isolated from cells or tissues using CelLyticM Lysis Reagent or RIPA buffer and concentration was determined by the BCA method according to manufacturer's instructions. Protein-matched samples (40 μg per lane) were separated by electrophoresis through 12% SDS-PAGE Tris·HCl gels. Wet transfer was used to transfer the proteins onto nitrocellulose membranes. The membranes were incubated for 1 h at room temperature in 5% nonfat dry milk in Tris-buffered saline-0.1% (vol/vol) Tween 20. The blots were then incubated at 4°C overnight with the appropriate antibody. The signal for the immunoreactive proteins was developed by using the appropriate secondary antibody and was visualized in a LICOR Odyssey CLx imaging system.
In vivo experiments.
Stock solutions of Escherichia coli LPS were prepared in saline. Mice received either vehicle (saline) or LPS (3,000 unit/g body wt, intratracheally) 24 h before receiving vehicle (10% DMSO in saline) or the Hsp90 inhibitor AUY922 (AUY; 10 μg/g body wt dissolved in 10% DMSO), intraperitoneally. Mice were euthanized 48 h later (i.e., 72 h after LPS) by cervical dislocation, and the lungs were flushed with 5 ml of ice-cold PBS (5 mM EDTA), excised, dipped in saline, blotted dry, quickly snap frozen in liquid nitrogen, crushed to powder in a prechilled mortar, and stored at −80°.
Densitometry/statistical analysis.
Image J software (National Institutes of Health) was used to perform densitometry of immunoblots. All data are expressed as mean values ± SE (standard error of mean). Student's t-test or one-way or two-way ANOVA with Bonferroni post hoc test was performed to determine statistically significant differences among groups. A value of P < 0.05 was considered significant. GraphPad Prism 4 (version 4.03, Graph Pad Software) was used for data analysis; n represents the number of experimental repeats.
RESULTS
p53 inhibition reduces endothelial barrier integrity.
HLMVEC were seeded on gold electrode arrays and were exposed to DMSO (vehicle) or 25 or 50 μM of the p53 expression inhibitor pifithrin. Figure 1A shows that pifithrin induced a concentration-dependent decrease in p53 expression. Furthermore, DMSO-treated cells maintained constant transendothelial resistance (TER) values, whereas cells exposed to 50 μM pifithrin exhibited a significant reduction in TER values (Fig. 1B).
Fig. 1.

Effects of p53 expression on endothelial barrier dysfunction. A: Western blot analysis of p53 expression after 4 h treatment with either vehicle (DMSO; VEH) or 25 μM or 50 μM pifithrin (PTH) in human lung microvascular endothelial cells (HLMVEC). Blot shown is representative of 3 independent experiments. Signal intensity of p53 was analyzed by densitometry. Protein levels were normalized to β-actin. *P < 0.05 vs. vehicle, **P < 0.01 vs. vehicle. Means ± SE. B: vehicle (DMSO) or PTH (25 μM, 50 μM) was added to the media of confluent HLMVEC monolayers at 0 h. PTH (50 μM) caused a reduction in transendothelial resistance (TER). ****P < 0.0001 vs. corresponding untreated cell group; n = 4 per group. Means ± SE. C: cells were pretreated for 4 h with either vehicle (DMSO) or 50 μM PTH and were exposed to LPS (1 EU/ml, at arrow). A gradual increase in endothelial permeability (reduced TER) was observed in LPS-treated cells, which was more pronounced in PTH-treated cells. ****P < 0.0001 vs. vehicle, ####P < 0.0001 vs. 50 μM PTH alone, $$P < 0.01 vs. VEH+LPS, &&P < 0.01 vs. VEH; n = 4 per group. Means ± SE. D: HLMVEC pretreated with either vehicle (DMSO) or 10 μM nutlin for 4 h were exposed to LPS (1 EU/ml, at arrow). LPS did not induce a significant increase in endothelial permeability (reduced TER) in nutlin-treated cells. However, vehicle-pretreated cells exhibited reduced TER values after LPS exposure. ****P < 0.0001 vs. VEH, n = 3 per group. Means ± SE (left). Western blot analysis of p53 expression in HLMVEC after 4 h treatment with either vehicle (DMSO) or 10 μM nutlin. Blot shown is representative of 3 independent experiments. Signal intensity of p53 was analyzed by densitometry. Protein levels were normalized to β-actin. *P < 0.05 vs. vehicle. Means ± SE. (right) E: HLMVEC were transfected with control (irrelevant) siRNA (siCtr) or p53 siRNA (si p53) (t = 0). p53 siRNA-treated cells exhibited reduced TER values after transfection. ****P < 0.0001 vs. siCtr, n = 4 per group. Means ± SE (left). Western blot analysis of p53 expression in HLMVEC 48 h after siCtr or sip53 transfection. Blot shown is representative of 3 independent experiments. Signal intensity of p53 was analyzed by densitometry. Protein levels were normalized to β-actin. *P < 0.05 vs. control siRNA. Means ± SE (right).
Effects of p53 inhibition by pifithrin on LPS-induced endothelial barrier dysfunction.
HLMVEC were seeded on gold electrode arrays and were exposed to either 10% DMSO (vehicle) or 50 μM pifithrin before a 4-h treatment with PBS (vehicle) or LPS (1 EU/ml). PBS-treated cells maintained constant TER values, in contrast to the pifithrin-treated cells, which exhibited reduced TER (Fig. 1C). LPS profoundly reduced TER in both vehicle and pifithrin-treated cells; however, the LPS-induced decrease in TER values was far greater in pifithrin-treated cells (Fig. 1C).
Effects of p53 induction by nutlin on endothelial barrier dysfunction.
Figure 1D, top right, demonstrates that p53 expression increased significantly in cells treated for 4 h with 10 μM nutlin. In additional experiments, HLMVEC were seeded on gold electrode arrays and were exposed to DMSO (vehicle) or 10 μM nutlin for 4 h before PBS (vehicle) or LPS (1 EU/ml) treatment. As shown in Fig. 1D, cells treated with PBS maintained stable TER values (Fig. 1E). The addition of LPS significantly reduced TER in vehicle-treated cells, but not in cells pretreated with the p53 inducer nutlin.
Effect of p53 silencing on HLMVEC TER.
Figure 1E, top right, demonstrates that p53 expression was significantly reduced by p53 silencing with a specific siRNA. In additional experiments, HLMVEC were seeded on gold electrode arrays and were exposed to control siRNA or p53 siRNA (t = 0). As shown in Fig. 1E, cells treated with control siRNA maintained stable TER values. On the other hand, cells subjected to p53 siRNA treatment exhibited significantly reduced TER values.
Suppression of p53 expression by LPS.
HLMVEC were exposed to either vehicle (PBS) or LPS (1 EU/ml) for 2 h. Cells exposed to LPS exhibited decreased p53 expression compared with the vehicle-treated cells (Fig. 2A).
Fig. 2.

Effects of LPS and 17-allyl-amino-demethoxy-geldanamycin (17-AAG) on p53 and p21 expression in HLMVEC. A: Western blot analysis of p53 levels in HLMVEC after 2 h treatment with vehicle or LPS. Blot shown is representative of 3 independent experiments. Signal intensity of p53 was analyzed by densitometry. Protein levels were normalized to β-actin. **P < 0.01 vs. vehicle. Means ± SE. B: Western blot analysis of p53 levels in HLMVEC after 4 h or 16 h treatment with 17-AAG; 10% DMSO was used as vehicle. Signal intensity of p53 was analyzed by densitometry. Protein levels were normalized to β-actin. ***P < 0.001 vs. vehicle. Means ± SE. C: Western blot analysis of p21 levels in HLMVEC after 2 h treatment with vehicle or LPS. Blot shown is representative of 3 independent experiments. Signal intensity of p21 was analyzed by densitometry. Protein levels were normalized to β-actin. *P < 0.05 vs. vehicle. Means ± SE. D: Western blot analysis of p21 levels in HLMVEC after 8 h treatment with either vehicle (10% DMSO) or 17-AAG. Signal intensity of p21 was analyzed by densitometry. Protein levels were normalized to β-actin. **P < 0.01 vs. vehicle. Means ± SE.
Induction of p53 expression by 17-AAG.
HLMVEC were treated with either vehicle (DMSO) or the Hsp90 inhibitor 17-AAG (1 μM) for 4 or 16 h. As shown in Fig. 2B, 17-AAG treatment for either time period significantly increased p53 expression compared with vehicle-treated cells.
Suppression of p21 expression by LPS.
HLMVEC were exposed to either vehicle (PBS) or LPS (1 EU/ml) for 2 h. Cells exposed to LPS demonstrated decreased expression of the p53 target, p21, compared with vehicle-treated cells (Fig. 2C).
Induction of p21 expression by 17-AAG.
HLMVEC were treated with either vehicle (DMSO) or the Hsp90 inhibitor 17-AAG (1 μM) for 8 h. As shown in Fig. 2D, p21 expression was significantly induced in 17-AAG treated cells.
Effects of LPS and AUY on p53 and p21 expression in mouse lung.
Mice received either vehicle or LPS (intratracheally) and were euthanized 24 h after LPS treatment. Lungs of LPS-treated mice exhibited reduced p53 expression compared with corresponding controls (Fig. 3A). In additional studies, mice received either vehicle or the Hsp90 inhibitor AUY intraperitoneally 24 h after vehicle or LPS (intratracheally) and tissues were analyzed 72 h after LPS. As shown in Fig. 3, lungs from LPS-treated mice exhibited significantly lower expression of p53, even after 72 h. AUY reversed the LPS-induced p53 downregulation (Fig. 3B) and induced p21 expression (Fig. 3C), in vivo.
Fig. 3.

Effects of LPS and 17-AAG on p53 and p21 expression in mouse lung. A: Western blot analysis of p53 levels in lungs retrieved from mice 24 h after treatment with LPS. Blot shown is representative of 3 experiments per group. Signal intensity of p53 was analyzed by densitometry. Protein levels were normalized to β-actin. *P < 0.05 vs. VEH. Means ± SE. B: Western blot analysis of p53 levels in lungs retrieved from mice 72 h after treatment with LPS or vehicle and posttreated (24 h after LPS) with AUY920 (AUY) or vehicle (10% DMSO). Blot shown is representative of 3 experiments per group. Signal intensity of p53 was analyzed by densitometry. Protein levels were normalized to β-actin. *P < 0.05 vs. LPS. Means ± SE. C: Western blot analysis of p21 levels in lungs retrieved from mice 72 h after treatment with LPS or vehicle and posttreated (24 h after LPS) with AUY or vehicle (10% DMSO). Blot shown is representative of 3 experiments per group. Signal intensity of p21 was analyzed by densitometry. Protein levels were normalized to β-actin. *P < 0.05, **P < 0.01 vs. vehicle. Means ± SE. D: Western blot analysis of MDM2 expression in lungs retrieved from mice 72 h after treatment with LPS or vehicle and posttreated (24 h after LPS) with AUY or vehicle (10% DMSO). Blot shown is representative of 3 experiments per group. Signal intensity of p53 was analyzed by densitometry. Protein levels were normalized to β-actin. *P < 0.05, **P < 0.01 vs. VEH. Means ± SE. E: Western blot analysis of MDMX levels in lungs retrieved from mice 72 h after treatment with LPS or vehicle and posttreated (24 h after LPS) with AUY or vehicle (10% DMSO). Blot shown is representative of 3 experiments per group. Signal intensity of p53 was analyzed by densitometry. Protein levels were normalized to β-actin. *P < 0.05 vs. VEH. Means ± SE.
Effects of LPS and AUY on MDM2 and MDMX expression in mouse lung.
Mice received either vehicle or the Hsp90 inhibitor AUY intraperitoneally 24 h after vehicle or LPS (intratracheally) and tissues were analyzed 72 h after LPS. As shown in Fig. 3, lungs from LPS-treated mice exhibited significantly higher expression of MDM2 and MDMX. AUY reversed the LPS-induced MDM2 (Fig. 3D) and MDMX (Fig. 3E) upregulation in vivo.
LPS reduces and 17-AAG increases the abundance of Hsp90/p53 complexes in HLMVEC.
HLMVEC were exposed to either vehicle, LPS (1 EU/ml) for 2 h, or 17-AAG for 8 h. LPS reduced and 17-AAG increased p53 expression (Fig. 4, top). Immunoblotting of samples that were immunoprecipitated with p53 antibody showed that LPS-treated cells contained less of the Hsp90/p53 complex than untreated controls. On the other hand, 17-AAG induced the abundance of that complex (Fig. 4, bottom left). However, when expressed as the ratio of p53 to Hsp90, to reflect the binding affinity of the components in the complex, neither LPS nor 17-AAG exhibited any effect.
Fig. 4.

17-AAG increases and LPS reduces the abundance of Hsp90/p53 complexes in HLMVEC. Top: Western blot analysis of p53 in HLMVEC treated with either vehicle (10% DMSO) or LPS for 2 h or 17-AAG for 8 h. Signal intensity of p53 was analyzed by densitometry. Protein levels were normalized to β-actin. *P < 0.05, **P < 0.01, vs. vehicle. Means ± SE. Bottom: Western blot analysis of samples at top, after immunoprecipitation (IP) with antibody against p53 and immunoblotting (IB) for p53 and Hsp90. Each blot is representative of 3 independent experiments. *P < 0.05, ****P < 0.0001, vs. vehicle. Means ± SE.
LPS increases the phosphorylation of MDM2 at Ser166. HLMVEC were treated with vehicle or 1 EU/ml LPS for 0.25 to 4 h. LPS induced Ser166 phosphorylation of the negative regulator of p53, MDM2, at each time point. Densitometric analysis of immunoblots revealed that maximal increase in pMDM2 occurred after 1 h exposure to LPS (Fig. 5A, top).
Fig. 5.
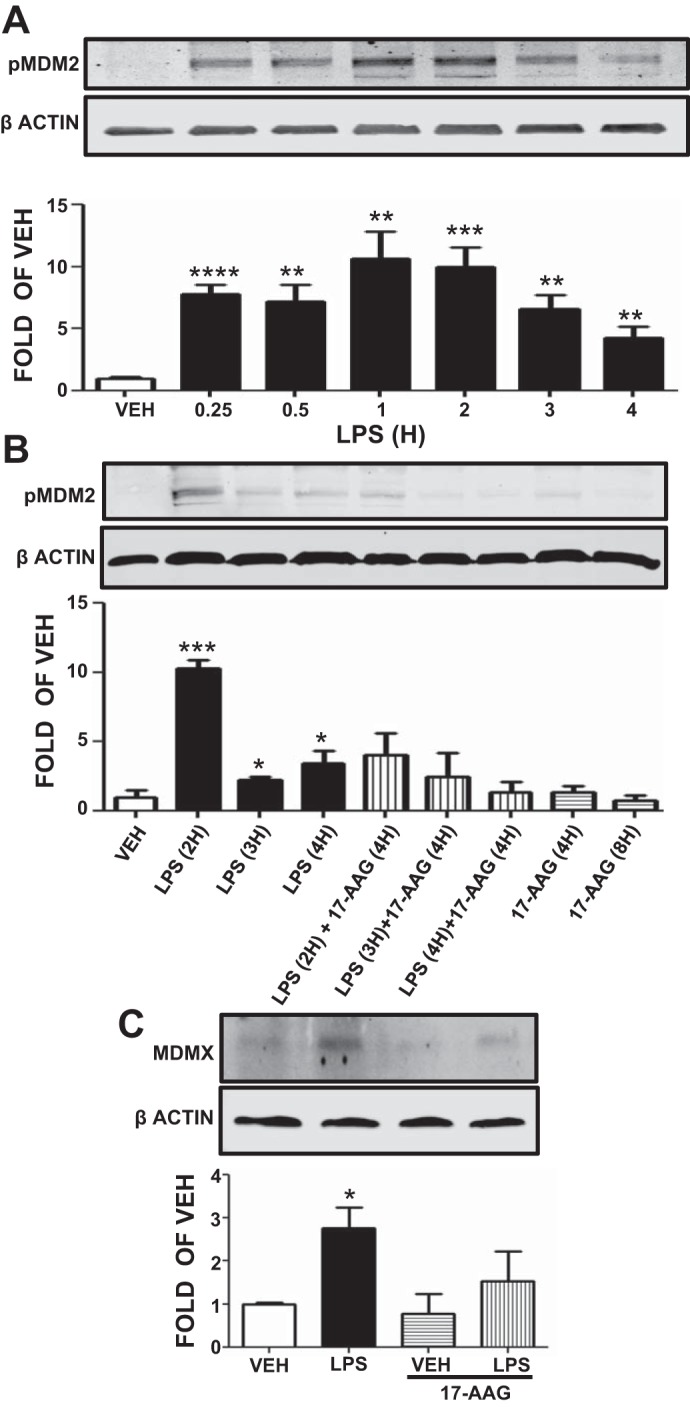
Effects of LPS and 17-AAG on MDM2 phosphorylation and MDMX expression. A: Western blot analysis of pMDM2 levels after treatment of HLMVEC with vehicle or LPS. Blot shown is representative of 4 independent experiments. Signal intensity of pMDM2 and β-actin was analyzed by densitometry. Protein levels were normalized to β-actin. **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. vehicle. Means ± SE. B: Western blot analysis of pMDM2 levels in HLMVEC treated with LPS or vehicle and pretreated with 17-AAG or vehicle (10% DMSO). Blot shown is representative of 3 independent experiments. Signal intensity of pMDM2 was analyzed by densitometry. Protein levels were normalized to β-actin. *P < 0.05, ***P < 0.001 vs. vehicle. Means ± SE. C: Western blot analysis of MDMX levels in HLMVEC treated with LPS or vehicle and pretreated with 17-AAG or vehicle (10% DMSO). Blot shown is representative of 3 independent experiments. Signal intensity of MDMX was analyzed by densitometry. Protein levels were normalized to β-actin. *P < 0.05 vs. vehicle. Means ± SE.
17-AAG reduces the LPS-induced MDM2 phosphorylation. HLMVEC were treated for 4 or 8 h with either vehicle (DMSO) or 17-AAG (1 μM) before LPS or vehicle treatment (2, 3, or 4 h). The results shown in Fig. 5B, demonstrate that LPS induced the phosphorylation of MDM2 and that 17-AAG was able to suppress that effect.
LPS induces and 17-AAG blocks MDMX expression. HLMVEC were treated with either vehicle (DMSO) or 17-AAG (1 μM) for 4 h before LPS (1 EU/ml) or vehicle treatment (2 h). Results shown in Fig. 5C demonstrate that LPS induced and 17-AAG blocked the expression of another p53 negative regulator, MDMX (Fig. 5C).
LPS induces and 17-AAG suppresses p53 phosphorylation. p53 phosphorylation increases its targeting to MDM2 and its degradation. HLMVEC were treated with vehicle, LPS (0.25–4 h), or 17-AAG (1 μM; 4 or 16 h). As shown in Fig. 6A, LPS induced p53 phosphorylation at S15 (Fig. 6, B and C) and S392 (Fig. 6, D and E), whereas 17-AAG decreased p53 phosphorylation (Fig. 6, A–D). The maximal effect of LPS on p53 phosphorylation occurred at 0.25 h (Fig. 6, B and D) or 2 h (Fig. 6, C and E). As shown before, p53 expression levels were reduced by LPS and induced by 17-AAG (Fig. 6, A and F).
Fig. 6.

LPS induces p53 phosphorylation. A: Western blot analysis of p-p53Ser15, p-p53Ser392, and p53 levels after treatment of HLMVEC with vehicle (10% DMSO), 1 EU/ml LPS, or 17-AAG. Blot shown is representative of 3 independent experiments. Signal intensity of p-p53Ser15 (B), p-p53Ser392 (C), and p53 (D) was analyzed by densitometry. Protein levels were normalized to p53 (B and C) or β-actin (C, E, and F). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. vehicle. Means ± SE.
17-AAG counteracts the LPS-induced p53 phosphorylation.
HLMVEC were pretreated with either vehicle or 17-AAG (1 μM) for 4 h before LPS (1 EU/ml, for 0.25 h) or vehicle treatment. LPS induced p(Ser15)p53 (Fig. 7B) and p(Ser392)p53 (Fig. 7D) expression. 17-AAG suppressed both (Fig. 7, B–E). Additionally, 17-AAG significantly suppressed the baseline phosphorylation at both Ser15 and Ser392
Fig. 7.

17-AAG suppresses the LPS-induced p53 phosphorylation. A: Western blot analysis of p-p53Ser15, p-p53Ser392, and p53 expression levels in HLMVEC treated with LPS or vehicle and pretreated with 17-AAG or vehicle (10% DMSO). Blot shown is representative of 4 independent experiments. Signal intensity of p-p53Ser15 (B and C) and p-p53392 (D and E) was analyzed by densitometry. Protein levels were normalized to p53 (B and D) or β (C and E) actin. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 vs. corresponding vehicle-treated cells. NS, nonsignificant. Means ± SE.
p53 negatively regulates RhoA activation.
RhoA activation is a major pathway leading to endothelial barrier dysfunction. We investigated whether the beneficial effects of p53 on endothelial barrier function involve suppression of RhoA activation. HLMVEC were treated for 4 h with vehicle, 50 μM pifithrin, or 10 μM nutlin prior to vehicle or LPS treatment (2 h). LPS induced RhoA activation. Nutlin reduced baseline RhoA activity and completely blocked LPS-induced RhoA activation. Conversely, pifithrin significantly increased the LPS-induced RhoA activation (Fig. 8A).
Fig. 8.
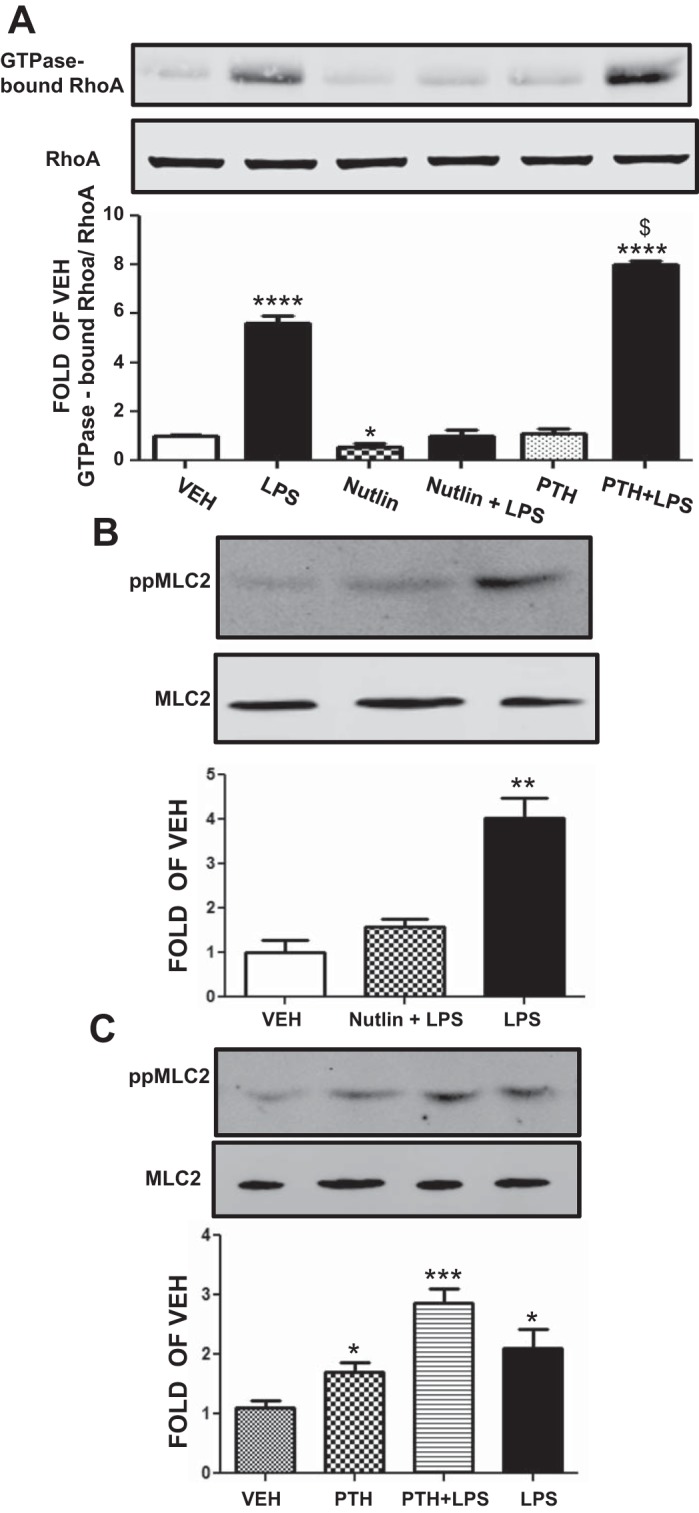
Effects of p53 manipulation on RhoA activity and MLC2 phosphorylation. A: Western blot analysis of RhoA activity in HLMVEC pretreated with vehicle (10% DMSO), PTH (4 h), or nutlin (4 h) prior to vehicle (saline) or LPS treatment (2 h). Blot shown is representative of 3 independent experiments. Signal intensity of GTPase bound RhoA was analyzed by densitometry. The GTPase-bound RhoA protein levels were normalized to total RhoA. *P < 0.05, ****P < 0.0001 vs. vehicle. $P < 0.001 vs. LPS-treated cells. Means ± SE. B: Western blot analysis of phosphorylated MLC2 (ppMLC2) levels in HLMVEC treated with LPS (2 h) or vehicle (saline) and pretreated with nutlin (4 h) or vehicle (10% DMSO). Blot shown is representative of 3 independent experiments. Signal intensity of ppMLC2 and MLC2 was analyzed by densitometry. Protein levels were normalized to MLC2. **P < 0.01 vs. vehicle. Means ± SE. C: Western blot analysis of ppMLC2 levels in HLMVEC treated with LPS (2 h) or vehicle (saline) and pretreated with PTH (4 h) or vehicle (10% DMSO). Blot shown is representative of 4 independent experiments. Signal intensity of ppMLC2 and MLC2 was analyzed by densitometry. Protein levels were normalized to MLC2. *P < 0.05, ***P < 0.001 vs. vehicle. Means ± SE.
p53 induction blocks the LPS-induced MLC2 phosphorylation.
HLMVEC were pretreated with either vehicle or nutlin for 4 h before LPS treatment. LPS induced MLC2 phosphorylation and nutlin pretreatment suppressed that induction (Fig. 8B).
p53 suppression potentiates the LPS-induced MLC2 phosphorylation.
HLMVEC were pretreated with either vehicle or pifithrin for 4 h before LPS or vehicle treatment. LPS induced MLC2 phosphorylation, which was further induced by pifithrin pretreatment (Fig. 8C).
DISCUSSION
Hsp90 is a major regulator of important physiological processes, through the stabilization and activation of various enzymes and transcriptions factors (8). Hsp90 inhibitors were initially developed to fight cancer, but recent reports suggest that they may also have a beneficial role in other diseases, since they possess strong anti-inflammatory properties (2, 3, 6).
p53 is an Hsp90 client protein that functions as a node in numerous intracellular pathways and regulates important biological activities from fertility and development to maintaining genomic stability and cell death (45). It has been recently suggested that it is also involved in the mediation of anti-inflammatory responses, since p53 can drive inflammatory cell apoptosis in vivo (25) and there is a reciprocal negative regulation between p53 and NF-κB (10, 18). Increased NF-κB activation results in decreased p53 activity and vice versa. Proinflammatory NF-κB-induced cytokines such as IL-6 and macrophage migration inhibitory factor reduce p53 transcriptional activity (23, 46, 68) and agents that downregulate NF-κB cause p53 activation (67). p53 is also known to suppress the cyclooxygenase 2 gene (58) and to antagonize pp60src-induced cell migration and proliferation in atherosclerosis (43). Additionally, neutrophils and macrophages from p53 knockout mice exhibit elevated responses to LPS stimulation, stronger induction of proinflammatory cytokines, and enhanced NF-κB DNA binding activity; the p53 knockouts are more susceptible to LPS-induced acute lung injury, compared with wild type (35). Autoimmune diseases, including collagen-induced arthritis (67) and experimental autoimmune encephalitis (48), are more severe in p53-deficient mice. Inflammatory cell infiltration into the lung and subsequent disruption of alveolar architecture caused by chronic exposure to bleomycin are markedly increased in p53-null mice and in transgenic mice expressing the mutant p53 in the lung, compared with wild-type mice (9, 15). Ionizing radiation induces faster and stronger invasion of inflammatory cells and fibroblasts into damaged tissues in p53-null mice than in wild type (29). Mice with a p53 P72R mutation have a markedly enhanced response to inflammatory challenges (11).
The present study reports for the first time that p53 expression levels are crucial for the maintenance of lung vascular integrity. Overexpression of p53 by nutlin, an inhibitor of the MDM2-p53 interaction (42), protected against LPS-induced lung barrier dysfunction. On the other hand, inhibition of p53 transcriptional activity and stability by pifithrin (28) resulted in the potentiation of LPS-induced barrier dysfunction. Pifithrin has been shown to also induce NF-κB activity, via the reciprocal regulation of NF-κB with p53 (49). Additionally, inhibition of the p53 gene expression by siRNA confirmed the importance of p53 on barrier integrity, since treated cells exhibited increased permeability (Fig. 1E). Furthermore, LPS was able to reduce the expression of p53 both in vitro and in vivo and that effect was blocked by Hsp90 inhibitors. We have employed two different Hsp90 inhibitors that represent two different generations of these compounds. 17-AAG is a geldanamycin derivative and it is the first Hsp90 inhibitor selected by for clinical use; AUY is a isoxazole derivative that is now being developed and is undergoing clinical trials in cancer patients (24). The key role of p53 in cellular defense against inflammatory stimuli was further demonstrated in a study showing that the activation of lung p53 by nutlin prevents and reverses pulmonary hypertension and that hypoxia-exposed p21-null mice exhibit exacerbated pulmonary hypertension compared with wild type (42).
We also investigated the effects of LPS and 17-AAG on p21 expression, since p21, the direct downstream target of p53, is involved in the vascular defense against pathogens. p21-null mice demonstrate an overactive inflammatory phenotype that is similar to that of p53-null mice and display increased susceptibility to endotoxic shock and increased serum levels of cytokines. Elevated NF-κB activity and secretion of cytokines was detected in LPS-stimulated p21-deficient mice and in human macrophages compared with similarly treated wild-type cells (55, 60). Our study shows that LPS reduces p21 expression in vitro, an effect that probably contributes to the LPS-induced endothelial barrier dysfunction. That effect was opposed by Hsp90 inhibitors (Figs. 2 and 3).
Furthermore, we show for the first time that Hsp90 forms a complex with p53 in HLMVEC and that the abundance, but not the affinity, of that complex in the cellular environment is influenced by both by LPS and 17-AAG (Fig. 4). When HLMVEC were treated with LPS the amount of the Hsp90-p53 complex was reduced, and this may be the cause of the lower p53 expression, as seen in Fig. 2A. On the other hand, 17-AAG induced the formation of that complex, which has been previously shown to partially stabilize p53 (21, 64, 65).
To further elucidate the mechanisms which regulate the p53 expression by LPS, we treated HLMVEC with LPS for various time points and evaluated the phosphorylation of the ubiquitin ligase MDM2, which plays a key role in the regulation of p53 stability by serving as its major negative regulator (31). LPS, an Akt activator (41), can significantly increase MDM2 phosphorylation, a posttranslational modification that is crucial for p53 degradation (40). Akt-mediated phosphorylation of MDM2 at Ser166 and Ser186 increases its interaction with p300, allowing MDM2-mediated ubiquitination and degradation of p53 (22, 39, 69). Phosphorylation of MDM2 also blocks its binding to p19ARF, thus increasing the degradation of p53 (17, 69).
To investigate the mechanisms by which 17-AAG induces p53 expression, we pretreated HLMVEC with 17-AAG prior to LPS treatment. 17-AAG suppressed the LPS-induced MDM2 phosphorylation. LPS also induced the expression of the p53 negative regulator MDMX (56), and this effect was opposed by 17-AAG pretreatment. p53 phosphorylation has been shown to regulate its activity (38). In the present work we evaluated the phosphorylation of p53 by LPS, since that posttranslational modification has been shown to accelerate the degradation of p53 through the polyubiquitination-proteasome pathway (19). LPS induced p53 phosphorylation at Ser15 and Ser392. On the other hand, 17-AAG suppressed both the basal p53 phosphorylation levels and the LPS-induced p53 phosphorylation at both sites. We thus conclude that 17-AAG inhibits Akt leading to reduced MDM2 phosphorylation, reduced p53 ubiquitination, degradation, and thus increased p53 survival.
The Rho family of GTPases regulates a wide variety of cellular processes, including apoptosis, cell cycle progression and migration. Although this family of protein is comprised of some 22 members, the best characterized are RhoA, RhoB, RhoC, Rac1, and Cdc42 and most of them are involved in the regulation of endothelial barrier function (7). RhoA is involved in the regulation of actomyosin contractility, cytokinesis, focal adhesion and cell polarity and has been shown to be activated by LPS (54, 57). In the present study we demonstrate that p53 negatively regulates the LPS-induced RhoA activation. Our results are in line with others' observations that p53 induction by nutlin reduces RhoA activity and that RhoA silencing increases p53 expression (37). Although pifithrin potentiated the LPS-induced RhoA activation, by itself it did not affect RhoA activity, even though it suppressed TER; this suggests that pifithrin may also affect endothelial barrier function through additional pathways. Loss of p53 has been also associated to the disruption of the RhoA signaling (12). Myosin light chain phosphorylation is a target of Rho kinase. Nutlin suppressed the LPS-induced MLC2 activation, and p53 inhibition potentiated this phosphorylation. MLC2 phosphorylation is crucial in the LPS-mediated endothelial barrier dysfunction (54).
Previous reports have suggested that p53 negatively modulates Rho GTPases (13, 14, 20) and restricts the Ras stimulation of RhoA as well as MLC2 phosphorylation (50). Furthermore, it was recently shown that, upon p53 loss, p190RhoGAP, a RhoA negative regulator, is inactivated, leading to RhoA activation (66). p53 can negatively regulate the expression of RhoA and Cdc42 effectors ROCK1, ROCK2, and MRCKa via transcriptional regulation of Notch1, which negatively regulates ROCK and MRCKa gene expression (33). Thus it has been suggested that p53 may function upstream of ROCK as a negative regulator of the Rho kinase activity (53).
Our study proposes that p53 possesses a protective role against LPS-induced lung endothelial barrier dysfunction. We suggest that LPS induced-Akt activation enhances pMDM2-mediated ubiquitination and degradation of p53 (47) as well as MDMX stabilization, which in turn results in p53 and p21 downregulation (36, 63). On the other hand, 17-AAG counteracts the LPS-induced p53 downregulation through MDMX and MDM2 degradation (62), as well as via Akt suppression (4). The 17-AAG-mediated p53 induction suppresses the pp60c-src activation (43) and induces p21 expression, which protects against LPS-induced responses, perhaps through negatively controlling NF-κB activation (60). Furthermore, our data suggest that p53 can negatively regulate LPS-induced RhoA activation. This working hypothesis is now depicted schematically in Fig. 9. In conclusion, our work reveals novel intracellular mechanisms responsible for the mediation of the anti-inflammatory role of Hsp90 in endothelial barrier dysfunction associated with acute lung injury and acute respiratory distress syndrome.
Fig. 9.
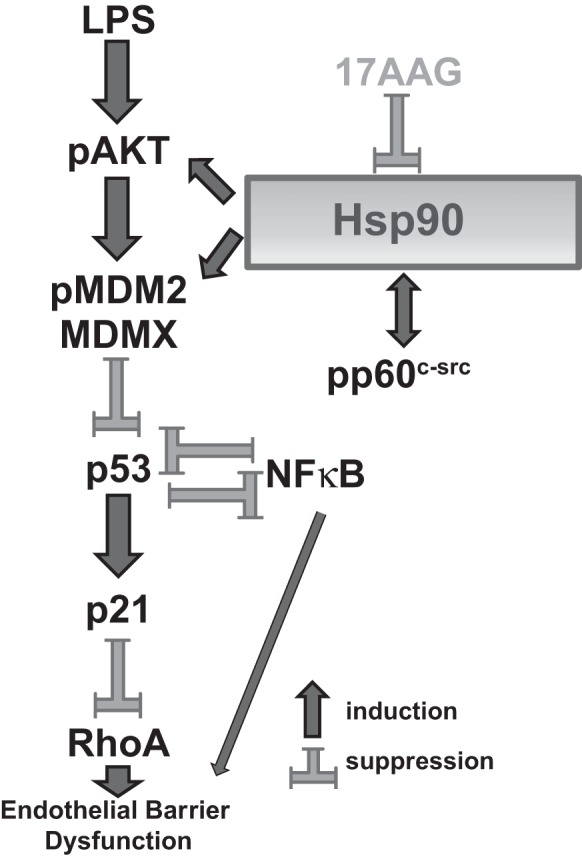
Working hypothesis. See discussion for detailed description.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grants HL101902 and HL93460. A portion of 17-AAG was provided by the National Cancer Institute.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
N.B. and J.D.C. conception and design of research; N.B., C.D., C.B., A.J., and G.T. performed experiments; N.B. and C.D. analyzed data; N.B., C.D., and J.D.C. interpreted results of experiments; N.B. prepared figures; N.B. drafted manuscript; N.B., C.D., C.B., A.J., G.T., and J.D.C. approved final version of manuscript; C.B., A.J., and J.D.C. edited and revised manuscript.
REFERENCES
- 1.Antonov A, Snead C, Gorshkov B, Antonova GN, Verin AD, Catravas JD. Heat shock protein 90 inhibitors protect and restore pulmonary endothelial barrier function. Am J Respir Cell Mol Biol 39: 551–559, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barabutis N, Catravas JD. Anti-inflammatory activity of Hsp90 inhibitors in the human vasculature. Med Surg Urol 2: e104, 2012. [Google Scholar]
- 3.Barabutis N, Handa V, Dimitropoulou C, Rafikov R, Snead C, Kumar S, Joshi A, Thangjam G, Fulton D, Black SM, Patel V, Catravas JD. LPS induces pp60c-src-mediated tyrosine phosphorylation of Hsp90 in lung vascular endothelial cells and mouse lung. Am J Physiol Lung Cell Mol Physiol 304: L883–L893, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem 277: 39858–39866, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Catravas JD, Snead C, Dimitropoulou C, Chang AS, Lucas R, Verin AD, Black SM. Harvesting, identification and barrier function of human lung microvascular endothelial cells. Vascul Pharmacol 52: 175–181, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chatterjee A, Snead C, Yetik-Anacak G, Antonova G, Zeng J, Catravas JD. Heat shock protein 90 inhibitors attenuate LPS-induced endothelial hyperpermeability. Am J Physiol Lung Cell Mol Physiol 294: L755–L763, 2008. [DOI] [PubMed] [Google Scholar]
- 7.Croft DR, Olson MF. Transcriptional regulation of Rho GTPase signaling. Transcription 2: 211–215, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med 16: 528–536, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis DW, Weidner DA, Holian A, McConkey DJ. Nitric oxide-dependent activation of p53 suppresses bleomycin-induced apoptosis in the lung. J Exp Med 192: 857–869, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev 246: 379–400, 2012. [DOI] [PubMed] [Google Scholar]
- 11.Frank AK, Leu JI, Zhou Y, Devarajan K, Nedelko T, Klein-Szanto A, Hollstein M, Murphy ME. The codon 72 polymorphism of p53 regulates interaction with NF-κB and transactivation of genes involved in immunity and inflammation. Mol Cell Biol 31: 1201–1213, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gadea G, de Toledo M, Anguille C, Roux P. Loss of p53 promotes RhoA-ROCK-dependent cell migration and invasion in 3D matrices. J Cell Biol 178: 23–30, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gadea G, Lapasset L, Gauthier-Rouviere C, Roux P. Regulation of Cdc42-mediated morphological effects: a novel function for p53. EMBO J 21: 2373–2382, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gadea G, Roger L, Anguille C, de Toledo M, Gire V, Roux P. TNFalpha induces sequential activation of Cdc42- and p38/p53-dependent pathways that antagonistically regulate filopodia formation. J Cell Sci 117: 6355–6364, 2004. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh S, Mendoza T, Ortiz LA, Hoyle GW, Fermin CD, Brody AR, Friedman M, Morris GF. Bleomycin sensitivity of mice expressing dominant-negative p53 in the lung epithelium. Am J Respir Crit Care Med 166: 890–897, 2002. [DOI] [PubMed] [Google Scholar]
- 16.Grandel U, Grimminger F. Endothelial responses to bacterial toxins in sepsis. Crit Rev Immunol 23: 267–299, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Grossman SR, Perez M, Kung AL, Joseph M, Mansur C, Xiao ZX, Kumar S, Howley PM, Livingston DM. p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol Cell 2: 405–415, 1998. [DOI] [PubMed] [Google Scholar]
- 18.Gudkov AV, Gurova KV, Komarova EA. Inflammation and p53: a tale of two stresses. Genes Cancer 2: 503–516, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gully CP, Velazquez-Torres G, Shin JH, Fuentes-Mattei E, Wang E, Carlock C, Chen J, Rothenberg D, Adams HP, Choi HH, Guma S, Phan L, Chou PC, Su CH, Zhang F, Chen JS, Yang TY, Yeung SC, Lee MH. Aurora B kinase phosphorylates and instigates degradation of p53. Proc Natl Acad Sci USA 109: E1513–E1522, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo F, Gao Y, Wang L, Zheng Y. p19Arf-p53 tumor suppressor pathway regulates cell motility by suppression of phosphoinositide 3-kinase and Rac1 GTPase activities. J Biol Chem 278: 14414–14419, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Hagn F, Lagleder S, Retzlaff M, Rohrberg J, Demmer O, Richter K, Buchner J, Kessler H. Structural analysis of the interaction between Hsp90 and the tumor suppressor protein p53. Nat Struct Mol Biol 18: 1086–1093, 2011. [DOI] [PubMed] [Google Scholar]
- 22.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature 387: 296–299, 1997. [DOI] [PubMed] [Google Scholar]
- 23.Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ, Beach DH. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med 190: 1375–1382, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta 1823: 742–755, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin Y, Hofseth AB, Cui X, Windust AJ, Poudyal D, Chumanevich AA, Matesic LE, Singh NP, Nagarkatti M, Nagarkatti PS, Hofseth LJ. American ginseng suppresses colitis through p53-mediated apoptosis of inflammatory cells. Cancer Prev Res (Phila) 3: 339–347, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joshi AD, Dimitropoulou C, Thangjam G, Snead C, Feldman S, Barabutis N, Fulton D, Hou Y, Kumar S, Patel V, Gorshkov B, Verin AD, Black SM, Catravas JD. Heat shock protein 90 inhibitors prevent LPS-induced endothelial barrier dysfunction by disrupting RhoA signaling. Am J Respir Cell Mol Biol 50: 170–179, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaplan KB, Li R. A prescription for ‘stress’—the role of Hsp90 in genome stability and cellular adaptation. Trends Cell Biol 22: 576–583, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285: 1733–1737, 1999. [DOI] [PubMed] [Google Scholar]
- 29.Komarova EA, Kondratov RV, Wang K, Christov K, Golovkina TV, Goldblum JR, Gudkov AV. Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic injury, but protects from gastro-intestinal syndrome in mice. Oncogene 23: 3265–3271, 2004. [DOI] [PubMed] [Google Scholar]
- 30.Komarova EA, Krivokrysenko V, Wang K, Neznanov N, Chernov MV, Komarov PG, Brennan ML, Golovkina TV, Rokhlin OW, Kuprash DV, Nedospasov SA, Hazen SL, Feinstein E, Gudkov AV. p53 is a suppressor of inflammatory response in mice. FASEB J 19: 1030–1032, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Lane D, Levine A. p53 Research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol 2: a000893, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lane DP. Cancer. p53, guardian of the genome. Nature 358: 15–16, 1992. [DOI] [PubMed] [Google Scholar]
- 33.Lefort K, Mandinova A, Ostano P, Kolev V, Calpini V, Kolfschoten I, Devgan V, Lieb J, Raffoul W, Hohl D, Neel V, Garlick J, Chiorino G, Dotto GP. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev 21: 562–577, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer 9: 749–758, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu G, Park YJ, Tsuruta Y, Lorne E, Abraham E. p53 Attenuates lipopolysaccharide-induced NF-kappaB activation and acute lung injury. J Immunol 182: 5063–5071, 2009. [DOI] [PubMed] [Google Scholar]
- 36.Lopez-Pajares V, Kim MM, Yuan ZM. Phosphorylation of MDMX mediated by Akt leads to stabilization and induces 14-3-3 binding. J Biol Chem 283: 13707–13713, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma J, Xue Y, Cui W, Li Y, Zhao Q, Ye W, Zheng J, Cheng Y, Ma Y, Li S, Han T, Miao L, Yao L, Zhang J, Liu W. Ras homolog gene family, member A promotes p53 degradation and vascular endothelial growth factor-dependent angiogenesis through an interaction with murine double minute 2 under hypoxic conditions. Cancer 118: 4105–4116, 2012. [DOI] [PubMed] [Google Scholar]
- 38.Maclaine NJ, Hupp TR. The regulation of p53 by phosphorylation: a model for how distinct signals integrate into the p53 pathway. Aging (Albany NY) 1: 490–502, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA 98: 11598–11603, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moll UM, Petrenko O. The MDM2-p53 interaction. Mol Cancer Res 1: 1001–1008, 2003. [PubMed] [Google Scholar]
- 41.Monick MM, Carter AB, Robeff PK, Flaherty DM, Peterson MW, Hunninghake GW. Lipopolysaccharide activates Akt in human alveolar macrophages resulting in nuclear accumulation and transcriptional activity of beta-catenin. J Immunol 166: 4713–4720, 2001. [DOI] [PubMed] [Google Scholar]
- 42.Mouraret N, Marcos E, Abid S, Gary-Bobo G, Saker M, Houssaini A, Dubois-Rande JL, Boyer L, Boczkowski J, Derumeaux G, Amsellem V, Adnot S. Activation of lung p53 by Nutlin-3a prevents and reverses experimental pulmonary hypertension. Circulation 127: 1664–1676, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mukhopadhyay UK, Eves R, Jia L, Mooney P, Mak AS. p53 suppresses Src-induced podosome and rosette formation and cellular invasiveness through the upregulation of caldesmon. Mol Cell Biol 29: 3088–3098, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muller L, Schaupp A, Walerych D, Wegele H, Buchner J. Hsp90 regulates the activity of wild type p53 under physiological and elevated temperatures. J Biol Chem 279: 48846–48854, 2004. [DOI] [PubMed] [Google Scholar]
- 45.Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 25: 304–317, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Prey J, Crighton D, Martin AG, Vousden KH, Fearnhead HO, Ryan KM. p53-mediated induction of Noxa and p53AIP1 requires NFkappaB. Cell Cycle 9: 947–952, 2010. [DOI] [PubMed] [Google Scholar]
- 47.Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K, Masuyama N, Gotoh Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem 277: 21843–21850, 2002. [DOI] [PubMed] [Google Scholar]
- 48.Okuda Y, Okuda M, Bernard CC. Regulatory role of p53 in experimental autoimmune encephalomyelitis. J Neuroimmunol 135: 29–37, 2003. [DOI] [PubMed] [Google Scholar]
- 49.Pal S, Bhattacharjee A, Ali A, Mandal NC, Mandal SC, Pal M. Chronic inflammation and cancer: potential chemoprevention through nuclear factor kappa B and p53 mutual antagonism. J Inflamm 11: 23, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pankova K, Rosel D, Novotny M, Brabek J. The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell Mol Life Sci 67: 63–71, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park SJ, Borin BN, Martinez-Yamout MA, Dyson HJ. The client protein p53 adopts a molten globule-like state in the presence of Hsp90. Nat Struct Mol Biol 18: 537–541, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem 75: 271–294, 2006. [DOI] [PubMed] [Google Scholar]
- 53.Qin Q, Baudry M, Liao G, Noniyev A, Galeano J, Bi X. A novel function for p53: regulation of growth cone motility through interaction with Rho kinase. J Neurosci 29: 5183–5192, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rafikov R, Dimitropoulou C, Aggarwal S, Kangath A, Gross C, Pardo D, Sharma S, Jezierska-Drutel A, Patel V, Snead C, Lucas R, Verin A, Fulton D, Catravas JD, Black SM. Lipopolysaccharide-induced lung injury involves the nitration-mediated activation of RhoA. J Biol Chem 289: 4710–4722, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scatizzi JC, Mavers M, Hutcheson J, Young B, Shi B, Pope RM, Ruderman EM, Samways DS, Corbett JA, Egan TM, Perlman H. The CDK domain of p21 is a suppressor of IL-1beta-mediated inflammation in activated macrophages. Eur J Immunol 39: 820–825, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shadfan M, Lopez-Pajares V, Yuan ZM. MDM2 and MDMX: alone and together in regulation of p53. Transl Cancer Res 1: 88–89, 2012. [PMC free article] [PubMed] [Google Scholar]
- 57.Shimizu S, Tahara M, Ogata S, Hashimoto K, Morishige K, Tasaka K, Murata Y. Involvement of nuclear factor-kB activation through RhoA/Rho-kinase pathway in LPS-induced IL-8 production in human cervical stromal cells. Mol Hum Reprod 13: 181–187, 2007. [DOI] [PubMed] [Google Scholar]
- 58.Subbaramaiah K, Altorki N, Chung WJ, Mestre JR, Sampat A, Dannenberg AJ. Inhibition of cyclooxygenase-2 gene expression by p53. J Biol Chem 274: 10911–10915, 1999. [DOI] [PubMed] [Google Scholar]
- 59.Thangjam GS, Dimitropoulou C, Joshi AD, Barabutis N, Shaw MC, Kovalenkov Y, Wallace CM, Fulton DJ, Patel V, Catravas JD. Novel mechanism of attenuation of LPS-induced NF-kappaB activation by the heat shock protein 90 inhibitor, 17-N-allylamino-17-demethoxygeldanamycin, in human lung microvascular endothelial cells. Am J Respir Cell Mol Biol 50: 942–952, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trakala M, Arias CF, Garcia MI, Moreno-Ortiz MC, Tsilingiri K, Fernandez PJ, Mellado M, Diaz-Meco MT, Moscat J, Serrano M, Martinez AC, Balomenos D. Regulation of macrophage activation and septic shock susceptibility via p21(WAF1/CIP1). Eur J Immunol 39: 810–819, 2009. [DOI] [PubMed] [Google Scholar]
- 61.Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 10: 537–549, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vaseva AV, Yallowitz AR, Marchenko ND, Xu S, Moll UM. Blockade of Hsp90 by 17AAG antagonizes MDMX and synergizes with Nutlin to induce p53-mediated apoptosis in solid tumors. Cell Death Dis 2: e156, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wade M, Wang YV, Wahl GM. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol 20: 299–309, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Walerych D, Gutkowska M, Klejman MP, Wawrzynow B, Tracz Z, Wiech M, Zylicz M, Zylicz A. ATP binding to Hsp90 is sufficient for effective chaperoning of p53 protein. J Biol Chem 285: 32020–32028, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walerych D, Kudla G, Gutkowska M, Wawrzynow B, Muller L, King FW, Helwak A, Boros J, Zylicz A, Zylicz M. Hsp90 chaperones wild-type p53 tumor suppressor protein. J Biol Chem 279: 48836–48845, 2004. [DOI] [PubMed] [Google Scholar]
- 66.Xia M, Land H. Tumor suppressor p53 restricts Ras stimulation of RhoA and cancer cell motility. Nat Struct Mol Biol 14: 215–223, 2007. [DOI] [PubMed] [Google Scholar]
- 67.Yamanishi Y, Boyle DL, Pinkoski MJ, Mahboubi A, Lin T, Han Z, Zvaifler NJ, Green DR, Firestein GS. Regulation of joint destruction and inflammation by p53 in collagen-induced arthritis. Am J Pathol 160: 123–130, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 352: 345–347, 1991. [DOI] [PubMed] [Google Scholar]
- 69.Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol 3: 973–982, 2001. [DOI] [PubMed] [Google Scholar]