

Abstract
Autophagy is one of the major causes of drug resistance. For example, the angiogenesis inhibitor bevacizumab shows only transient and short-term therapeutic effects, whereas long-term therapeutic benefits are rarely observed, probably due to hypoxia-induced autophagy. Nitric oxide (NO) is an important molecule with multiple functions, and it has recently been reported to function as a regulator of autophagy. Therefore, a reasonable therapeutic strategy for overcoming drug resistance by NO would involve it being directly delivered to the tumor. Here, we investigated the inhibitory effect of NO on autophagy by using a macromolecular NO donor S-nitrosated human serum albumin (SNO-HSA) with a high degree of NO loading and tumor targeting potential. In colon 26 (C26) cells, SNO-HSA significantly suppressed hypoxia-induced autophagy by inhibiting the phosphorylation of JNK1 and the expression of its downstream molecule Beclin1. The effect of SNO-HSA was also confirmed in vivo by combining it with Bev. In C26-bearing mice, significant suppression of tumor growth as well as lung metastasis was achieved in the combination group compared to the SNO-HSA or bevacizumab alone group. Similar to the in vitro experiments, the immunostaining of tumor tissues clearly showed that SNO-HSA inhibited the autophagy of tumor cells induced by bevacizumab treatment. In addition to other known antitumor effects of SNO-HSA, that is, the induction of apoptosis and the inhibition of multidrug efflux pumps, these data may open alternate strategies for cancer chemotherapy by taking advantage of the ability of SNO-HSA to suppress autophagy-mediated drug resistance and enhance the efficacy of chemotherapy.
Keywords: Autophagy, bevacizumab, drug resistance, nitric oxide, SNO-HSA
Cancer remains one of the major causes of human death in most advanced countries. In early-stage cases, the best treatment of cancers is surgical removal, especially for those confined to a limited area and without metastasis. Chemotherapy is an alternate major method for controlling cancer. However, conventional chemotherapy, which usually uses low molecular weight drugs, is far from successful. One major cause of this problem is a lack of tumor selectivity, resulting in severe adverse effects that limit the use of such approaches. As a result, many efforts have been made to increase tumor selectivity, that is, molecular target drugs1 and, more importantly, macromolecular drug-based tumor tissue targeting by taking advantage of the enhanced permeability and retention (EPR) effect.2–4 Accordingly, increasing numbers of tumor-targeted anticancer drugs are currently available and some are now in clinical trials, and many others are in preclinical stages.1,3–6
Another major problem hampering the efficacy of many anticancer drugs is the acquisition of drug resistance. Many anticancer drugs show decreased therapeutic effects after several cycles of drug treatment. Regarding the mechanisms of drug resistance, multidrug efflux pumps such as P-glycoprotein and ABC transporters are known to be involved.7–9 It has also recently been reported that autophagy is significantly associated with the process of drug resistance.10–15 Autophagy is the basic catabolic mechanism of cells to degrade unnecessary or dysfunctional cellular components through the actions of lysosomes,16 which can ensure cellular survival during starvation by maintaining cellular energy levels.16 Tumor cells may thus use this mechanism to maintain their growth under severe environments, for example, hypoxia and glucose starvation, thus affecting the therapeutic effects of anticancer drugs or inducing drug resistance. For example, bevacizumab (Bev), an inhibitor of angiogenesis, which is clinically used for the treatment of metastatic colorectal cancer, non-small-cell lung cancer, glioblastoma, and metastatic kidney cancer, shows relatively good short-term therapeutic effects, that is, disrupted and decreased tumor vasculature and suppression of tumor growth. However, the decreased tumor vasculature/blood supply generates hypoxic conditions in the tumor which, in turn, induces autophagy, resulting in the regrowth of the tumor. As a result, no long-term therapeutic benefits can be achieved.10–15 Clinical trials using autophagy inhibitors with conventional anticancer drugs further support the importance of controlling autophagy.17–20
Nitric oxide (NO) is a widely known signal molecule with versatile functions in many biological and pathological processes.21,22 The effect of NO is dose-dependent; low or moderate concentrations of NO are important for cell growth and signal transductions, but high or excessive amounts of NO may induce apoptosis of cells.21,22 More recently, NO was also reported to downregulate autophagy.23 The delivery of NO to a tumor may thus become a new therapeutic strategy for overcoming drug resistance through the inhibition of autophagy.
In our laboratory, we previously developed a macromolecular NO donor in which NO was chemically bound (6.6 mol/mol) to the SH group of human serum albumin (HSA), namely S-nitrosated HSA (SNO-HSA).24,25 Because of its macromolecular nature, SNO-HSA showed more selective accumulation in tumor and thus the release of NO in tumor tissue based on the EPR effect,26,27 the excessively levels of NO generated in tumor tissues thus triggered the apoptosis of tumor cells, resulting in a marked delay in tumor growth in vivo, suggesting the potential of SNO-HSA as a new anticancer agent.24–27 Along this line, in the present study, to further investigate the potential of SNO-HSA as an antitumor molecule, we focused on the effect of NO on autophagy, which was examined in a mouse colon cancer cell line, colon 26 (C26), and its therapeutic potential was further examined when used in combination with Bev.
Materials and Methods
Materials
Human serum albumin of mean molecular weight of 66 500 was generously provided by the Chemo-Sero-Therapeutic Research Institute (Kumamoto, Japan). Isoamyl nitrite and diethylenetriamine pentaacetate were purchased from Wako Pure Chemical Industries (Osaka, Japan). Paraformaldehyde and anti-light chain 3 (LC3) rabbit polyclonal antibody were purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies (goat anti-human) against JNK1, phosphorylated JNK1 (p-JNK1), Beclin1, CD31, hypoxia inducible factor-1α (HIF-1α), and vascular endothelial growth factor (VEGF) were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). Griess reagents (sulfanilamide, N-(1-naphthyl) ethylenediamine dihydrochloride) and 5,5′-dithiobis-2-nitrobenzoic acid were purchased from Nacalai Tesque (Kyoto, Japan). Bevacizumab was kindly provided by Chugai Pharmaceutical Co. Ltd. (Tokyo, Japan) All other reagents were of reagent grade and were used without further purification.
Synthesis of SNO-HSA
Preparation of SNO-HSA was carried out according to the method reported previously.24,25 In a typical run, 150 μM HSA was dissolved in 0.1 M potassium phosphate buffer (pH 7.8) with 0.5 mM diethylenetriamine pentaacetate, then 3 mM Traut's reagent (Pierce Biotechnology, Rockford, IL, USA) was added to react for 1 h at 25°C, after which isoamyl nitrite (15 mM) was added and allowed to further react for 3 h at 37°C. The resulting SNO-HSA was then lyophilized and stored at −80°C.
Quantification of S-nitrosated proteins
S-nitrosated proteins were quantified by the Griess method by HPLC (YMC-pack Diol-120, 300 × 8 mm; Eicom, Kyoto, Japan) as reported previously.28
Cell culture
Mouse colon cancer cells C26 were donated by the Institute of Development, Aging and Cancer of Tohoku University (Sendai, Miyagi, Japan). The cells were cultured in PRMI-1640 medium (Sigma-Aldrich) with 10% FBS at 37°C in a 5% CO2/95% air atmosphere.
Immunofluorescence study of LC3 in cells treated with SNO-HSA
To investigate the effect of SNO-HSA on autophagy, an immunofluorescence study was carried out using an autophagy marker, microtubule-associated protein 1 light chain 3 LC3.29 In a typical experiment, C26 cells were seeded in an 8-well imaging chamber (5 × 104 cells/well) containing different concentrations of SNO-HSA, and the cells were incubated for 24 h. The cells were collected and fixed by treatment with 4% paraformaldehyde, following by a blocking process using 0.1% Triton X-100 in PBS (−) and then 1% BSA and 0.1% Tween 20 in PBS (−) (30 min each at room temperature). The cells were then treated with an anti-LC3 antibody for 2 h at room temperature according to the manufacturer's instructions. After treatment with a secondary antibody (anti-rabbit IgG–Alexa555) for 1 h at room temperature, the cells were washed with PBS (−) twice, resuspended in PBS (−), and seeded in an imaging chamber. The fluorescence was quantified using a Guava easyCyte Flow Cytometry system (Millipore, Bedford, MA, USA).
Details of other experimental procedures are given in Document S1.
Results
Effect of SNO-HSA on autophagy
The effect of SNO-HSA on autophagy was first investigated in C26 cells by detecting the autophagy marker LC3, which was quantified as the fluorescence intensity/cell by a FACS method. As shown in Figure1, SNO-HSA significantly inhibited LC3 expression, in a dose-dependent manner at concentrations up to 0.32 μM, whereas higher concentrations did not induce further inhibition. The concentrations of SNO-HSA in the following studies were thus in the range of 0.5–500 nM.
Fig 1.
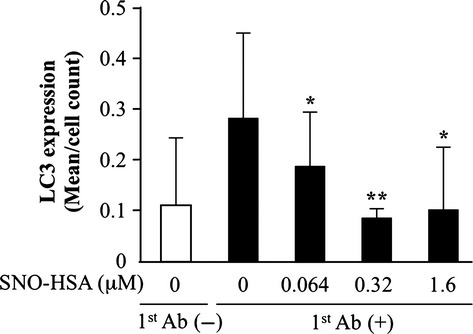
Effect of nitric oxide donor S-nitrosated human serum albumin (SNO-HSA) on autophagy. Colon cancer C26 cells were incubated with different concentrations (μM) of SNO-HSA for 24 h. The cells were then treated with (1st Ab (+)) or without (1st Ab (−)) an antimicrotubule-associated protein 1 light chain 3 (LC3) antibody. Fluorescence was quantified using the Guava easyCyte Flow Cytometry system. Results are given as means ± SD. *P < 0.05, **P < 0.01 versus 1st Ab alone (n = 3).
Hypoxia-induced autophagy and inhibitory activity of SNO-HSA
Hypoxia is a well-known condition that induces autophagy. We investigated autophagy under hypoxic conditions in C26 cells and examined the effect of SNO-HSA on this process. To evaluate autophagic activity more accurately, a Western blot analysis of LC3 was carried out. LC3 is a cytosolic protein that is distributed ubiquitously in mammalian tissues and cultured cells. During autophagy, the cytosolic form of LC3 (LC3-I) is conjugated to phosphatidylethanolamine to form an LC3–phosphatidylethanolamine conjugate (LC3-II), which is recruited to autophagosomal membranes. Thus, the extent of LC3 conversion (LC3-I to LC3-II) by immunoblot analysis is widely used to monitor autophagy.30
The results are shown in Figure2. Although hypoxia significantly increased autophagy activity in C26 cells (increased LC3-II/LC3-I), the activity was dramatically dose-dependently suppressed by SNO-HSA. More importantly, in contrast to previous reports in which another NO donor was used,23 relatively low concentrations of SNO-HSA (0.5–500 nM vs 100–500 μM) resulted in a significant inhibition of autophagy, probably due to the stability and sustained release of NO from SNO-HSA.24,25
Fig 2.
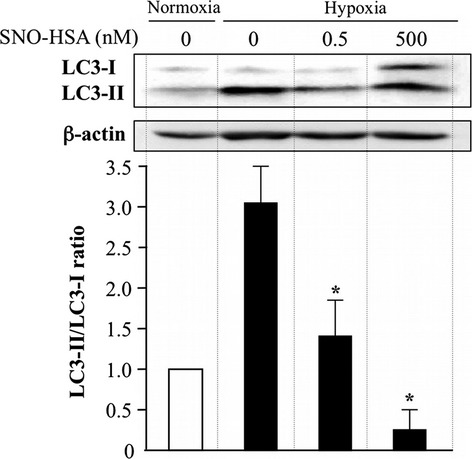
Western blot analysis of the cytosolic form of antimicrotubule-associated protein 1 light chain 3 (LC3-I) and the LC3–phosphatidylethanolamine conjugate (LC3-II) and the inhibitory activity of nitric oxide donor S-nitrosated human serum albumin (SNO-HSA). Colon cancer C26 cells were incubated under hypoxic conditions with 0.5 and 500 nM SNO-HSA for 24 h. The cell lysates were used to carry out Western blotting for LC3 and β-actin. The density of the bands was quantitatively analyzed using NIH Image J Software and they were standardized by β-actin. Results are given as the means ± SD. *P < 0.05 versus hypoxia group (n = 4).
Mechanisms involved in the inhibitory effect of SNO-HSA on autophagy
Regarding the molecular mechanisms responsible for the NO-induced inhibition of autophagy, it is known that NO inhibits autophagy mostly through the JNK1 and mTOR pathways, and Beclin1, a downstream molecule of JNK1 is also reported as an important signal molecule in hypoxia-induced autophagy.23 We then investigated the involvement of JNK1 as well as Beclin1 in the autophagy inhibitory effect of SNO-HSA using C26 cells. Similar to the findings in Figure2(b), JNK1—especially p-JNK1—was upregulated during hypoxia, and this upregulation was significantly inhibited by SNO-HSA treatment (Fig.3a). Clearer results were observed for Beclin1; hypoxia induced an almost twofold increase in Beclin1 expression, whereas it was remarkably reversed by SNO-HSA in a dose-dependent manner (Fig.3b). All of the above data are consistent with the autophagy inhibition by SNO-HSA shown in Figure2, suggesting that SNO-HSA suppresses autophagy, at least in part, through the JNK1–Beclin1 pathway, by the generation of NO. This was supported by the suppression of hypoxia-induced autophagy using sc-200635 and curcumin, JNK1 inhibitors (Fig. S1).
Fig 3.
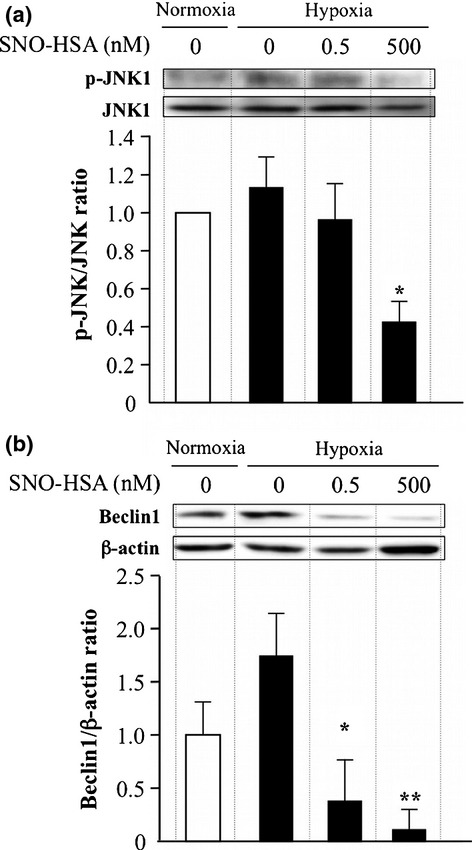
Mechanisms involved in the inhibitory effect of nitric oxide donor S-nitrosated human serum albumin (SNO-HSA) on autophagy in vitro. Colon cancer C26 cells were incubated under conditions of hypoxia with 0.5 and 500 nM SNO-HSA for 24 h. The cell lysates were used to carry out Western blotting for JNK1 and phosphorylated JNK1 (p-JNK) (a) and Beclin1 (b). The protein band that reacted immunologically with the antibody was visualized using an enhanced chemiluminescence system. Phosphorylated JNK1 was standardized by JNK1. Results are given as means ± SD. *P < 0.05, **P < 0.01 versus hypoxia group (n = 3–4).
Enhanced in vivo antitumor effect of Bev by SNO-HSA
To further investigate the potential of SNO-HSA on inhibiting autophagy and thus enhancing chemotherapeutic effects, Bev, an angiogenesis inhibitor that is known to show drug resistance by inducing hypoxia and thus autophagy,10–15 was used in combination with SNO-HSA, and in vivo therapeutic studies were carried out using a C26 mouse tumor model. Because we previously reported that SNO-HSA itself induced apoptosis in tumors at high concentrations,24,25 a low dose of SNO-HSA (66.5 mg/kg) at which no induction of apoptosis was observed (Fig. S2) was applied.
As shown in Figure4(a), treatment with Bev or SNO-HSA alone showed weak suppression of tumor growth, however, a significant delay in tumor growth was achieved when they were combined. More importantly, a remarkable decrease in the numbers of metastatic nodules in the lung was found as the result of this combination therapy, whereas this decrease was not found when Bev was given alone (Fig.4b).
Fig 4.
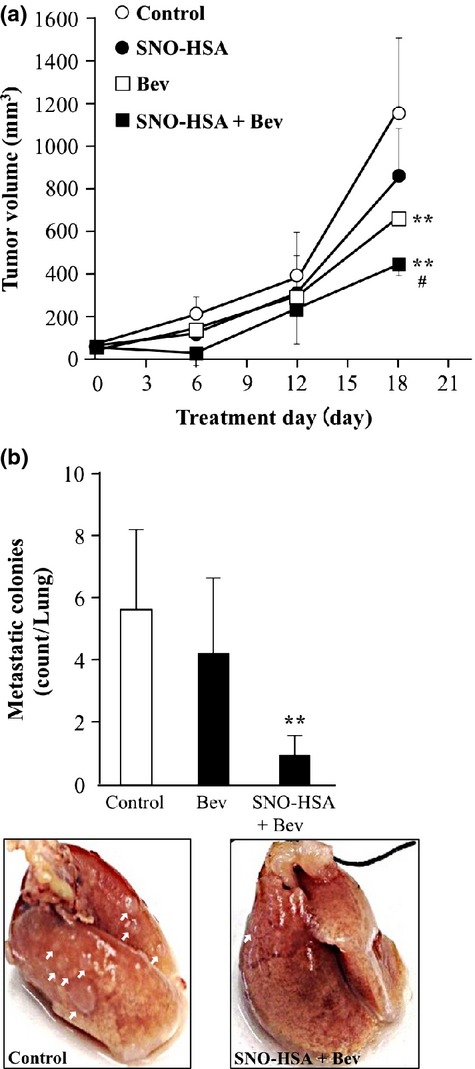
Enhanced in vivo antitumor effect of bevacizumab (Bev) by nitric oxide donor S-nitrosated human serum albumin (SNO-HSA). (a) In C26 tumor-bearing mice (n = 4–9), SNO-HSA was injected i.v. through the tail vein, followed by Bev given i.p. Treatment was carried every 3 days for a total of six times. Results are for the following 18 days and are given as means ± SD. **P < 0.01 versus control. #P < 0.05 versus Bev alone. (b) At day 18, mice were killed, lungs were collected, and metastatic nodules on the surface of the lung were counted. Results are given as means ± SD. **P < 0.01 versus control.
Effect of SNO-HSA/Bev combination therapy on angiogenesis, hypoxia, and autophagy in tumor tissues in vivo
The antitumor mechanisms involved in SNO-HSA/Bev combination therapy were then examined focusing on angiogenesis, hypoxia, and autophagy using the vascular markers CD31, HIF-1α, and LC3 immunofluorescence staining, respectively. The tumor tissue sections were divided artificially into two parts, in which the inner third was considered as the core with more necrosis, less angiogenesis, and more hypoxia, and the remaining two-thirds as peripheral tissue, which usually has a better blood supply and shows rapid tumor growth.31
As expected, in the peripheral regions of the tumor, more active angiogenesis/vasculature was observed compared to that in the core, as evidenced by CD31 staining (Fig.5a); the angiogenesis inhibitor Bev largely decreased the angiogenesis of tumors, especially in the peripheral of tumor (Fig.5a). A more significant suppression of tumor angiogenesis was observed when Bev was used in combination with SNO-HSA (Fig.5a). These results were also supported by the amount of VEGF in tumors, where the combination group showed a more significant decrease compared to the Bev alone group (Fig. S3).
Fig 5.

Effect of nitric oxide donor S-nitrosated human serum albumin (SNO-HSA)/bevacizumab (Bev) combination therapy on angiogenesis, hypoxia, and autophagy in tumor tissues. Expression of CD31 (a), hypoxia-inducible factor-1α (HIF-1α) (b), antimicrotubule-associated protein 1 light chain 3 (LC3) (c), and TUNEL (d) in tumor tissues after SNO-HSA and/or Bev treatment was detected immunohistochemically in paraffin-embedded sections using anti-CD31, anti-HIF-1α, and anti-LC3 antibodies, with the corresponding fluorescence-labeled secondary antibodies. TUNEL staining with an in situ apoptosis detection kit, according to the manufacturer's instructions, was used to investigate apoptosis. Fluorescence was then detected by fluorescence microscopy.
In contrast, the core of tumors showed apparent hypoxia compared to peripheral areas, and treatment with Bev greatly induced hypoxia in the peripheral region; the combination therapy improved the hypoxic conditions in both cases, and a more significant effect was found in the peripheral region (Fig.5b).
In parallel with these findings and consistent with the in vitro studies described above (Figs.2), apparent autophagy as indicated by LC3 staining was detected in the untreated control and Bev-treated group, which was remarkably suppressed in the presence of SNO-HSA, in both the core and peripheral regions of the tumor (Fig.5c). Bev treatment induced apoptosis in tumor areas and endothelial cells, but this low dose of SNO-HSA had no effect on Bev-induced apoptosis in the tumor and endothelial cells in this study (Fig.5d). However, a high dose of SNO-HSA induced apoptosis in the tumor area and in endothelial cells (Fig. S2). These findings further support the involvement of autophagy inhibition in the enhancement of the chemotherapeutic effect by SNO-HSA.
Discussion
Nitric oxide is a critical molecule in carcinogenesis and tumor progression, being involved in many cancer-associated events including angiogenesis, apoptosis, the cell cycle, tumor invasion, and metastasis.32 However, it has only recently been considered to be a potential antitumor agent when overproduced in a tumor.33 Therapeutic strategies using NO for cancer treatment are currently being investigated and developed, by manipulating its in vivo production and the exogenous delivery of this molecule.
In this context, we developed a NO donor using albumin as the NO carrier, SNO-HSA, which showed a high NO loading and sustained NO release.24,25 Using this NO delivery system, we found a remarkable induction of apoptosis in tumor cells, not only in in vitro studies, but in in vivo solid tumor models.24,25 In addition, because NO functions as a vascular mediator, it enhances the EPR effect and thus tumor drug delivery, especially for macromolecular drugs.4,34,35 By selectively delivering NO into tumors, SNO-HSA significantly augmented the EPR effect, and thus greatly improved the chemotherapeutic effect of macromolecular anticancer drugs.27 This finding indicates that SNO-HSA may not only become an anticancer drug by itself, but it may also greatly enhance the tumor delivery and therapeutic effect of macromolecular anticancer drugs by minimizing the drug resistance and augmenting the EPR effect.
Thus, one major strategy of SNO-HSA in improving the chemotherapeutic effects of Bev is the benefit of the EPR effect, namely that NO enhances the tumor delivery of Bev, a macromolecule that results in a significantly increased antitumor effect (Fig.4). However, in this study, we also found another possible mechanism of NO involving its chemotherapeutic effect: inhibition of autophagy. Autophagy in C26 cells was inhibited by SNO-HSA, not only in normoxic conditions (Fig.1), but also in hypoxic conditions (Fig.2). These data were also translated in vivo, in which SNO-HSA significantly improved the hypoxic conditions caused by Bev treatment, and suppressed autophagy in C26 solid tumor cells (Fig.5), resulting in a significant antitumor effect in vivo by combining Bev and SNO-HSA (Fig.4a). One issue that should be noted in this study is that relatively low concentrations of SNO-HSA (i.e., 0.5–500 nM in vitro and 66.5 mg/kg (<10 μM) in vivo) did not induce apoptosis in tumor cells (Fig. S2) but sufficiently suppressed autophagy. These concentrations are ∼1000 times lower than that reported in previous published works using a commercial low molecular weight NO donor,23 suggesting the applicability of SNO-HSA for therapeutic gain as a NO donor. Another concern is that the reversal of hypoxia to normoxia, accompanying autophagy by SNO-HSA, is probably not due to the proangiogenic effect of NO. Indeed, the angiogenesis in tumors that were destroyed by Bev were not improved, but, rather, further deteriorated (Fig.5). Considering the dual functions of NO on angiogenesis, that is, a proangiogenic state at low concentrations and an antiangiogenic at high concentrations,36 these data suggest that, although SNO-HSA was applied at low dose, the NO concentrations in tumor may gradually increase, eventually reaching relatively high levels because of the tumor targeted accumulation and sustained release of NO.
It should also be noted that autophagy is a dichotomous phenomenon. It is not only involved in cell survival but also in cell death, depending upon the amount of degradation product by autophagy and cell type. Autophagy can promote cell survival by removing old or damaged cellular organelles, which are frequently used by cancer cells to supply deprived nutrients induced by therapeutic agents. However, above a certain level, NO can also induce cell death, namely autophagy-mediated cell death. Tripathi et al. showed that NO, when applied as a gas, decreased the viability of MCF7 breast cancer cells, through an autophagy-mediated pathway.37 Because relatively high concentrations of NO were used in that study, at 2.5 mM/min, in the presence of normoxic oxygen compared to that used in our present study (500 nM). The effect of NO on autophagy may be a concentration-dependent phenomenon. In addition, certain types of tumor cells may be sensitive to NO-induced autophagy-mediated cell death, and some may prefer the autophagy inhibitory effect of NO, which needs to be further investigated. However, these findings also suggest new insights into how to modulate the effect of NO on autophagy appropriately under different conditions, with the objective of developing more effective cancer therapies.
Regarding the mechanisms responsible for the impairment of autophagy by NO, it appears to involve inhibiting the activity of the S-nitrosation substrates JNK1 and IKKβ.23 The former involves a reduction in Bcl-2 phosphorylation and an interaction with Beclin1; the latter involves a reduction in AMPK phosphorylation, leading to mTORC1 activation.23 This notion was partly confirmed in the present study. Namely, SNO-HSA significantly decreased the extent of JNK1 phosphorylation and the expression of its downstream molecule Beclin1 during hypoxia-induced autophagy, which occurred in parallel with the events of autophagy (Fig.3).
An interesting and surprising finding in this study is that the combination therapy of SNO-HSA/Bev significantly suppressed lung metastasis of C26 solid tumor cells in a remarkable manner, to a far greater extent than autophagy parameters (Fig.4b). However, neither SNO-HSA nor Bev alone significantly inhibited tumor metastasis (Fig.4b). In colon cancer patients, it has been shown that patients with a lower expression of both HIF-1α and VEGF experienced significantly lower rates of metastasis to lymph nodes as well as other organs.38,39 In this context, we previously reported that SNO-HSA significantly lowered the expression of HIF-1α.26 Similar results were also observed in the present study, and, more importantly, the combination of SNO-HSA/Bev showed a more prominent effect (Fig.5b). Moreover, the Bev-triggered decrease of VEGF was further enhanced when used in combination with SNO-HSA (Fig. S3). These findings strongly suggest that the use of a combination of SNO-HSA/Bev has considerable potential for a therapeutic strategy for suppressing tumor metastasis, but further investigations using other types of metastatic models will be needed to confirm this conclusion. In addition, as shown in Figures4 and 5, SNO-HSA can inhibit not only hypoxia-induced autophagy but also drug-inducible autophagy (including inhibitors of angiogenesis), indicating that other combination therapies are possible, in which other inhibitors of angiogenesis and targeting endothelium drugs could be used.
Taken together, in the present study we showed the effect of NO on autophagy inhibition by using a tumor-targeted NO donor, SNO-HSA. This autophagy inhibitory activity of NO may be related to the enhanced antitumor effect of the angiogenesis inhibitor Bev that is known to develop drug resistance by inducing hypoxia and thus autophagy. Moreover, the combination therapy of SNO-HSA/Bev resulted in a significant decrease in lung metastasis. These findings, along with our previous findings showing the effect of SNO-HSA on inducing tumor cell apoptosis and overcoming drug resistance by inhibiting P-glycoprotein,24–26 strongly suggest that SNO-HSA has potential for use in cancer chemotherapy with multiple anticancer mechanisms, and that this system warrants further investigation.
Disclosure Statement
The authors have no conflict of interest.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1. Effect of treatment with JNK inhibitors (sc-200635 and curcumin) on expression of phosphorylated JNK1 (p-JNK1) and autophagy induced by hypoxia.
Fig. S2. Antiapoptotic activity of nitric oxide donor S-nitrosated human serum albumin (SNO-HSA) in vivo.
Fig. S3. Effect of treatment with bevacizumab (Bev) or/and nitric oxide donor S-nitrosated human serum albumin (SNO-HSA) on tumor vascular endothelial growth factor (VEGF) concentration.
Doc. S1. Supplementary experimental procedures.
References
- Sawyers C. Targeted cancer therapy. Nature. 2004;432:294–7. doi: 10.1038/nature03095. [DOI] [PubMed] [Google Scholar]
- Matsumura Y, Maeda H. A new concept for macromolecular thera-peutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–92. [PubMed] [Google Scholar]
- Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65:71–9. doi: 10.1016/j.addr.2012.10.002. [DOI] [PubMed] [Google Scholar]
- Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63:136–51. doi: 10.1016/j.addr.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discov. 2003;2:347–60. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- Matsumura Y, Kataoka K. Preclinical and clinical studies of anticancer agent-incorporating polymer micelles. Cancer Sci. 2009;100:572–9. doi: 10.1111/j.1349-7006.2009.01103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holohan C, Van Schaeybroeck S, Longley DB, et al. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- Ling V. Multidrug resistance: molecular mechanisms and clinical relevance. Cancer Chemother Pharmacol. 1997;40:3–8. doi: 10.1007/s002800051053. [DOI] [PubMed] [Google Scholar]
- Jones PM, George AM. The ABC transporter structure and mechanism: perspectives on recent research. Cell Mol Life Sci. 2004;61:682–99. doi: 10.1007/s00018-003-3336-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vredenburgh JJ, Desjardins A, Herndon JE, II, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13:1253–9. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- Bergers G, Hanahan D. Modes of resistance to antiangiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK. Antiangiogenic therapy for cancer: current and emerging concepts. Oncology. 2005;19:7–16. [PubMed] [Google Scholar]
- Saltz LB, Lenz HJ, Kindler HL, et al. Randomized phase II trial of cetuximab, bevacizumab, and irinotecan compared with cetuximab and bevacizumab alone in irinotecanrefractory colorectal cancer: the BOND-2 study. J Clin Oncol. 2007;25:4557–61. doi: 10.1200/JCO.2007.12.0949. [DOI] [PubMed] [Google Scholar]
- Shojaei F, Ferrara N. Antiangiogenic therapy for cancer: an update. Cancer J. 2007;13:345–8. doi: 10.1097/PPO.0b013e31815a7b69. [DOI] [PubMed] [Google Scholar]
- Kindler HL, Niedzwiecki D, Hollis D, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303) J Clin Oncol. 2010;28:3617–22. doi: 10.1200/JCO.2010.28.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvakumaran M, Amaravadi RK, Vasilevskaya IA, et al. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin Cancer Res. 2013;19:2995–3007. doi: 10.1158/1078-0432.CCR-12-1542. [DOI] [PubMed] [Google Scholar]
- Augustine MK, Choi MD, Stefan W, et al. Autophagy in human health and disease. N Engl J Med. 2013;368:651–62. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- Hu YL, DeLay M, Jahangiri A, et al. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012;72:1773–83. doi: 10.1158/0008-5472.CAN-11-3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–7. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culotta E, Koshland DE., Jr NO news is good news. Science. 1992;258:1862–5. doi: 10.1126/science.1361684. [DOI] [PubMed] [Google Scholar]
- Maeda H, Akaike T. Nitric oxide and oxygen radicals in infection, inflammation, and cancer. Biochemistry (Mosc) 1998;63:854–65. [PubMed] [Google Scholar]
- Sarkar S, Korolchuk VI, Renna M, et al. Complex inhibitory effects of nitric oxide on autophagy. Mol Cell. 2011;43:19–32. doi: 10.1016/j.molcel.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama N, Nakajou K, Komori H, et al. Design and evaluation of S-nitrosylated human serum albumin as a novel anticancer drug. J Pharmacol Exp Ther. 2008;325:69–76. doi: 10.1124/jpet.107.132100. [DOI] [PubMed] [Google Scholar]
- Katayama N, Nakajou K, Ishima Y, et al. Nitrosylated human serum albumin (SNO-HSA) induces apoptosis in tumor cells. Nitric Oxide. 2010;22:259–65. doi: 10.1016/j.niox.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Ishima Y, Hara M, Kragh-Hansen U, et al. Elucidation of the therapeutic enhancer mechanism of poly-S-nitrosated human serum albumin against multidrug-resistant tumor in animal models. J Control Release. 2012;164:1–7. doi: 10.1016/j.jconrel.2012.10.003. [DOI] [PubMed] [Google Scholar]
- Ishima Y, Chen D, Fang J, et al. S-Nitrosated human serum albumin dimer is not only a novel anti-tumor drug but also a potentiator for anti-tumor drugs with augmented EPR effects. Bioconjug Chem. 2012;23:264–71. doi: 10.1021/bc2005363. [DOI] [PubMed] [Google Scholar]
- Akaike T, Inoue K, Okamoto T, et al. Nanomolar quantification and identification of various nitrosothiols by high performance liquid chromatography coupled with flow reactors of metals and Griess reagent. J Biochem. 1997;122:459–66. doi: 10.1093/oxfordjournals.jbchem.a021774. [DOI] [PubMed] [Google Scholar]
- Ito A, Katoh F, Kataoka TR, et al. A role for heterologous gap junctions between melanoma and endothelial cells in metastasis. J Clin Invest. 2000;105:1189–97. doi: 10.1172/JCI8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- Yoshizawa Y, Ogawara KI, Fushimi A, et al. Deeper penetration into tumor tissues and enhanced in vivo antitumor activity of liposomal paclitaxel by pretreatment with angiogenesis inhibitor SU5416. Mol Pharm. 2012;9:3486–94. doi: 10.1021/mp300318q. [DOI] [PubMed] [Google Scholar]
- Choudhari SK, Chaudhary M, Bagde S, et al. Nitric oxide and cancer: a review. World J Surg Oncol. 2013;11:118. doi: 10.1186/1477-7819-11-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike T, Maeda H. Nitric oxide and virus infection. Immunology. 2000;101:300–8. doi: 10.1046/j.1365-2567.2000.00142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Akaike T, Maeda H. Modulation of enhanced vascular permeability in tumors by a bradykinin antagonist, a cyclooxygenase inhibitor, and a nitric oxide scavenger. Cancer Res. 1998;58:159–65. [PubMed] [Google Scholar]
- Seki T, Fang J, Maeda H. Enhanced delivery of macromolecular antitumor drugs to tumors by nitroglycerin application. Cancer Sci. 2009;100:2426–30. doi: 10.1111/j.1349-7006.2009.01323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridnour LA, Isenberg JS, Espey MG, et al. Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1. Proc Natl Acad Sci U S A. 2005;102:13147–52. doi: 10.1073/pnas.0502979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi DN, Chowdhury R, Trudel LJ, et al. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc Natl Acad Sci U S A. 2013;110:E2950–7. doi: 10.1073/pnas.1307736110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Jin M, Xu H, et al. Clinicopathologic significance of HIF-1α, CXCR4, and VEGF expression in colon cancer. Clin Dev Immunol. 2010;2010:537531. doi: 10.1155/2010/537531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda T, Okuyama H, Nishizawa Y, et al. Hypoxia inducible factor-1α is necessary for invasive phenotype in Vegf-deleted islet cell tumors. Sci Rep. 2012;2:494. doi: 10.1038/srep00494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Effect of treatment with JNK inhibitors (sc-200635 and curcumin) on expression of phosphorylated JNK1 (p-JNK1) and autophagy induced by hypoxia.
Fig. S2. Antiapoptotic activity of nitric oxide donor S-nitrosated human serum albumin (SNO-HSA) in vivo.
Fig. S3. Effect of treatment with bevacizumab (Bev) or/and nitric oxide donor S-nitrosated human serum albumin (SNO-HSA) on tumor vascular endothelial growth factor (VEGF) concentration.
Doc. S1. Supplementary experimental procedures.