

To The Editor
Hereditary non-polyposis colorectal cancer (HNPCC), also known as Lynch Syndrome, is the most common inherited colorectal cancer susceptibility syndrome. First described in 1966 by Dr. Henry Lynch with the description of two large Midwest kindred, HNPCC is due to germline mutations in MLH1, MSH2, MSH6 and PMS2, which result in a deficiency in DNA mismatch repair. HNPCC is not only associated with an increased risk for colorectal cancer but also extra-colonic cancers including endometrium, ovarian, pancreas and brain. By instituting routine screening of all newly diagnosed colorectal adenocarcinomas for microsatellite instability, health systems have increased the yield of identifying patients with HNPCC allowing for appropriate genetic counseling and screening.1 Although the common extracolonic malignancies associated with HNPCC have been identified, there are an increasing number of case reports of established HNPCC patients with neuroendocrine tumors (NETs), raising the possibility that this type of tumor could represent another extracolonic manifestation for which these patients are at increased risk. The increased incidence much like in the general population may be due to the increased use of cross sectional imaging for screening purposes.
Case Report
We report a case of a pheochromocytoma developing in a patient known to have HNPCC. The patient is a 52 year old female with a family history of early onset colon cancer, pancreatic cancer and endometrial cancer. Despite her family history, the consideration of HNPCC did not arise until a skin lesion was removed from her nose in 2011 at age 49 with pathology showing a sebaceous adenoma and immunohistochemistry (IHC) revealing absent staining for MSH2. Sebaceous adenomas are strongly associated with Muir-Torre, a variant of HNPCC. Genetic testing was performed showing a pathogenic mutation of p.Q462X in MSH2 (c. 1384 C>T).2 Endoscopic ultrasound was performed to screen the pancreas which was normal but an incidental adrenal mass was identified. The lesion was non-secretory on laboratory testing and the patient underwent a laparoscopic adrenalectomy. Pathology was consistent with a pheochromocytoma. IHC staining was performed on the pheochromocytoma tissue and this revealed an absence of both MSH2 and MSH6 expression (Figure 1). Microsatellite studies also were performed and the tumor was microsatellite instable. Additionally, the patient had genetic testing with the Ambry Genetics PGLNext panel (genes in which inherited mutations are associated with an increased risk of pheochromocytoma), screening for both mutations and large genomic rearrangements of MAX, NF1, RET, SDHA, SDHAF2, SDHB, SDHC, SDHD, TMEM127 and VHL, which was negative.
Figure 1.
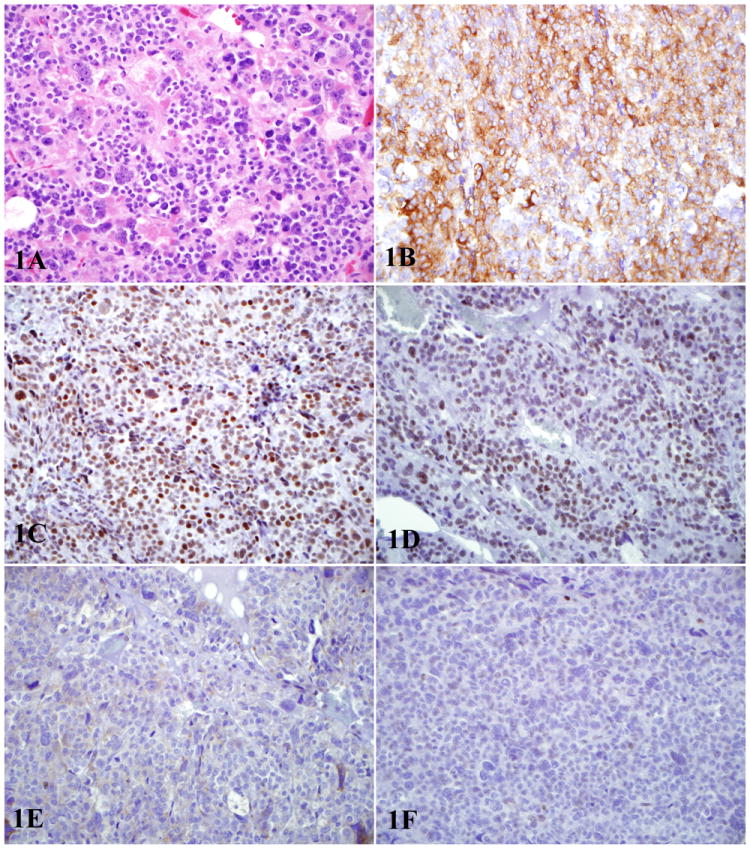
1A: Hematoxylin and eosin stained section of pheochromocytoma with marked cytologic atypia 1B: Tumor shows diffuse cytoplasmic staining for synaptophysin 1C: Tumor demonstrates presence of mismatch repair proteins MLH-1 1D: Tumor demonstrates presence of PMS-2 1E: Tumor shows loss of mismatch repair proteins MSH-2 (1E) 1F: Tumor shows loss of mismatch repair proteins MSH-6 (1F).
* All photos at 400X.
The case presented above adds to the growing case series reported in the literature of neuroendocrine tumors occurring in the setting of the HNPCC. The patient presented had a mutation in the DNA mismatch repair protein, MSH2. MSH2 mutation is well established to play a role in human carcinogenesis by allowing for DNA repair defects and is seen in 40% of patients with HNPCC. While the MSH2 mutation has been shown to markedly increase the risk of colon cancer and the associated other HNPCC malignancies,3 no abnormalities in MSH2 protein levels have been demonstrated in PNET4 or small bowel carcinoids.5
To date, only one other case of pheochromoctytoma in the setting of HNPCC has been reported in the literature. In that case only MSH6 expression was absent. In our patient both MSH2 and MSH6 were absent. This patient has an established germline MSH2 mutation and both her HNPCC associated lesion, the sebaceous adenoma, and the pheochromocytoma lacked staining for MSH2 and MSH6 indicating a high likelihood that this was not a sporadic NET but rather an extra-colonic manifestation of the HNPCC. In many of the prior case reports of NET-associated tumors in the HNPCC, there has remained a question of whether the NET was actually a metastatic lesion from the primary colonic adenocarcinoma with eventual neuroendocrine differentiation. Our case is unique in that our patient has never been diagnosed with a colonic adenocarcinoma but was diagnosed via family history, genetic testing and the phenotypic presentation of Muir-Torre syndrome. In addition, pheochromocytomas are traditionally associated with hypermethylation of MSH2 in both the sporadic and MEN2 types rather than with an absence of MSH26 as we describe here.
While NETs traditionally develop in the absence of a mutation in the mismatch repair structure, the case reported above and those in the literature with demonstratable MSH2 mutations add evidence that NETs can be seen as an extracolonic manifestation of the Lynch Syndrome.
Footnotes
The Authors Declare No Conflict of Interest
Contributor Information
Brian P. Riff, Fellow in Gastroenterology, University of Pennsylvania, Philadelphia, Pennyslvania.
Bryson Katona, Fellow in Gastroenterology, University of Pennsylvania, Philadelphia, Pennyslvania.
Myra Wilkerson, Division of Pathology and Laboratory Medicine, Geisinger Health System, Wilkes-Barre, Pennsylvania.
Katherine L. Nathanson, Associate Professor of Medicine and Translational Medicine & Human Genetics, University of Pennsylvania, Philadelphia, Pennsylvania.
David C. Metz, Professor of Medicine, Division of Gastroenterology, University of Pennsylvania.
References
- 1.Hampel Heather, et al. Screening for the HNPCC (hereditary nonpolyposis colorectal cancer) New England Journal of Medicine. 2005;352(18):1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 2.Ligtenberg Marjolijn JL, et al. Heritable somatic methylation and inactivation of MSH2 in families with HNPCC due to deletion of the 3′ exons of TACSTD1. Nature genetics. 2008;41(1):112–117. doi: 10.1038/ng.283. [DOI] [PubMed] [Google Scholar]
- 3.Vasen HFA, et al. MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. Journal of Clinical Oncology. 2001;19(20):4074–4080. doi: 10.1200/JCO.2001.19.20.4074. [DOI] [PubMed] [Google Scholar]
- 4.Arnason Thomas, et al. Loss of expression of DNA mismatch repair proteins is rare in pancreatic and small intestinal neuroendocrine tumors. Archives of Pathology & Laboratory Medicine. 2011;135(12):1539–1544. doi: 10.5858/arpa.2010-0560-OA. [DOI] [PubMed] [Google Scholar]
- 5.Kidd Mark, et al. Microsatellite instability and gene mutations in transforming growth factor-beta type II receptor are absent in small bowel carcinoid tumors. Cancer. 2005;103(2):229–236. doi: 10.1002/cncr.20750. [DOI] [PubMed] [Google Scholar]
- 6.Dammann R, et al. Frequent promoter methylation of tumor-related genes in sporadic and men2-associated pheochromocytomas. Experimental and clinical endocrinology & diabetes. 2005;113(01):1–7. doi: 10.1055/s-2004-830522. [DOI] [PubMed] [Google Scholar]