

Abstract
Oxidative stress is increased in metabolic syndrome and type 2 diabetes mellitus (T2DM) and this appears to underlie the development of cardiovascular disease, T2DM and diabetic complications. Increased oxidative stress appears to be a deleterious factor leading to insulin resistance, dyslipidemia, β-cell dysfunction, impaired glucose tolerance and ultimately leading to T2DM. Chronic oxidative stress, hyperglycemia and dyslipidemia are particularly dangerous for β-cells from lowest levels of antioxidant, have high oxidative energy requirements, decrease the gene expression of key β-cell genes and induce cell death. If β-cell functioning is impaired, it results in an under production of insulin, impairs glucose stimulated insulin secretion, fasting hyperglycemia and eventually the development of T2DM.
Keywords: Insulin resistance, Dyslipidemia, Type 2 diabetes mellitus, Oxidative stress
Core tip: Oxidative stress is underling in the development of cardiovascular disease, type 2 diabetes mellitus (T2DM) and diabetic complications. Increased oxidative stress appears to be a deleterious factor leading to insulin resistance, dyslipidemia, β-cell dysfunction, impaired glucose tolerance and ultimately leading to T2DM.
INTRODUCTION
Aerobic life uses oxygen to oxidize (metabolism) food substrates (carbon- and hydrogen-rich) to obtain the heat energy and chemical essential for life. When we oxidize molecules with oxygen, the oxygen molecule itself becomes reduced and forms intermediates. In eukaryotic cells, reactive oxygen species (ROS) are always produced as the consequence of regular physiological metabolism[1]. These ROS (pro-oxidants) productions are counter-balanced by cellular antioxidant defense mechanisms in the normal physiological conditions. ROS define as diverse chemical that have reactive properties are capable to accommodate or donate electrons (e-) to the broad range of biological molecules. Normally, the production and neutralization of ROS are balance with antioxidants in a living system and does not cause any oxidative damage, determines as physiological state[2]. The imbalance between these prooxidants and antioxidants in the living organism system to determine as oxidative stress state, brings to cellular disruption and damage[3]. The free-radical can attack polyunsaturated fatty acids oxidation in physiological systems known as lipid peroxidation. Lipid peroxidation is an autocatalytic free radical mediated destructive process whereby poly-unsaturated fatty acids in cell membranes undergo degradation to form lipid hydroperoxides[4,5]. By-products of lipid peroxidation such as conjugated dienes and malondialdehyde (MDA) are increased in the patients with obesity, metabolic syndrome and type 2 diabetes mellitus (T2DM). Carbohydrates, lipids, proteins and DNA are the targets of oxidative stress modification biomolecules generally as the principal of ROS induced cellular damage. Therefore, these ROS modified biomolecules are used as oxidative stress markers both in vivo and in vitro measurement. Recent study suggests that ROS may act as the mechanical link of salt sensitive hypertension, over nutrition and high fat diet, metabolic syndrome and T2DM animal models[6]. ROS levels are increased in obesity, especially in abdominal obesity which is the major component of metabolic syndrome and it can be reduced by weight loss[7]. Many studies demonstrated that increased oxidative stress is associated with insulin resistance pathogenesis by insulin signals inhibition and adipokines dysregulation[8,9]. In animal studies, oxidative stress enhances insulin resistance. The evidence suggested that angiotensin II (Ang II) infused rats required the increased glucose load to maintain normal glucose levels during hyperinsulinemic clamp to stimulate ROS production[10]. Thus, ROS may also contribute and accelerate the insulin resistance development in insulin-targeted organs of the over nutrition and the excess salt individuals.
In the large general population studies demonstrated that insulin resistance is multifactorial[11,12] and the genetic component[11,13,14]. Insulin resistance most often precedes in many years before the onset of T2DM. Insulin resistance and the consequence of declined of insulin secretion are the principle of the T2DM pathogenesis[11,12,15,16]. The late complications of diabetes have been associated and implicated in their etiology with oxidative stress[17-19]. The influence of oxidative stress on insulin resistance, dyslipidemia, abnormal lipoprotein production and the pathophysiology of T2DM by using in vivo, in vitro and animal models data on these effects were also included in this review.
ROS
Oxygen exists in air known as oxygen molecule (O2) or dioxygen. Oxygen on the surface of earth appeared in significant amounts approximately 2.5 × 109 years ago. It was created by the photosynthetic activity of plants and microorganisms (blue green algae). Increased atmospheric oxygen concentration was followed by the ozone layer formation in the stratosphere. Both oxygen and ozone layer were filters against the solar ultraviolet radiation reaching surface of the Earth. In eukaryotic cells, ROS is produced as the consequence of the normal aerobic physiological metabolism[1]. These ROS levels are counter-balanced with the cellular antioxidants in the normal physiological conditions. ROS define as diverse chemical that have reactive properties are capable to accommodate or donate electrons (e-) to the broad range of biological molecules. These species includeinstability radicals arise from an unpaired e-. Existence of the presence of oxygen and the aerobic organisms on the earth is possible[20].
O2 + e + H+ → HO2• (hydroperoxyl radical)
HO2• → H+ + O2•- (superoxide radical)
O2•- + 2H+ + e → H2O2 (hydrogen peroxide)
H2O2 + e → OH- + OH• (hydroxyl radical)
However, these molecules are also played an adverse role in the biological systems as oxidative stress. At the steady state of the living systems, oxygen metabolism always produce oxygen-derived free radicals such as superoxide O2•-, hydroxyl OH•, alkoxyl RO•, peroxyl RO2•, peroxynitrite ONOO- and oxygen-derived non-radicals such as hydrogen peroxide H2O2, hypochlorous acid HOCl and hypobromous acid HOBr. Both free radicals and non-radicals groups are the important factors of the oxidative stress mediated cellular damages[21]. Normally, the neutralization of ROS productions by cellular antioxidant defense mechanisms are determine as the physiological state and do not cause any oxidative damage[2]. The imbalance of the ROS production and antioxidants defense system in the living systems caused oxidative stress brings to cellular function disruption and damage[3].
This imbalance occurs due to over production of ROS and reduction of the antioxidant defense mechanisms. The electron transport chain in mitochondrial, peroxisomes and cytochrome P450 system are the most important sources of ROS production (involves in O2•- production)[22]. Moreover, various enzymes can be accelerated ROS production such as cyclooxygenases[23,24], xanthine oxidase[25], uncoupled nitric oxide synthases (NOS)[26-28] and NADPH oxidases[29]. Drugs such as doxorubicin[30,31], cisplatin, acetaminophen[32-34] and nimesulide[35]. Heavy metals (Fe, Cd, Pb, Hg) as the toxic substances [36-39], acrolein, chloroform, carbon tetrachloride[40], tertiary butyl hydroperoxide[41-44], environmental pollutants (oxides of nitrogen, SO2, CO2), xenobiotics, UV irradiation and the other factors induce ROS overproduction.
In metabolic disorders assist the increased ROS production in the physiological system such as obesity, insulin resistance and diabetes mellitus[45-48]. In Figure 1 summarized of obesity and metabolic syndrome elevate in oxidative stress. Superoxide radical (O2•-), hydroxyl radical (OH•) and hydrogen peroxide (H2O2) are the three major ROS in physiological organisms[49]. Superoxide radical (O2•-) acts as the parent ROS molecules caused from the one electron reduction of oxygen molecule by electron transport chain enzymes in mitochondrial such as enzymes in cytochrome P450, cyclooxygenase and NADPH oxidase. Various reactions of enzymes and non-enzymes system further convert these ROS molecules to hydroxyl radical (OH•), peroxynitrite ion (ONOO-) and hyperchlorous acid (HOCl). For example superoxide dismutase converts O2•- to H2O2 by the dismutase reaction[50,51].
Figure 1.
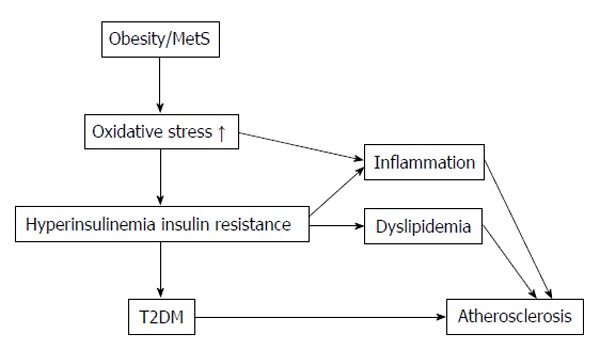
Summarized of obesity and metabolic syndrome elevate in oxidative stress. T2DM: Type 2 diabetes mellitus; MetS: Metabolic syndrome.
Elevated ROS molecules caused the cellular macromolecules damage such as lipids[52], proteins[53] and nucleic acids[54]. In the anti-oxidants system of the living system, possess own antioxidant defense mechanisms[55] includes enzymes and non-enzyme molecules such as SOD, catalase (CAT) and glutathione peroxidases (GPx). Enzyme SOD catalyzes O2•- conversion to H2O2, while CAT converts H2O2 to H2O and O2. For reduction of two peroxide molecules use non-enzymatic glutathione (GSH; reduced and oxidized forms), reduced glutathione (GSH) and GPx catalyze to produce oxidized glutathione (GSSG) and water[56]. Various enzymes play the important combination roles in the series of antioxidant defense systems such as glutathione reductase, glutathione S-transferase, and glutathione disulfide (GSSG).
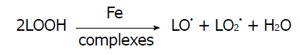
ROS production is identified as endogenous and exogenous source. UV exposure and xenobiotic agents has been shown to generate these ROS[57]. In fact, dietary is the major source of these oxidant compounds, especially in animal fat as the source of high lipid peroxides[58]. ROS may also be derived from the general biochemical reactions in living organism to generate ROS as by-products or end products. In the transition heavy metals such as iron (Fe2+) and copper (Cu+) are pose the oxidative stress production, especially in Fe2+ may cause autooxidation to cause O2•- generation and/or interaction with H2O2 can generate OH• via the Fenton and Harber Weiss reactions[59]. Fenton chemical reaction may also causes lipid peroxides generation and propagation[60].
Auto oxidation of Fe2+:
Fe2+ + O2 → Fe3+ + O2•-
Fenton reaction:
H2O2 + Fe2+ → Fe3+ + OH- + OH•
Haber-Weiss reaction:
Math 1
Math 1.
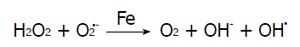
Math(A1).
The major cellular oxidative stress is come from mitochondrial respiration. Heart, brain, kidney, liver and skeletal muscle are the effective oxygen consumption organs is converted oxygen to O2•-, approximate releasing 0.1%-0.2% while the liver is converted oxygen to O2•-, approximately releasing 2%[61]. Electron transport chain complex of the mitochondrial has been sourced to O2•- generation and have been estimated upto 107 ROS molecules per mitochondria per day[62].
In the enzymatic systems of xanthine oxidase generated via xanthine dehydrogenase, which utilize oxygen molecule as e- acceptor during catabolism of xanthine. Xanthine oxidase is the generator of O2•-, H2O2[63] and OH• producer[64], highly expressed in epithelial, injured and diseased tissues as shown in Figure 2. Xanthine oxidase has been involved to peroxynitrite (OONO-) and nitric oxide (NO) productions through nitrite reduction[65,66]. Intracellular nitric oxide synthases (NOS) catalyze L-arginine to form citrulline and NO. Endothelial NOS and neuronal NOS are activated by calcium-induced calmodulin binding to produce NO levels[67]. Inducible NOS (iNOS) has also calmodulin bound molecule. It may rapid and chronic expression in many cell types such as smooth muscle cells, hepatocytes and macrophages. INOS is induced by the many inflammatory cytokines [tumor necrosis factor-α (TNF-α), interleukin-6 and growth factors] regulation at the transcriptional level, results in micromolar NO production[67]. INOS can poduce O2•- and OONO- when lower in L-arginine substrate[68].
Figure 2.
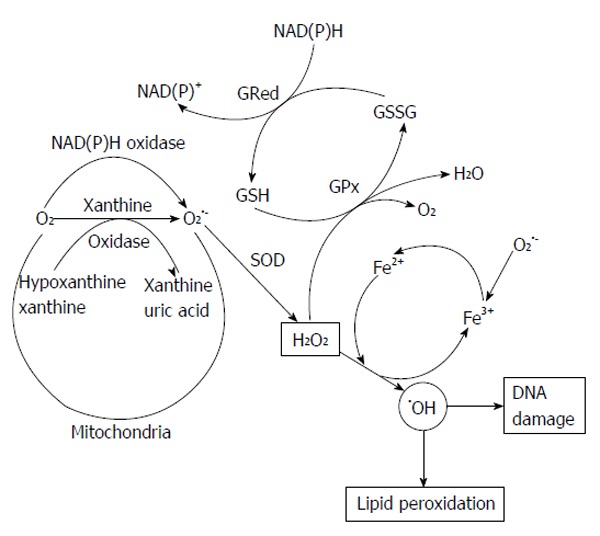
Increased oxidative stress by xanthine oxidase. NADH: Nicotinamide adenine dinucleotide.
LIPID PEROXIDATION
Fats and oils oxidized with characteristic changes in texture, color, taste and odor. This process, known as rancidity, was chemically defined in the 1940s as an autoxidative free-radical chain reaction[69]. The most powerful oxidant formed in biological systems is hydroxyl radical. It can attack any biological molecule. The initiation step of lipid peroxidation occurred when hydroxyl radicals attack to polyunsaturated fatty acids, to cause the free-radical polyunsaturated fatty acids oxidation in biological systems. Lipid peroxidation is autocatalytic lipid hydroperoxides radical production mediated poly-unsaturated fatty acids in cell membranes destruction and degradation process[4,5]. Conjugated dienes and MDA, by-products of lipid peroxidation are increased in the circulation of obesity, metabolic syndrome and T2DM patients.
First-peroxidation chain initiation, results from the attack by any species to reduce a hydrogen atom from methylene (-CH2-) group of polyunsaturated fatty acid or membrane. Because one hydrogen atom contains one electron, reduction leaves an unpaired electron on the carbon of -CH-, double bond in the fatty acid weakens the C-H bonds on the carbon atom adjacent to the other double bond and facilitates it removal. Then, the polyunsaturated fattyacid chains in lipids membrane are sensitive to cause lipid peroxidation. The carbon-centered radical forms a conjugated diene by the molecular rearrangement (Figure 3), which combines with oxygen to form a peroxyl radical that able to reduce a hydrogen atom from another fatty acid to start a chain reaction. Peroxidation continues to use up the polyunsaturated fatty acid substrate unless the chain-breaking antioxidant (vitamin E) agent is added to terminate the chain reaction. The three stages of lipid peroxidation are initiation, propagation and termination. Hydroxyl radical (•OH), alkoxyl radical (RO•), peroxyl radical (ROO•), and HO2• species can abstract the first hydrogen atom of polyunsaturated fatty acid but not H2O2 or O2•-[70]. Variety of lipid hydroperoxides and cyclic peroxides are the end products of the chain reaction. Lipid peroxides are stable molecules in the physiological temperatures. Lipid peroxides decomposition is catalyzed by transition heavy metals. For example, iron ion-active complexes present in circulating can participate in the Fenton reaction to promote lipid peroxide decomposition. Hemoglobin and the cytochromes molecules can also facilitate peroxide decomposition, although they do not directly catalyze Fenton chemistry. However, hemeproteins can release chelatable iron that can participate in Fenton chemistry[71]. Ferritin and hemosiderin are effective at stimulating lipid peroxidation and catalase is weakly effective, caused problems to use catalase as a probe for H2O2 in lipid peroxidation systems[72].
Figure 3.
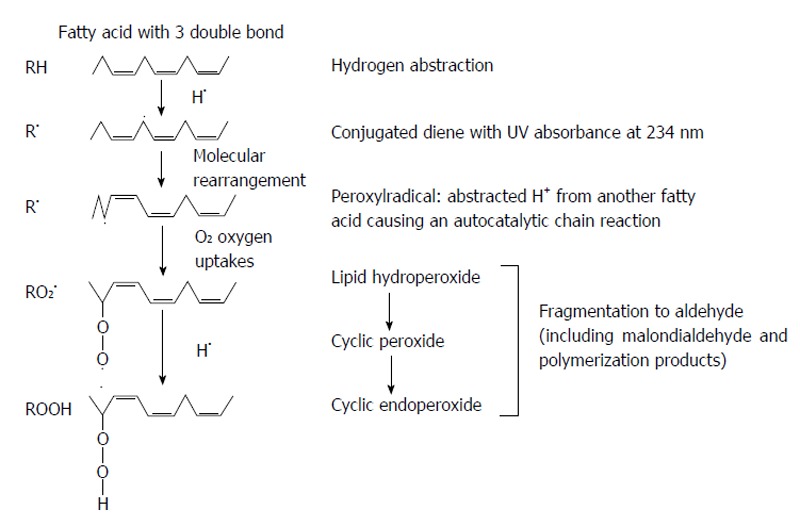
The chain reaction of lipid peroxidation.
Reduced heavy metal [Fe+2, Cu+] react with lipid peroxides (LOOH) to alkoxyl radical or Cu+ react with LOOH to alkoxyl radical.
LOOH + Mn+ → LO• + M(n+1)+ + OH-
In the reaction oxidized-heavy metals [Fe+3, Cu+2] slowly react with LOOH to produce alkoxyl and peroxyl radicals. Both peroxyl and alkoxyl radicals initiate the chain reaction by reducing hydrogen atoms (Figure 3). The fixed oxidation metals ions can affect the rate of lipid peroxidation (Ca2+, Pb2+ and Al3+ ions). Lipid peroxidation accelerates by the iron salts stimulation result in the membrane structure changes and important implications for environmental toxicology[73].
Rawls et al[74] demonstrated that singlet O2• is formed during the lipid peroxidation degradation and might contribute to cause more initiation in the chain reaction. Initiation in the first-chain initiation should be used as lipid peroxide decomposition reactions to start the new chain reaction. Iron ions and ferrous ions are free radicals[55], can act in electron transfer reactions with oxygen molecule. Then, the presence of iron ions can promote the hydroxyl radicals formation by Fenton reaction. Bielski et al[75] demonstrated that the •OH radical production in any source can initiate lipid peroxidation reaction.
LH + OH• → L• + H2O
Superoxide-dependent Fenton reaction (superoxide resulting H2O2 and reducing Fe3+ to Fe2+) did not demonstrate any substantial involvement of the hydroxyl radical in liposomal peroxidation systems as detected by the scavengers action[76]. Hydroxyl radicals in the systems can be measured by spin trapping[77] or deoxyribose degradation measurements[76] but do not contribute to the lipid peroxidation rate[76]. The addition of iron ion in any preparations can stimulate peroxidation reaction by lipid hydroperoxide degradation to generate peroxyl (LO2•) and alkoxyl (LO•) radicals.
Math 2
Math 2.
Math(A1).
The rate constant of the reaction when ferrous ions are reacted as 1.5 × 103 /mol/L per second[78], which is higher than the rate reaction constant of ferrous ions with H2O2 reaction (76 /mol/L per second)[79]. The iron ions stimulate lipid peroxidation by the lipid degradation reactions from the present of abundant hydroperoxide.
Iron or copper in a biological system attach to biological molecules at the specific location of OH radicals formation to cause lipid, protein and DNA damage. On lipid membrane, the propagation step of lipid peroxidation reactions does not proceedes further until the reaction reach the protein portion. Thus, lipid peroxidation in vivo causes proteins membrane damage[80,81]. This damage has more biologically important than those lipids membrane damage. Cells also contain mechanisms for recognizing and removing oxidative modified proteins[80,81].
OXIDATIVE STRESS
Oxidative stress occurs at the molecular level as the cellular event when increased ROS overwhelm the antioxidant defense capabilities systems. Oxidative stress was defines as the increasing ROS production, vary in intensities, the different cellular locations and may be occurred either acutely or chronically[82]. Oxidative damage to macromolecules including carbohydrates, proteins, lipids and DNA typically viewed as increased ROS induced cellular damage to cause the irreversible macromolecules modifications. Therefore, the by-products of these oxidative modified biomolecules are used as oxidative stress biomarkers in vivo and in vitro. Many research studies demonstrated the association of oxidative stress and the pathogenesis of insulin resistance via insulin signals inhibition and adipocytokines dysregulation[8,9]. Oxidative stress biomarkers included MDA[83], 4-hydroxy-2-nonenal and isoprostanes species[84], protein carbonyls, 3-nitrotyrosine, hydroperoxides, protein oxidation products[85], glycation end products, carbohydrate modifications[86] and 8-hydroxy-2′-deoxyguanosine (8-OH-dG), an oxidized DNA product[84].
Assaying lipid peroxidation
The lipid peroxidation contributes to the pathogenesis of atherosclerosis. It is occurred in the blood vessel walls and does not occur from low density lipoproteins (LDL) in circulation[87,88]. LDL can enter to the blood vessel walls. The modified LDL (oxidized LDL) may escape from the scavenger recognition receptors and back to the circulation. Therefore, this circulating LDL peroxidation is a potentially useful biomarker of lipid peroxidation in circulation. Indeed, this assay is used for the demonstration of in vivo antioxidants inhibit the effects of lipid peroxidation[89,90].
Thiobarbituric acid-reactive substance
MDA from the oxidative polyunsaturated fatty acids (PUFA) degradation is determined by the reaction of thiobarbituric acid (TBA) with MDA to generate the stable end product of MDA-TBA adduct[91-95]. This MDA free radical has been demonstrated as a causative of the atherosclerosis pathogenesis[96,97], aging[98], cancer[99] and Alzheimer’s disease[100,101]. Serum MDA levels have been used as the lipid peroxidation biomarker and indicator of free radical damage[37,83,102]. MDA, the three-carbon dialdehyde, can exist in many forms in the aqueous circulation. This method was used the reaction of MDA with TBA and heated under acidic conditions but the TBA can react with many chemical species such as proteins, phospholipids, aldehydes, amino acid and nucleic acids[103,104]. One MDA molecule reacts with TBA two molecules to form a stable pink to red chromophore that absorbs maximally at 532 nm[105] or fluorescence detection. This chromophore is termed thiobarbituric acid reacting substances. Elevated MDA levels in T2DM patients are associated with cardiovascular disease risk[83].
Isoprostanes
The most valuable of lipid peroxidation biomarker in the biological system is the isoprostanes, elevated from the PUFA peroxidation[106-113]. Isoprostanes identified as free form and the most are esterified to lipids in circulation. Isoprostanes can be analyzed by mass spectrometry techniques, so that can easily be detected in human body fluids[108,109,112,113]. Isoprostanes appear to turn over rapidly in metabolized and excreted[108,109]. Isoprostanes and their metabolites detection in urine may be the useful biomarker for lipid peroxidation[113]. Isoprostanes assay have focused on the F2-isoprostanes measurement, which elevate from the arachidonic acid peroxidation[109]. Elevation of F2-isoprostanes levels have been shown in conditions of the cardiovascular disease, diabetes development[114,115], cigarette smoking[111,116,117], hyperhomocysteinaemia[118] and hypercholesterolaemia[110,119]. F2-isoprostane levels have also been shown to decrease by antioxidants supplementation both in animal models and humans subjects[120-124].
Oxidative stress in metabolic syndrome
The components of metabolic syndrome consist with abdominal obesity, dyslipidemia, hypertension and diabetes[125,126]. It is the major modern lifestyle complication cause from physical inactivity and overeating and associated with the increased risk of cardiovascular diseases, hypertension and T2DM that summarized in Figure 4.
Figure 4.
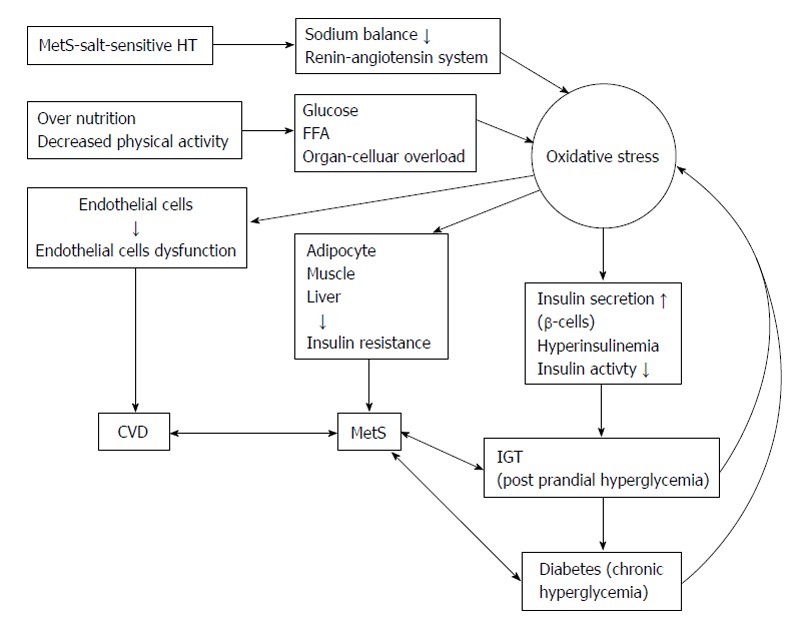
Summarized the increasing reactive oxygen species in obesity, metabolic syndrome and salt sensitive hypertension. FFA: Free fatty acid; MetS: Metabolic syndrome; HT: Hypertension; IGT: Impaired glucose tolerance.
Over nutrition and oxidative stress: In metabolism of glucose through glycolysis and tricarboxylic acid (TCA) cycle to generate nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) as the electron donors. In over nutrition, the excessive glucose occur and a large amount of glucose is oxidized in the glycolysis and TCA cycle to increase NADH and FADH2 generation in electron transport chain of mitochondrial and increased superoxide generation[127]. The excessive of free fatty acids (FFAs) leads to increase FFA-oxidation and acetyl coenzyme A (CoA) oxidation in TCA cycle generate the NADH and FADH2 electron donors as glucose oxidation results in mitochondrial ROS overproduction[127]. Furthermore, NADPH oxidase in the plasma membrane can convert oxygen molecule to superoxide radical and involve in ROS nutrient-based generation. In adipocytes, ROS is generated by in fused with FFAs, treatment with NADPH oxidase inhibitor can block this ROS generation. This indicates that NADPH oxidase involves in fatty acids ROS generation[8]. Palmitate can activate diacylglycerol synthesis and protein kinase C (PKC) leading to activate NADPH oxidase[128]. Thus, over accumulated fat result in the increased fatty acids oxidation and lead to activate NADPH oxidase (in local or remotely cells) to cause ROS over production in over nutrition or obesity (Figure 4). Conversely, calorie restriction may be associated with normal physiological system[129] and may involve in normal cellular redox state[130]. In aged animals models treated with antioxidant agents or hypocaloric diets led to ameliorate in oxidative stress status and tissue function[131,132]. Treatment with resveratrol, a polyphenol reduced atherosclerosis and diabetes development[133]. These studies demonstrate that nutrition is associated with increased or decreased redox status and over nutrition result to increase oxidative stress to contribute pathogenesis of atherosclerosis, cancer and other diseases.
Oxidative stress in adipose tissue: Increased fat accumulation in human has been associated with oxidative stress biomarkers[134]. Similarly, obese mice were significantly higher oxidative stress levels in circulation[8]. Moreover, lipid peroxidation and H2O2 levels were increased in adipose tissue[8]. These mean that adipose tissue may the major source of ROS production and can be released to the circulation potentially affecting various distance organs functions and damage (Figure 4).
Increased NADPH oxidase expression in adipose tissue associated with increased oxidative stress levels. Increased mRNA expression was found in adipose tissue of obese mice[8]. Increased ROS generation in lipid accumulation and further elevating ROS generation with FFA treatment were found in 3T3-L1 adipocytes cultured[8]. These ROS generation processes can be blocked by NADPH oxidase inhibitors, apocynin or diphenyleneiodonium. Many studies suggest that NADPH oxidase induces adipocytes ROS production[8]. Moreover, obese mice ameliorated hyperinsulinemia, hypertriglyceridemia, hyperglycemia and hepatic steatosis by supplementation with apocynin[8]. These data demonstrate that NADPH oxidase increase ROS production in obesity and metabolic syndrome may play the important roles in the atherosclerosis, T2DM and cancer pathogenesis. Adipose tissue tries to increase antioxidant enzymes levels to against ROS over production. However, these antioxidant enzymes activity and expression are decreased in adipose tissue[8,135-137]. Then, increased ROS-production enzymes and decreased antioxidant enzymes may cause oxidative stress in obese and metabolic syndrome.
Oxidative stress and salt-sensitive hypertension: As in mention above, ROS levels are increased in obesity and can be ameliorated by weight loss[7]. Obese rats induced by refined sugar or high fat diet leading to ROS overproduction and increase oxidative stress[6,138]. Many research evidences suggest that metabolic syndrome was associated with the salt-sensitive hypertension. ROS play the roles as mechanical link of metabolic syndrome and salt-sensitive hypertension[125,126], which itself leads to ROS overproduction[139-142]. Salt restriction in hypertensive obesity was more effective reduction in blood pressure than in hypertensive non-obesity patients, and weight loss in obesity and salt sensitive hypertensive patients caused the successful of blood pressure reduction[143]. Salt-sensitive hypertensive patients were significantly more prevalent in metabolic syndrome patients than without metabolic syndrome[144]. Oxidative stress in abdominal adipocytes due to increase adipocytokines secretion such as TNF-α, angiotensinogen, non-esterified fatty acids[126]. Interestingly, infused Ang II-rats disturbed sodium balance to cause ROS overproduction in salt-sensitive rats[139-141]. Moreover, in salt-sensitive hypertensive patients are also increased 8-isoprostane levels[142]. Thus, ROS may the underling pathogenesis of diseases in metabolic syndrome, obese and non-obese intake excessive salt as the salt-sensitive hypertensive patients.
In high-renin patients (non-modulating salt sensitive hypertension) had elevated the homeostasis model assessment of insulin resistance (HOMA-IR) levels[145]. Insalt-sensitive hypertensive non-obesity patients had significantly lower insulin sensitivity than in non-salt-sensitive hypertensive patients[146]. Insulin resistance caused salt-sensitive hypertensive obesity and/or metabolic syndrome patients[125]. Increased renal ROS overproduction may increase the salt sensitive hypertension[147]. Then, increased renal oxidative stress may contribute to cause salt-sensitive hypertension development. Moreover, ROS overproduction in vascular endothelial cells suppresses the NO-dependent vasodilation[148] and may play the role in the salt-sensitive hypertension development.
Oxidative stress in type 2 diabetes
Many research studies demonstrated that T2DM patients have increased ROS production-induced higher oxidative damage in the circulation and also have reduced antioxidant defenses mechanisms[149-152]. Increased ROS production in T2DM patients is thought to activate many detrimental pathways including hexosamine pathways, advanced glycation end-products (AGEs) formation, and PKCβ1/2[127]. Hyperglycemia condition can induce oxidative stress by several mechanisms such as glucose autoxidation, polyol pathway, AGE formation and PKCβ1/2 kinase. Elevated free fatty acids, leptin and other circulating factors in T2DM patients may also contribute to cause ROS overproduction. Figure 5 demonstrates the association of increased ROS production with atherosclerosis and sources of ROS generations in T2DM patients.
Figure 5.
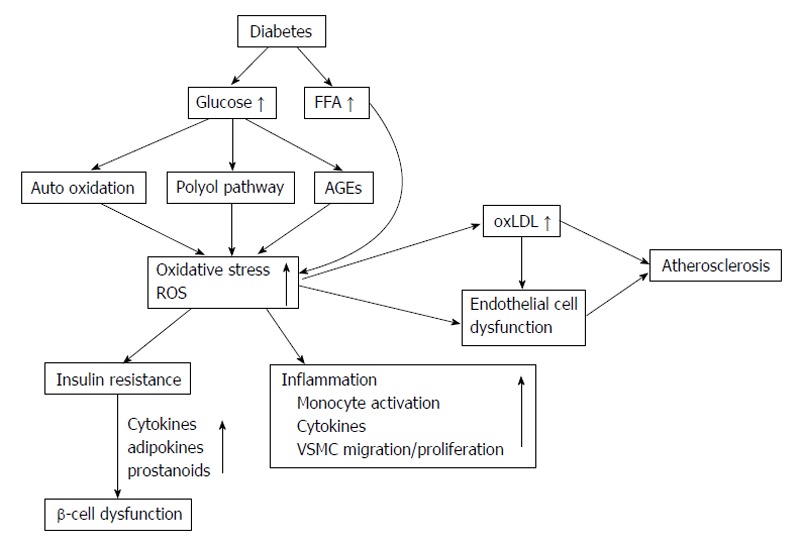
Summarizes the reactive oxygen species associations with atherosclerosis and sources of reactive oxygen species production in type 2 diabetes. oxLDL: Oxidized low density lipoprotein; FFA: Free fatty acids; AGEs: Advanced glycation end-products; VSMC: Vascular smooth muscle cells; ROS: Reactive oxygen species.
Glucose autoxidation
Hyperglycemia due to cause increased glucose metabolism leading to increase NADH and FADH2 overproduction, which are used by the electron transport chain of mitochondria to generate ATP[153]. NADH overproduction can cause the higher proton gradient production in mitochondria. These electrons are transferred to oxygen to produce higher superoxide[154]. The NADH dehydrogenase of the complex I ubiquinone oxidoreductase and complex III cytochrome c reductase are the two main site of superoxide production via the electron transport chain[155].
The polyol pathway
Oxidative stress increased in circulation of T2DM patients from the polyol pathway. ROS was generated by two enzymes: (1) Aldose reductase in the reaction use NADPH to change glucose to sorbitol. Sorbitol production is a minor reaction in normal physiological conditions. However, 30%-35% of glucose in T2DM conditions is metabolized by polyol pathway[156]. In the condition of sorbitol overproduction, the availability of NADPH is reduced this reflect to reduce glutathione regeneration and NOS synthase activity to cause increased oxidative stress[153]; and (2) Sorbitol dehydrogenase in the second step oxidizes sorbitol to fructose concomitant with NADH overproduction. Increased NADH may be used by NADH oxidases to increase superoxide production[157] include in mitochondrial over superoxide production.
PKCβ1/2
Many structures and biochemical components changed in the circulation of T2DM patients were caused from PKCβ1/2 activation via diacylglycerol leading to cause dysfunction in endothelial contractility and permeability, hemodynamics (retinal blood flow) changes, extracellular matrix protein synthesis, VEGF production and intracellular signaling in the vascular[128,158,159].
Non-enzymatic glycation
Glycation end-product is the binding of ketone or aldehyde groups of glucose with the free amino groups of proteins leading Schiff bases formation without enzymes, then to form the Amadori product and rearrangements of the structure to the irreversible AGEs in the final[160,161]. AGEs has been demonstrated in atherosclerotic lesions and their tissue of T2DM patients and increased AGEs levels associated with severity of the diseases[162]. Moreover, binding of AGEs to specific cell surface receptor for AGE can activate intracellular redox signaling and subsequent to activate the expression of redox-sensitive transcription factors and inflammatory mediator[163-165].
Inflammation
Oxidative stress is the major factor underlying in the CVD, insulin resistance and T2DM pathogenesis. These may explain by the presence of the inflammation conditions. Now, inflammation recognized as the one manifestation of oxidative stress[166] and can be generate the inflammatory mediators including adhesion molecules and interleukins to induce oxidative stress[166]. The concept of atherosclerosis is an inflammatory disease now well established. This chronic inflammation may be involved in the insulin resistance and T2DM pathogenesis[167]. Recent clinical research indicates that sub-clinical inflammation may impact in the development and progression of diabetic complications[165,168]. Moreover, excessive FFA and glucose induce inflammation effect through oxidative stress and reduced antioxidants[169]. Interestingly, the subclinical pro-inflammatory state observed in many pathogenesis conditions such as atherosclerosis, aging, T2DM and cancer, is caused from mitochondrial ROS overgeneration[170].
Other sources of oxidative stress in diabetes
Non-esterified FFAs are elevated in T2DM patients[171]. These excessive FFAs enter the citric acid cycle to generate acetyl-CoA to receive NADH overproduction to cause mitochondrial superoxide over production. In humans, infused FFA has been shown increased lipid peroxidation by elevated isoprostanes marker levels[172,173]. Adipocytokine, leptin is secreted from the adipocytes to act on the central nervous system to decrease food intake. It reflects all effects on the vascular smooth muscle cells, endothelial cells, macrophages and monocytes[174]. Leptin levels are increased and associated with cardiovascular disease in T2DM patients[175-177]. In culture of endothelial cells incubated with leptin to cause ROS production[178,179].
Antioxidants
Regulation of the cellular redox status is depends on the rate of ROS counterbalance and elimination from the enzymatic and/or non-enzymatic antioxidants. Superoxide is converted by SOD to H2O2 and O2 molecule. There are 3 isoforms of SOD such as cytosolic Cu/Zn SOD (SOD1), mitochondrial Mn-SOD (SOD2) and extracellular SOD (SOD3). Catalase, the heme metalloenzyme is expressed in peroxisomes, mitochondria, cytoplasm and nucleus. H2O2 is catalyzed by catalase to oxygen and water[180]. While glutathione peroxidase the selenoprotein, was found in both intracellular and extracellular. Glutathione peroxidase has a highly sensitive function for lipid peroxides degradation, converses H2O2 to water by using the thiol group of glutathione[181]. Their H2O2 detoxification plays the important roles to prevent lipid peroxidation production and regulation of the cellular redox status[182]. The glutathione system, thioredoxin peroxidase is key enzyme to regulate the cellular levels of thiol/disulfide while the production of antioxidant enzymes is regulated by the redox-cellular transcription factors[183]. For example, the expressed transcription factor NF-E2 related factor in the cytosolic is interrupted binding with Keap-1 as the responsible to increase oxidative stress and translocate to the nucleus for initiation of the transcription of the various antioxidant enzymes[184] as the strategy to develop many class of antioxidant, anti-inflammatory, and anticancer agents. Reduction in non-enzymatic antioxidants, thiol glutathione and thioredoxin are the major dysregulation of the cellular redox status[185]. The cellular redox status is reflected by the reduction of glutathione (GSH), oxidized glutathione (GSSG) ratio (or GSH:GSSG ratio), ascorbic acid, tocopherols and methionine and cysteine amino acids. Exogenous herbal antioxidants compounds in dietary foods include flavanoids, anthocyanins and polyphenolics act as ROS scavenging[186,187]. The direct interaction of ROS with non-enzymatic antioxidants is based on chemical structure properties. In free radicals participate in 1e- oxidation while non-radical species was 2e- oxidation. For example, O2•- and OH• radicals react with the ascorbic acid and thiols. While the OH• more activity and instability react with methionine and tocopherols. H2O2 and the non-radical may react with thiols and methionine, and the OONO- discriminate to react with thiols, ascorbic acid, tocopherols and methionine[188].
Oxidative stress induces insulin resistance
Oxidative stress plays the major role in the association with the insulin resistance pathogenesis by insulin signals disruption and adipocytokines dysregulation[8,9]. In rat models, oxidative stress enhances insulin resistance. The evidence suggested that Ang II infused rats required the increased glucose infusion to maintain euglycemia during hyperinsulinemic clamp to stimulate ROS production[10]. For this example, Ang II-infused rats were caused insulin resistance from the suppression on insulin-induced glucose uptake in skeletal muscle and increased in oxidative stress biomarkers in this animal experiment. In experimental model, superoxide dismutase and tempol can reduce the insulin resistance. Many evidences indicated that ROS overproduction may induce insulin resistance and confirmed by the supplementation of antioxidant tempol to cause insulin resistance amelioration in Ren-2 transgenic rats[189]. Insulin-target organs of the obese and diabetic KKAy mice were stimulated and caused ROS over production (skeletal muscle, liver and adipose tissue)[8] and to cause insulin resistance in these organs. High fat-fed mice found ROS overproduction in liver and adipose tissue of these obese mices to induce insulin resistance[190]. Many research studies suggested that antioxidant agents decreased plasma insulin, glucose, triglycerides levels and ameliorate insulin resistance in KKAy mice with no weight loss[8]. Antioxidant coenzyme Q10 supplementation can ameliorate the increased insulin levels in circulation of SHR/cp rats[191]. As mention above, in over nutrition, the excessive glucose occur and a large amount of glucose is metabolized in the glycolysis and TCA cycle leading to increased NADH and FADH2 production in electron transport chain of mitochondrial and increased superoxide production[127]. In aged animals models treated with antioxidant agents or hypocaloric diets led to ameliorate in oxidative stress status and tissue function[131,132].
Insulin resistance
In general population, insulin resistance precede in many years before onset of T2DM and it is also multifactorial[11,12] such as genetic component[11,13]. Insulin resistance and reduction in insulin production are the major characteristics of the T2DM pathogenesis[11,12,14-16]. Modern lifestyle, physical inactivity, abdominal obesity and excessive of adipokines can cause insulin resistance[11,15]. In early stage, normal glucose tolerance is preserved by compensation hyperinsulinemia. About 25% of non-diabetic subject cause insulin resistance in the same ranges that found in T2DM patients[12]. Insulin resistance continuous increases and/or decreases in insulin secretory compensation responses, the deterioration into impaired glucose tolerance occurred. Increased glucose, FFA and insulin levels lead to ROS overproduction, increased oxidative stress and activate stress transduction factor pathways. This can cause insulin activity inhibition and secretion to accelerate the onset of T2DM as shown in Figure 6.
Figure 6.
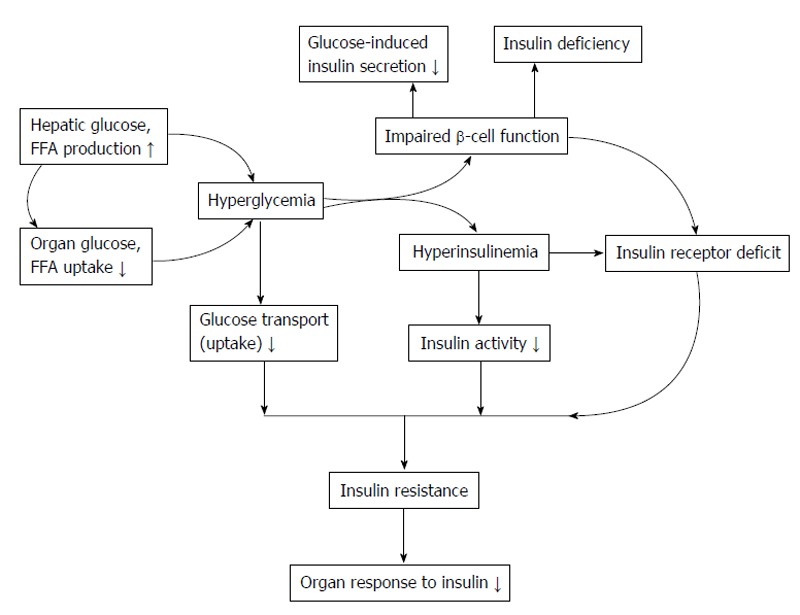
Insulin resistancedevelopment and consequence of β-cell dysfunction. FFA: Free fatty acid.
Oxidative stress has been demonstrated the implication and association in the late complications of diabetes mellitus[17,18] as in the schematic of Figure 5. Many studies have demonstrated ROS overproduction and increased oxidative stress to insulin resistance[192-194]. Both in vitro studies and in animal models demonstrated that α-lipoic acid (LA), antioxidant agent increase insulin sensitivity[194-196]. In clinical trials, supplementation with vitamin C, vitamin E, glutathione increases insulin sensitivity in both insulin-resistance and T2DM patients[197,198]. LA act as insulin sensitizer agent, it increased insulin sensitivity about approximately 25% and approximately 20% higher than metformin and rosiglitazone, respectively[199,200]. Oral supplementation with LA formulation for 6 wk decreased circulating fructosamine levels[201] and increase insulin sensitivity[202] in T2DM patients and the other studies have confirmed 2.5 mmol/L of LA to cause GLUT4 activation and translocation[203-205].
Because insulin resistance occurred before chronic hyperglycemia development[12], that difference from insulin resistance in the pre-diabetic state result from oxidative stress activation by increased glucose levels. However, obesity demonstrated the strong association with insulin resistance. In this regard, the mediator of oxidative stress-induced insulin resistance of the pre-diabetic state might be from the adipocyte-derived factor such as TNF-α[206], leptin[207], FFAs[208-210] and resistin[211]. However, the FFAs elevations are associated with insulin resistance and obesity[208,209,212]. Many studies found that increased FFA levels decrease insulin sensitivity, as in the Randle hypothesis[210] and insulin-signaling inhibition[212]. The increased fasting FFA levels are significantly correlated with decreased reduced/oxidized glutathione ratio in T2DM patients[190]. Elevated FFA concentrations cause mitochondrial dysfunction such as uncouplers of oxidative phosphorylation in mitochondria[213] and increased superoxide production[214]. These caused the exacerbated situation from FFAs induce oxidative stress and reduce intracellular glutathione caused impaired endogenous antioxidant defenses[190,215,216]. Supplementation with glutathione improves insulin sensitivity and β-cell function by the restoration of redox status in T2DM patients and healthy subjects[217].
FFA mediated the nuclear factor-κB (NF-κB) activation, as the consequence of FFAs increased ROS overproduction and glutathione reduction[216,218-220] and also linked to FFA-activated PKC-θ [221] to caused NF-κB activation[222]. Vitamin E supplementation inhibits the FFA-induced NF-κB activation[216] indicated that FFAs act as pro-inflammatory agent effects the alteration of the cellular redox status.
The HOMA-IR was proposed by Matthews et al[223] that can be used to estimate insulin resistance and insulin sensitivity in individuals. HOMA-IR is easy to calculate and no more laborious technique. HOMA-IR method derives from the mathematic calculation from fasting plasma insulin and glucose concentrations.
Oxidative stress and β-cells dysfunction
Increased circulating glucose levels stimulate the β-Cells function by sensing and secreting of insulin in appropriate amount[224] and as the target of oxidative stress. The processes are complex and depend on many factors[16]. The critical glucose metabolism in mitochondrial is the importance linking stimulus the insulin secretion[224-226]. Therefore, mitochondria damage and markedly blunt insulin secretion is also occur by the ability of oxidative stress (H2O2)[226]. Many studies in T2DM patients have suggested that chronic exposure to high glucose and/or high FFA levels impaired β-cells function and β-cells dysfunction[16,227]. Because β-Cells are lower in antioxidant enzymes levels (superoxide dismutase, catalase and glutathione peroxidase) and higher sensitive to oxidative stress[228]. Oxidative stress exposure to β-cells activated the increased p21 cyclin-dependent kinase inhibitor production, decreased insulin mRNA, ATP and calcium flux reductions in mitochondria and cytosol to cause apoptosis[226]. Glucose or methyl succinate can stimulate insulin secretion and inhibit by response to K+ within 30 min[226]. The results indicate that mitochondria in β-cells involved in the processes of glucose induced insulin secretion are affected by increased oxidative stress. Lipid peroxidation, oxidative stress products exposed to islets, inhibited insulin secretion and also caused glucose oxidation[229]. Conversely, antioxidants can protect β-cell against the toxicity of oxidative stress, AGEs production and inhibit NF-κB activation[230-234]. These antioxidants are N-acetyl cysteine (NAC), α-phenyl-tert butylnitrone, aminoguanidine and zinc. Recent research study evaluated β-cells function after over expression of glutamine. Hexosamine over production resulted from the deterioration of insulin signaling of glucose-stimulated insulin secretion. Fructose-6-phosphate amidotransferase is the rate-limiting enzyme increase in hexosamine pathway[235], coincident with increased H2O2 production[235] that can ameliorate by NAC supplementation.
β-cells glucose-induced toxicity
West[19] demonstrated that insulin secretion in T2DM patients improved by the reduction of hyperglycemia with diet, insulin or sulfonylureas. On the other hand, in healthy normal, high glucose infused as a clamp reduces insulin secretion[236]. In the study of long term culture of HIT-T15 and/or βTC-6 cells demonstrated that increased glucose levels cause decreased insulin secretion, insulin mRNA and decreased binding of transcription factors[237,238]. Thus, glucose toxicity, the concept of the condition of hyperglycaemia itself can decrease insulin secretion which implies the irreversible damage to cellular components of β-cells[239]. Generally in β-cells, excessive glucose oxidation and metabolism will always cause to ROS over production. Superoxide dismutase and catalase are normally as the detoxified antioxidant enzymes. β-Cells are low amount of these antioxidant enzymes and also low in glutathione peroxidase, a redox-regulating enzyme[240]. Then, hyperglycaemia condition leads to increase ROS production and accumulation in β-cells and subsequent of cellular components damage. Pancreas duodenum homeobox-1 is an insulin promoter activity regulator was loss leading to β-cell dysfunction[240]. Supplementation with NAC and/or aminoguanidine can ameliorate the glucotoxic effects on insulin gene activity[230], reduced insulin levels and increased insulin mRNA and insulin sensitivity[230].
β-cells lipid-induced toxicity
Lipotoxicity to β-cells concept, elevation of non-esterified fatty acids concentrations in diabetic and non-diabetic obese patients, result of the enhanced adipocyte lipolysis. In the presence of the excessive fatty acid oxidation in β-cells is caused increased long-chain acyl CoA accumulation leading to inhibite β-cells function[241]. This process is as an integral part of the normal insulin secretory function. This long-chain acyl CoA can inhibit the insulin secretory function by opening β-cell K+-sensitive ATP channels. In the second mechanism, in long-term culture of β-cells formulas with FFAs can effect the potential reduction on mitochondrial membrane and uncoupler proteins-2 over expression to cause the K+-sensitive ATP channels opening which lead to decreased ATP production and insulin secretion[242,243]. Third mechanism, β-cells apoptosis might possess from triglyceride or fatty acid induced ceramide synthesis and/or nitric oxide production. Thus, impaired insulin secretion and β-cell dysfunction strongly associated with the FFA-stimulated ROS overproduction[244].
β-cells combined glucose/lipid toxicity
Elevation of glucose and FFA levels are the major characteristic of T2DM patients. This combination is the major β-cells toxicity and require the maximize protection. In culture cells of islets or HIT cells were exposed to high concentrations of glucose and FFA levels. There was decrease in insulin-gene activity and insulin mRNA[245]. In the study of islets co-culture with high glucose and palmitate levels caused impaired insulin signaling of the glucose-stimulated insulin secretion[244]. Recent studies have confirmed that β-cells lipotoxicity is the concurrent status as the amplifying effect mediated by glucose toxicity in hyperglycemia condition[246,247].
Dyslipidemia
Insulin resistance and T2DM are characterized by dyslipidemia one major risk factor for cardiovascular disease. Lipid triad is the complex metabolic milieu associated with dyslipidaemia[248] comprise with hypertriglyceridemia, low levels of high-density lipoprotein cholesterol (HDL-C) and the appearance of small, dense, LDL (sdLDL) - and caused excessive post prandial lipemia[249,250]. Diabetic dyslipidemia caused from the disturbance of lipid metabolism, an early event cardiovascular complications development and was preceded in T2DM patients by several years[249-253]. Indeed, insulin resistance status in both with and without T2DM patients was display qualitatively similar lipid abnormalities[250]. The different components of diabetic dyslipidemia are closely linked to each other metabolically[249-253] and are initiated by the elevation of triglyceriderich very LDL (VLDL) from hepatic over production[249,251]. It is the key importance mechanisms to elucidate the over production of VLDL involved in diabetic dyslipidemia[249].
In insulin resistance state, decrease insulin function and lack of insulin inhibits lipolysis leads to increase FFAs generation of and lower lipoprotein lipase activity. This occurs after meal consumption, generates a chylomicron remnant rich in TG[254], caused elevated hepatic FFAs and VLDL TG-rich particles secretion. These processes affects HDL-C metabolism through the interchange with TG-rich lipoproteins via cholesteryl ester transfer protein to produce HDL particles containing high TG concentrations. These HDL-TG particles were hydrolyzed with hepatic lipase to TG and HDL. This HDL becomes smaller and less antiatherogenic activity, easily to remove from the circulation by the kidneys. Moreover, insulin resistance in T2DM patients associated with endothelial dysfunction led to increase risk of CVD[255]. The most atherogenic subfractions of sdLDL are elevated in circulation of obesity individuals, as a key feature in association with elevated triglyceride and low HDL cholesterol. Elevated sdLDL concentrations are also founded in abdominal obesity subjects and demonstrated greater myocardial risk.The mechanisms are related to excess accumulation of abdominal adipose tissues, elevated total cholesterol and LDL-C and related to high saturated-fat consumption, weight gain and obesity.
Dyslipidemia is commonly occurred in T2DM patients and might play the major role in accelerated macrovascular atherosclerotic disease and increased CVD risk in T2DM patients[256]. Dyslipidemia in T2DM patients as lipids triad is characterized by increased insulin levels, hypertriglyceridemia, low HDL-C levels and increased sdLDL-particles (independent of LDL-cholesterol) and increased TG-rich remnant lipoprotein (TGRLs) concentrations[257,258]. In this manner, low HDL-C levels associated with hyperinsulinemia or insulin resistance and insulin signaling for insulin-mediated glucose disposal[259] characterized by higher fasting plasma glucose and insulin levels. Then, these major changes associated with the insulin resistance syndrome are increased TGRLs and decreased HDL-C levels. Thus, in dyslipidemia, using the lipoprotein concentration ratios are associated with insulin resistance and increased CVD risk conditions. Lipoprotein ratios might be useful to identify insulin resistance individuals even different in fasting glucose or insulin levels. Obesity, metabolic syndrome, and T2DM may also show the same dyslipidemia characteristic[12,257,259] and measuring TG, HDL-C, TC/HDL-C and TG/HDL-C ratio in circulation may also use as insulin resistance estimation. For example, these TG, HDL-C, TC/HDL-C and TG/HDL-C ratio are independently associated with insulin levels, insulin resistance and CVD risk[258,260,261].
Lipoprotein ratios: In description above, the major change is increased TGRLs and decreased HDL-C levels are associated with insulin resistance syndrome. Insulin plays the important role in TG metabolism, in normal condition TGRLs particles reduces synthesis by the distinct pathways when compared with VLDL particles synthesis[249,258]. Insulin fails to suppress VLDL particles synthesis[262]. Insulin resistance is significantly associated with increased lipid synthesis in the liver, increased FFAs flow to the liver and decreased VLDL particles clearance resulting in increased VLDL levels in the circulation[251]. Thus, dyslipidemia (as lipoprotein ratios) may associate with insulin resistance and increased CVD risk. On this basis, waist circumference, LDL-C, TG levels, insulin resistance and the CVD risk are estimated[263]. The major features of dyslipidemia are determined by hypertriglyceridemia, low HDL-C levels and slightly high or normal LDL-C levels with altered composition. Hypertriglyceridemia is indicate as elevated atherogenic chylomicron and VLDL remnant and associated with increased CVD risk[264,265]. These phenomenons demonstrated the problems of VLDL and HDL levels but not the LDL levels and concurrent with increased insulin levels. Low HDL-C level is associated with the hyperinsulinemia and/or insulin resistance and insulin signaling for insulin-mediated glucose disposal[259]. All of these features are associated with coronary heart disease risk in obesity, metabolic syndrome and T2DM patients. The TC/HDL-C, TG/HDL-C ratios and non-HDL-C (as TC - HDL-C) were used as surrogate markers for insulin levels and insulin resistance estimation. In Tangvarasittichai et al[258] study suggests that TC/HDL-C, TG/HDL-C ratios and non-HDL-C can be used as markers of insulin levels, insulin resistance and CVD risk factor[258,263]. The highest % sensitivity and % specificity cut-off points corresponding to the TC/HDL-C, TG/HDL-C ratios and non-HDL-C are 3.58, 2.48 and 130.4, respectively[258]. Because of TC/HDL-C, TG/HDL-C ratios and non-HDL-C are easily calculated and ordered with every lipid profiles available to the clinician and no costs addition. The cut-off value of these ratios in Tangvarasittichai et al[258] study was lower than the results from Western populations[266-268]. Then, insulin resistance was significantly predicted by these markers. For atherosclerotic risk assessment in obesity, metabolic syndrome and T2DM patients requires more attention to lipid screening.
Development of T2DM from insulin resistance
Insulin resistance often occurs with T2DM but is insufficient for the T2DM development. β-cells dysfunction are important event for the T2DM development and progression. In early stage of insulin resistance, β-cells increase the secretory function try to compensate and control hyperglycemia. In Pima Indian population study caused acute insulin response dysfunction or decreased β-cell responses was found during the normal glucose tolerance state in individuals who eventually progressed from normal glucose tolerance to impaired glucose tolerance or T2DM when compared with individuals who persisted in the state of normal glucose tolerance[269]. There was evidence of early defects in glucose disposal by decreased insulin sensitivity before the development of glucose intolerance state, although output of circulating glucose did not increase until the progression from impaired glucose tolerance to T2DM revealed. Interestingly, individuals who demonstrated transient glucose intolerance but were able to recover and to reach normal glucose tolerance and did not show the early secretory defect observed in progressed individuals[269]. β-cells failure or dysfunction occurred as the results of the combination of increased oxidative stress, glucose and lipids accumulation to cause glucotoxicity and lipotoxicity to β-cells to progress increased apoptosis and loss of the insulin granule secretory components expression[270].
T2DM
The World Health Organization updated the prevalence of T2DM estimated by the year 2025 those 30.3 million people in the United States and total of 380 million people worldwide will be diagnosed as DM[271]. By the year 2050, those 45.6 million Americans will be diagnosed as DM[272]. T2DM is associated with obesity, sedentary lifestyle and lack of exercise in the aging population. There are a number of gene abnormalities related to T2DM, that showed significant differences exist in the abnormalities gene associated with T2DM among the various ethnic populations, such as African Americans, Asians and Europids[273,274]. The contribution of any one of these genes to T2DM is small and total aggregate of all described genes accounts for < 15% of the predisposition[273,275]. It is typically diagnosed in patients older than 30 years with overweight or obesity and positive in family history of T2DM. However, insulin resistance may occur and develop in many years before diagnosed as T2DM[276]. Figure 7 summarized the etiology of the T2DM pathogenesis.
Figure 7.
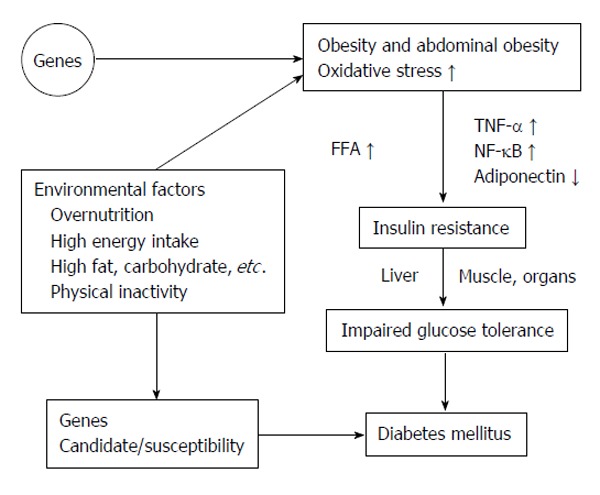
Summarized the etiology of the type 2 diabetes mellitus pathogenesis. FFA: Free fatty acid; NF-κB: Nuclear factor-κB; TNF-α: Tumor necrosis factor α.
Patients are diagnosed as T2DM when plasma glucose levels reach at the diagnostic criteria (Table 1). These T2DM patients are at high risk for microvascular complications (e.g., nephropathy, retinopathy and neuropathy) and macrovascular complications (e.g., peripheral vascular disease, cerebrovascular disease and cardiovascular disease). T2DM patients with good controlled plasma glucose levels demonstrated to delay the progression of microvascular and macrovascular complications[271,277].
Table 1.
Type 2 diabetes mellitus and glucose levels for diagnostic criteria1
| Glucose management test | Range | Diagnosis |
| Fasting plasma glucose (mg/dL) (at least 8 h fast) | ≥ 126 | Diabetes mellitus |
| 100-125 | Impaired fasting glucose | |
| ≤ 99 | Normal | |
| 2-h oral glucose tolerance test of 75 g glucose load (mg/dL) WITH | ≥ 200 | Diabetes |
| Random screening with common symptoms of diabetes (polyuria, | 140-199 | Impaired glucose tolerance |
| polydipsia, weight loss, etc.) | ≤ 139 | Normal |
| Hemoglobin A1c (%) | ≥ 6.5 | Diabetes |
| 5.7-6.4 | Prediabetes/high risk | |
| ≤ 5.7 | Normal |
1All tests in diabetes range must be repeated after 24 h, to be confirmed diagnosis.
MANAGEMENT OF DYSLIPIDEMIA AND HYPERGLYCEMIA IN T2DM PATIENTS
Fasting serum lipids profile should be determined annually in T2DM patients as in the recommendation by the American Diabetes Association (ADA)[278]. ADA recommended for the satisfied lipids profile level as low-risk by LDL-C < 100 mg/dL (2.6 mmol/L), triglycerides < 150 mg/dL (1.7 mmol/L) and HDL-C > 50 mg/dL (1.3 mmol/L)[276].
Treatment
Lifestyle interventions: The American Diabetes Association and the American Heart Association recommend that increased physical activity and lifestyle modifications should be advised for all T2DM patients[278,279]. Combination with such interventions included nutrition therapy or supplementation, weight loss and non-smoking. These have been help T2DM patients to receive better controlled their lipid concentrations. Nutrition interventions and supplementations should be designed according to the condition of T2DM individuals such as diabetes status, age, other comorbidities and avoidance to intake transfat, saturated fat, cholesterol and should increase intake of fiber (fiber in oats, legumes, citrus), omega-3 fatty acids and plant stanols/sterols[278]. Glycemic control can also modify circulating triglycerides levels, especially in T2DM patients with hypertriglyceridemia and poor glycemic control[278].
Pharmacological interventions of dyslipidemia
There are many pharmacological classes available for dyslipidemia treatment.
Statins: Statins inhibit enzyme 3-hydroxy-3-methylglutaryl CoA reductase suppress cholesterol synthesis and increase number and activity of LDL-receptor. Statins are effective drug for lowering LDL-cholesterol, raising HDL-C and reducing TG levels. There are seven pharmaceutical forms of statins including lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, rosuvastatin and pitavastatin available in the market. Statins also have the other pharmacodynamic actions such as vascular inflammation reduction, immune suppression, improved endothelial function, platelet aggregability, enhanced fibrinolysis, antithrombotic action, increase neovascularization in ischemic tissue and stabilization of atherosclerotic plaques[280].
Fibrates
Fibrates control the lipid metabolism by mediated through peroxisome proliferator-activated receptors-α activation, stimulation of β-oxidation of fatty acids in peroxisomes and mitochondria to cause lowering fatty acid and triglycerides levels in circulation. The first drug of this class is Clofibrate. Eventually, the revolution in lipid-lowering drugs research discover of many other fibrate drugs such as fenofibrate, bezafibrate, gemfibrozil and ciprofibrate. These drugs demonstrated the adverse effect to cause hepatomegaly and tumor formation in the liver of rodents. Then, they had restricted for the widely use in humans. Gemfibrozil and fenofibrate are Food and Drug Administration (FDA)-approved for lipid lowering drugs due to milder effect on peroxisome proliferation.
Nicotinic acid
Long term study of the coronary drug project demonstrated that niacin is the effective drug to increase HDL-C levels and reduced CVD events[281] in a non-diabetic subjects. Niacin cause adverse effects on the glycemic control levels in T2DM patients. In high doses treatment with niacin may increase blood glucose levels. The modest doses of 750-2000 mg/d of niacin are significantly increased HDL-C levels and decreased LDL-C, triglyceride levels and accompanied with modest changes in glucose levels for diabetes therapy[282,283]. However, there is no evidence for the CVD outcomes reduction with niacin supplementation in T2DM patients.
Antihyperglycemic drugs: The standard care for T2DM patients is mainly in controlled blood glucose levels by using glycemic lowering drugs and concomitant with controlled diet and increased physical activity. With proper controlled and managed these contributors such as circulating glucose levels, hemoglobin A1c, lifestyle modifications, these can be effectively controlled and reduced the progression and complications disease. In general, only approximately 50% to 60% of T2DM patients have achieved their glycemic goals[284]. There are many reasons for poor control of T2DM including medication efficacy, adverse effects, access to medications and health care education, poor adherence, lack of lifestyle changes and no physical activity. Now a day, more pharmacologicals for T2DM treatment have been approved for use. There are 12 classes of antihyperglycemic drugs FDA-approved in the United States[285] such as sulfonylureas, meglitinides, thiazolidinediones, dipeptidyl peptidase-4 (DPP-4) inhibitors, biguanides, sodium glucose transporter 2 inhibitors, α-glucosidase inhibitors, amylin analogues and glucagon-like peptide-1 (GLP-1) receptor agonists. These are insulin analogues. Metformin is one of the most commonly prescribed medications for T2DM management. Metformin treatment ameliorate the insulin resistance especially in liver and skeletal muscle but less effect in adipose tissue[286,287], decreased inflammatory response, improved glycemic control[288,289] and enhance β-cell function in T2DM patients by increased insulin sensitivity and glucotoxicity reduction[290]. Metformin reduces fatty acid oxidation in adipose tissue[291], increased GLUT4 translocation in muscle and adipose tissues by activated enzyme adenosine monophosphate kinase and reduced gluconeogenesis in liver[292-295]. There are many developed non-conventional drugs to improve glycemic control such as Cycloset is used together with diet and exercise to treat type 2 diabetes. Cycloset is not for treating type 1 diabetes. Welchol is a non-absorbed, polymeric form, lipid-lowering and glucose-lowering agent for oral administration. Welchol is a high-capacity bile acid-binding molecule. Afrezza Inhalation Powder is the FDA approved the inhalation form of insulin. The new drug is not a substitute for long-acting insulin and use as the combination with conventional long-acting insulin drug for both types of diabetes and many drugs are in the late clinical trials state.
There are new medications and treatments were identified from the FDA, they are in the clinical trials or waiting for approval treatment in dyslipidemia, obesity and T2DM[296]. Recent research study reports that metformin treatment cause metabolic effects to increase GLP-1 concentration in the circulation[297,298]. GLP-1 is an incretin generated from the transcription product of the proglucagon gene. Incretin is a signaling polypeptide contained with 30-amino acid. GLP-1 secretion by ileal L-cells is not depend on the presence of nutrients in the small intestine and responsible for stimulated insulin secretion to limit glucose elevations with the higher efficacy at high glucose levels[276,299]. Elevated GLP-1 secretion might possibly cause increased glucose absorption in the distal segments of small intestine.
Incretins are the gastrointestinal hormone secreted from the intestine and stomach responsible for oral food intake and stimulated the secretion of insulin during meals in healthy peoples[276]. Two major incretin molecules are (1) GLP-1; and (2) Glucose-dependent insulinotopic peptide knows as gastric inhibitory polypeptide (GIP) and to neutralize stomach acid to protect the small intestine and no therapeutic efficacy in T2DM. GLP-1 has lower glucose levels by stimulated insulinproduction and increased glucose metabolism in adipose tissue and muscle. GLP-1 promote the pancreatic β-cells proliferation, reduce apoptosis, increase cardiac chronotropic, inotropic activity, decreases glucagon secretion, reduces glucose production, increase appetite suppression for food intake reduction and slow gastric emptying[271,276,299]. GLP-1 is degraded by enzyme DPP-4 and this enzyme does not inhibit by metformin[298]. The prevention of GLP-1degradation by DPP-4 is one method to increase the effects of GLP-1. DPP-4 inhibitor drugs inhibit the glucagon secretion which in turn increases secretion of insulin to decrease blood glucose levels and decreases gastric emptying. The FDA-approved the DPP-4 inhibitor drugs including sitagliptin (Januvia), alogliptin (Nesina), saxagliptin (Onglyza), linagliptin (Tradjenta), anagliptin, vildagliptin, teneligliptin, gemigliptin and dutogliptin. The adverse effects are dose-dependent to cause headache, vomiting, nausea, nasopharyngitis, hypersensitivity and other conditions. Other side effects of exenatide (GLP-1 agonist) note for abdominal pain, acid stomach, diarrhea, altered renal function, weight loss, dysgeusia, belching and cause pruritus, urticaria and rash reactions at the injection site.
CONCLUSION
In this present review has described the detrimental effects from chemicals and biochemicals reaction, metals, medications, over nutrition, obesity and diseases in oxidative stress, insulin resistance development and the progression of T2DM and the progression of diabetic complications and organ dysfunctions. Oxidative stress played underling associated with the pathogenesis of diseases, leading to increases risk of insulin resistance, dyslipidemia, elevated blood pressure, metabolic syndrome, inflammation and endothelial dysfunction. This reviewed support the oxidative stress contribution of the multifactorial etiology of oxidative stress and insulin resistance in the whole body. ROS act as the signal transduction factor and plays the important role in oxidative stress-mediated downstream signaling pathways and enhances the cell death. Furthermore, risk for several chronic diseases development associated with oxidative stress and metabolic syndrome including T2DM, hypertension, arthritis, congestive heart failure, chronic renal failure, cancer and Alzheimer’s. These diseases may be substantially reduced by dietary modifications, increased physical activity and antioxidant drugs ameliorated oxidative stress. The therapeutic approaches target on oxidative stress may delay or prevent the progression and onset of diseases. Then, antioxidants supplementation may curtail the progression and onset of the metabolic disease complications. Antioxidant interventions, an importance goal of future clinical investigations should be implementation and to improve oral bioavailability targeted to the oxidant overproduction site. Lifestyle change remains the best prevention and therapeutic approach to oppose the increasing epidemic of cardiovascular diseases, obesity, hypertension, dyslipidemia and T2DM. Finally, the connection between oxidative stress, insulin resistance, dyslipidemia, inflammation, life style, atherosclerosis and diabetes as demonstrated in the schematic in Figure 8.
Figure 8.
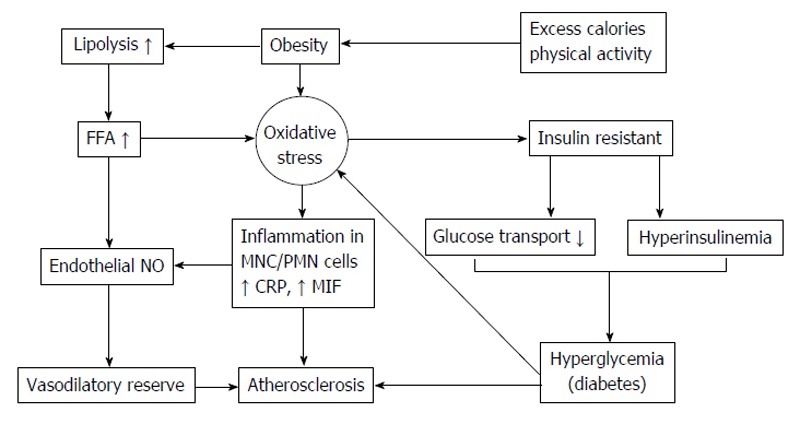
Connection between life style, oxidative stress, insulin resistance, inflammation and atherosclerosis. FFA: Free fatty acid; NO: Nitric oxide; MNC: Mononuclear cells; PMN: Polymorpho nuclear cells; CRP: C-reactive protein; MIF: Migration inhibitory factor.
Footnotes
P- Reviewer: Sicari R, Soare A S- Editor: Ji FF L- Editor: A E- Editor: Liu SQ
Conflict-of-interest: The author declares that there are no conflicts of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: September 4, 2014
First decision: November 14, 2014
Article in press: January 12, 2015
References
- 1.Cossarizza A, Ferraresi R, Troiano L, Roat E, Gibellini L, Bertoncelli L, Nasi M, Pinti M. Simultaneous analysis of reactive oxygen species and reduced glutathione content in living cells by polychromatic flow cytometry. Nat Protoc. 2009;4:1790–1797. doi: 10.1038/nprot.2009.189. [DOI] [PubMed] [Google Scholar]
- 2.Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191:421–427. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sies H. Oxidative stress: oxidants and antioxidants. Exp Physiol. 1997;82:291–295. doi: 10.1113/expphysiol.1997.sp004024. [DOI] [PubMed] [Google Scholar]
- 4.Freeman BA, Crapo JD. Biology of disease: free radicals and tissue injury. Lab Invest. 1982;47:412–426. [PubMed] [Google Scholar]
- 5.Slater TF. Free-radical mechanisms in tissue injury. Biochem J. 1984;222:1–15. doi: 10.1042/bj2220001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dobrian AD, Davies MJ, Schriver SD, Lauterio TJ, Prewitt RL. Oxidative stress in a rat model of obesity-induced hypertension. Hypertension. 2001;37:554–560. doi: 10.1161/01.hyp.37.2.554. [DOI] [PubMed] [Google Scholar]
- 7.Vincent HK, Taylor AG. Biomarkers and potential mechanisms of obesity-induced oxidant stress in humans. Int J Obes (Lond) 2006;30:400–418. doi: 10.1038/sj.ijo.0803177. [DOI] [PubMed] [Google Scholar]
- 8.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 10.Ogihara T, Asano T, Ando K, Chiba Y, Sakoda H, Anai M, Shojima N, Ono H, Onishi Y, Fujishiro M, et al. Angiotensin II-induced insulin resistance is associated with enhanced insulin signaling. Hypertension. 2002;40:872–879. doi: 10.1161/01.hyp.0000040262.48405.a8. [DOI] [PubMed] [Google Scholar]
- 11.Dedoussis GV, Kaliora AC, Panagiotakos DB. Genes, diet and type 2 diabetes mellitus: a review. Rev Diabet Stud. 2007;4:13–24. doi: 10.1900/RDS.2007.4.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reaven GM. Insulin resistance: the link between obesity and cardiovascular disease. Med Clin North Am. 2011;95:875–892. doi: 10.1016/j.mcna.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 13.Kahn CR, Vicent D, Doria A. Genetics of non-insulin-dependent (type-II) diabetes mellitus. Annu Rev Med. 1996;47:509–531. doi: 10.1146/annurev.med.47.1.509. [DOI] [PubMed] [Google Scholar]
- 14.Unger RH. Reinventing type 2 diabetes: pathogenesis, treatment, and prevention. JAMA. 2008;299:1185–1187. doi: 10.1001/jama.299.10.1185. [DOI] [PubMed] [Google Scholar]
- 15.Kahn CR. Banting Lecture. Insulin action, diabetogenes, and the cause of type II diabetes. Diabetes. 1994;43:1066–1084. doi: 10.2337/diab.43.8.1066. [DOI] [PubMed] [Google Scholar]
- 16.Grodsky GM. The importance of rapid insulin secretion: revisited. Diabetes Technol Ther. 1999;1:259–260. doi: 10.1089/152091599317161. [DOI] [PubMed] [Google Scholar]
- 17.Rösen P, Nawroth PP, King G, Möller W, Tritschler HJ, Packer L. The role of oxidative stress in the onset and progression of diabetes and its complications: a summary of a Congress Series sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes Metab Res Rev. 2001;17:189–212. doi: 10.1002/dmrr.196. [DOI] [PubMed] [Google Scholar]
- 18.Nishikawa T, Edelstein D, Brownlee M. The missing link: a single unifying mechanism for diabetic complications. Kidney Int Suppl. 2000;77:S26–S30. doi: 10.1046/j.1523-1755.2000.07705.x. [DOI] [PubMed] [Google Scholar]
- 19.West IC. Radicals and oxidative stress in diabetes. Diabet Med. 2000;17:171–180. doi: 10.1046/j.1464-5491.2000.00259.x. [DOI] [PubMed] [Google Scholar]
- 20.Farrugia G, Balzan R. Oxidative stress and programmed cell death in yeast. Front Oncol. 2012;2:64. doi: 10.3389/fonc.2012.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J. Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem. 2004;266:37–56. doi: 10.1023/b:mcbi.0000049134.69131.89. [DOI] [PubMed] [Google Scholar]
- 22.Narayanan D, Xi Q, Pfeffer LM, Jaggar JH. Mitochondria control functional CaV1.2 expression in smooth muscle cells of cerebral arteries. Circ Res. 2010;107:631–641. doi: 10.1161/CIRCRESAHA.110.224345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Didion SP, Hathaway CA, Faraci FM. Superoxide levels and function of cerebral blood vessels after inhibition of CuZn-SOD. Am J Physiol Heart Circ Physiol. 2001;281:H1697–H1703. doi: 10.1152/ajpheart.2001.281.4.H1697. [DOI] [PubMed] [Google Scholar]
- 24.Niwa K, Haensel C, Ross ME, Iadecola C. Cyclooxygenase-1 participates in selected vasodilator responses of the cerebral circulation. Circ Res. 2001;88:600–608. doi: 10.1161/01.res.88.6.600. [DOI] [PubMed] [Google Scholar]
- 25.Kinugawa S, Huang H, Wang Z, Kaminski PM, Wolin MS, Hintze TH. A defect of neuronal nitric oxide synthase increases xanthine oxidase-derived superoxide anion and attenuates the control of myocardial oxygen consumption by nitric oxide derived from endothelial nitric oxide synthase. Circ Res. 2005;96:355–362. doi: 10.1161/01.RES.0000155331.09458.A7. [DOI] [PubMed] [Google Scholar]
- 26.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dikalova AE, Góngora MC, Harrison DG, Lambeth JD, Dikalov S, Griendling KK. Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via eNOS uncoupling. Am J Physiol Heart Circ Physiol. 2010;299:H673–H679. doi: 10.1152/ajpheart.00242.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santhanam AV, d’Uscio LV, Smith LA, Katusic ZS. Uncoupling of eNOS causes superoxide anion production and impairs NO signaling in the cerebral microvessels of hph-1 mice. J Neurochem. 2012;122:1211–1218. doi: 10.1111/j.1471-4159.2012.07872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das J, Roy A, Sil PC. Mechanism of the protective action of taurine in toxin and drug induced organ pathophysiology and diabetic complications: a review. Food Funct. 2012;3:1251–1264. doi: 10.1039/c2fo30117b. [DOI] [PubMed] [Google Scholar]
- 31.Das J, Ghosh J, Manna P, Sil PC. Taurine suppresses doxorubicin-triggered oxidative stress and cardiac apoptosis in rat via up-regulation of PI3-K/Akt and inhibition of p53, p38-JNK. Biochem Pharmacol. 2011;81:891–909. doi: 10.1016/j.bcp.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 32.Ghosh J, Das J, Manna P, Sil PC. Acetaminophen induced renal injury via oxidative stress and TNF-alpha production: therapeutic potential of arjunolic acid. Toxicology. 2010;268:8–18. doi: 10.1016/j.tox.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 33.Sarkar K, Sil PC. Attenuation of Acetaminophen-Induced Hepatotoxicity In Vivo and In Vitro by a 43-kD Protein Isolated from the Herb Cajanus indicus L. Toxicol Mech Methods. 2007;17:305–315. doi: 10.1080/15376510601031919. [DOI] [PubMed] [Google Scholar]
- 34.Sarkar K, Sil PC. Cajanus indicus leaf protein: Beneficial role in experimental organ pathophysiology. A review. Pathophysiology. 2011;18:295–303. doi: 10.1016/j.pathophys.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 35.Chatterjee M, Sil PC. Protective role ofPhyllanthus niruri against nimesulide induced hepatic damage. Indian J Clin Biochem. 2007;22:109–116. doi: 10.1007/BF02912892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhattacharyya S, Ghosh J, Sil PC. Iron induces hepatocytes death via MAPK activation and mitochondria-dependent apoptotic pathway: beneficial role of glycine. Free Radic Res. 2012;46:1296–1307. doi: 10.3109/10715762.2012.712690. [DOI] [PubMed] [Google Scholar]
- 37.Kayankarnna W, Thessomboon D, Niyomtam S, Pingmuangkaew P, Nunthawarasilp P, Tangvarasittichai S. Elevated cadmium exposure associated with oxidative stress and oxidative DNA damage in population of cadmium-contaminated area. IJTPR. 2013;5:102–108. [Google Scholar]
- 38.Pal PB, Pal S, Das J, Sil PC. Modulation of mercury-induced mitochondria-dependent apoptosis by glycine in hepatocytes. Amino Acids. 2012;42:1669–1683. doi: 10.1007/s00726-011-0869-3. [DOI] [PubMed] [Google Scholar]
- 39.Pal PB, Sinha K, Sil PC. Mangiferin, a natural xanthone, protects murine liver in Pb(II) induced hepatic damage and cell death via MAP kinase, NF-κB and mitochondria dependent pathways. PLoS One. 2013;8:e56894. doi: 10.1371/journal.pone.0056894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jia L, Liu Z, Sun L, Miller SS, Ames BN, Cotman CW, Liu J. Acrolein, a toxicant in cigarette smoke, causes oxidative damage and mitochondrial dysfunction in RPE cells: protection by (R)-alpha-lipoic acid. Invest Ophthalmol Vis Sci. 2007;48:339–348. doi: 10.1167/iovs.06-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roy A, Sil PC. Tertiary butyl hydroperoxide induced oxidative damage in mice erythrocytes: Protection by taurine. Pathophysiology. 2012;19:137–148. doi: 10.1016/j.pathophys.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 42.Bhattacharya S, Chatterjee S, Manna P, Das J, Ghosh J, Gachhui R, Sil PC. Prophylactic role of D-Saccharic acid-1,4-lactone in tertiary butyl hydroperoxide induced cytotoxicity and cell death of murine hepatocytes via mitochondria-dependent pathways. J Biochem Mol Toxicol. 2011;25:341–354. doi: 10.1002/jbt.20393. [DOI] [PubMed] [Google Scholar]
- 43.Ghosh A, Mandal AK, Sarkar S, Das N. Hepatoprotective and neuroprotective activity of liposomal quercetin in combating chronic arsenic induced oxidative damage in liver and brain of rats. Drug Deliv. 2011;18:451–459. doi: 10.3109/10717544.2011.577110. [DOI] [PubMed] [Google Scholar]
- 44.Sarkar MK, Sil PC. Prevention of tertiary butyl hydroperoxide induced oxidative impairment and cell death by a novel antioxidant protein molecule isolated from the herb, Phyllanthus niruri. Toxicol In Vitro. 2010;24:1711–1719. doi: 10.1016/j.tiv.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 45.Bhattacharya S, Manna P, Gachhui R, Sil PC. D-saccharic acid 1,4-lactone protects diabetic rat kidney by ameliorating hyperglycemia-mediated oxidative stress and renal inflammatory cytokines via NF-κB and PKC signaling. Toxicol Appl Pharmacol. 2013;267:16–29. doi: 10.1016/j.taap.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 46.Rashid K, Bhattacharya S, Sil PC. Protective role of D-saccharic acid-1,4-lactone in alloxan induced oxidative stress in the spleen tissue of diabetic rats is mediated by suppressing mitochondria dependent apoptotic pathway. Free Radic Res. 2012;46:240–252. doi: 10.3109/10715762.2011.650694. [DOI] [PubMed] [Google Scholar]
- 47.Manna P, Ghosh J, Das J, Sil PC. Streptozotocin induced activation of oxidative stress responsive splenic cell signaling pathways: protective role of arjunolic acid. Toxicol Appl Pharmacol. 2010;244:114–129. doi: 10.1016/j.taap.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 48.Das J, Vasan V, Sil PC. Taurine exerts hypoglycemic effect in alloxan-induced diabetic rats, improves insulin-mediated glucose transport signaling pathway in heart and ameliorates cardiac oxidative stress and apoptosis. Toxicol Appl Pharmacol. 2012;258:296–308. doi: 10.1016/j.taap.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 49.Matés JM, Segura JA, Alonso FJ, Márquez J. Oxidative stress in apoptosis and cancer: an update. Arch Toxicol. 2012;86:1649–1665. doi: 10.1007/s00204-012-0906-3. [DOI] [PubMed] [Google Scholar]
- 50.Chrissobolis S, Faraci FM. The role of oxidative stress and NADPH oxidase in cerebrovascular disease. Trends Mol Med. 2008;14:495–502. doi: 10.1016/j.molmed.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller AA, Budzyn K, Sobey CG. Vascular dysfunction in cerebrovascular disease: mechanisms and therapeutic intervention. Clin Sci (Lond) 2010;119:1–17. doi: 10.1042/CS20090649. [DOI] [PubMed] [Google Scholar]
- 52.Biliński T, Litwińska J, Błaszczyński M, Bajus A. Superoxide dismutase deficiency and the toxicity of the products of autooxidation of polyunsaturated fatty acids in yeast. Biochim Biophys Acta. 1989;1001:102–106. doi: 10.1016/0005-2760(89)90312-3. [DOI] [PubMed] [Google Scholar]
- 53.Cabiscol E, Piulats E, Echave P, Herrero E, Ros J. Oxidative stress promotes specific protein damage in Saccharomyces cerevisiae. J Biol Chem. 2000;275:27393–27398. doi: 10.1074/jbc.M003140200. [DOI] [PubMed] [Google Scholar]
- 54.Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gutteridge JM, Halliwell B. Comments on review of Free Radicals in Biology and Medicine, second edition, by Barry Halliwell and John M. C. Gutteridge. Free Radic Biol Med. 1992;12:93–95. doi: 10.1016/0891-5849(92)90062-l. [DOI] [PubMed] [Google Scholar]
- 56.Savaskan NE, Ufer C, Kühn H, Borchert A. Molecular biology of glutathione peroxidase 4: from genomic structure to developmental expression and neural function. Biol Chem. 2007;388:1007–1017. doi: 10.1515/BC.2007.126. [DOI] [PubMed] [Google Scholar]
- 57.Djordjević VB. Free radicals in cell biology. Int Rev Cytol. 2004;237:57–89. doi: 10.1016/S0074-7696(04)37002-6. [DOI] [PubMed] [Google Scholar]
- 58.Addis PB. Occurrence of lipid oxidation products in foods. Food Chem Toxicol. 1986;24:1021–1030. doi: 10.1016/0278-6915(86)90283-8. [DOI] [PubMed] [Google Scholar]
- 59.Rada B, Hably C, Meczner A, Timár C, Lakatos G, Enyedi P, Ligeti E. Role of Nox2 in elimination of microorganisms. Semin Immunopathol. 2008;30:237–253. doi: 10.1007/s00281-008-0126-3. [DOI] [PubMed] [Google Scholar]
- 60.Gutteridge JM. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin Chem. 1995;41:1819–1828. [PubMed] [Google Scholar]
- 61.Tahara EB, Navarete FD, Kowaltowski AJ. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic Biol Med. 2009;46:1283–1297. doi: 10.1016/j.freeradbiomed.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 62.Richter C. Do mitochondrial DNA fragments promote cancer and aging? FEBS Lett. 1988;241:1–5. doi: 10.1016/0014-5793(88)81018-4. [DOI] [PubMed] [Google Scholar]
- 63.Kelley EE, Khoo NK, Hundley NJ, Malik UZ, Freeman BA, Tarpey MM. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic Biol Med. 2010;48:493–498. doi: 10.1016/j.freeradbiomed.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Granger DN, Rutili G, McCord JM. Superoxide radicals in feline intestinal ischemia. Gastroenterology. 1981;81:22–29. [PubMed] [Google Scholar]
- 65.Godber BL, Doel JJ, Durgan J, Eisenthal R, Harrison R. A new route to peroxynitrite: a role for xanthine oxidoreductase. FEBS Lett. 2000;475:93–96. doi: 10.1016/s0014-5793(00)01639-2. [DOI] [PubMed] [Google Scholar]
- 66.Vorbach C, Harrison R, Capecchi MR. Xanthine oxidoreductase is central to the evolution and function of the innate immune system. Trends Immunol. 2003;24:512–517. doi: 10.1016/s1471-4906(03)00237-0. [DOI] [PubMed] [Google Scholar]
- 67.Luiking YC, Engelen MP, Deutz NE. Regulation of nitric oxide production in health and disease. Curr Opin Clin Nutr Metab Care. 2010;13:97–104. doi: 10.1097/MCO.0b013e328332f99d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xia Y, Zweier JL. Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc Natl Acad Sci USA. 1997;94:6954–6958. doi: 10.1073/pnas.94.13.6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Farmer EH, Sutton DA. The course of autoxidation reactions in polyisoprenes and allied compounds: V. Observations on fish-oil acids. J Chem Soc. 1943;24:122–125. [Google Scholar]
- 70.Halliwell B, Gutteridge JM. Lipid peroxidation in brain homogenates: the role of iron and hydroxyl radicals. J Neurochem. 1997;69:1330–1331. doi: 10.1046/j.1471-4159.1997.69031330.x. [DOI] [PubMed] [Google Scholar]
- 71.Gutteridge JM. Iron promoters of the Fenton reaction and lipid peroxidation can be released from haemoglobin by peroxides. FEBS Lett. 1986;201:291–295. doi: 10.1016/0014-5793(86)80626-3. [DOI] [PubMed] [Google Scholar]
- 72.Gutteridge JM, Beard AP, Quinlan GJ. Superoxide-dependent lipid peroxidation. Problems with the use of catalase as a specific probe for fenton-derived hydroxyl radicals. Biochem Biophys Res Commun. 1983;117:901–907. doi: 10.1016/0006-291x(83)91681-9. [DOI] [PubMed] [Google Scholar]
- 73.Gutteridge JM. Free radicals in disease processes: a compilation of cause and consequence. Free Radic Res Commun. 1993;19:141–158. doi: 10.3109/10715769309111598. [DOI] [PubMed] [Google Scholar]
- 74.Rawls HR, Van Santen PJ. Singlet oxygen: a possible source of the original hydroperoxides in fatty acids. Ann NY Acad Sci. 1970;171:135–137. [Google Scholar]
- 75.Bielski BH, Arudi RL, Sutherland MW. A study of the reactivity of HO2/O2- with unsaturated fatty acids. J Biol Chem. 1983;258:4759–4761. [PubMed] [Google Scholar]
- 76.Gutteridge JM. The role of superoxide and hydroxyl radicals in phospholipid peroxidation catalysed by iron salts. FEBS Lett. 1982;150:454–458. doi: 10.1016/0014-5793(82)80788-6. [DOI] [PubMed] [Google Scholar]
- 77.Schaich KM, Borg DC. Solvent effects in the spin trapping of lipid oxyl radicals. Free Radic Res Commun. 1990;9:267–278. doi: 10.3109/10715769009145685. [DOI] [PubMed] [Google Scholar]
- 78.Garnier-Suillerot A, Tose L, Paniago E. Kinetic and mechanism of vesicle lipoperoxide decomposition by Fe(II) Biochim Biophys Acta. 1984;794:307–312. [Google Scholar]
- 79.Hardwick TJ. The rate constant of the reaction between ferrous ions and hydrogen peroxide in acid solution. Can J Chem. 1957;35:428–436. [Google Scholar]
- 80.Davies KJ. Protein damage and degradation by oxygen radicals. I. general aspects. J Biol Chem. 1987;262:9895–9901. [PubMed] [Google Scholar]
- 81.Wolff SP, Dean RT. Fragmentation of proteins by free radicals and its effect on their susceptibility to enzymic hydrolysis. Biochem J. 1986;234:399–403. doi: 10.1042/bj2340399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 83.Tangvarasittichai S, Poonsub P, Tangvarasittichai O, Sirigulsatien V. Serum levels of malondialdehyde in type 2 diabetes mellitus Thai subjects. Siriraj Med J. 2009;61:20–23. [Google Scholar]
- 84.Moreira PI, Sayre LM, Zhu X, Nunomura A, Smith MA, Perry G. Detection and localization of markers of oxidative stress by in situ methods: application in the study of Alzheimer disease. Methods Mol Biol. 2010;610:419–434. doi: 10.1007/978-1-60327-029-8_25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rahmanto AS, Morgan PE, Hawkins CL, Davies MJ. Cellular effects of peptide and protein hydroperoxides. Free Radic Biol Med. 2010;48:1071–1078. doi: 10.1016/j.freeradbiomed.2010.01.025. [DOI] [PubMed] [Google Scholar]
- 86.Hunt JV, Dean RT, Wolff SP. Hydroxyl radical production and autoxidative glycosylation. Glucose autoxidation as the cause of protein damage in the experimental glycation model of diabetes mellitus and ageing. Biochem J. 1988;256:205–212. doi: 10.1042/bj2560205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rosenfeld ME. Inflammation, lipids, and free radicals: lessons learned from the atherogenic process. Semin Reprod Endocrinol. 1998;16:249–261. doi: 10.1055/s-2007-1016285. [DOI] [PubMed] [Google Scholar]
- 88.Steinberg D. Lewis A. Conner Memorial Lecture. Oxidative modification of LDL and atherogenesis. Circulation. 1997;95:1062–1071. doi: 10.1161/01.cir.95.4.1062. [DOI] [PubMed] [Google Scholar]
- 89.Esterbauer H, Puhl H, Dieber-Rotheneder M, Waeg G, Rabl H. Effect of antioxidants on oxidative modification of LDL. Ann Med. 1991;23:573–581. doi: 10.3109/07853899109150520. [DOI] [PubMed] [Google Scholar]
- 90.Porkkala-Sarataho EK, Nyyssönen MK, Kaikkonen JE, Poulsen HE, Hayn EM, Salonen RM, Salonen JT. A randomized, single-blind, placebo-controlled trial of the effects of 200 mg alpha-tocopherol on the oxidation resistance of atherogenic lipoproteins. Am J Clin Nutr. 1998;68:1034–1041. doi: 10.1093/ajcn/68.5.1034. [DOI] [PubMed] [Google Scholar]
- 91.Horton AA, Fairhurst S. Lipid peroxidation and mechanisms of toxicity. Crit Rev Toxicol. 1987;18:27–79. doi: 10.3109/10408448709089856. [DOI] [PubMed] [Google Scholar]
- 92.Mihara M, Uchiyama M. Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal Biochem. 1978;86:271–278. doi: 10.1016/0003-2697(78)90342-1. [DOI] [PubMed] [Google Scholar]
- 93.Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 94.Rankinen T, Hietanen E, Väisänen S, Lehtiö M, Penttilä I, Bouchard C, Rauramaa R. Relationship between lipid peroxidation and plasma fibrinogen in middle-aged men. Thromb Res. 2000;99:453–459. doi: 10.1016/s0049-3848(00)00271-1. [DOI] [PubMed] [Google Scholar]
- 95.Pasaoglu H, Sancak B, Bukan N. Lipid peroxidation and resistance to oxidation in patients with type 2 diabetes mellitus. Tohoku J Exp Med. 2004;203:211–218. doi: 10.1620/tjem.203.211. [DOI] [PubMed] [Google Scholar]
- 96.Cavalca V, Cighetti G, Bamonti F, Loaldi A, Bortone L, Novembrino C, De Franceschi M, Belardinelli R, Guazzi MD. Oxidative stress and homocysteine in coronary artery disease. Clin Chem. 2001;47:887–892. [PubMed] [Google Scholar]
- 97.Witztum JL. The oxidation hypothesis of atherosclerosis. Lancet. 1994;344:793–795. doi: 10.1016/s0140-6736(94)92346-9. [DOI] [PubMed] [Google Scholar]
- 98.Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis. 2000;21:361–370. doi: 10.1093/carcin/21.3.361. [DOI] [PubMed] [Google Scholar]
- 99.Niedernhofer LJ, Daniels JS, Rouzer CA, Greene RE, Marnett LJ. Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J Biol Chem. 2003;278:31426–31433. doi: 10.1074/jbc.M212549200. [DOI] [PubMed] [Google Scholar]
- 100.Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med. 1989;320:915–924. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 101.Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol Aging. 1998;19:33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 102.Staruchova M, Collins AR, Volkovova K, Mislanová C, Kovacikova Z, Tulinska J, Kocan A, Staruch L, Wsolova L, Dusinska M. Occupational exposure to mineral fibres. Biomarkers of oxidative damage and antioxidant defence and associations with DNA damage and repair. Mutagenesis. 2008;23:249–260. doi: 10.1093/mutage/gen004. [DOI] [PubMed] [Google Scholar]
- 103.Nair V, Cooper CS, Vietti DE, Turner GA. The chemistry of lipid peroxidation metabolites: crosslinking reactions of malondialdehyde. Lipids. 1986;21:6–10. doi: 10.1007/BF02534294. [DOI] [PubMed] [Google Scholar]
- 104.Requena JR, Fu MX, Ahmed MU, Jenkins AJ, Lyons TJ, Thorpe SR. Lipoxidation products as biomarkers of oxidative damage to proteins during lipid peroxidation reactions. Nephrol Dial Transplant. 1996;11 Suppl 5:48–53. doi: 10.1093/ndt/11.supp5.48. [DOI] [PubMed] [Google Scholar]
- 105.Knight JA, Pieper RK, McClellan L. Specificity of the thiobarbituric acid reaction: its use in studies of lipid peroxidation. Clin Chem. 1988;34:2433–2438. [PubMed] [Google Scholar]
- 106.Praticò D, Iuliano L, Mauriello A, Spagnoli L, Lawson JA, Rokach J, Maclouf J, Violi F, FitzGerald GA. Localization of distinct F2-isoprostanes in human atherosclerotic lesions. J Clin Invest. 1997;100:2028–2034. doi: 10.1172/JCI119735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mallat Z, Philip I, Lebret M, Chatel D, Maclouf J, Tedgui A. Elevated levels of 8-iso-prostaglandin F2alpha in pericardial fluid of patients with heart failure: a potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation. 1998;97:1536–1539. doi: 10.1161/01.cir.97.16.1536. [DOI] [PubMed] [Google Scholar]
- 108.Lawson JA, Rokach J, FitzGerald GA. Isoprostanes: formation, analysis and use as indices of lipid peroxidation in vivo. J Biol Chem. 1999;274:24441–24444. doi: 10.1074/jbc.274.35.24441. [DOI] [PubMed] [Google Scholar]
- 109.Roberts LJ, Morrow JD. The generation and actions of isoprostanes. Biochim Biophys Acta. 1997;1345:121–135. doi: 10.1016/s0005-2760(96)00162-2. [DOI] [PubMed] [Google Scholar]
- 110.Reilly MP, Praticò D, Delanty N, DiMinno G, Tremoli E, Rader D, Kapoor S, Rokach J, Lawson J, FitzGerald GA. Increased formation of distinct F2 isoprostanes in hypercholesterolemia. Circulation. 1998;98:2822–2828. doi: 10.1161/01.cir.98.25.2822. [DOI] [PubMed] [Google Scholar]
- 111.Mori TA, Croft KD, Puddey IB, Beilin LJ. An improved method for the measurement of urinary and plasma F2-isoprostanes using gas chromatography-mass spectrometry. Anal Biochem. 1999;268:117–125. doi: 10.1006/abio.1998.3037. [DOI] [PubMed] [Google Scholar]
- 112.Praticò D. F(2)-isoprostanes: sensitive and specific non-invasive indices of lipid peroxidation in vivo. Atherosclerosis. 1999;147:1–10. doi: 10.1016/s0021-9150(99)00257-9. [DOI] [PubMed] [Google Scholar]
- 113.Li H, Lawson JA, Reilly M, Adiyaman M, Hwang SW, Rokach J, FitzGerald GA. Quantitative high performance liquid chromatography/tandem mass spectrometric analysis of the four classes of F(2)-isoprostanes in human urine. Proc Natl Acad Sci USA. 1999;96:13381–13386. doi: 10.1073/pnas.96.23.13381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gopaul NK, Anggård EE, Mallet AI, Betteridge DJ, Wolff SP, Nourooz-Zadeh J. Plasma 8-epi-PGF2 alpha levels are elevated in individuals with non-insulin dependent diabetes mellitus. FEBS Lett. 1995;368:225–229. doi: 10.1016/0014-5793(95)00649-t. [DOI] [PubMed] [Google Scholar]
- 115.Davì G, Ciabattoni G, Consoli A, Mezzetti A, Falco A, Santarone S, Pennese E, Vitacolonna E, Bucciarelli T, Costantini F, et al. In vivo formation of 8-iso-prostaglandin f2alpha and platelet activation in diabetes mellitus: effects of improved metabolic control and vitamin E supplementation. Circulation. 1999;99:224–229. doi: 10.1161/01.cir.99.2.224. [DOI] [PubMed] [Google Scholar]
- 116.Morrow JD, Frei B, Longmire AW, Gaziano JM, Lynch SM, Shyr Y, Strauss WE, Oates JA, Roberts LJ. Increase in circulating products of lipid peroxidation (F2-isoprostanes) in smokers. Smoking as a cause of oxidative damage. N Engl J Med. 1995;332:1198–1203. doi: 10.1056/NEJM199505043321804. [DOI] [PubMed] [Google Scholar]
- 117.Bachi A, Zuccato E, Baraldi M, Fanelli R, Chiabrando C. Measurement of urinary 8-Epi-prostaglandin F2alpha, a novel index of lipid peroxidation in vivo, by immunoaffinity extraction/gas chromatography-mass spectrometry. Basal levels in smokers and nonsmokers. Free Radic Biol Med. 1996;20:619–624. doi: 10.1016/0891-5849(95)02087-x. [DOI] [PubMed] [Google Scholar]
- 118.Voutilainen S, Morrow JD, Roberts LJ, Alfthan G, Alho H, Nyyssönen K, Salonen JT. Enhanced in vivo lipid peroxidation at elevated plasma total homocysteine levels. Arterioscler Thromb Vasc Biol. 1999;19:1263–1266. doi: 10.1161/01.atv.19.5.1263. [DOI] [PubMed] [Google Scholar]
- 119.Davi G, Alessandrini P, Mezzetti A, Minotti G, Bucciarelli T, Costantini F, Cipollone F, Bon GB, Ciabattoni G, Patrono C. In vivo formation of 8-Epi-prostaglandin F2 alpha is increased in hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1997;17:3230–3235. doi: 10.1161/01.atv.17.11.3230. [DOI] [PubMed] [Google Scholar]
- 120.Praticò D, Tangirala RK, Rader DJ, Rokach J, FitzGerald GA. Vitamin E suppresses isoprostane generation in vivo and reduces atherosclerosis in ApoE-deficient mice. Nat Med. 1998;4:1189–1192. doi: 10.1038/2685. [DOI] [PubMed] [Google Scholar]
- 121.Schnackenberg CG, Wilcox CS. Two-week administration of tempol attenuates both hypertension and renal excretion of 8-Iso prostaglandin f2alpha. Hypertension. 1999;33:424–428. doi: 10.1161/01.hyp.33.1.424. [DOI] [PubMed] [Google Scholar]
- 122.Palmer AM, Thomas CR, Gopaul N, Dhir S, Anggård EE, Poston L, Tribe RM. Dietary antioxidant supplementation reduces lipid peroxidation but impairs vascular function in small mesenteric arteries of the streptozotocin-diabetic rat. Diabetologia. 1998;41:148–156. doi: 10.1007/s001250050883. [DOI] [PubMed] [Google Scholar]
- 123.Laight DW, Kengatharan KM, Gopaul NK, Anggård EE, Carrier MJ. Investigation of oxidant stress and vasodepression to glyceryl trinitrate in the obese Zucker rat in vivo. Br J Pharmacol. 1998;125:895–901. doi: 10.1038/sj.bjp.0702132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.England T, Beatty E, Rehman A, Nourooz-Zadeh J, Pereira P, O’Reilly J, Wiseman H, Geissler C, Halliwell B. The steady-state levels of oxidative DNA damage and of lipid peroxidation (F2-isoprostanes) are not correlated in healthy human subjects. Free Radic Res. 2000;32:355–362. doi: 10.1080/10715760000300351. [DOI] [PubMed] [Google Scholar]
- 125.Fujita T. Insulin resistance and salt-sensitive hypertension in metabolic syndrome. Nephrol Dial Transplant. 2007;22:3102–3107. doi: 10.1093/ndt/gfm409. [DOI] [PubMed] [Google Scholar]
- 126.Fujita T. Aldosterone in salt-sensitive hypertension and metabolic syndrome. J Mol Med (Berl) 2008;86:729–734. doi: 10.1007/s00109-008-0343-1. [DOI] [PubMed] [Google Scholar]
- 127.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 128.Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 129.Anderson RM, Weindruch R. The caloric restriction paradigm: implications for healthy human aging. Am J Hum Biol. 2012;24:101–106. doi: 10.1002/ajhb.22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rebrin I, Kamzalov S, Sohal RS. Effects of age and caloric restriction on glutathione redox state in mice. Free Radic Biol Med. 2003;35:626–635. doi: 10.1016/s0891-5849(03)00388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Grattagliano I, Portincasa P, Cocco T, Moschetta A, Di Paola M, Palmieri VO, Palasciano G. Effect of dietary restriction and N-acetylcysteine supplementation on intestinal mucosa and liver mitochondrial redox status and function in aged rats. Exp Gerontol. 2004;39:1323–1332. doi: 10.1016/j.exger.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 132.Cocco T, Sgobbo P, Clemente M, Lopriore B, Grattagliano I, Di Paola M, Villani G. Tissue-specific changes of mitochondrial functions in aged rats: effect of a long-term dietary treatment with N-acetylcysteine. Free Radic Biol Med. 2005;38:796–805. doi: 10.1016/j.freeradbiomed.2004.11.034. [DOI] [PubMed] [Google Scholar]
- 133.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Keaney JF, Larson MG, Vasan RS, Wilson PW, Lipinska I, Corey D, Massaro JM, Sutherland P, Vita JA, Benjamin EJ. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol. 2003;23:434–439. doi: 10.1161/01.ATV.0000058402.34138.11. [DOI] [PubMed] [Google Scholar]
- 135.Okuno Y, Matsuda M, Kobayashi H, Morita K, Suzuki E, Fukuhara A, Komuro R, Shimabukuro M, Shimomura I. Adipose expression of catalase is regulated via a novel remote PPARgamma-responsive region. Biochem Biophys Res Commun. 2008;366:698–704. doi: 10.1016/j.bbrc.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 136.Okuno Y, Matsuda M, Miyata Y, Fukuhara A, Komuro R, Shimabukuro M, Shimomura I. Human catalase gene is regulated by peroxisome proliferator activated receptor-gamma through a response element distinct from that of mouse. Endocr J. 2010;57:303–309. doi: 10.1507/endocrj.k09e-113. [DOI] [PubMed] [Google Scholar]
- 137.Kobayashi H, Matsuda M, Fukuhara A, Komuro R, Shimomura I. Dysregulated glutathione metabolism links to impaired insulin action in adipocytes. Am J Physiol Endocrinol Metab. 2009;296:E1326–E1334. doi: 10.1152/ajpendo.90921.2008. [DOI] [PubMed] [Google Scholar]
- 138.Roberts CK, Barnard RJ, Sindhu RK, Jurczak M, Ehdaie A, Vaziri ND. Oxidative stress and dysregulation of NAD(P)H oxidase and antioxidant enzymes in diet-induced metabolic syndrome. Metabolism. 2006;55:928–934. doi: 10.1016/j.metabol.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 139.Meng S, Roberts LJ, Cason GW, Curry TS, Manning RD. Superoxide dismutase and oxidative stress in Dahl salt-sensitive and -resistant rats. Am J Physiol Regul Integr Comp Physiol. 2002;283:R732–R738. doi: 10.1152/ajpregu.00346.2001. [DOI] [PubMed] [Google Scholar]
- 140.Kido M, Ando K, Oba S, Fujita T. Renoprotective effect of pravastatin in salt-loaded Dahl salt-sensitive rats. Hypertens Res. 2005;28:1009–1015. doi: 10.1291/hypres.28.1009. [DOI] [PubMed] [Google Scholar]
- 141.Wang H, Shimosawa T, Matsui H, Kaneko T, Ogura S, Uetake Y, Takenaka K, Yatomi Y, Fujita T. Paradoxical mineralocorticoid receptor activation and left ventricular diastolic dysfunction under high oxidative stress conditions. J Hypertens. 2008;26:1453–1462. doi: 10.1097/HJH.0b013e328300a232. [DOI] [PubMed] [Google Scholar]
- 142.Laffer CL, Bolterman RJ, Romero JC, Elijovich F. Effect of salt on isoprostanes in salt-sensitive essential hypertension. Hypertension. 2006;47:434–440. doi: 10.1161/01.HYP.0000202480.06735.82. [DOI] [PubMed] [Google Scholar]
- 143.Rocchini AP, Key J, Bondie D, Chico R, Moorehead C, Katch V, Martin M. The effect of weight loss on the sensitivity of blood pressure to sodium in obese adolescents. N Engl J Med. 1989;321:580–585. doi: 10.1056/NEJM198908313210905. [DOI] [PubMed] [Google Scholar]
- 144.Uzu T, Kimura G, Yamauchi A, Kanasaki M, Isshiki K, Araki S, Sugiomoto T, Nishio Y, Maegawa H, Koya D, et al. Enhanced sodium sensitivity and disturbed circadian rhythm of blood pressure in essential hypertension. J Hypertens. 2006;24:1627–1632. doi: 10.1097/01.hjh.0000239299.71001.77. [DOI] [PubMed] [Google Scholar]
- 145.Sanchez RA, Masnatta LD, Pesiney C, Fischer P, Ramirez AJ. Telmisartan improves insulin resistance in high renin nonmodulating salt-sensitive hypertensives. J Hypertens. 2008;26:2393–2398. doi: 10.1097/HJH.0b013e328312677e. [DOI] [PubMed] [Google Scholar]
- 146.Giner V, Coca A, de la Sierra A. Increased insulin resistance in salt sensitive essential hypertension. J Hum Hypertens. 2001;15:481–485. doi: 10.1038/sj.jhh.1001216. [DOI] [PubMed] [Google Scholar]
- 147.Dobrian AD, Schriver SD, Lynch T, Prewitt RL. Effect of salt on hypertension and oxidative stress in a rat model of diet-induced obesity. Am J Physiol Renal Physiol. 2003;285:F619–F628. doi: 10.1152/ajprenal.00388.2002. [DOI] [PubMed] [Google Scholar]
- 148.Annuk M, Zilmer M, Fellström B. Endothelium-dependent vasodilation and oxidative stress in chronic renal failure: impact on cardiovascular disease. Kidney Int Suppl. 2003;(84):S50–S53. doi: 10.1046/j.1523-1755.63.s84.2.x. [DOI] [PubMed] [Google Scholar]
- 149.Martín-Gallán P, Carrascosa A, Gussinyé M, Domínguez C. Biomarkers of diabetes-associated oxidative stress and antioxidant status in young diabetic patients with or without subclinical complications. Free Radic Biol Med. 2003;34:1563–1574. doi: 10.1016/s0891-5849(03)00185-0. [DOI] [PubMed] [Google Scholar]
- 150.Varvarovská J, Racek J, Stozický F, Soucek J, Trefil L, Pomahacová R. Parameters of oxidative stress in children with Type 1 diabetes mellitus and their relatives. J Diabetes Complications. 2003;17:7–10. doi: 10.1016/s1056-8727(01)00228-8. [DOI] [PubMed] [Google Scholar]
- 151.Seghrouchni I, Drai J, Bannier E, Rivière J, Calmard P, Garcia I, Orgiazzi J, Revol A. Oxidative stress parameters in type I, type II and insulin-treated type 2 diabetes mellitus; insulin treatment efficiency. Clin Chim Acta. 2002;321:89–96. doi: 10.1016/s0009-8981(02)00099-2. [DOI] [PubMed] [Google Scholar]
- 152.VanderJagt DJ, Harrison JM, Ratliff DM, Hunsaker LA, Vander Jagt DL. Oxidative stress indices in IDDM subjects with and without long-term diabetic complications. Clin Biochem. 2001;34:265–270. doi: 10.1016/s0009-9120(01)00204-1. [DOI] [PubMed] [Google Scholar]
- 153.Bonnefont-Rousselot D. Glucose and reactive oxygen species. Curr Opin Clin Nutr Metab Care. 2002;5:561–568. doi: 10.1097/00075197-200209000-00016. [DOI] [PubMed] [Google Scholar]
- 154.Ceriello A, Motz E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscler Thromb Vasc Biol. 2004;24:816–823. doi: 10.1161/01.ATV.0000122852.22604.78. [DOI] [PubMed] [Google Scholar]
- 155.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 156.Ramana KV, Chandra D, Srivastava S, Bhatnagar A, Srivastava SK. Nitric oxide regulates the polyol pathway of glucose metabolism in vascular smooth muscle cells. FASEB J. 2003;17:417–425. doi: 10.1096/fj.02-0722com. [DOI] [PubMed] [Google Scholar]
- 157.Morré DM, Lenaz G, Morré DJ. Surface oxidase and oxidative stress propagation in aging. J Exp Biol. 2000;203:1513–1521. doi: 10.1242/jeb.203.10.1513. [DOI] [PubMed] [Google Scholar]
- 158.Shams N, Ianchulev T. Role of vascular endothelial growth factor in ocular angiogenesis. Ophthalmol Clin North Am. 2006;19:335–344. doi: 10.1016/j.ohc.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 159.Bhisitkul RB. Vascular endothelial growth factor biology: clinical implications for ocular treatments. Br J Ophthalmol. 2006;90:1542–1547. doi: 10.1136/bjo.2006.098426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Stitt AW. The role of advanced glycation in the pathogenesis of diabetic retinopathy. Exp Mol Pathol. 2003;75:95–108. doi: 10.1016/s0014-4800(03)00035-2. [DOI] [PubMed] [Google Scholar]
- 161.Basta G, Schmidt AM, De Caterina R. Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res. 2004;63:582–592. doi: 10.1016/j.cardiores.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 162.Nakamura Y, Horii Y, Nishino T, Shiiki H, Sakaguchi Y, Kagoshima T, Dohi K, Makita Z, Vlassara H, Bucala R. Immunohistochemical localization of advanced glycosylation end products in coronary atheroma and cardiac tissue in diabetes mellitus. Am J Pathol. 1993;143:1649–1656. [PMC free article] [PubMed] [Google Scholar]
- 163.Yamagishi S. Role of advanced glycation end products (AGEs) and receptor for AGEs (RAGE) in vascular damage in diabetes. Exp Gerontol. 2011;46:217–224. doi: 10.1016/j.exger.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 164.Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS, Pinsky D, Stern D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem. 1994;269:9889–9897. [PubMed] [Google Scholar]
- 165.Schmidt AM, Hori O, Chen JX, Li JF, Crandall J, Zhang J, Cao R, Yan SD, Brett J, Stern D. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest. 1995;96:1395–1403. doi: 10.1172/JCI118175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Roebuck KA. Oxidant stress regulation of IL-8 and ICAM-1 gene expression: differential activation and binding of the transcription factors AP-1 and NF-kappaB (Review) Int J Mol Med. 1999;4:223–230. doi: 10.3892/ijmm.4.3.223. [DOI] [PubMed] [Google Scholar]
- 167.Hu FB, Stampfer MJ. Is type 2 diabetes mellitus a vascular condition? Arterioscler Thromb Vasc Biol. 2003;23:1715–1716. doi: 10.1161/01.ATV.0000094360.38911.71. [DOI] [PubMed] [Google Scholar]
- 168.Kaul K, Hodgkinson A, Tarr JM, Kohner EM, Chibber R. Is inflammation a common retinal-renal-nerve pathogenic link in diabetes? Curr Diabetes Rev. 2010;6:294–303. doi: 10.2174/157339910793360851. [DOI] [PubMed] [Google Scholar]
- 169.Esposito K, Nappo F, Marfella R, Giugliano G, Giugliano F, Ciotola M, Quagliaro L, Ceriello A, Giugliano D. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation. 2002;106:2067–2072. doi: 10.1161/01.cir.0000034509.14906.ae. [DOI] [PubMed] [Google Scholar]
- 170.Lane N. A unifying view of ageing and disease: the double-agent theory. J Theor Biol. 2003;225:531–540. doi: 10.1016/s0022-5193(03)00304-7. [DOI] [PubMed] [Google Scholar]
- 171.Steinberg HO, Baron AD. Vascular function, insulin resistance and fatty acids. Diabetologia. 2002;45:623–634. doi: 10.1007/s00125-002-0800-2. [DOI] [PubMed] [Google Scholar]
- 172.Lopes HF, Morrow JD, Stojiljkovic MP, Goodfriend TL, Egan BM. Acute hyperlipidemia increases oxidative stress more in African Americans than in white Americans. Am J Hypertens. 2003;16:331–336. doi: 10.1016/s0895-7061(03)00041-4. [DOI] [PubMed] [Google Scholar]
- 173.Stojiljkovic MP, Lopes HF, Zhang D, Morrow JD, Goodfriend TL, Egan BM. Increasing plasma fatty acids elevates F2-isoprostanes in humans: implications for the cardiovascular risk factor cluster. J Hypertens. 2002;20:1215–1221. doi: 10.1097/00004872-200206000-00036. [DOI] [PubMed] [Google Scholar]
- 174.Peelman F, Waelput W, Iserentant H, Lavens D, Eyckerman S, Zabeau L, Tavernier J. Leptin: linking adipocyte metabolism with cardiovascular and autoimmune diseases. Prog Lipid Res. 2004;43:283–301. doi: 10.1016/j.plipres.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 175.Chan WB, Ma RC, Chan NN, Ng MC, Lee ZS, Lai CW, Tong PC, So WY, Chan JC. Increased leptin concentrations and lack of gender difference in Type 2 diabetic patients with nephropathy. Diabetes Res Clin Pract. 2004;64:93–98. doi: 10.1016/j.diabres.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 176.Wauters M, Considine RV, Yudkin JS, Peiffer F, De Leeuw I, Van Gaal LF. Leptin levels in type 2 diabetes: associations with measures of insulin resistance and insulin secretion. Horm Metab Res. 2003;35:92–96. doi: 10.1055/s-2003-39054. [DOI] [PubMed] [Google Scholar]
- 177.Reilly MP, Iqbal N, Schutta M, Wolfe ML, Scally M, Localio AR, Rader DJ, Kimmel SE. Plasma leptin levels are associated with coronary atherosclerosis in type 2 diabetes. J Clin Endocrinol Metab. 2004;89:3872–3878. doi: 10.1210/jc.2003-031676. [DOI] [PubMed] [Google Scholar]
- 178.Yamagishi SI, Edelstein D, Du XL, Kaneda Y, Guzmán M, Brownlee M. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J Biol Chem. 2001;276:25096–25100. doi: 10.1074/jbc.M007383200. [DOI] [PubMed] [Google Scholar]
- 179.Bouloumie A, Marumo T, Lafontan M, Busse R. Leptin induces oxidative stress in human endothelial cells. FASEB J. 1999;13:1231–1238. [PubMed] [Google Scholar]
- 180.Yamamoto K, Völkl A, Hashimoto T, Fahimi HD. Catalase in guinea pig hepatocytes is localized in cytoplasm, nuclear matrix and peroxisomes. Eur J Cell Biol. 1988;46:129–135. [PubMed] [Google Scholar]
- 181.Lu SC. Regulation of hepatic glutathione synthesis: current concepts and controversies. FASEB J. 1999;13:1169–1183. [PubMed] [Google Scholar]
- 182.Zhang P, Liu B, Kang SW, Seo MS, Rhee SG, Obeid LM. Thioredoxin peroxidase is a novel inhibitor of apoptosis with a mechanism distinct from that of Bcl-2. J Biol Chem. 1997;272:30615–30618. doi: 10.1074/jbc.272.49.30615. [DOI] [PubMed] [Google Scholar]
- 183.Galter D, Mihm S, Dröge W. Distinct effects of glutathione disulphide on the nuclear transcription factor kappa B and the activator protein-1. Eur J Biochem. 1994;221:639–648. doi: 10.1111/j.1432-1033.1994.tb18776.x. [DOI] [PubMed] [Google Scholar]
- 184.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- 185.Thomas JA, Poland B, Honzatko R. Protein sulfhydryls and their role in the antioxidant function of protein S-thiolation. Arch Biochem Biophys. 1995;319:1–9. doi: 10.1006/abbi.1995.1261. [DOI] [PubMed] [Google Scholar]
- 186.Shirwaikar A, Shirwaikar A, Rajendran K, Punitha IS. In vitro antioxidant studies on the benzyl tetra isoquinoline alkaloid berberine. Biol Pharm Bull. 2006;29:1906–1910. doi: 10.1248/bpb.29.1906. [DOI] [PubMed] [Google Scholar]
- 187.Ulrich-Merzenich G, Zeitler H, Vetter H, Kraft K. Synergy research: vitamins and secondary plant components in the maintenance of the redox-homeostasis and in cell signaling. Phytomedicine. 2009;16:2–16. doi: 10.1016/j.phymed.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 188.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 189.Blendea MC, Jacobs D, Stump CS, McFarlane SI, Ogrin C, Bahtyiar G, Stas S, Kumar P, Sha Q, Ferrario CM, et al. Abrogation of oxidative stress improves insulin sensitivity in the Ren-2 rat model of tissue angiotensin II overexpression. Am J Physiol Endocrinol Metab. 2005;288:E353–E359. doi: 10.1152/ajpendo.00402.2004. [DOI] [PubMed] [Google Scholar]
- 190.Matsuzawa-Nagata N, Takamura T, Ando H, Nakamura S, Kurita S, Misu H, Ota T, Yokoyama M, Honda M, Miyamoto K, et al. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism. 2008;57:1071–1077. doi: 10.1016/j.metabol.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 191.Kunitomo M, Yamaguchi Y, Kagota S, Otsubo K. Beneficial effect of coenzyme Q10 on increased oxidative and nitrative stress and inflammation and individual metabolic components developing in a rat model of metabolic syndrome. J Pharmacol Sci. 2008;107:128–137. doi: 10.1254/jphs.fp0072365. [DOI] [PubMed] [Google Scholar]
- 192.Paolisso G, Giugliano D. Oxidative stress and insulin action: is there a relationship? Diabetologia. 1996;39:357–363. doi: 10.1007/BF00418354. [DOI] [PubMed] [Google Scholar]
- 193.Rudich A, Kozlovsky N, Potashnik R, Bashan N. Oxidant stress reduces insulin responsiveness in 3T3-L1 adipocytes. Am J Physiol. 1997;272:E935–E940. doi: 10.1152/ajpendo.1997.272.5.E935. [DOI] [PubMed] [Google Scholar]
- 194.Maddux BA, See W, Lawrence JC, Goldfine AL, Goldfine ID, Evans JL. Protection against oxidative stress-induced insulin resistance in rat L6 muscle cells by mircomolar concentrations of alpha-lipoic acid. Diabetes. 2001;50:404–410. doi: 10.2337/diabetes.50.2.404. [DOI] [PubMed] [Google Scholar]
- 195.Rudich A, Tirosh A, Potashnik R, Khamaisi M, Bashan N. Lipoic acid protects against oxidative stress induced impairment in insulin stimulation of protein kinase B and glucose transport in 3T3-L1 adipocytes. Diabetologia. 1999;42:949–957. doi: 10.1007/s001250051253. [DOI] [PubMed] [Google Scholar]
- 196.Packer L, Kraemer K, Rimbach G. Molecular aspects of lipoic acid in the prevention of diabetes complications. Nutrition. 2001;17:888–895. doi: 10.1016/s0899-9007(01)00658-x. [DOI] [PubMed] [Google Scholar]
- 197.Teachey MK, Taylor ZC, Maier T, Saengsirisuwan V, Sloniger JA, Jacob S, Klatt MJ, Ptock A, Kraemer K, Hasselwander O, et al. Interactions of conjugated linoleic acid and lipoic acid on insulin action in the obese Zucker rat. Metabolism. 2003;52:1167–1174. doi: 10.1016/s0026-0495(03)00145-8. [DOI] [PubMed] [Google Scholar]
- 198.Evans JL, Goldfine ID. Alpha-lipoic acid: a multifunctional antioxidant that improves insulin sensitivity in patients with type 2 diabetes. Diabetes Technol Ther. 2000;2:401–413. doi: 10.1089/15209150050194279. [DOI] [PubMed] [Google Scholar]
- 199.Inzucchi SE, Maggs DG, Spollett GR, Page SL, Rife FS, Walton V, Shulman GI. Efficacy and metabolic effects of metformin and troglitazone in type II diabetes mellitus. N Engl J Med. 1998;338:867–872. doi: 10.1056/NEJM199803263381303. [DOI] [PubMed] [Google Scholar]
- 200.Mayerson AB, Hundal RS, Dufour S, Lebon V, Befroy D, Cline GW, Enocksson S, Inzucchi SE, Shulman GI, Petersen KF. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes. 2002;51:797–802. doi: 10.2337/diabetes.51.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Evans JL, Heymann CJ, Goldfine ID, Gavin LA. Pharmacokinetics, tolerability, and fructosamine-lowering effect of a novel, controlled-release formulation of alpha-lipoic acid. Endocr Pract. 2002;8:29–35. doi: 10.4158/EP.8.1.29. [DOI] [PubMed] [Google Scholar]
- 202.Ansar H, Mazloom Z, Kazemi F, Hejazi N. Effect of alpha-lipoic acid on blood glucose, insulin resistance and glutathione peroxidase of type 2 diabetic patients. Saudi Med J. 2011;32:584–588. [PubMed] [Google Scholar]
- 203.Estrada DE, Ewart HS, Tsakiridis T, Volchuk A, Ramlal T, Tritschler H, Klip A. Stimulation of glucose uptake by the natural coenzyme alpha-lipoic acid/thioctic acid: participation of elements of the insulin signaling pathway. Diabetes. 1996;45:1798–1804. doi: 10.2337/diab.45.12.1798. [DOI] [PubMed] [Google Scholar]
- 204.Konrad D, Somwar R, Sweeney G, Yaworsky K, Hayashi M, Ramlal T, Klip A. The antihyperglycemic drug alpha-lipoic acid stimulates glucose uptake via both GLUT4 translocation and GLUT4 activation: potential role of p38 mitogen-activated protein kinase in GLUT4 activation. Diabetes. 2001;50:1464–1471. doi: 10.2337/diabetes.50.6.1464. [DOI] [PubMed] [Google Scholar]
- 205.Ramrath S, Tritschler HJ, Eckel J. Stimulation of cardiac glucose transport by thioctic acid and insulin. Horm Metab Res. 1999;31:632–635. doi: 10.1055/s-2007-978811. [DOI] [PubMed] [Google Scholar]
- 206.Hotamisligil GS, Spiegelman BM. Tumor necrosis factor alpha: a key component of the obesity-diabetes link. Diabetes. 1994;43:1271–1278. doi: 10.2337/diab.43.11.1271. [DOI] [PubMed] [Google Scholar]
- 207.Cohen B, Novick D, Rubinstein M. Modulation of insulin activities by leptin. Science. 1996;274:1185–1188. doi: 10.1126/science.274.5290.1185. [DOI] [PubMed] [Google Scholar]
- 208.McGarry JD. Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes. 2002;51:7–18. doi: 10.2337/diabetes.51.1.7. [DOI] [PubMed] [Google Scholar]
- 209.Boden G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes. 1997;46:3–10. [PubMed] [Google Scholar]
- 210.Randle PJ, Kerbey AL, Espinal J. Mechanisms decreasing glucose oxidation in diabetes and starvation: role of lipid fuels and hormones. Diabetes Metab Rev. 1988;4:623–638. doi: 10.1002/dmr.5610040702. [DOI] [PubMed] [Google Scholar]
- 211.Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 212.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest. 2000;106:171–176. doi: 10.1172/JCI10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wojtczak L, Schönfeld P. Effect of fatty acids on energy coupling processes in mitochondria. Biochim Biophys Acta. 1993;1183:41–57. doi: 10.1016/0005-2728(93)90004-y. [DOI] [PubMed] [Google Scholar]
- 214.Bakker SJ, IJzerman RG, Teerlink T, Westerhoff HV, Gans RO, Heine RJ. Cytosolic triglycerides and oxidative stress in central obesity: the missing link between excessive atherosclerosis, endothelial dysfunction, and beta-cell failure? Atherosclerosis. 2000;148:17–21. doi: 10.1016/s0021-9150(99)00329-9. [DOI] [PubMed] [Google Scholar]
- 215.Toborek M, Hennig B. Fatty acid-mediated effects on the glutathione redox cycle in cultured endothelial cells. Am J Clin Nutr. 1994;59:60–65. doi: 10.1093/ajcn/59.1.60. [DOI] [PubMed] [Google Scholar]
- 216.Hennig B, Meerarani P, Ramadass P, Watkins BA, Toborek M. Fatty acid-mediated activation of vascular endothelial cells. Metabolism. 2000;49:1006–1013. doi: 10.1053/meta.2000.7736. [DOI] [PubMed] [Google Scholar]
- 217.Paolisso G, Di Maro G, Pizza G, D’Amore A, Sgambato S, Tesauro P, Varricchio M, D’Onofrio F. Plasma GSH/GSSG affects glucose homeostasis in healthy subjects and non-insulin-dependent diabetics. Am J Physiol. 1992;263:E435–E440. doi: 10.1152/ajpendo.1992.263.3.E435. [DOI] [PubMed] [Google Scholar]
- 218.Dichtl W, Nilsson L, Goncalves I, Ares MP, Banfi C, Calara F, Hamsten A, Eriksson P, Nilsson J. Very low-density lipoprotein activates nuclear factor-kappaB in endothelial cells. Circ Res. 1999;84:1085–1094. doi: 10.1161/01.res.84.9.1085. [DOI] [PubMed] [Google Scholar]
- 219.Hennig B, Meerarani P, Toborek M, McClain CJ. Antioxidant-like properties of zinc in activated endothelial cells. J Am Coll Nutr. 1999;18:152–158. doi: 10.1080/07315724.1999.10718843. [DOI] [PubMed] [Google Scholar]
- 220.Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001;276:16683–16689. doi: 10.1074/jbc.M011695200. [DOI] [PubMed] [Google Scholar]
- 221.Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci N, Lee D, Goodyear LJ, Kraegen EW, White MF, Shulman GI. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes. 1999;48:1270–1274. doi: 10.2337/diabetes.48.6.1270. [DOI] [PubMed] [Google Scholar]
- 222.Coudronniere N, Villalba M, Englund N, Altman A. NF-kappa B activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-theta. Proc Natl Acad Sci USA. 2000;97:3394–3399. doi: 10.1073/pnas.060028097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 224.Meglasson MD, Matschinsky FM. Pancreatic islet glucose metabolism and regulation of insulin secretion. Diabetes Metab Rev. 1986;2:163–214. doi: 10.1002/dmr.5610020301. [DOI] [PubMed] [Google Scholar]
- 225.Malaisse WJ. Physiology, pathology and pharmacology of insulin secretion: recent acquisitions. Diabetes Metab. 1997;23 Suppl 3:6–15. [PubMed] [Google Scholar]
- 226.Maechler P, Jornot L, Wollheim CB. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J Biol Chem. 1999;274:27905–27913. doi: 10.1074/jbc.274.39.27905. [DOI] [PubMed] [Google Scholar]
- 227.Robertson RP, Harmon JS. Diabetes, glucose toxicity, and oxidative stress: A case of double jeopardy for the pancreatic islet beta cell. Free Radic Biol Med. 2006;41:177–184. doi: 10.1016/j.freeradbiomed.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 228.Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes. 1997;46:1733–1742. doi: 10.2337/diab.46.11.1733. [DOI] [PubMed] [Google Scholar]
- 229.Miwa I, Ichimura N, Sugiura M, Hamada Y, Taniguchi S. Inhibition of glucose-induced insulin secretion by 4-hydroxy-2-nonenal and other lipid peroxidation products. Endocrinology. 2000;141:2767–2772. doi: 10.1210/endo.141.8.7614. [DOI] [PubMed] [Google Scholar]
- 230.Tanaka Y, Gleason CE, Tran PO, Harmon JS, Robertson RP. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci USA. 1999;96:10857–10862. doi: 10.1073/pnas.96.19.10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ho E, Bray TM. Antioxidants, NFkappaB activation, and diabetogenesis. Proc Soc Exp Biol Med. 1999;222:205–213. doi: 10.1046/j.1525-1373.1999.d01-137.x. [DOI] [PubMed] [Google Scholar]
- 232.Tajiri Y, Möller C, Grill V. Long-term effects of aminoguanidine on insulin release and biosynthesis: evidence that the formation of advanced glycosylation end products inhibits B cell function. Endocrinology. 1997;138:273–280. doi: 10.1210/endo.138.1.4851. [DOI] [PubMed] [Google Scholar]
- 233.Ho E, Chen G, Bray TM. Supplementation of N-acetylcysteine inhibits NFkappaB activation and protects against alloxan-induced diabetes in CD-1 mice. FASEB J. 1999;13:1845–1854. [PubMed] [Google Scholar]
- 234.Ho E, Chen G, Bray TM. Alpha-phenyl-tert-butylnitrone (PBN) inhibits NFkappaB activation offering protection against chemically induced diabetes. Free Radic Biol Med. 2000;28:604–614. doi: 10.1016/s0891-5849(99)00271-3. [DOI] [PubMed] [Google Scholar]
- 235.Kaneto H, Xu G, Song KH, Suzuma K, Bonner-Weir S, Sharma A, Weir GC. Activation of the hexosamine pathway leads to deterioration of pancreatic beta-cell function through the induction of oxidative stress. J Biol Chem. 2001;276:31099–31104. doi: 10.1074/jbc.M104115200. [DOI] [PubMed] [Google Scholar]
- 236.Boden G, Ruiz J, Kim CJ, Chen X. Effects of prolonged glucose infusion on insulin secretion, clearance, and action in normal subjects. Am J Physiol. 1996;270:E251–E258. doi: 10.1152/ajpendo.1996.270.2.E251. [DOI] [PubMed] [Google Scholar]
- 237.Robertson RP, Zhang HJ, Pyzdrowski KL, Walseth TF. Preservation of insulin mRNA levels and insulin secretion in HIT cells by avoidance of chronic exposure to high glucose concentrations. J Clin Invest. 1992;90:320–325. doi: 10.1172/JCI115865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Poitout V, Olson LK, Robertson RP. Chronic exposure of betaTC-6 cells to supraphysiologic concentrations of glucose decreases binding of the RIPE3b1 insulin gene transcription activator. J Clin Invest. 1996;97:1041–1046. doi: 10.1172/JCI118496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Yki-Järvinen H. Glucose toxicity. Endocr Rev. 1992;13:415–431. doi: 10.1210/edrv-13-3-415. [DOI] [PubMed] [Google Scholar]
- 240.Robertson RP, Harmon J, Tran PO, Tanaka Y, Takahashi H. Glucose toxicity in beta-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes. 2003;52:581–587. doi: 10.2337/diabetes.52.3.581. [DOI] [PubMed] [Google Scholar]
- 241.Robertson RP, Harmon J, Tran PO, Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes. 2004;53 Suppl 1:S119–S124. doi: 10.2337/diabetes.53.2007.s119. [DOI] [PubMed] [Google Scholar]
- 242.Lameloise N, Muzzin P, Prentki M, Assimacopoulos-Jeannet F. Uncoupling protein 2: a possible link between fatty acid excess and impaired glucose-induced insulin secretion? Diabetes. 2001;50:803–809. doi: 10.2337/diabetes.50.4.803. [DOI] [PubMed] [Google Scholar]
- 243.Segall L, Lameloise N, Assimacopoulos-Jeannet F, Roche E, Corkey P, Thumelin S, Corkey BE, Prentki M. Lipid rather than glucose metabolism is implicated in altered insulin secretion caused by oleate in INS-1 cells. Am J Physiol. 1999;277:E521–E528. doi: 10.1152/ajpendo.1999.277.3.E521. [DOI] [PubMed] [Google Scholar]
- 244.Carlsson C, Borg LA, Welsh N. Sodium palmitate induces partial mitochondrial uncoupling and reactive oxygen species in rat pancreatic islets in vitro. Endocrinology. 1999;140:3422–3428. doi: 10.1210/endo.140.8.6908. [DOI] [PubMed] [Google Scholar]
- 245.Jacqueminet S, Briaud I, Rouault C, Reach G, Poitout V. Inhibition of insulin gene expression by long-term exposure of pancreatic beta cells to palmitate is dependent on the presence of a stimulatory glucose concentration. Metabolism. 2000;49:532–536. doi: 10.1016/s0026-0495(00)80021-9. [DOI] [PubMed] [Google Scholar]
- 246.Poitout V, Robertson RP. Minireview: Secondary beta-cell failure in type 2 diabetes--a convergence of glucotoxicity and lipotoxicity. Endocrinology. 2002;143:339–342. doi: 10.1210/endo.143.2.8623. [DOI] [PubMed] [Google Scholar]
- 247.Harmon JS, Gleason CE, Tanaka Y, Poitout V, Robertson RP. Antecedent hyperglycemia, not hyperlipidemia, is associated with increased islet triacylglycerol content and decreased insulin gene mRNA level in Zucker diabetic fatty rats. Diabetes. 2001;50:2481–2486. doi: 10.2337/diabetes.50.11.2481. [DOI] [PubMed] [Google Scholar]
- 248.Grundy SM. Hypertriglyceridemia, atherogenic dyslipidemia, and the metabolic syndrome. Am J Cardiol. 1998;81:18B–25B. doi: 10.1016/s0002-9149(98)00033-2. [DOI] [PubMed] [Google Scholar]
- 249.Taskinen MR. Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia. 2003;46:733–749. doi: 10.1007/s00125-003-1111-y. [DOI] [PubMed] [Google Scholar]
- 250.Ginsberg HN, Zhang YL, Hernandez-Ono A. Metabolic syndrome: focus on dyslipidemia. Obesity (Silver Spring) 2006;14 Suppl 1:41S–49S. doi: 10.1038/oby.2006.281. [DOI] [PubMed] [Google Scholar]
- 251.Adiels M, Olofsson SO, Taskinen MR, Borén J. Diabetic dyslipidaemia. Curr Opin Lipidol. 2006;17:238–246. doi: 10.1097/01.mol.0000226115.97436.c0. [DOI] [PubMed] [Google Scholar]
- 252.Vergès B. New insight into the pathophysiology of lipid abnormalities in type 2 diabetes. Diabetes Metab. 2005;31:429–439. doi: 10.1016/s1262-3636(07)70213-6. [DOI] [PubMed] [Google Scholar]
- 253.Packard CJ. Triacylglycerol-rich lipoproteins and the generation of small, dense low-density lipoprotein. Biochem Soc Trans. 2003;31:1066–1069. doi: 10.1042/bst0311066. [DOI] [PubMed] [Google Scholar]
- 254.Coppack SW, Evans RD, Fisher RM, Frayn KN, Gibbons GF, Humphreys SM, Kirk ML, Potts JL, Hockaday TD. Adipose tissue metabolism in obesity: lipase action in vivo before and after a mixed meal. Metabolism. 1992;41:264–272. doi: 10.1016/0026-0495(92)90269-g. [DOI] [PubMed] [Google Scholar]
- 255.Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- 256.Garg A, Grundy SM. Management of dyslipidemia in NIDDM. Diabetes Care. 1990;13:153–169. doi: 10.2337/diacare.13.2.153. [DOI] [PubMed] [Google Scholar]
- 257.Sheu WH, Shieh SM, Fuh MM, Shen DD, Jeng CY, Chen YD, Reaven GM. Insulin resistance, glucose intolerance, and hyperinsulinemia. Hypertriglyceridemia versus hypercholesterolemia. Arterioscler Thromb. 1993;13:367–370. doi: 10.1161/01.atv.13.3.367. [DOI] [PubMed] [Google Scholar]
- 258.Tangvarasittichai S, Poonsub P, Tangvarasittichai O. Association of serum lipoprotein ratios with insulin resistance in type 2 diabetes mellitus. Indian J Med Res. 2010;131:641–648. [PubMed] [Google Scholar]
- 259.Laws A, Reaven GM. Evidence for an independent relationship between insulin resistance and fasting plasma HDL-cholesterol, triglyceride and insulin concentrations. J Intern Med. 1992;231:25–30. doi: 10.1111/j.1365-2796.1992.tb00494.x. [DOI] [PubMed] [Google Scholar]
- 260.Miller GJ, Miller NE. Plasma-high-density-lipoprotein concentration and development of ischaemic heart-disease. Lancet. 1975;1:16–19. doi: 10.1016/s0140-6736(75)92376-4. [DOI] [PubMed] [Google Scholar]
- 261.Hokanson JE, Austin MA. Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J Cardiovasc Risk. 1996;3:213–219. [PubMed] [Google Scholar]
- 262.Malmström R, Packard CJ, Caslake M, Bedford D, Stewart P, Yki-Järvinen H, Shepherd J, Taskinen MR. Defective regulation of triglyceride metabolism by insulin in the liver in NIDDM. Diabetologia. 1997;40:454–462. doi: 10.1007/s001250050700. [DOI] [PubMed] [Google Scholar]
- 263.Stampfer MJ, Sacks FM, Salvini S, Willett WC, Hennekens CH. A prospective study of cholesterol, apolipoproteins, and the risk of myocardial infarction. N Engl J Med. 1991;325:373–381. doi: 10.1056/NEJM199108083250601. [DOI] [PubMed] [Google Scholar]
- 264.Sacks FM, Alaupovic P, Moye LA, Cole TG, Sussex B, Stampfer MJ, Pfeffer MA, Braunwald E. VLDL, apolipoproteins B, CIII, and E, and risk of recurrent coronary events in the Cholesterol and Recurrent Events (CARE) trial. Circulation. 2000;102:1886–1892. doi: 10.1161/01.cir.102.16.1886. [DOI] [PubMed] [Google Scholar]
- 265.Wilson PW, Kannel WB, Anderson KM. Lipids, glucose intolerance and vascular disease: the Framingham Study. Monogr Atheroscler. 1985;13:1–11. [PubMed] [Google Scholar]
- 266.Jeppesen J, Facchini FS, Reaven GM. Individuals with high total cholesterol/HDL cholesterol ratios are insulin resistant. J Intern Med. 1998;243:293–298. doi: 10.1046/j.1365-2796.1998.00301.x. [DOI] [PubMed] [Google Scholar]
- 267.Li C, Ford ES, Meng YX, Mokdad AH, Reaven GM. Does the association of the triglyceride to high-density lipoprotein cholesterol ratio with fasting serum insulin differ by race/ethnicity? Cardiovasc Diabetol. 2008;7:4. doi: 10.1186/1475-2840-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.McLaughlin T, Reaven G, Abbasi F, Lamendola C, Saad M, Waters D, Simon J, Krauss RM. Is there a simple way to identify insulin-resistant individuals at increased risk of cardiovascular disease? Am J Cardiol. 2005;96:399–404. doi: 10.1016/j.amjcard.2005.03.085. [DOI] [PubMed] [Google Scholar]
- 269.Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest. 1999;104:787–794. doi: 10.1172/JCI7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Muoio DM, Newgard CB. Mechanisms of disease: Molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:193–205. doi: 10.1038/nrm2327. [DOI] [PubMed] [Google Scholar]
- 271.Nicholson G, Hall GM. Diabetes mellitus: new drugs for a new epidemic. Br J Anaesth. 2011;107:65–73. doi: 10.1093/bja/aer120. [DOI] [PubMed] [Google Scholar]
- 272.Boyle JP, Thompson TJ, Gregg EW, Barker LE, Williamson DF. Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul Health Metr. 2010;8:29. doi: 10.1186/1478-7954-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Morris AP, Voight BF, Teslovich TM, Ferreira T, Segrè AV, Steinthorsdottir V, Strawbridge RJ, Khan H, Grallert H, Mahajan A, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Palmer ND, McDonough CW, Hicks PJ, Roh BH, Wing MR, An SS, Hester JM, Cooke JN, Bostrom MA, Rudock ME, et al. A genome-wide association search for type 2 diabetes genes in African Americans. PLoS One. 2012;7:e29202. doi: 10.1371/journal.pone.0029202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Bonnefond A, Froguel P, Vaxillaire M. The emerging genetics of type 2 diabetes. Trends Mol Med. 2010;16:407–416. doi: 10.1016/j.molmed.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 276.Tahrani AA, Bailey CJ, Del Prato S, Barnett AH. Management of type 2 diabetes: new and future developments in treatment. Lancet. 2011;378:182–197. doi: 10.1016/S0140-6736(11)60207-9. [DOI] [PubMed] [Google Scholar]
- 277.Handelsman Y, Mechanick JI, Blonde L, Grunberger G, Bloomgarden ZT, Bray GA, Dagogo-Jack S, Davidson JA, Einhorn D, Ganda O, et al. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for developing a diabetes mellitus comprehensive care plan. Endocr Pract. 2011;17 Suppl 2:1–53. doi: 10.4158/ep.17.s2.1. [DOI] [PubMed] [Google Scholar]
- 278.Haffner SM. Management of dyslipidemia in adults with diabetes. Diabetes Care. 2003;26 Suppl 1:S83–S86. doi: 10.2337/diacare.26.2007.s83. [DOI] [PubMed] [Google Scholar]
- 279.Tangvarasittichai S, Lertsinthai P, Taechasubamorn P, Veerapun O, Tangvarasittichai O. Effect of Moderate-Intensity Exercise Training on Body Weight, Serum Uric Acid, Serum hs-CRP, and Insulin Sensitivity in Type 2Diabetic Patients. Siriraj Med J. 2009;61:310–313. [Google Scholar]
- 280.Mills EJ, Wu P, Chong G, Ghement I, Singh S, Akl EA, Eyawo O, Guyatt G, Berwanger O, Briel M. Efficacy and safety of statin treatment for cardiovascular disease: a network meta-analysis of 170,255 patients from 76 randomized trials. QJM. 2011;104:109–124. doi: 10.1093/qjmed/hcq165. [DOI] [PubMed] [Google Scholar]
- 281.Canner PL, Berge KG, Wenger NK, Stamler J, Friedman L, Prineas RJ, Friedewald W. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol. 1986;8:1245–1255. doi: 10.1016/s0735-1097(86)80293-5. [DOI] [PubMed] [Google Scholar]
- 282.Elam MB, Hunninghake DB, Davis KB, Garg R, Johnson C, Egan D, Kostis JB, Sheps DS, Brinton EA. Effect of niacin on lipid and lipoprotein levels and glycemic control in patients with diabetes and peripheral arterial disease: the ADMIT study: A randomized trial. Arterial Disease Multiple Intervention Trial. JAMA. 2000;284:1263–1270. doi: 10.1001/jama.284.10.1263. [DOI] [PubMed] [Google Scholar]
- 283.Grundy SM, Vega GL, McGovern ME, Tulloch BR, Kendall DM, Fitz-Patrick D, Ganda OP, Rosenson RS, Buse JB, Robertson DD, et al. Efficacy, safety, and tolerability of once-daily niacin for the treatment of dyslipidemia associated with type 2 diabetes: results of the assessment of diabetes control and evaluation of the efficacy of niaspan trial. Arch Intern Med. 2002;162:1568–1576. doi: 10.1001/archinte.162.14.1568. [DOI] [PubMed] [Google Scholar]
- 284.Centers for Disease Control and Prevention. [Accessed 2014 June 15] Available from: http: www.cdc.gov.
- 285.Qaseem A, Humphrey LL, Sweet DE, Starkey M, Shekelle P. Oral pharmacologic treatment of type 2 diabetes mellitus: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2012;156:218–231. doi: 10.7326/0003-4819-156-3-201202070-00011. [DOI] [PubMed] [Google Scholar]
- 286.Giannarelli R, Aragona M, Coppelli A, Del Prato S. Reducing insulin resistance with metformin: the evidence today. Diabetes Metab. 2003;29:6S28–6S35. doi: 10.1016/s1262-3636(03)72785-2. [DOI] [PubMed] [Google Scholar]
- 287.Staels B. Metformin and pioglitazone: Effectively treating insulin resistance. Curr Med Res Opin. 2006;22 Suppl 2:S27–S37. doi: 10.1185/030079906X112732. [DOI] [PubMed] [Google Scholar]
- 288.Bulcão C, Ribeiro-Filho FF, Sañudo A, Roberta Ferreira SG. Effects of simvastatin and metformin on inflammation and insulin resistance in individuals with mild metabolic syndrome. Am J Cardiovasc Drugs. 2007;7:219–224. doi: 10.2165/00129784-200707030-00007. [DOI] [PubMed] [Google Scholar]
- 289.Fidan E, Onder Ersoz H, Yilmaz M, Yilmaz H, Kocak M, Karahan C, Erem C. The effects of rosiglitazone and metformin on inflammation and endothelial dysfunction in patients with type 2 diabetes mellitus. Acta Diabetol. 2011;48:297–302. doi: 10.1007/s00592-011-0276-y. [DOI] [PubMed] [Google Scholar]
- 290.Ferner RE, Rawlins MD, Alberti KG. Impaired beta-cell responses improve when fasting blood glucose concentration is reduced in non-insulin-dependent diabetes. Q J Med. 1988;66:137–146. [PubMed] [Google Scholar]
- 291.Perriello G, Misericordia P, Volpi E, Santucci A, Santucci C, Ferrannini E, Ventura MM, Santeusanio F, Brunetti P, Bolli GB. Acute antihyperglycemic mechanisms of metformin in NIDDM. Evidence for suppression of lipid oxidation and hepatic glucose production. Diabetes. 1994;43:920–928. doi: 10.2337/diab.43.7.920. [DOI] [PubMed] [Google Scholar]
- 292.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, Ianculescu AG, Yue L, Lo JC, Burchard EG, et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest. 2007;117:1422–1431. doi: 10.1172/JCI30558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Kim YD, Park KG, Lee YS, Park YY, Kim DK, Nedumaran B, Jang WG, Cho WJ, Ha J, Lee IK, et al. Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes. 2008;57:306–314. doi: 10.2337/db07-0381. [DOI] [PubMed] [Google Scholar]
- 296.Food and Drug Administration. [Accessed 2014 June 9] Available from: http: //www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm.
- 297.Lindsay JR, Duffy NA, McKillop AM, Ardill J, O’Harte FP, Flatt PR, Bell PM. Inhibition of dipeptidyl peptidase IV activity by oral metformin in Type 2 diabetes. Diabet Med. 2005;22:654–657. doi: 10.1111/j.1464-5491.2005.01461.x. [DOI] [PubMed] [Google Scholar]
- 298.Sinha Roy R, Bergeron R, Zhu L, He H, Jiang G, Liu F, Lyons K, Pryor K, Yao J, Zhang BB, et al. Metformin is a GLP-1 secretagogue, not a dipeptidyl peptidase-4 inhibitor. Diabetologia. 2007;50(suppl 1):S284. [Google Scholar]
- 299.Sisson EM. Liraglutide: clinical pharmacology and considerations for therapy. Pharmacotherapy. 2011;31:896–911. doi: 10.1592/phco.31.9.896. [DOI] [PubMed] [Google Scholar]