

Abstract
Malignant gliomas can be counted to the most devastating tumors in humans. Novel therapies do not achieve significant prolonged survival rates. The cancer cells have an impact on the surrounding vital tissue and form tumor zones, which make up the tumor microenvironment. We investigated the effects of sunitinib, a small molecule multitargeted receptor tyrosine kinase inhibitor, on constituents of the tumor microenvironment such as gliomas, astrocytes, endothelial cells, and neurons. Sunitinib has a known anti-angiogenic effect. We found that sunitinib normalizes the aberrant tumor-derived vasculature and reduces tumor vessel pathologies (i.e. auto-loops). Sunitinib has only minor effects on the normal, physiological, non-proliferating vasculature. We found that neurons and astrocytes are protected by sunitinib against glutamate-induced cell death, whereas sunitinib acts as a toxin towards proliferating endothelial cells and tumor vessels. Moreover, sunitinib is effective in inducing glioma cell death. We determined the underlying pathways by which sunitinib operates as a toxin on gliomas and found vascular endothelial growth factor receptor 2 (VEGFR2, KDR/Flk1) as the main target to execute gliomatoxicity. The apoptosis-inducing effect of sunitinib can be mimicked by inhibition of VEGFR2. Knockdown of VEGFR2 can, in part, foster the resistance of glioma cells to receptor tyrosine kinase inhibitors. Furthermore, sunitinib alleviates tumor-induced neurodegeneration. Hence, we tested whether temozolomide treatment could be potentiated by sunitinib application. Here we show that sunitinib can amplify the effects of temozolomide in glioma cells. Thus, our data indicate that combined treatment with temozolomide does not abrogate the effects of sunitinib. In conclusion, we found that sunitinib acts as a gliomatoxic agent and at the same time carries out neuroprotective effects, reducing tumor-induced neurodegeneration. Thus, this report uncovered sunitinib's actions on the brain tumor microenvironment, revealing novel aspects for adjuvant approaches and new clinical assessment criteria when applied to brain tumor patients.
Keywords: Angiogenesis modulators, cell death, cytoprotection, glioma, tyrosine kinase-type cell surface receptor
Malignant gliomas are the most fatal tumor entities of the central nervous system.1,2 Uncontrolled cell proliferation, diffuse tissue invasion, and a microenvironment characterized by the formation of abnormal vessels and neurodegenerative processes are some of the hallmarks of malignant gliomas. Although complete surgical resection seems improbable, advances in the fields of surgical resection, intraoperative imaging, and multimodal regimens of combined radiotherapy and chemotherapy have led to modestly increased overall survival rates in patients with malignant gliomas of approximately 20 months.3 In spite of these advances, malignant gliomas remain among the most devastating diseases and improving therapeutics is a highly anticipated and challenging goal in neurooncology.
Molecular targeting of oncoproteins has recently changed the standard of clinical practice for many solid tumors. The three most common genomically altered cell-surface receptor tyrosine kinases (RTKs), vascular endothelial cell growth factor receptor 2 (VEGFR2), platelet-derived growth factor receptor β (PDGFRβ), and c-KIT, are promising targets. Receptor tyrosine kinase inhibitors like imatinib and sunitinib or mAbs such as bevacizumab are already implemented in the treatment of cancer entities such as gastrointestinal stromal tumor (GIST), non-small-cell lung cancer, and renal cell carcinoma.4,5 Concerning malignant gliomas, it still has to be shown that treatment with small molecule inhibitors or mAbs has considerable survival benefit for patients.6–8 This raises the question of whether RTK inhibitors impact the tumor growth and whether the angiogenic process in the peritumoral zone is reached and affected. Moreover, it is important to determine the influence of RTK inhibitors on the tumor microenvironment.
To answer these questions, we investigated the effects of sunitinib, a 532-Da, orally applicable, small molecule multitargeted RTK inhibitor on the tumor microenvironment. Sunitinib was the ideal choice, for the potent anti-angiogenic and antitumoral effects of the drug are known. Additionally, we tested specific single-targeted RTK inhibitors (such as bryostatin, imatinib, orantinib, salirasib, SU1498, vandetanib, and wortmannin) that block particular single receptors and downstream targets commonly affected by sunitinib.9 Sunitinib is a rationally designed drug aimed at inhibiting members of the split-kinase domain family of RTKs. The bioavailability of sunitinib is high with a half-life of 40–60 h. It has a very high efficacy on angiogenically active RTKs, VEGFR2/KDR/Flk1, PDGFRα, and PDGFRβ. Sunitinib also inhibits KIT, FLT3, CRF1R, and RET, which are known to be involved in oncogenic signaling in different malignant tumor entities.10–12 In 2006, sunitinib was approved by the US FDA for the treatment of metastatic renal cell carcinoma and imatinib-resistant GIST.
In this study, we analyzed the impact of sunitinib on the brain tumor microenvironment. We found that sunitinib acts specifically on aberrant tumor vessels and induces endothelial cell death. Sunitinib is selectively gliomatoxic, an effect that can be mimicked by VEGFR2 inhibition and can be amplified by combined treatment regimens with temozolomide. Our study reveals that sunitinib has no toxic effects on astrocytes and neurons and does not impede astrocyte cell growth or neuronal survival. Moreover, sunitinib seems to operate neuroprotectively and alleviates glutamate-induced cell stress.
Materials and Methods
Cell culture
Rodent glioma cell line F98 and the human glioma cell line U87 were obtained from ATCC (Wesel, Germany). The human glioma cells U251 and T98G were kindly provided by Drs. Yvonne Rübner and Rainer Fietkau (Erlangen, Germany). Murine hippocampal neuronal cell line HT22 was kindly provided by Drs. C. Behl and D. Schubert (Mainz, Germany and La Jolla, CA, USA). All cell lines were cultured under standard humidified conditions (37°C, 5% CO2) with DMEM (Biochrom, Berlin, Germany) supplemented with 10% FBS (Biochrom), 1% penicillin/streptomycin (Biochrom), and 1% glutamax (Darmstadt, Gibco/Invitrogen, CA, USA). Cells were passaged at approximately 80% confluence. Cells were scraped off or trypsinized after washing in PBS. After centrifugation (150 g for 5 min), 500 000 cells were plated out in culture flasks.
Primary cell culture
Primary astrocytes were prepared from up to six days old rats.13,14 The removed brains were minced, trypsinized, and filtered twice. The isolated astrocytes were cultured with DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% glutamax under humidified conditions (37°C, 5% CO2). Rat brain endothelial cells (RBEC) were prepared from up to 1-month-old rats according to the astrocyte preparation protocol as described.13 After removing the leptomeninges from the brain, microvessels were isolated out of the grey matter. Microvessels were prepared by digesting with collagenase and DNAse, and subsequently plated out in collagen-coated culture flasks maintained in EC medium.
Drug preparation
Sunitinib malate (SU-11248; see Chow and Eckhardt, 2007 for chemical structure)15 and temozolomide were purchased from Sigma-Aldrich (Taufkirchen, Germany). For ex vivo and in vitro assays, sunitinib was solubilized in sterile water to a dilution concentration of 10 mM. Temozolomide was dissolved in DMSO at 300 mM and working concentrations were prepared with PBS. Imatinib, orantinib (SU6668), vandetanib, and wortmannin were purchased from Selleck Chemicals (Selleckchem, Munich, Germany), bryostatin and SU1498 were from Merck (Darmstadt, Germany), and salirasib was purchased from Cayman Chemical Company (Ann Arbor, MI, USA). All inhibitors were diluted under sterile conditions with DMSO to a suggested dilution concentration of 100 mM. The final working solutions had a maximal DMSO concentration of 0.2%.
Vascular organotypic brain slice cultures
Brain slice cultures were prepared and maintained as previously described.16,17 Six- to nine-day-old Wistar rats (Charles River, Boston, MA, USA) were decapitated; brains were removed and kept under ice-cold conditions. Frontal lobes and cerebellum were dissected of the hemispheres and the remaining brain was cut into 350-μm-thick horizontal slices with a vibratome (VT1000S; Leica, Bensheim, Germany). Thereafter, hippocampal brain slices were transferred onto culture plate insert membrane dishes (pore size 0.4 μm; Greiner BioOne, Frickenhausen, Germany) and subsequently transferred into 6-well culture dishes (GreinerBioOne). Brain slices were cultured in humidified conditions (35°C, 5% CO2) with 1.2 mL culture medium per well (MEM–Hanks' balanced salt solution (HBSS), 2:1, 25% normal horse serum, 2% L-glutamine, 2.64 mg/mL glucose, 100 U/mL penicillin, 0.1 mg/mL streptomycin, 10 μg/mL insulin–transferrin–sodium selenite supplement, and 0.8 μg/mL vitamin C). The medium was changed on the first day after preparation and from that time on every other day over a course of 7 days. To monitor neurodegeneration and cell death, propidium iodide (PI) staining was carried out every other day during the full medium exchange.13 On the second day in culture, 10 000 tumor cells in a concentration of 100 000 cells per 1 μL culture medium were implanted onto the hippocampal cortex of the brain slices. Starting from the third day in culture, the brain slices were treated with sunitinib at concentrations of 1–20 μM. For controlling tumor-induced effects we applied the cell death and angiogenesis analysis on sham operated brain slices. These controls showed similar results compared to untreated controls. Moreover, within tumor-implanted brain slices, regions far away from the tumor provided reliable controls for distinguishing tumor-induced effects from technical impacts.
Cell proliferation analysis and toxicity assays
Cell proliferation assays were carried out according to Eyüpoglu et al.18 Proliferation was measured using the MTT assay. Briefly, cells were plated at a density of 2000 cells/cm² in 96-well plates and incubated under standard conditions for several days. On the second day, various dosages of sunitinib (1, 5, 10, and 20 μM) were administered and on the fourth day 10 mM glutamic acid was added. At measure point on the fifth day, cells were incubated with MTT solution (Roth, Karlsruhe, Germany) (5 mg/mL) for 4 h at 37°C, 5% CO2. Cells were lysed with 100 μL isopropanol + 0.1 N HCl and thereafter optical density was measured with an SLT Spectra microplate reader (Crailsheim, Germany) using Tecan XFluor4 software.14 For PI staining (Molecular Probes, Leiden, The Netherlands) we used a final concentration of 1 μg/mL out of a stock containing 5 mg/mL. After 20 min of incubation, excess dye was washed out by a pre-warmed complete medium exchange.
Statistical analysis
Quantitative data from experiments were obtained as stated in the figure legends. Analysis was carried out using the unpaired Student's t-test (Excel; Microsoft Unterschleissheim, Germany).14 Data were obtained from at least three independent experiments. The level of significance was set at P < 0.05. Error bars represent ± SD.
Results
Sunitinib does not alter brain vessels and specifically impacts tumor angiogenesis
First, we investigated the impact of sunitinib on normal brain vasculature. In physiological processes (e.g. during development or reorganizational processes) angiogenesis is active in a transient mode as a self-limiting process whereas in tumor-induced angiogenesis an angiogenic switch is constantly turned on.19 In sunitinib-treated brain tissue the vascular density and vessel diameters remained unchanged compared to untreated brain vessels (Fig.1a). Next, we analyzed the architecture of tumor angiogenesis in glioma infiltrated brain slices. In untreated tumor-implanted brain slices we found a high density of vessels and a branched microvessel network in the peritumoral zone or tumor zone 2 according to the glioma tumor zone model (Fig.1b).3 Treatment of the brain tissue with various concentrations of sunitinib ranging from 1 to 10 μM led to a significant decrease in vessel numbers (Fig.1b,c). However, the average vessel diameter was not affected by sunitinib (Fig.1d). Tumor vessels are known to have abnormalities showing a tortuous, saccular, and disorganized structure of the vasculature (Fig.1e).20 Interestingly, sunitinib treatment revealed a certain normalization of tumor vessel structures with disappearance of these morphological pathologies (Fig.1e). Through these investigations we found that sunitinib treatment reduces tumor-associated vessel pathologies and normalizes the vasculature (Fig.1e).
Fig 1.
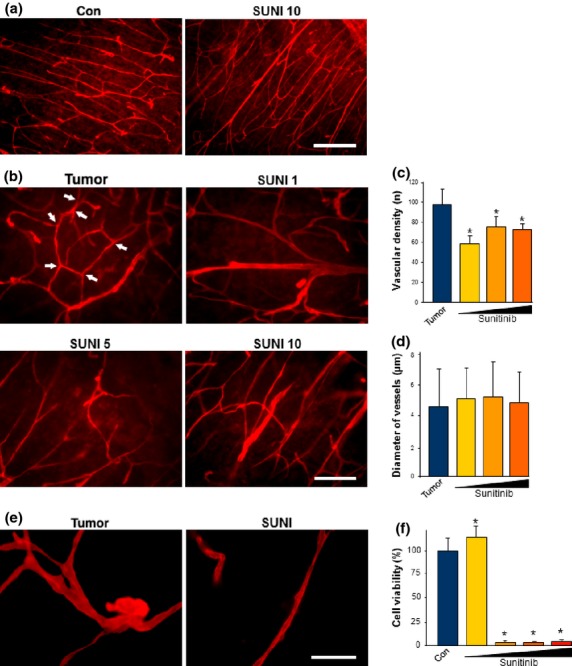
Sunitinib impacts tumor vessels, normalizes vascular morphology, and induces endothelial cell death. (a) Sunitinib does not affect vascular morphology in normal brain tissue. Brain slices in culture were treated with the solvent only as the control (Con) or with 10 μM sunitinib (SUNI 10). After 72 h of incubation vessels were analyzed by laminin immunostaining (red). Scale bar = 200 μm. (b) Sunitinib treatment reduces tumor-induced angiogenesis in glioma implanted brain slices. F98 glioma cells were implanted in brain slices; after 7 days vessels were evaluated (shown in red). Tumor-implanted brains without sunitinib treatment served as controls (Tumor). Sunitinib was given in concentrations of 1 μM (SUNI 1), 5 μM (SUNI 5), and 10 μM (SUNI 10) for 72 h. Arrows mark typical tumor vessel aberrations found in the untreated group. Scale bar = 100 μm. (c, d) Quantification of vascular density, vessel branching, and diameters. Means ± SD are given. *P < 0.05, Student's t-test (n = 68). (e) Gliomas induce vascular alterations such as atypical tortous and disorganized vessels (Tumor). Sunitinib treatment led to normalization of these vascular alterations and the vascular patterns reveal a physiological phenotype (SUNI). Scale bar = 40 μm. (f) Purified rat brain endothelial cells were treated with various concentration of sunitinib. Endothelial cell proliferation assessed by MTT assay revealed that sunitinib was highly toxic at concentrations of 5 μM (dark yellow column), 10 μM (orange column), and 20 μM (red column). Sunitinib at 1 μM (yellow column) did not affect endothelial cells. Quantification is given as means ± SD. *P < 0.05, Student's t-test (n = 73).
Next, we analyzed the direct effect of sunitinib on vessel-forming cells. For this, we isolated endothelial cells directly from 1-month-old rats (RBEC) and cultured them under endothelial cell-specific conditions. As hypothesized from the tumor–vessel studies, RBECs were highly vulnerable to sunitinib. When treated with various concentrations (1–20 μM), cell viability analysis showed that sunitinib is already toxic to proliferating endothelial cells at a concentration of 5 μM (Fig.1f).
Sunitinib impedes glioma cell growth and reduces tumor-induced neurodegeneration
We investigated the effects of sunitinib on malignant gliomas. For this we used various established glioma cells. We tested rat gliomas (F98) and treated these cells for 72 h with sunitinib. Interestingly, at a concentration of 5 μM, glioma cells started to change their morphology (Fig.2a). The glioma cells appeared bigger in cell diameter and the cell bodies were revealed to be swollen. Starting from concentrations of 10 μM sunitinib, glioma cells showed rounded cells and massive cell death (Fig.2a). Quantitative evaluation of this effect showed that sunitinib reduced cell viability of F98 glioma cells in a dose-dependent manner (Fig.2b). We also investigated the effects of sunitinib on various human glioma cell lines such as T98G, U87, and U251 (Fig.2c–e). Sunitinib was effective in impairing cell growth in all of the tested human glioma cells (Fig.2c–e). Of note, sunitinib was also efficient in inducing cell death in primary Glioblastoma multiforme tissue derived from patients of the neurosurgical university clinics of Erlangen-Nuremberg (Fig. S1).
Fig 2.
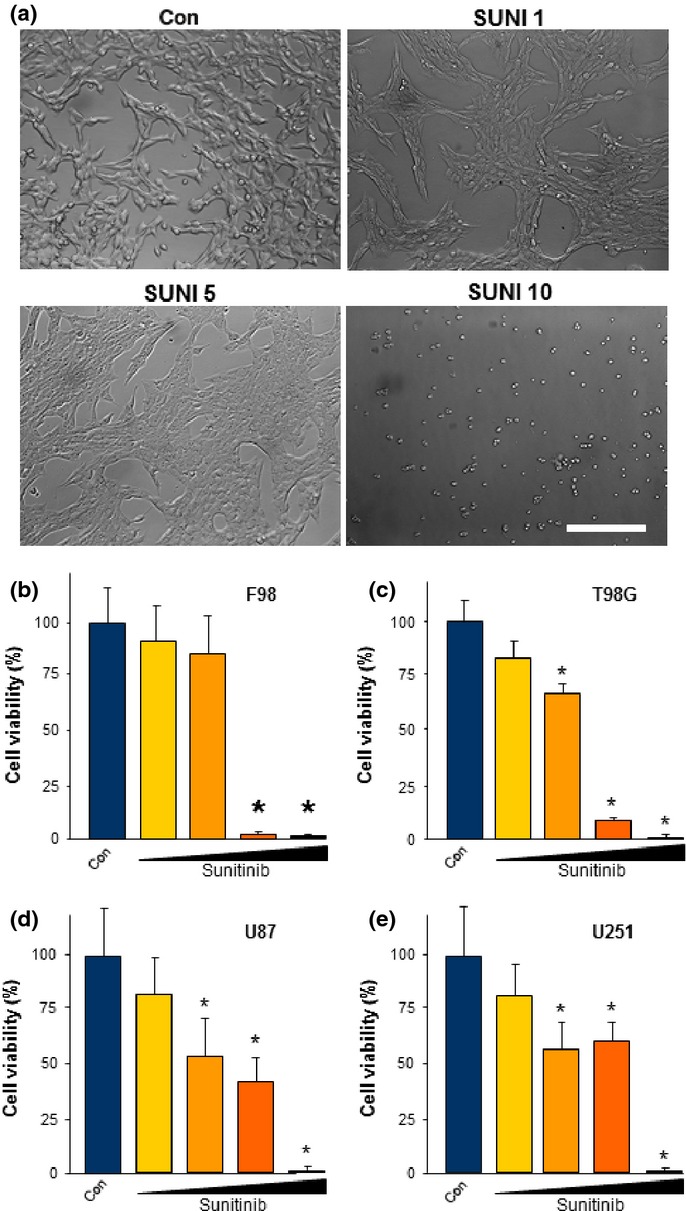
Sunitinib induces cell death in malignant gliomas. Cell survival was monitored in human as well as rodent malignant glioma cells and cell death was evaluated. (a) Representative images of rodent glioma cells (F98) cultured at various concentrations of sunitinib: 1 μM (SUNI 1), 5 μM (SUNI 5), and 10 μM (SUNI 10). Note that glioma morphology was already altered at 5 μM SUNI. Scale bar = 50 μm. Con, control. (b) Quantification of cell survival after sunitinib treatment. Sunitinib acted in a dose-dependent manner on glioma cells. Control, blue column; 1 μM sunitinib, yellow column; 5 μM sunitinib, orange column; 10 μM sunitinib, red column; 20 μM sunitinib, purple column. (c–e) Sunitinib is cytotoxic towards various human glioma cells. Dose–response curves of sunitinib in human T98G (c), human U87 (d), and human U251 (e) glioma cells. Cell growth is given in relation to untreated controls (Con; blue columns). Sunitinib was given at 1 μM (yellow column), 5 μM (dark yellow column), 10 μM (orange column), and 20 μM (red column) concentrations. Quantification is given for at least n = 12. Values are given as mean ± SD with controls set as 100%. Differences were considered statistically significant at *P < 0.05 (two-sided Student's t-test).
These data led to the question whether sunitinib has a general toxic effect on proliferating cells or whether the observed effects are specific for gliomas and endothelial cells. To answer this, we facilitated primary rat astrocytes and treated these cells with various concentrations of sunitinib. We also used the glutamate toxicity paradigm as a cell death reference system, which plays an important role in tumor-induced cell death.21,22 Monitoring of cell death revealed that glutamate treatment is highly toxic towards astrocytes compared to untreated controls (Fig.3a). Notably, treatment with different concentrations of sunitinib revealed only minor effects on proliferation (Fig.3b).
Fig 3.
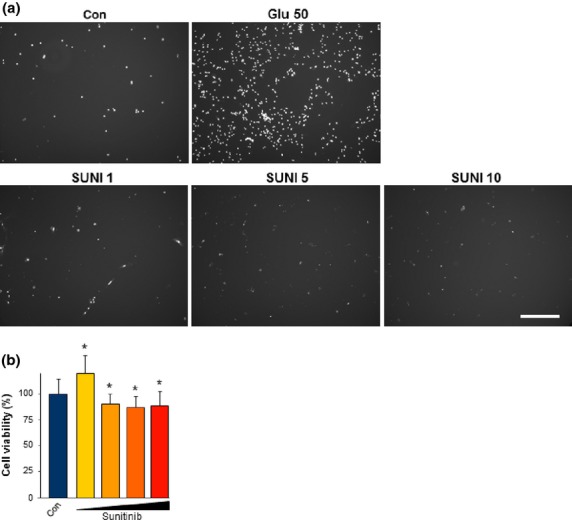
Sunitinib does not affect astrocytes at low concentrations. (a) Representative images of primary rat astrocytes treated with various concentrations of sunitinib. Primary rat astrocytes were treated with sunitinib at 1 μM (SUNI 1), 5 μM (SUNI 5), 10 μM (SUNI 10), or 20 μM (SUNI 20) and cell death was monitored (white signal, propidium iodide). Treatment with different levels of sunitinib did not increase cell death. Glutamate treatment (50 mM; Glu 50) served as a control for cell death. Scale bar = 250 μm. Con, untreated control. (b) Quantification of cell viability revealed that sunitinib at higher concentrations has minor effects on primary astrocytes. Quantification is given for n = 3 per group. Values are given as mean ± SD with controls (Con) set as 100%. Differences were considered statistically significant with *P < 0.05 (two-sided Student's t-test).
Next, we investigated the effects of sunitinib on tumor growth within the organotypic microenvironment. For this, glioma cells were implanted into brain tissue slices and subsequently treated with sunitinib. Tumor growth was measured over a course of 7 days (Fig.4). Low dosages of sunitinib at 5 and 10 μM led to an almost steady tumor bulk, indicating that tumor cell proliferation and progression were significantly decreased compared to untreated tumors (controls) (Fig.4a). At high concentrations, the gliomatoxic effects of sunitinib evident, resulting in impaired tumor growth to the extent of almost vanished tumor bulks (Fig.4a,b).
Fig 4.
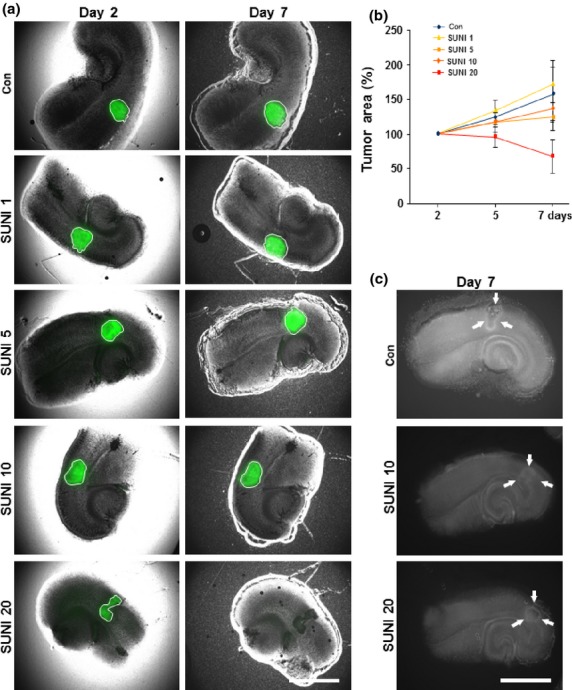
Sunitinib is gliomatoxic within the brain tumor microenvironment. Organotypic brain slices (ex vivo) with glioma tumor implantation were cultured with sunitinib in doses of 1 μM (SUNI 1), 5 μM (SUNI 5), 10 μM (SUNI 10), or 20 μM (SUNI 20). (a) Representative images from tumor-implanted brain slices treated with sunitinib. Sunitinib treatment induced tumor cell death in glioma implanted brain slices. F98 glioma cells (green) were implanted in brain slices and the size of the tumor bulk was documented for 7 days using fluorescent microscopic imaging. Images are shown from day 2 and 7 after tumor implantation. Scale bar = 1000 μm. Con, untreated control. (b) Quantification of tumor growth in brain slices. The dimensions of the tumor bulks of the various groups were measured and compared to untreated controls (Con). Low concentrations of sunitinib (5 and 10 μM) led to stunted tumor growth. Sunitinib at a concentration of 20 μM resulted in diminished tumor bulk. Means ± SD are given. *P < 0.05, Student's t-test (n = 40). (c) Sunitinib induced glioma cell death and reduced tumor-induced neurodegeneration. F98 glioma cells were implanted in brain slices and cell death was evaluated (shown as white signal). Scale bar = 1000 μm.
We further investigated the impact of sunitinib treatment on tumor-induced cell death and neuronal integrity. In untreated control slices tumor-induced neurodegeneration could be monitored in peritumoral areas (Fig.4c). However, following sunitinib treatment this cell death zone was reduced and the overall cell death stain appeared lower compared to untreated gliomas (Fig.4c). Together, our data revealed that sunitinib is specifically toxic towards gliomas and, in addition, sunitinib exerts neuroprotective effects within the tumor microenvironment.
We then investigated the pathway causing the cytotoxic effects of sunitinib. In principle, sunitinib can affect glioma cells, pericytes, and endothelial cells within the tumor microenvironment (Fig. S2a). As sunitinib is a multi-RTK inhibitor with different molecular targets and downstream pathways, we continued our investigations with single-targeted RTK and kinase inhibitors with comparable molecular weights (Fig. S2b, Table S1). For this approach we applied all inhibitors at concentrations at and above IC90 to the F98 malignant glioma cell line. Protein kinase C (PKC) isoenzymes are recognized as potential targets against cancer cells. Bryostatins are modulators of PKC comparable to diacylglycerols. Beside a small tumor-promoting component, the tumor inhibiting effects gain the upper hand: bryostatin mainly activates PKCδ, a tumor suppressor.23 Nevertheless, proliferation assays showed that bryostatin did not significantly affect glioma proliferation or cell survival in the range of 1–20 μM, where sunitinib is effective (Figs5a,S3a). Furthermore, we used imatinib, a small molecule RTK inhibitor of c-KIT used in the treatment for Philadelphia chromosome (t(9,22)(q34;q11))-positive chronic myeloid leukemia inhibiting BCR-abl24 and in the therapy of GIST. Imatinib used in various concentrations (1, 5, 10, and 20 μM) had no effect on glioma cell growth (Figs5b,S3). Next, we tested the Ras-inhibitor salirasib (trans-farnesylthiosalicylic acid). Salirasib is known to compete with activated Ras for binding to Ras-escort proteins.25 In the cell viability assay, salirasib decreased the proliferation of glioma cells significantly at high concentrations of 20 μM, whereas at lower concentrations salirasib had no effect (Fig.5c). Activation of phosphatidylinositol 3-kinase (PI3K) or the PI3K/Akt pathway is found in up to 80% of malignant gliomas.26 Wortmannin is a specific inhibitor of the PI3K pathway. Interestingly, wortmannin reduced the glioma cell proliferation rate significantly (up to 30%) in a dose-dependent manner (Figs5d,S3). However, the growth inhibition efficacy of wortmannin was low in comparison to sunitinib. For targeting PDGFR we used orantinib (SU6668), as an established competitive inhibitor of ATP and autophosphorylation.27,28 Various concentrations of orantinib were facilitated without lowering glioma proliferation significantly (Figs5e,S4).
Fig 5.
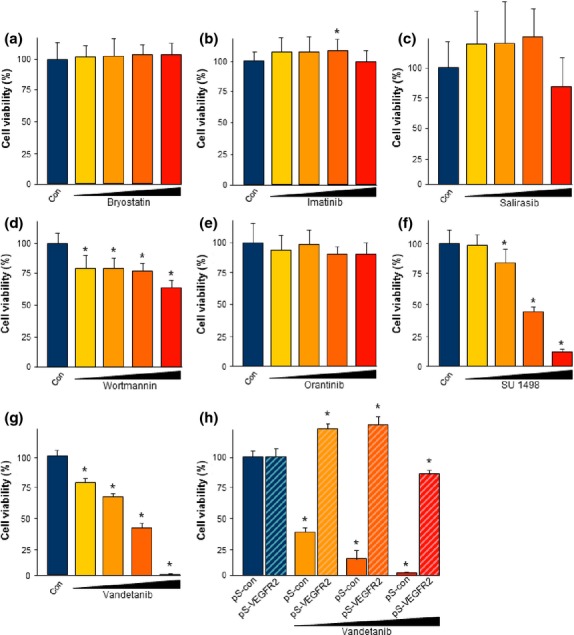
Sunitinib-induced gliomatoxicity is primarily mediated through phosphatidylinositol 3-kinase (PI3K) and vascular endothelial growth factor receptor 2 (VEGFR2) inhibition. Molecular targets of sunitinib-induced gliomatoxicity were tested with specific inhibitors. Glioma cells were treated with various receptor tyrosine kinase inhibitors at the corresponding IC90 and above and tumor cell viability was evaluated by MTT assay. (a) Bryostatin treatment on glioma cells. Bryostatin is a modulator of protein kinase C and was not effective on glioma cell viability. (b) Imatinib is an inhibitor of the c-Kit pathway and did not reduce glioma cell survival. Moreover, even at high concentrations imatinib did not inhibit proliferation of glioma cells. (c) Ras inhibitor salirasib decreased glioma cell proliferation only at a high concentration of 20 μM but had no effect at lower concentrations. (d) Wortmannin, an inhibitor of PI3K, affected glioma cell proliferation in a concentration-dependent manner. (e) Orantinib, a platelet-derived growth factor receptor β inhibitor, did not affect glioma cell proliferation. (f) Tyrphostin SU1498 (SU) specifically blocks VEGFR2. The efficacy of glioma cell growth inhibition by SU1498 closely resembled the dose-dependent effects of sunitinib. (g) Vandetanib, another specific inhibitor of VEGFR2, was efficient in glioma growth inhibition comparable to the extent induced by sunitinib. Quantification is given for n = 12 per group. Values are given as mean ± SD with controls (Con) set as 100%. Differences were considered statistically significant at *P < 0.05 (two-sided Student's t-test). (h) Glioma cells were transfected with either scrambled siRNAs (pS-con) or siRNAs directed against VEGFR2 (pS-VEGFR2). Vandetanib treatment of pS-con transfected cells resulted in high cell death at low dosages: 1 μM, dark yellow; 5 μM, orange; and 10 μM, red. RNAi-mediated silencing of VEGFR2 made glioma cells insensitive towards VEGF receptor tyrosine kinase inhibitor vandetanib (striped columns).
We then investigated specific inhibitors of VEGFR2 (KDR/Flk-1). Known to be important in mediating angiogenic or anti-angiogenic effects, VEGFR2 plays a vital role during tumor progression and the angiogenic switch. For the experiments we used two clinically promising inhibitors of VEGFR2, namely SU1498 and vandetanib. Both were similarly effective in inducing glioma cell death (Fig.5f,g). Glioma cells were vulnerable to VEGFR inhibition at already moderate dosages and application of 20 μM SU1498 and vandenatib led to abolished glioma growth below 10% compared to untreated controls (Fig.5f,g).
To confirm these pharmacological findings we investigated the genetic interference with VEGFR2 expression. For this we transfected siRNAs specifically directed against VEGFR2 into glioma cells and monitored their responsiveness towards pharmacological VEGFR2 inhibitors. Interestingly, vandetanib appeared to be ineffective in VEGFR2 knockdown in gliomas whereas in wild-type gliomas vandetanib induced massive cell death (Fig.5h). Taken together, we can conclude from the findings that the newly discovered VEGFR2-dependent gliomatoxic effect of sunitinib can be corroborated with inhibitors directed against VEGFR2. These data also indicate that the efficacy of sunitinib on different glioma cells results from synergistic coactions and concerted activities of multiple kinases or orphan kinase targets as well.
Sunitinib protects from excitotoxic cell death and is neuroprotective
The initial experiments on brain tissue and sunitinib administration already revealed a beneficial effect for neurons. To gain additional information, we tested whether sunitinib has the properties to protect neurons after an insult. As a common cell stress protocol for neurons and glial cells, we used glutamate excitotoxicity and oxitotoxicity. Neuronal cells treated with high concentrations of glutamate showed increased cell death as revealed by PI staining (Fig.6a,b). The excitotoxic effect of glutamate was alleviated by treatment with sunitinib (Fig.6a). Cell viability assays confirmed the excitotoxic-neutralizing and neuroprotective effects (Fig.6b).
Fig 6.
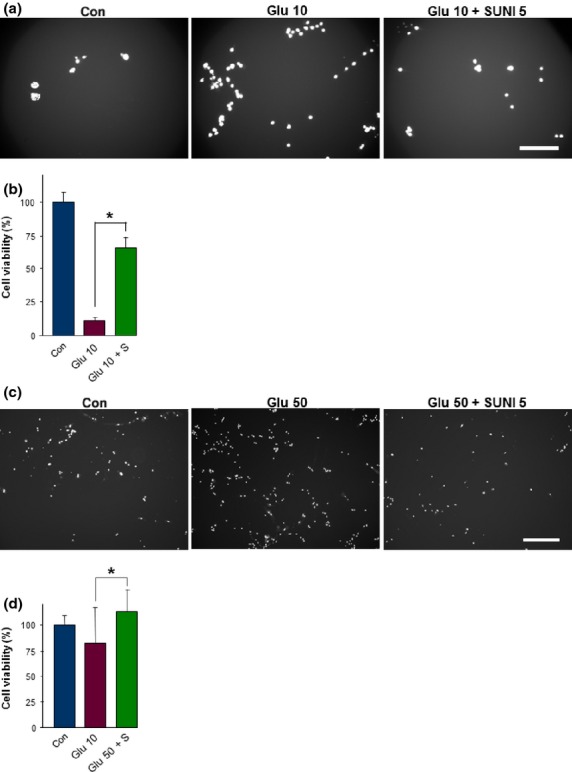
Sunitinib alleviates glutamate-induced neurotoxicity and prevents gliotoxicity. Neuronal cells and primary rat astrocytes were treated with toxic concentrations of glutamate and subsequently with sunitinib. Cell death and cell survival were monitored. (a) Representative images of neuronal cell death under control conditions (Con, untreated), glutamate treatment (10 mM; Glu 10), and 10 mM glutamate treatment plus 5 μM sunitinib (Glu 10 + SUNI 5). Propidium iodide staining revealed dead cells (white signal). Scale bar = 250 μm. (b) Quantification of neuronal cell death after excitotoxic treatment with glutamate. Black column, untreated controls (Con); purple column, glutamate-treated neurons; green column, glutamate and sunitinib-treated neurons. (c) Representative images of cell death in primary astrocytes under control conditions (Con, untreated), glutamate treatment (50 mM; Glu 50), and 50 mM glutamate treatment plus 5 μM sunitinib (Glu 50 + SUNI 5). Propidium iodide staining revealed dead cells (white signal). Note the reduced numbers of dead cells after sunitinib treatment. Scale bar = 500 μm. (d) Quantification of glutamate-induced gliotoxicity. Quantification is given for at least n ;=3 per group. Values are given as mean ± SD with controls (Con) set as 100%. Differences were considered statistically significant at *P < 0.05 (two-sided Student's t-test).
We also tested the probable cell stress-protective effects of sunitinib on primary rat astrocytes. Astrocytes were stressed with high glutamate levels and cell death was monitored. We observed massive cell death after glutamate application (Fig.6c). Astrocytes pre-treated with sunitinib withstood glutamate-induced cell death and survival rates were comparable to untreated controls (Fig.6c,d).
In a further step we analyzed neuronal cell death in brain tissue that had retained its organotypical organization. The brain tissue was treated continuously with sunitinib at 1, 5, or 10 μM over a course of 7 days. Cell death was monitored using PI staining. Control brain slices revealed only basal cell death level within the cortex and in the hippocampus (Fig.7a). As a positive control of apoptosis we induced cell death with sulfasalazine,29 which led to massive cell death (Fig.7a). Conversely, brain slices treated with sunitinib showed reduced cell death levels below the range of untreated controls (Fig.7). Thus, in addition to its anti-angiogenic and gliomatoxic effects, we uncovered that sunitinib exerts neuroprotective effects on brain tissue slices.
Fig 7.
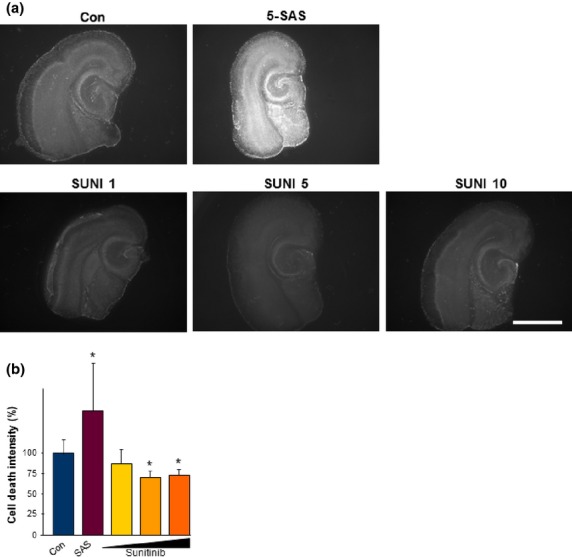
Sunitinib is neuroprotective on brain tissue. (a) Representative images of organotypic brain slices monitored for cell death (propidium iodide staining, white signal). Controls were left untreated (Con). Sulfasalazine (5-SAS) was used to induce massive cell death. Sunitinib was applied to the brain slices at 1 μM (SUNI 1), 5 μM (SUNI 5), and 10 μM (SUNI 10). Scale bar = 1000 μm. (b) Cell death intensity was quantified with Biophotonics–ImageJ and for statistical analysis the t-test was applied. Evaluation of cell death intensity showed a significant neuroprotective effect in brain tissue when treated with 5 or 10 μM sunitinib. Quantification is given for at least n ;=3 per group. Values are given as mean ± SD with controls set as 100%. Differences were considered statistically significant at *P < 0.05 (two-sided Student's t-test).
We next ask the question whether sunitinib might interfere with standard chemotherapeutics applied to glioblastoma patients. As anti-angiogenic and cytotoxic treatment regimens are an appealing approach for malignant gliomas, we tested a combined therapy with temozolomide. Temozolomide is a standard chemotherapeutic in the management of malignant gliomas.3,30 Interestingly, temozolomide reduced rodent and human glioma cell proliferation significantly at 100 μM (Fig.8). However, the growth inhibition efficacy of temozolomide was low in comparison to sunitinib (Fig.8). Next, we combined both agents and added sunitinib 1 day after temozolomide treatment. These data show that sunitinib could significantly amplify the cytotoxic effects of temozolomide in glioma cells (Fig.8).
Fig 8.

Sunitinib amplifies the toxic effects of temozolomide. Combined treatment regimen with a proved and currently clinically applied chemotherapeutic temozolomide (100 μM; t100) with sunitinib (2.5 μM; s2.5) on rodent (a) and human (b) glioma cells. (a) Representative images of rodent F98 glioma cells monitored for cell morphology (light microscopy, upper row) and cell death (propidium iodide staining, white signal; bottom row). Controls (Con) were treated with the same solvent and final concentration as for temozolomide (t100). Scale bar = 200 μm. Right, cells treated solely with temozolomide for 1 day; 2.5 μM sunitinib was added to the cells the following day. Quantification is given for n = 24. Values are given as mean ± SD with controls set as 100%. Differences were considered statistically significant at *P < 0.05 (two-sided Student's t-test). (b) Representative images of human U87 glioma cells monitored for cell morphology (light microscopy, upper row) and cell death (propidium iodide staining, white signal; bottom row). Scale bar = 200 μm. Right, cells were treated solely with temozolomide for 1 day; 5 μM sunitinib was added the next day. Quantification is given for n = 24. Values are given as mean ± SD with controls set as 100%. Differences were considered statistically significant with *P < 0.05 (two-sided Student's t-test).
Discussion
The goal of this study was to determine the impact of sunitinib on the tumor microenvironment with focus on particular cellular constituents of the brain. This study was especially motivated by the heterogeneous design and outcomes of recent clinical trials with RTK targeting small molecule inhibitors in brain tumors.31–35
We identified the toxicity profile of sunitinib on various components of the brain. A notable finding was that sunitinib acts with high toxicity on the proliferating endothelium and tumor vessels. Fully differentiated and integrated vessels are not affected. Endothelial cells in vitro are present in an active proliferating state with common signaling programs found in tumor-dependent angiogenesis.36–38 Our data are further supported by the finding that vessel abnormalities in tumors are reversed to a normalized morphology after sunitinib treatment. However, sunitinib did not lead to the degradation of vessels, indicating its context-dependent specificity and efficacy. Pro-angiogenic factors such as vascular endothelial growth factor A and platelet-derived growth factor are involved in tumor-induced angiogenesis and overactivity of these factors results in imbalances of pro- and anti-angiogenic factors. Sunitinib seems to restore this balance to a physiological level.
We found that sunitinib has a highly toxic potential on human glioma cells. Starting at a sunitinib concentration of 5 μM induced apoptotic cell death in gliomas. These data have also been confirmed in isolated glioma tissue from neurosurgical patients. Previous studies revealed sunitinib as an effective agent to inhibit cell growth and invasion of glioblastoma multiforme oncospheres and the GL15 cell line.39,40 Findings of amplified and mutated expression of Kit, PDGFR, and VEGFR2 in malignant gliomas6,41,42 give the rationale to target cell-surface RTKs. Malignant gliomas in patients frequently show overexpression and co-amplification of these three cell-surface receptors.43 However, experimental studies as well as clinical trials uncovered that small molecule RTK inhibitor responses are not strictly associated with these mutations.34,44 This paved the way for pan-receptor tyrosine kinase inhibitors such as sunitinib.
It is significant that sunitinib affects various RTKs independent of their expression levels. The concerted inhibition of RTKs in a synergistic manner could also raise the power of the efficacy. Sunitinib may also target orphan RTKs in addition to VEGFR2, PDGFR, and c-Kit. We investigated this by deciphering the contribution of each cell-surface receptor and downstream target kinase of sunitinib. We exposed gliomas to concentrations equal to the reported IC90 levels and higher. Cell death analyses revealed that SU1468 and vandetanib were as potent in dispatching gliomas as sunitinib, thus glioma cell death could be induced solely through inhibition of VEGFR2. Our results showing VEGFR2 to be a central cytotoxic target in gliomas were corroborated by experiments with other VEGFR2 inhibitors such as cediranib (AZD2171) and carboxantinib.45–47 In fact, VEGFR inhibition within the tumor microenvironment revealed further pathophysiological processes.48. Cediranib-treated patients show reduced brain edema formation, a common complication in malignant gliomas.49 This finding indicates that cell-surface RTK inhibitors have possible pleiotropic effects on the tumor and its environment. In this context the glial and neuroprotective effects are of relevance. The finding that sunitinib concentrations that have been toxic to gliomas but did not affect normal brain tissue supports the concept of context-dependent VEGFR2 signaling.
Hence, sunitinib treatment was effective in improving neuronal cell survival under standard culture conditions. Corroborating studies reported supportive effects of sunitinib on embryonic cortical neurons, cerebellar neurons, neuronal cell lines, and astrocytes when compared to standard culture conditions.40,50,51 We went one step further and tested the impact of sunitinib under cell stress and apoptosis-inducing conditions. Our experiments revealed that sunitinib alleviates glutamate-induced cell death in neurons and astrocytes. As glioma-induced neurodegeneration and edema formation are essential factors in the progression of this disease,52–54 agents active on these processes are essential for multimodal approaches. In particular, neuronal cell death and edema-preventing agents are rarely available in clinical use. For instance, brain edema, which is the major cause for brain tumor death due to herniation, has been treated for over 40 years with glucocorticoid derivatives such as dexamethasone. However, recent studies indicate that the mainly clinically used glucocorticoid analogue dexamethasone even fosters cell proliferation in some glioma cells.13 Thus, sunitinib actions may be monitored beyond simple survival rates by implementing other assessment criteria. Complication rates, edema progression, and responsiveness to other therapeutics in multimodal approaches are additional relevant parameters for quality of life and overall tumor management.3
Clinical studies on high grade glioma patients and recurrent glioblastoma reported no prolongation of progression-free survival within 6 months in a comparison of bevacizumab-resistant and naïve patients treated with sunitinib only.34,35 Recently, it has been shown that acquired resistance to sunitinib in human glioma cells involves various phosphoproteins and, in particular, activated phospholipase C-γ1.55 A crucial difference to the experimental research is the fact that recurrent malignant gliomas are preselected due to the pre-use of cytotoxic chemotherapeutics and thus can be regarded as a special entity of malignant gliomas.56,57 We tested whether the combination of sunitinib with standard chemotherapeutics has any value for antitumor therapy. For this we used temozolomide as a first-line chemotherapeutic agent in gliomas.3 We found that sunitinib treatment does not reduce the efficacy of temozolomide. Moreover, we found that a temozolomide–sunitinib combination does add antitumor efficacy to monotherapy, thus opening another option for multimodal approaches. A recent study showed that sequential application of temozolomide and sunitinib does not enhance antitumor efficacy compared to single agent therapy, whereas a combinatory application of both drugs was potent on tumor volume.58 Altogether, these findings indicate that sunitinib can be combined with cytotoxic drugs such as alkylating agents to reinforce antitumor efficacy.
Another aspect is the monitoring of clinical benefits such as peritumoral edema formation as well as efficacy and responsiveness of multimodal agents during sunitinib treatment. Critical for all RTK inhibitor approaches is the question whether they should be used as first-line monotherapy, adjuvant, or neo-adjuvant therapy. We and others have provided evidence in favor of a combinatory approach for sunitinib. In addition, it should be clarified how effective sunitinib and other RTK inhibitors are when delivered to the tumor and the tumor microenvironment passing the blood–brain barrier. For clinical applications there are some hints pointing at the limitations of sunitinib and vandetanib usage in patients. Although we found effective concentrations of 5–10 μM for the neuroprotective and gliomatoxic actions, current treatment regimens use 35–50 mg sunitinib orally, a dose that results in median plasma levels of approximately 0.1 μM.4 As there are many therapeutic routes for treating malignant gliomas, such as intraventricular, intraneoplastic, and wafer applications into the resection cavity, elevated levels of sunitinib in the tumor bed are reachable.59,60
In conclusion, our findings uncovered protective effects of sunitinib on neurons and astrocytes after glutamate-induced damage. Our results show that sunitinib is effective in alleviating tumor-induced neurodegeneration and tumor progression. Clinical evaluation of the affected parameters with optimized delivery routes and additional trials using in vivo models may be considered in the near future.
Acknowledgments
We thank all members of the Cell Biology and Neurooncology Laboratory for excellent technical support and critical discussions and inputs during the course of experimentations. We cordially thank Katrin Ebert and Teja Groemer (Department of Psychiatry, Erlangen, Germany) for help with the hippocampal neuron preparation. Christine Mühe and Johannes Kornhuber (Department of Psychiatry, Erlangen, Germany) are gratefully acknowledged for providing access and assistance with the qRT-PCR software procedures. We thank Yvonne Rübner and Rainer Fietkau (Department of Radiation Oncology, Erlangen, Germany) for providing us with the T98G and U251 cells. Matthias Zenkel and Ulrike Schlötzer-Schrehardt (Departement of Ophtalmology, Erlangen, Germany) are acknowledged for continuous reagents exchange and methods discussions.
Disclosure Statement
The authors declare no conflict of interest.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1.Sunitinib is gliomatoxic against primary glioblastoma cells.
Fig. S2.Molecular targets of sunitinib and other receptor tyrosine kinase inhibitors on endothelial and glioma cells.
Fig. S3.Toxicity profiles of various kinase inhibitors on malignant gliomas.
Fig. S4.Extended dose–response analysis in glioma cells.
Table S1.Details and comparison of various kinase inhibitors used in this study.
References
- Kohler BA, Ward E, McCarthy BJ, et al. Annual report to the nation on the status of cancer, 1975–2007, featuring tumors of the brain and other nervous system. J Natl Cancer Inst. 2011;103(9):714–36. doi: 10.1093/jnci/djr077. May 4; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62(4):220–41. doi: 10.3322/caac.21149. Jul–Aug; [DOI] [PubMed] [Google Scholar]
- Eyupoglu IY, Buchfelder M, Savaskan NE. Surgical resection of malignant gliomas-role in optimizing patient outcome. Nat Rev Neurol. 2013;9(3):141–51. doi: 10.1038/nrneurol.2012.279. Mar; [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Bukowski RM. Targeted therapy for metastatic renal cell carcinoma. J Clin Oncol. 2006;24(35):5601–8. doi: 10.1200/JCO.2006.08.5415. Dec 10; [DOI] [PubMed] [Google Scholar]
- Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–38. doi: 10.1016/S0140-6736(06)69446-4. Oct 14; [DOI] [PubMed] [Google Scholar]
- De Witt Hamer PC. Small molecule kinase inhibitors in glioblastoma: a systematic review of clinical studies. Neuro Oncol. 2010;12:304–16. doi: 10.1093/neuonc/nop068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razis E, Selviaridis P, Labropoulos S, et al. Phase II study of neoadjuvant imatinib in glioblastoma: evaluation of clinical and molecular effects of the treatment. Clin Cancer Res. 2009;15(19):6258–66. doi: 10.1158/1078-0432.CCR-08-1867. Oct 1; [DOI] [PubMed] [Google Scholar]
- Chakravarti A, Wang M, Robins HI, et al. RTOG 0211: a phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int J Radiat Oncol Biol Phys. 2013;85(5):1206–11. doi: 10.1016/j.ijrobp.2012.10.008. Apr 1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre S, Demetri G, Sargent W, Raymond E. Molecular basis for sunitinib efficacy and future clinical development. Nat Rev Drug Discov. 2007;6(9):734–45. doi: 10.1038/nrd2380. Sep; [DOI] [PubMed] [Google Scholar]
- Naoe T, Kiyoi H. Normal and oncogenic FLT3. Cell Mol Life Sci. 2004;61(23):2932–8. doi: 10.1007/s00018-004-4274-x. Dec; [DOI] [PubMed] [Google Scholar]
- Phay JE, Shah MH. Targeting RET receptor tyrosine kinase activation in cancer. Clin Cancer Res. 2010;16(24):5936–41. doi: 10.1158/1078-0432.CCR-09-0786. Dec 15; [DOI] [PubMed] [Google Scholar]
- Kogan M, Fischer-Smith T, Kaminsky R, Lehmicke G, Rappaport J. CSF-1R up-regulation is associated with response to pharmacotherapy targeting tyrosine kinase activity in AML cell lines. Anticancer Res. 2012;32(3):893–9. Mar; [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Sehm T, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan NE. Dexamethasone alleviates tumor-associated brain damage and angiogenesis. PLoS ONE. 2014;9(4):e93264. doi: 10.1371/journal.pone.0093264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehm T, Fan Z, Weiss R, et al. The impact of dietary isoflavonoids on malignant brain tumors. Cancer Med. 2014;3(4):865–77. doi: 10.1002/cam4.265. Aug; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007;25(7):884–96. doi: 10.1200/JCO.2006.06.3602. Mar 1; [DOI] [PubMed] [Google Scholar]
- Eyupoglu IY, Savaskan NE, Brauer AU, Nitsch R, Heimrich B. Identification of neuronal cell death in a model of degeneration in the hippocampus. Brain Res Brain Res Protoc. 2003;11:1–8. doi: 10.1016/s1385-299x(02)00186-1. [DOI] [PubMed] [Google Scholar]
- Hock SW, Fan Z, Buchfelder M, Eyupoglu IY, Savaskan NE. Brain tumor-induced angiogenesis: approaches and bioassays. In: Lichtor T, editor. Evolution of the Molecular Biology of Brain Tumors and the Therapeutic Implications. InTech; 2013. ISBN: 978-953-51-0989-1, doi: 105772/53182 Available from URL: http://wwwintechopencom/books/evolution-of-the-molecular-biology-of-brain-tumors-and-the-therapeutic-implications/brain-tumor-induced-angiogenesis-approaches-and-bioassays. [Google Scholar]
- Eyupoglu IY, Hahnen E, Buslei R, et al. Suberoylanilide hydroxamic acid (SAHA) has potent anti-glioma properties in vitro, ex vivo and in vivo. J Neurochem. 2005;93(4):992–9. doi: 10.1111/j.1471-4159.2005.03098.x. May; [DOI] [PubMed] [Google Scholar]
- Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86(3):353–64. doi: 10.1016/s0092-8674(00)80108-7. Aug 9; [DOI] [PubMed] [Google Scholar]
- Vakoc BJ, Lanning RM, Tyrrell JA, et al. Three-dimensional microscopy of the tumor microenvironment in vivo using optical frequency domain imaging. Nat Med. 2009;15(10):1219–23. doi: 10.1038/nm.1971. Oct; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savaskan NE, Seufert S, Hauke J, Trankle C, Eyupoglu IY, Hahnen E. Dissection of mitogenic and neurodegenerative actions of cystine and glutamate in malignant gliomas. Oncogene. 2011;30(1):43–53. doi: 10.1038/onc.2010.391. Jan 6; [DOI] [PubMed] [Google Scholar]
- Buckingham SC, Campbell SL, Haas BR, et al. Glutamate release by primary brain tumors induces epileptic activity. Nat Med. 2011;17(10):1269–74. doi: 10.1038/nm.2453. Oct; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie K, Yanagita RC, Nakagawa Y. Challenges to the development of bryostatin-type anticancer drugs based on the activation mechanism of protein kinase Cdelta. Med Res Rev. 2012;32(3):518–35. doi: 10.1002/med.20220. May; [DOI] [PubMed] [Google Scholar]
- Deininger MW, Druker BJ. Specific targeted therapy of chronic myelogenous leukemia with imatinib. Pharmacol Rev. 2003;55(3):401–23. doi: 10.1124/pr.55.3.4. Sep; [DOI] [PubMed] [Google Scholar]
- Rotblat B, Ehrlich M, Haklai R, Kloog Y. The Ras inhibitor farnesylthiosalicylic acid (Salirasib) disrupts the spatiotemporal localization of active Ras: a potential treatment for cancer. Methods Enzymol. 2008;439:467–89. doi: 10.1016/S0076-6879(07)00432-6. [DOI] [PubMed] [Google Scholar]
- Koul D, Shen R, Kim YW, et al. Cellular and in vivo activity of a novel PI3K inhibitor, PX-866, against human glioblastoma. Neuro Oncol. 2010;12(6):559–69. doi: 10.1093/neuonc/nop058. Jun; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta M, Kawabata T, Yamamoto M, et al. TSU68, an antiangiogenic receptor tyrosine kinase inhibitor, induces tumor vascular normalization in a human cancer xenograft nude mouse model. Surg Today. 2009;39:1046–53. doi: 10.1007/s00595-009-4020-y. [DOI] [PubMed] [Google Scholar]
- Laird AD, Vajkoczy P, Shawver LK, et al. SU6668 is a potent antiangiogenic and antitumor agent that induces regression of established tumors. Cancer Res. 2000;60(15):4152–60. Aug 1; [PubMed] [Google Scholar]
- Linares V, Alonso V, Domingo JL. Oxidative stress as a mechanism underlying sulfasalazine-induced toxicity. Expert Opin Drug Saf. 2011;10(2):253–63. doi: 10.1517/14740338.2011.529898. Mar; [DOI] [PubMed] [Google Scholar]
- Batich KA, Sampson JH. Standard of care and future pharmacological treatment options for malignant glioma: an urgent need for screening and identification of novel tumor-specific antigens. Expert Opin Pharmacother. 2014;15(14):2047–61. doi: 10.1517/14656566.2014.947266. Oct; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stommel JM, Kimmelman AC, Ying H, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318(5848):287–90. doi: 10.1126/science.1142946. Oct 12; [DOI] [PubMed] [Google Scholar]
- Sathornsumetee S, Reardon DA, Desjardins A, Quinn JA, Vredenburgh JJ, Rich JN. Molecularly targeted therapy for malignant glioma. Cancer. 2007;110(1):13–24. doi: 10.1002/cncr.22741. Jul 1; [DOI] [PubMed] [Google Scholar]
- Bielen A, Perryman L, Box GM, et al. Enhanced efficacy of IGF1R inhibition in pediatric glioblastoma by combinatorial targeting of PDGFRalpha/beta. Mol Cancer Ther. 2011;10(8):1407–18. doi: 10.1158/1535-7163.MCT-11-0205. Aug; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyns B, Sadones J, Chaskis C, et al. Phase II study of sunitinib malate in patients with recurrent high-grade glioma. J Neurooncol. 2011;103(3):491–501. doi: 10.1007/s11060-010-0402-7. Jul; [DOI] [PubMed] [Google Scholar]
- Kreisl TN, Smith P, Sul J, et al. Continuous daily sunitinib for recurrent glioblastoma. J Neurooncol. 2013;111(1):41–8. doi: 10.1007/s11060-012-0988-z. Jan; [DOI] [PubMed] [Google Scholar]
- Jiang J, Yan M, Mehta JL, Hu C. Angiogenesis is a link between atherosclerosis and tumorigenesis: role of LOX-1. Cardiovasc Drugs Ther. 2011;25:461–8. doi: 10.1007/s10557-011-6343-3. [DOI] [PubMed] [Google Scholar]
- Jimenez B, Volpert OV. Mechanistic insights on the inhibition of tumor angiogenesis. J Mol Med (Berl) 2001;78(12):663–72. doi: 10.1007/s001090000178. [DOI] [PubMed] [Google Scholar]
- Jubb AM, Browning L, Campo L, et al. Expression of vascular Notch ligands Delta-like 4 and Jagged-1 in glioblastoma. Histopathology. 2012;60(5):740–7. doi: 10.1111/j.1365-2559.2011.04138.x. Apr; [DOI] [PubMed] [Google Scholar]
- Joshi AD, Loilome W, Siu IM, Tyler B, Gallia GL, Riggins GJ. Evaluation of tyrosine kinase inhibitor combinations for glioblastoma therapy. PLoS ONE. 2012;7(10):e44372. doi: 10.1371/journal.pone.0044372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bouard S, Herlin P, Christensen JG, et al. Antiangiogenic and anti-invasive effects of sunitinib on experimental human glioblastoma. Neuro Oncol. 2007;9(4):412–23. doi: 10.1215/15228517-2007-024. Oct; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor GS, O'Rourke DM. Receptor tyrosine kinase signaling in gliomagenesis: pathobiology and therapeutic approaches. Cancer Biol Ther. 2003;2:330–42. doi: 10.4161/cbt.2.4.507. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8. doi: 10.1038/nature07385. Oct 23; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Brugieres L, Pacquement H, Le Deley MC, et al. Single-drug vinblastine as salvage treatment for refractory or relapsed anaplastic large-cell lymphoma: a report from the French Society of Pediatric Oncology. J Clin Oncol. 2009;27(30):5056–61. doi: 10.1200/JCO.2008.20.1764. Oct 20; [DOI] [PubMed] [Google Scholar]
- Martinho O, Silva-Oliveira R, Miranda-Goncalves V, et al. In vitro and in vivo analysis of RTK inhibitor efficacy and identification of its novel targets in glioblastomas. Transl Oncol. 2013;6:187–96. doi: 10.1593/tlo.12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KV, Chang JP, Parachoniak CA, et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell. 2012;22(1):21–35. doi: 10.1016/j.ccr.2012.05.037. Jul 10; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakes FM, Chen J, Tan J, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10(12):2298–308. doi: 10.1158/1535-7163.MCT-11-0264. Dec; [DOI] [PubMed] [Google Scholar]
- Hamerlik P, Lathia JD, Rasmussen R, et al. Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J Exp Med. 2012;209(3):507–20. doi: 10.1084/jem.20111424. Mar 12; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor TT, Sorensen AG, di Tomaso E, et al. AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11(1):83–95. doi: 10.1016/j.ccr.2006.11.021. Jan; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez A, Tripathy D, Yin X, Luo J, Martinez JM, Grammas P. Sunitinib enhances neuronal survival in vitro via NF-kappaB-mediated signaling and expression of cyclooxygenase-2 and inducible nitric oxide synthase. J Neuroinflammation. 2013;10:93. doi: 10.1186/1742-2094-10-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W, Zhang ZJ, Hu SQ, et al. Sunitinib produces neuroprotective effect via inhibiting nitric oxide overproduction. CNS Neurosci Ther. 2014;20:244–52. doi: 10.1111/cns.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savaskan NE, Heckel A, Hahnen E, et al. Small interfering RNA-mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat Med. 2008;14(6):629–32. doi: 10.1038/nm1772. Jun; [DOI] [PubMed] [Google Scholar]
- Lee SG, Kim K, Kegelman TP, et al. Oncogene AEG-1 promotes glioma-induced neurodegeneration by increasing glutamate excitotoxicity. Cancer Res. 2011;71(20):6514–23. doi: 10.1158/0008-5472.CAN-11-0782. Oct 15; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald DP, Subramanian P, Deshpande M, et al. Opposing effects of pigment epithelium-derived factor on breast cancer cell versus neuronal survival: implication for brain metastasis and metastasis-induced brain damage. Cancer Res. 2012;72(1):144–53. doi: 10.1158/0008-5472.CAN-11-1904. Jan 1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Lv H, Mazloom AR, Xu H, Ma'ayan A, Gallo JM. Activation of alternate prosurvival pathways accounts for acquired sunitinib resistance in U87MG glioma xenografts. J Pharmacol Exp Ther. 2012;343(2):509–19. doi: 10.1124/jpet.112.196097. Nov; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterhof NK, de Rooi JJ, Kros M, et al. Molecular subtypes of glioma identified by genome-wide methylation profiling. Genes Chromosom Cancer. 2013;52:665–74. doi: 10.1002/gcc.22062. [DOI] [PubMed] [Google Scholar]
- Sintupisut N, Liu PL, Yeang CH. An integrative characterization of recurrent molecular aberrations in glioblastoma genomes. Nucleic Acids Res. 2013;41(19):8803–21. doi: 10.1093/nar/gkt656. Jul 31; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabanka M, Bruenner J, Parmaksiz G, et al. Combined temozolomide and sunitinib treatment leads to better tumour control but increased vascular resistance in O6-methylguanine methyltransferase-methylated gliomas. Eur J Cancer. 2013;49(9):2243–52. doi: 10.1016/j.ejca.2013.02.019. Jun; [DOI] [PubMed] [Google Scholar]
- Panciani PP, Fontanella M, Tamagno I, et al. Stem cells based therapy in high grade glioma: why the intraventricular route should be preferred? J Neurosurg Sci. 2012;56:221–9. [PubMed] [Google Scholar]
- Aoki T, Nishikawa R, Sugiyama K, et al. A multicenter phase I/II study of the BCNU implant (Gliadel((R)) Wafer) for Japanese patients with malignant gliomas. Neurol Med Chir. 2014;54(4):290–301. doi: 10.2176/nmc.oa2013-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1.Sunitinib is gliomatoxic against primary glioblastoma cells.
Fig. S2.Molecular targets of sunitinib and other receptor tyrosine kinase inhibitors on endothelial and glioma cells.
Fig. S3.Toxicity profiles of various kinase inhibitors on malignant gliomas.
Fig. S4.Extended dose–response analysis in glioma cells.
Table S1.Details and comparison of various kinase inhibitors used in this study.