

Abstract
Romidepsin (FK228, depsipeptide) is a potent histone deacetylase (HDAC) inhibitor that has FDA approval for the treatment of cutaneous and peripheral T-cell lymphomas. We have previously reported that FK228 and its analogs have an additional activity as phosphatidylinositol 3-kinase (PI3K) inhibitors, and are defined as HDAC/PI3K dual inhibitors. Because a combination of an HDAC inhibitor and a PI3K inhibitor induces apoptosis in human cancer cells in a synergistic manner, development of an HDAC/PI3K dual inhibitor will provide an attractive novel drug for cancer therapy. Using structure-based optimization of the analogs, FK-A11 was identified as the most potent analog. FK-A11 inhibited phosphorylation of AKT and accelerated histone acetylation at lower concentrations, resulting in stronger cytotoxic effects than FK228 and the other analogs in human cancer cells. In this study, we have characterized the biochemical, biological and structural properties of FK228 analogs as PI3K inhibitors. First, FK-A11 is an ATP competitive PI3K inhibitor. Second, FK-A11 is a pan-p110 isoform inhibitor. Third, FK-A11 selectively inhibits PI3K among 22 common cellular kinases. Fourth, conformational changes of FK228 analogs by reduction of an internal disulfide bond have no effect on PI3K inhibitory activity, unlike HDAC inhibitory activity. Finally, molecular modeling of PI3K-FK228 analogs and analyses of the binding affinities identified the structure that defines potency for PI3K inhibitory activity. These results prove our concept that a series of FK228 analogs are HDAC/PI3K dual inhibitors. These findings should help in the development of FK228 analogs as novel HDAC/PI3K dual inhibitors.
Keywords: Drug development, dual inhibitor, HDAC, PI3K, romidepsin
Phosphatidylinositol 3-kinase (PI3K), an ubiquitously expressed lipid kinase, phosphorylates phosphatidylinositol-4,5-biphosphate (PIP2), generating phosphatidylinositol-3,4,5-triphosphate (PIP3).1 PIP3 serves as a second messenger in the signal transduction pathway,leading to cancer cell initiation, proliferation and survival.2 Class I PI3K is composed of a catalytic subunit, p110, and a regulatory subunit, p85 or p101. The catalytic subunit consists of four isoforms: p110α, β, γ and δ.3 In many human cancers, the PI3K pathway is frequently activated through gain of function mutations in the PIK3CA gene that encodes p110α.4 Therefore, the catalytic subunits of PI3K are considered to be potential drug targets for cancer therapy.5,6
In addition to genetic mutations, epigenetic changes, such as dysregulation of histone deacetylases (HDAC), contribute to cancer cell initiation and growth, by altering the cell phenotype and gene expression and by disturbing homeostasis.7 Thus, HDAC inhibitors are newly emerging drugs for cancer therapeutics.8,9 Romidepsin (FK228, depsipeptide) is an HDAC inhibitor with high inhibitory activity for class I HDAC.10 FK228 is a bicyclic depsipeptide that is structurally characteristic compared with other HDAC inhibitors. Reduction of the internal disulfide bond changes the conformation of FK228 to the open form, producing free sulfhydryl groups that can interact with the catalytic active pocket of HDAC.10 FK228 is approved by the FDA for the treatment of patients with cutaneous T-cell lymphoma or peripheral T-cell lymphoma.11,12
Recently, it has been reported that the combination of an HDAC inhibitor and a kinase inhibitor, such as epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor or PI3K inhibitor, overcomes kinase inhibitor resistance and induces apoptosis in human solid cancers in a synergistic manner.13–15 Therefore, development of inhibitors targeting both HDAC and PI3K would be advantageous as anticancer drug candidates. Indeed, a single molecule dual inhibitor of HDAC and PI3K is under investigation in a phase I clinical trial.16
We have previously demonstrated that FK228 and its analogs directly inhibit PI3K activity and potently induce apoptosis through HDAC/PI3K dual inhibition.17 In other words, FK228 and its analogs have been identified as novel HDAC/PI3K dual inhibitors. However, no additional finding of FK228 as a kinase inhibitor has been reported to date. Here, we describe the biochemical, biological and structural properties of FK228 and its analogs as PI3K inhibitors. These findings provide important clues for the development of more potent FK228 analogs as HDAC/PI3K dual inhibitors.
Materials and Methods
Reagents
LY294002 and suberoylanilide hydroxamic acid (SAHA) were purchased from Cayman Chemical Company (Ann-Arbor, MI, USA). Wortmannin was purchased from Millipore (Billerica, MA, USA). FK228 and its analogs were synthesized and provided by T. K. from Tohoku Pharmaceutical University.18
Kinase assay
PI3K (p110α/p85α) activity and other 20 common cellular kinase activities were evaluated by the mobility shift assay (Carna Biosciences, Kobe, Japan).19 Individual activity for the four p110 isoforms, α, β, γ and δ, was measured using an homogenous time-resolved fluorescence (HTRF) assay (PI3K assay kit; Millipore). Mammalian target of rapamaycin (mTOR) activity was measured using a K-LISA mTOR activity kit (Millipore). The detailed methods of each assay are described in Supplementary Document S1.
Enzyme kinetic assay
The PI3K assay for Lineweaver–Burk analysis was performed using an ADP-Glo assay kit (Promega, Madison, WI, USA). PI3K activity was assayed at five different ATP concentrations (5, 10, 25, 50 and 100 μM), with 40 nM PI3Kα, 25 μM PIP2 and FK-A11 (0, 2.5, 5 and 10 μM). A Lineweaver–Burk plot was developed by plotting 1/v (reaction velocity) against 1/[ATP] (ATP concentration).
HDAC activity assay
FK228 and its analogs, except for FK-A21 and FK-A22, were evaluated for HDAC inhibitory activity using the Screening Committee of Anticancer Drugs (SCADS). The HDAC inhibitory activities of FK-A21 and FK-A22 were evaluated using an HDAC inhibitor screening kit (Cayman Chemical Company) as described in Supplementary Document S1.
Cell lines
The human cancer cell lines used in this study included the prostate cancer cell line PC3 and the colorectal cancer cell line HCT116. PC3 was obtained from the Cell Resource Center (Institute of Development, Aging, and Cancer, Tohoku University, Sendai, Japan). HCT116 was purchased from the ATCC (Mannassas, VA, USA).
Western blot analysis
Western blotting was conducted as previously described,17 using antibodies described in Supplementary Document S1.
Cell proliferation assay
A cell proliferation assay was performed with a Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan) as previously described.17 Cells (8 × 103 per well) were seeded and incubated in 96-well plates for 24 h. Compounds were added to the cells and further cultured for 24 h at 37°C. The ratios of surviving cells to control cells treated with 0.5 or 1% DMSO were calculated.
Molecular modeling of the PI3K-FK228 analog and HDAC1-FK228 analog complexes
A computational docking simulation of PI3K and FK-A11 was carried out using the predicted PI3K-FK228 complex structure previously described.17 The structure of the PI3K (p110α H1047R/p85α)–wortmannin complex (Protein Data Bank ID: 3HHM) was used to obtain the PI3K template structure. The HDAC1 template structure was obtained from the protein data bank (ID: 4BXK). Conformational searches of docking complexes were performed using LMOD and OMEGA (OpenEye Scientific Software, Boston, MA, USA).20 Subsequently, the docking calculations were carried out using the calculation method FRED, to obtain Chemgauss 4 scores that suggest the binding affinities.21
Results
Search for novel FK228 analogs with potent HDAC/PI3K dual inhibitory activity in cell-free systems
In our previous study, we reported that FK-A5 was the most potent PI3K inhibitor identified among FK228 and its 10 analogs.17 From the perspective of chemical constitution, FK-A5 has a characteristic structure, possessing a benzyl group at the C7 position. To identify a more potent PI3K inhibitor, most of the additional 14 analogs (FK-A7, FK-A8, FK-A9, FK-A10, FK-A11, FK-A12, FK-A13, FK-A15, FK-A17, FK-A18, FK-A20, FK-A21, FK-A22 and SP-4) were synthesized by modification of the benzyl group at the C7 position. PI3K inhibitory activities were evaluated in a cell-free system (Fig.1). Among the newly synthesized analogs, FK-A11, FK-A12 and FK-A20 were more potent than the other analogs. In particular, FK-A11 was the most potent PI3K inhibitor, with activity exceeding that of both FK228 and FK-A5. The IC50 values against p110α of FK-A11, FK-A5 and FK228 were 6.7, 27.3 and 57.1 μM, respectively (Figs1 and 2a). FK-A11 has 4-fold and 8.5-fold stronger activity than FK-A5 and FK228, respectively. From a structural viewpoint, the benzyl group at the C7 position of FK-A5 was replaced by a naphthylmethyl group which consists of a fused pair of benzene rings in FK-A11 (Fig.1). In addition to the PI3K inhibitory activity, FK-A11 was also a more potent HDAC inhibitor, exceeding both FK228 and FK-A5. The IC50 values against HDAC1 of FK-A11, FK-A5 and FK228 were 0.64, 2.5 and 3.6 nM, respectively (Fig.1). FK-A12 and FK-A20 were weaker HDAC inhibitors than FK228, FK-A5 and FK-A11. Although FK-A11 and FK-A12 are stereoisomers with respect to the C7 position, their HDAC inhibitory activities were different. These results indicated that FK-A11 was the most potent HDAC/PI3K dual inhibitor among the FK228 analogs synthesized so far.
Fig 1.
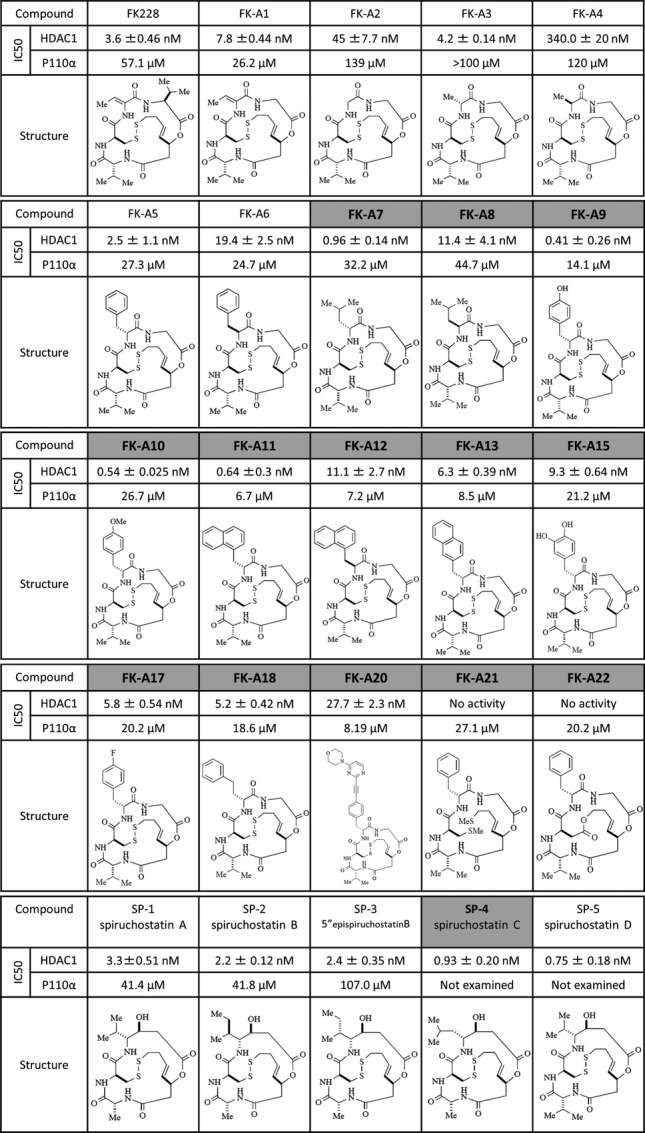
Chemical structures and IC50 values for p110α and HDAC1. The chemical structures of FK228 and the analogs are shown. The additional 14 analogs are written in bold letters on gray backgrounds. Their IC50 values for HDAC1 (mean ± SD) and for p110α are indicated. IC50 values for HDAC1 were assayed by Screening Committee of Anticancer Drugs (SCADS) and by using HDAC inhibitor screening kit. IC50 values were determined as the mean ± SD of the concentrations calculated from at least three independent dose-response curves. Inhibition of PI3K activity was evaluated by a mobility shift assay. Data are the mean of two experiments performed in duplicate. IC50 values were calculated from concentration versus % inhibition curves by fitting to a four-parameter logistic curve.
Fig 2.

Effects of FK228, FK-A5 and FK-A11 on inhibition of PI3K activity and cell proliferation. (a) Inhibition of PI3K activity was evaluated by a mobility shift assay. The activity in the control reaction was defined as 0% inhibition. Percent inhibition of each test reaction was calculated. The experiments were carried out in duplicate. The two independent percent inhibition values for each concentration were plotted. (b) Western blot analysis of phosphorylated AKT in PC3 cells. Cells were treated for 3 h with various concentrations of FK228, FK-A5 and FK-A11. (c) Western blot analysis of acetyl-histone H3 in PC3 cell. Cells were treated for 3 h with various concentrations of FK228, FK-A5 and FK-A11. (d) PC3 cell proliferation assay. Cells were treated for 24 h with DMSO, 50 μM LY294002 (LY), 2.5 μM SAHA, a combination of 50 μM LY and 2.5 μM SAHA, FK228, FK-A5 and FK-A11 at concentrations of 10 and 100 nM and 1 and 10 μM. The ratio of surviving cells to the control treated with 1% DMSO was calculated. (e) HCT116 cell proliferation assay. Cells were treated for 24 h with 0.5% DMSO as the control, 50 μM LY, 2.5 μM SAHA, a combination of 50 μM LY and 2.5 μM SAHA, FK228, FK-A5, FK-A11, FK-A12, FK-A21, SP-3 and FK-A3 at concentrations of 5, 50 and 500 nM and 5 μM.
Inhibitory effects of FK228 analogs on PI3K, HDAC and cell proliferation in human cancer cell lines
A prostate cancer cell line PC3 and a human colon cancer cell line HCT116 were cultured in the presence of FK228 analogs, an HDAC inhibitor SAHA, a PI3K inhibitor LY294002, or a combination of SAHA and LY294002. The data with PC3 cells showed that FK-A11 inhibited the phosphorylation of AKT, a downstream component of PI3K, at lower concentrations than that observed with FK228 and FK-A5 (Fig.2b). As for HDAC inhibition, FK-A11 increased acetylated histones at nM-range concentrations similar to FK228 and FK-A5 (Fig.2c). In cell proliferation assay in PC3 cells, FK-A11 exhibited stronger cytotoxic effects than any other FK228 analog and the combination of SAHA and LY294002 (Fig.2d). Furthermore, to elucidate the contribution of HDAC/PI3K dual inhibition to the cytotoxic effects, the effects of some FK228 analogs were compared. FK-A11 and FK-A12 are stereoisomers with similar PI3K inhibitory activity but different HDAC inhibitory activity. FK-A11 has 17.3-fold stronger HDAC inhibitory activity than FK-A12 (Fig.1). In comparison of the cytotoxic effects, FK-A12 showed weaker cytotoxic effects than FK-A11 at the lower concentrations, 10 and 100 nM (Fig.2d). The cytotoxic effects of FK-A11 and FK-A12 were similar at the concentrations of 1 and 10 μM (Fig.2d). In addition, SP-3 and FK-A3 with weaker PI3K inhibitory activity than FK228, and with similar HDAC inhibitory activity to FK228 have been evaluated. Both SP-3 and FK-A3 did not potentiate cytotoxic effects even at concentrations of 1 and 10 μM (Fig.2d). Similar cytotoxic effects of FK228 analogs were observed in HCT116 cells that were more sensitive to HDAC inhibitors than PC3 cells (Fig.2e). These results indicate that PI3K inhibition clearly potentiates the cytotoxic effect of HDAC inhibition, and that FK-A11 is the most potent inhibitor. FK-A21 with PI3K inhibitory activity and without HDAC inhibitory activity exhibited no cytotoxic effect (Fig.2d,e). Moreover, FK-A21 did not inhibit AKT phosphorylation in cells in spite of PI3K inhibitory activity confirmed in the cell free mobility shift assay (Suppl. Fig. S1). DMSO had no toxicity on cell proliferation in both cell lines.
FK-A11, an ATP competitive PI3K inhibitor
We previously reported that FK228 binds to the ATP-binding pocket of p110α based on a study with a docking model. Furthermore, this model is supported by the fact that the PI3K inhibitory activity of FK228 was weakened at a high concentration of ATP in an in vitro assay.17 We also predicted that FK-A5 would bind to the ATP-binding pocket of p110α.22 To investigate whether FK-A11 is an ATP competitive PI3K inhibitor, an in vitro ATP competition assay and a docking simulation were performed. In the in vitro assay, the PI3K activity was measured at various concentrations of ATP in the absence and presence of increasing concentrations of FK-A11. Lineweaver–Burk plot analysis showed that FK-A11 changed Km (the Michaelis constant) without affecting Vmax (the max velocity), indicating the apparent mode of competitive inhibition with ATP (Fig.3). In the docking model, FK-A11 was predicted to bind to the ATP-binding site of p110α, similarly to FK228 and FK-A5. FK-A11 occupied the ATP-binding pocket of p110α with its naphthalene moiety localized to a deep portion of the pocket (Fig.4a–c). These results indicated that FK-A11 is an ATP competitive PI3K inhibitor.
Fig 3.
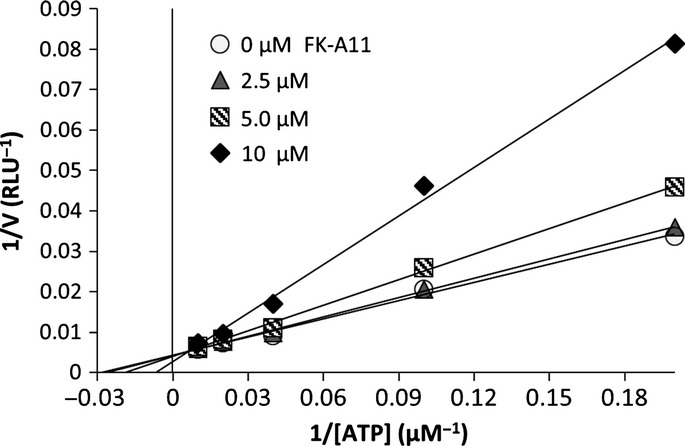
Lineweaver–Burk plot of the inhibition of PI3K activity with FK-A11. PI3K activity assay was performed at five concentrations of ATP (5, 10, 25, 50 and 100 μM), 40 nM PI3Kα and 25 μM PIP2 in the absence or presence of increasing concentrations of FK-A11. 1/v (reaction velocity) was plotted versus 1/[ATP] (ATP concentration).
Fig 4.
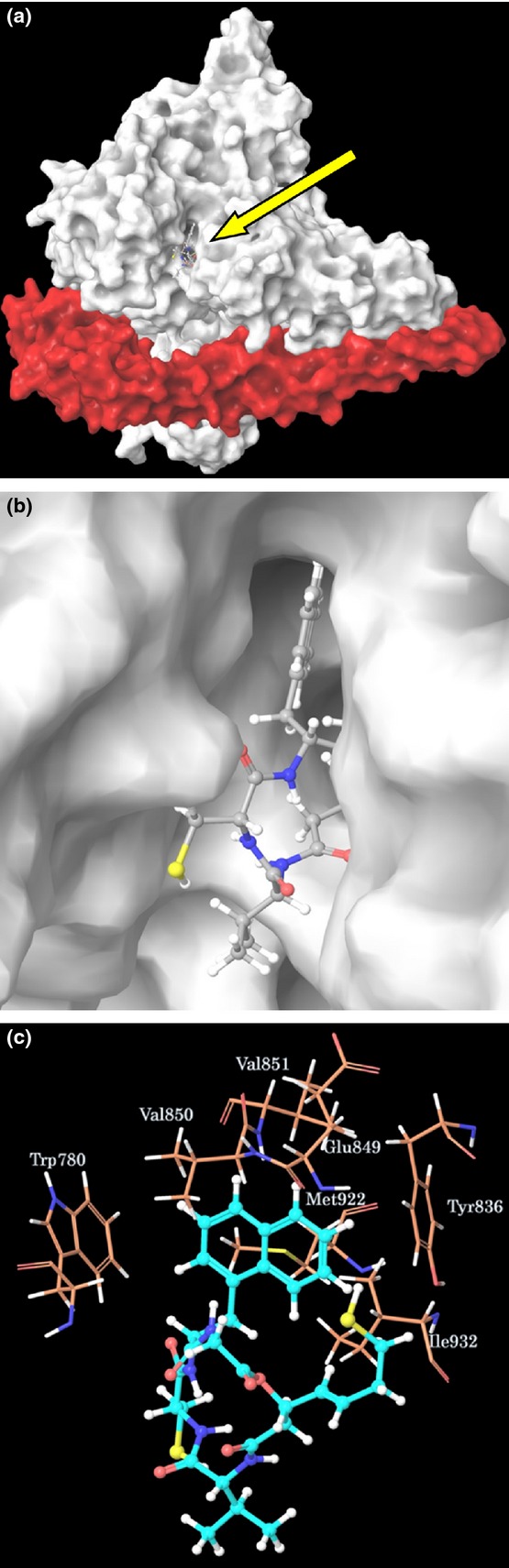
Molecular modeling of PI3K-FK-A11 complex. (a) Molecular surface structure. White and red components represent p110α and p85α, respectively. FK-A11 in the complex is indicated by the yellow arrow. (b) The enlarged picture around FK-A11 in the ATP-binding pocket. (c) Hydrophobic amino acids around the naphthalene ring of FK-A11 in the PI3K-FK-A11 complex.
FK-A11, a selective PI3K kinase inhibitor
To elucidate the kinase selectivity, the inhibitory activities of FK-A11 for 21 kinases were assessed using a mobility shift assay. In addition, inhibition of mTOR activity was measured by using a K-LISA mTOR activity kit (Millipore). At a concentration of 10 μM, FK-A11 inhibited PI3K by 81%, whereas it inhibited the other 21 kinases by only 0–32.3% (Table1). FK-A11 showed no mTOR inhibitory activity at 10 μM (Table1), and very weak inhibitory activity even at higher concentrations of 50 and 100 μM (Suppl. Fig. S2). These results indicated that FK-A11 is a selective PI3K inhibitor.
Table 1.
Inhibitory activities of FK-A11 for 22 kinases
| Kinase | % inhibition at 10 μM FK-A11 |
|---|---|
| ABL | 12.6 |
| ALK | 13.6 |
| EML4-ALK | 0.3 |
| EGFR | 3.4 |
| FGFR1 | 15.3 |
| FLT3 | 4.7 |
| IGF1R | −3.8 |
| JAK3 | 2.6 |
| MET | 0.3 |
| PDGFRα | 5.4 |
| SRC | −1.5 |
| AKT1 | 0.4 |
| BRAF | 19.9 |
| Erk1 | 5.3 |
| Erk2 | 6.5 |
| MAP2K1(MEK) | 30.9 |
| MAP3K1 | 32.3 |
| MAP3K2 | 18.8 |
| p38α | 7.1 |
| RAF1 | 20.5 |
| PI3K(p110α) | 81 |
| mTOR | −7.6 |
FK-A11, a pan-PI3K inhibitor
The catalytic subunit of class I PI3K, p110 has four isoforms: α, β, γ and δ.3 To investigate whether FK-A11 inhibits each isoform, an HTRF assay was performed. FK-A11 inhibited all four p110 isoforms similar to wortmannin, a pan-PI3K inhibitor (Fig.5). The percent inhibition of each isoform, α, β, γ and δ by 3 μM FK-A11 was 41.2, 17.1, 10.5 and 32.9%, respectively (Fig.5b). These results indicated that FK-A11 is a pan-PI3K inhibitor.
Fig 5.
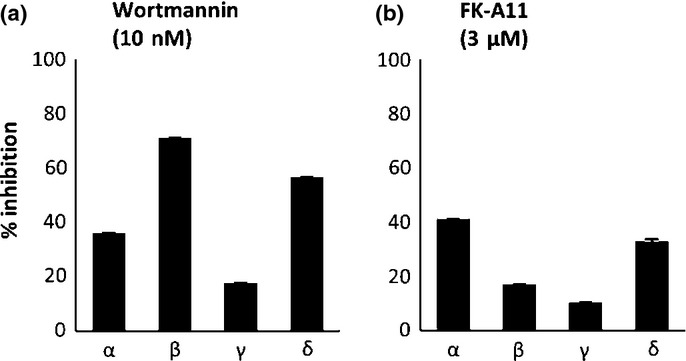
Inhibition profiles of FK-A11 for p110α isoforms. Enzyme activities of p110 isoforms, α, β, γ and δ, were measured using a homogenous time-resolved fluorescence (HTRF) assay. 10 nM wortmannin (a) or 3 μM FK-A11 (b) was incubated with a solution containing each PI3K isoform protein, 10 μM PIP2, 25 μM ATP and 2 mM DTT. Percent inhibition of the enzyme activity by the compounds was calculated with the following formula. Percent inhibition = [(readout value of inhibited activity – that of normal activity)/(readout value of biotinylated-PIP3 – that of normal activity)] × 100.
Effects of conformational changes of FK228 analogs on PI3K inhibitory activity
Reducing activity, such as that generated by glutathione reductase, changes FK228 to an open form, exposing the free sulfhydryl group that is involved in HDAC inhibition.10 To investigate whether the conformational change by reduction is also related to PI3K inhibition, FK-A21 and FK-A22 were designed and synthesized based on the FK-A5 structure (Fig.1). Because both FK-A21 and FK-A22 lack the internal disulfide bond, their conformations should not be affected by reduction. FK-A21 always retains the open form because the disulfide bond is replaced with two methylthio groups. In contrast, FK-A22 always retains the closed form because it has a lactone substructure. Both FK-A21 and FK-A22 did not inhibit HDAC activity because they lack free sulfhydryl groups (Fig.1). In contrast, the IC50 values of FK-A21 and FK-A22 for PI3K were approximately equivalent to that of FK-A5 (Fig.1). These results indicated that the conformational change induced by reduction does not affect PI3K inhibitory activity, and that the active site of FK228 analogs for PI3K inhibition is different from that for HDAC inhibition.
Evaluation of the binding affinities of FK228, FK-A5 or FK-A11 for PI3K and its drug-resistant mutants using computational docking simulations
To investigate whether FK-A11 interacts with PI3K similar to FK228 and FK-A5, Chemgauss 4 scores that indicate the intensity of the binding affinity were calculated in computational docking simulations. The scores showed that FK-A11 has a higher affinity for PI3K than FK228 and FK-A5, in accordance with the results of an in vitro kinase assay (Table2 and Fig.2a). Drug resistant PI3K mutants have been discovered by PIK3CA mutagenesis in yeast. The mutants were I800L and I800M with substitution of isoleucine located in the ATP-binding pocket of p110α.23 Computational docking simulations of the two PI3K mutants and FK-A11 were carried out, so that Chemgauss 4 scores showed reduced affinities of FK-A11 for these mutants compared with wild type p110α (Table2). These results indicated that stronger PI3K inhibition by FK-A11 is based on stronger binding to the ATP-binding pocket of PI3K.
Table 2.
Binding affinity of FK228 and its analogs to PI3K based on computational docking simulations using FRED program
| PI3K-ligand complex | Chemgauss 4 score† |
|---|---|
| PI3K–FK228 | −4.33 |
| PI3K–FK-A5 | −5.2 |
| PI3K–FK-A11 | −8.95 |
| PI3K(p110α I800L)–FK-A11 | −2.17 |
| PI3K(p110α I800M)–FK-A11 | −1.41 |
Lower score means higher affinity.
Evaluation of the binding affinities of FK228 and FK-A11 for HDAC1 using computational docking simulations
We generated molecular docking models of HDAC1-FK228 (data not shown) and HDAC1-FK-A11 complexes (Fig.6). Subsequently calculated Chemgauss 4 scores showed higher binding affinity of FK-A11 than that of FK228 for HDAC1 (Table3). In a molecular model of HDAC1-FK-A11 complex, the free sulfhydryl group of FK-A11 interacted with the active-site zinc. Moreover, the naphthylmethyl group of FK-A11 interacted with Tyr204 or Cys273 in HDAC1 (Fig.6).
Fig 6.
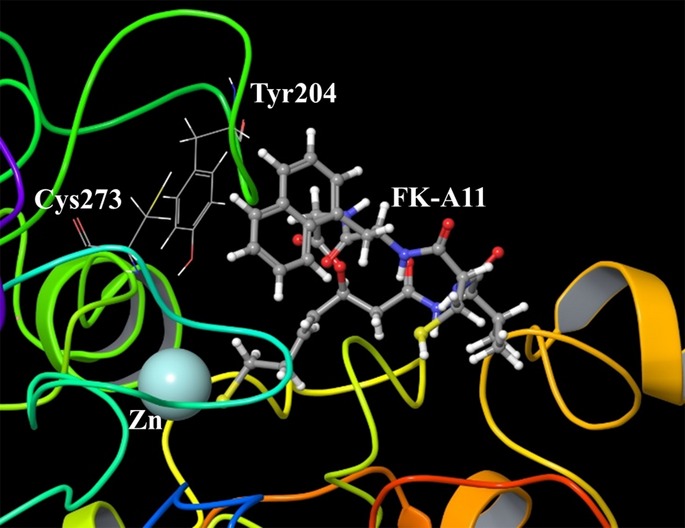
Molecular modeling of HDAC1-FK-A11 complex. The enlarged picture around the active-site zinc and FK-A11. The sulfhydryl group of FK-A11 interacted with zinc. The naphthylmethyl group of FK-A11 was located closely to Tyr204 and Cys273.
Table 3.
Binding affinity of FK228 and FK-A11 to HDAC1 based on computational docking simulations using FRED program
| HDAC1-ligand complex | Chemgauss 4 score† |
|---|---|
| HDAC1–FK228 | 0.198 |
| HDAC1–FK-A11 | −1.771 |
Lower score means higher affinity.
Discussion
We have previously identified FK228 and its analogs as HDAC/PI3K dual inhibitors.17 In this study, we investigated the biochemical, biological and structural properties of FK228 analogs, focusing on FK-A11, from the viewpoint of the kinase inhibitory activities. In search for novel analogs, we identified FK-A11 as the most potent HDAC/PI3K dual inhibitor. FK-A11 inhibits PI3K with 8.5-fold more potency, and inhibits HDAC1 with 5.6-fold more potency than FK228. As a kinase inhibitor, FK-A11 highly selectively inhibited PI3K among 22 kinases tested (Table1). Based on pharmacokinetic properties and isoform selectivity for the ATP-binding site, PI3K inhibitors have been classified into the following three groups.6,24 The first group of inhibitors are pan-PI3K inhibitors, which are able to bind to all class I PI3K and include LY294002, wortmannin, GDC-0941 and ZSTK474.24–26 The second group of inhibitors are isoform-specific inhibitors which have been developed to overcome the toxicity of a pan-PI3K inhibitor and include idelalisib (CAL-101), which targets p110δ.27 The third group of inhibitors is PI3K/mTOR dual inhibitors that share similar structures in the ATP-binding sites of PI3K and mTOR. FK-A11 inhibited all four of the class I PI3K isoforms (Fig.5). In contrast, FK-A11 did not inhibit mTOR (Table2). Therefore, FK228 analogs are classified as pan-PI3K inhibitors.
Considerable interest has centered on the effect of reduction on PI3K inhibitory activity, because reductase activity changes FK228 to an open structure that is active for HDAC inhibition.10 To elucidate the effect of reduction, we synthesized FK-A21 and FK-A22 with conformations that are not affected by reduction. Both compounds inhibited PI3K, but did not inhibit HDAC as expected (Fig.1). Despite the PI3K inhibitory activity confirmed in the cell free mobility shift assay, FK-A21 exhibited no activity in cells (Fig.2 and Suppl. Fig. S1). We assumed that this depends on the instability of FK-A21 in cells: as previously reported, the reduced open form of FK228 increases the instability in cells.10 Our results indicate that the potential structural site for PI3K inhibition is different from that for HDAC inhibition.
All previously described PI3K inhibitors bind to the ATP-binding site of the p110.28–30 FK-A11 is also an ATP competitive PI3K inhibitor based on the results of the ATP competition assay and the docking simulation (Figs3 and 4). In the docking simulations, FK-A11 has higher affinity to PI3K than FK228 and FK-A5 (Table2). This result correlates with the potency of PI3K inhibitory activity. The surface of the ATP-binding pocket in p110α is surrounded by hydrophobic residues, and there is some space created by the hydrophobic residues around the naphthalene ring of FK-A11 (Fig.4c). The interaction between the aromatic rings of the ligand and the hydrophobic aromatic residues of the internal binding pocket, the so-called π–π staking interaction, may play an important role in allowing for higher affinity. The naphthalene ring of FK-A11 could approach closer to the hydrophobic aromatic residues of the ATP-binding pocket, and could confer a stronger π–π stacking interaction than the phenyl ring of FK-A5. Similarly, in docking simulations for HDAC1, FK-A11 had higher affinity than FK228 (Table3). The naphthalene ring of FK-A11 might contribute to higher affinity not only for PI3K but also for HDAC1 (Fig.6).
When compared to combination therapy involving several drugs, a single dual target inhibitor is expected to have advantages, such as a simpler pharmacokinetic property, lower toxicity, higher compliance and more efficient clinical benefit.16 Therefore, development of the dual target inhibitor will provide an attractive novel drug for cancer therapy. Because the combination of an HDAC inhibitor and a PI3K inhibitor induces apoptosis in a synergistic manner, development of an HDAC/PI3K dual inhibitor is especially attractive. We anticipate that our investigation might prompt ideas for the further development of novel analogs as HDAC/PI3K dual inhibitors.
Acknowledgments
This work was supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT) KAKENHI grant numbers 24300339, 25830110 and 26460034, and the Gonryo Medical Foundation.
Disclosure Statement
Chikashi Ishioka received research grants from Chugai Pharmaceutical Co., Ltd., Taiho Pharmaceutical Co., Ltd., Daiichi-Sankyo Co., Ltd. and Merck Serono Co., Ltd. The other authors have no conflict of interest to declare.
Supporting Information
Additional supporting information may be found in the online version of this article:
Fig. S1. Western blot analysis of phosphorylated AKT and acetyl-histones in PC3 cells treated with FK-A5 or FK-A21.
Fig. S2. The effect of FK-A11 on inhibition of mTOR activity.
Doc. S1 Detailed materials and methods.
References
- Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- Vogt PK, Gymnopoulos M, Hart JR. PI 3-kinase and cancer: changing accents. Curr Opin Genet Dev. 2009;19:12–7. doi: 10.1016/j.gde.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini M, Ciraolo E, Gulluni F, Hirsch E. Targeting PI3K in cancer: any good news? Front Oncol. 2013;3:108. doi: 10.3389/fonc.2013.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30–9. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27:5459–68. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- Furumai R, Matsuyama A, Kobashi N, et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002;62:4916–21. [PubMed] [Google Scholar]
- Bertino EM, Otterson GA. Romidepsin: a novel histone deacetylase inhibitor for cancer. Expert Opin Investig Drugs. 2011;20:1151–8. doi: 10.1517/13543784.2011.594437. [DOI] [PubMed] [Google Scholar]
- Mummaneni P, Shord SS. Epigenetics and oncology. Pharmacotherapy. 2014;34:495–505. doi: 10.1002/phar.1408. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Takeuchi S, Yamada T, et al. EGFR-TKI resistance due to BIM polymorphism can be circumvented in combination with HDAC inhibition. Cancer Res. 2013;73:2428–34. doi: 10.1158/0008-5472.CAN-12-3479. [DOI] [PubMed] [Google Scholar]
- Chakraborty AR, Robey RW, Luchenko VL, et al. MAPK pathway activation leads to Bim loss and histone deacetylase inhibitor resistance: rationale to combine romidepsin with an MEK inhibitor. Blood. 2013;121:4115–25. doi: 10.1182/blood-2012-08-449140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka T, Yogosawa S, Yamada T, Kitawaki J, Sakai T. Combination of a novel HDAC inhibitor OBP-801/YM753 and a PI3K inhibitor LY294002 synergistically induces apoptosis in human endometrial carcinoma cells due to increase of Bim with accumulation of ROS. Gynecol Oncol. 2013;129:425–32. doi: 10.1016/j.ygyno.2013.02.008. [DOI] [PubMed] [Google Scholar]
- Qian C, Lai CJ, Bao R, et al. Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling. Clin Cancer Res. 2012;18:4104–13. doi: 10.1158/1078-0432.CCR-12-0055. [DOI] [PubMed] [Google Scholar]
- Saijo K, Katoh T, Shimodaira H, Oda A, Takahashi O, Ishioka C. Romidepsin (FK228) and its analogs directly inhibit phosphatidylinositol 3-kinase activity and potently induce apoptosis as histone deacetylase/phosphatidylinositol 3-kinase dual inhibitors. Cancer Sci. 2012;103:1994–2001. doi: 10.1111/cas.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita K, Kikuchi T, Watanabe K, et al. Total synthesis of the bicyclic depsipeptide HDAC inhibitors spiruchostatins A and B, 5″-epi-spiruchostatin B, FK228 (FR901228) and preliminary evaluation of their biological activity. Chemistry. 2009;15:11174–86. doi: 10.1002/chem.200901552. [DOI] [PubMed] [Google Scholar]
- Kitagawa D, Yokota K, Gouda M, et al. Activity-based kinase profiling of approved tyrosine kinase inhibitors. Genes Cells. 2013;18:110–22. doi: 10.1111/gtc.12022. [DOI] [PubMed] [Google Scholar]
- Kolossvary I, Guida WC. Low mode search. An efficient, automated computational method for conformational analysis: application to cyclic and acyclic alkanes and cyclic peptides. J Am Chem Soc. 1996;118:5011–9. [Google Scholar]
- McGann MR, Almond HR, Nicholls A, Grant JA, Brown FK. Gaussian docking functions. Biopolymers. 2003;68:76–90. doi: 10.1002/bip.10207. [DOI] [PubMed] [Google Scholar]
- Oda A, Saijo K, Ishioka C, et al. Predicting the structures of complexes between phosphoinositide 3-kinase (PI3K) and romidepsin-related compounds for the drug design of PI3K/histone deacetylase dual inhibitors using computational docking and the ligand-based drug design approach. J Mol Graph Model. 2014;54:46–53. doi: 10.1016/j.jmgm.2014.08.007. [DOI] [PubMed] [Google Scholar]
- Zunder ER, Knight ZA, Houseman BT, Apsel B, Shokat KM. Discovery of drug-resistant and drug-sensitizing mutations in the oncogenic PI3K isoform p110 alpha. Cancer Cell. 2008;14:180–92. doi: 10.1016/j.ccr.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinleye A, Avvaru P, Furqan M, Song Y, Liu D. Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. J Hematol Oncol. 2013;6:88. doi: 10.1186/1756-8722-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkes AJ, Ahmadi K, Alderton WK, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51:5522–32. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- Yaguchi S, Fukui Y, Koshimizu I, et al. Antitumor activity of ZSTK474, a new phosphatidylinositol 3-kinase inhibitor. J Natl Cancer Inst. 2006;98:545–56. doi: 10.1093/jnci/djj133. [DOI] [PubMed] [Google Scholar]
- Lannutti BJ, Meadows SA, Herman SE, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117:591–4. doi: 10.1182/blood-2010-03-275305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker EH, Pacold ME, Perisic O, et al. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell. 2000;6:909–19. doi: 10.1016/s1097-2765(05)00089-4. [DOI] [PubMed] [Google Scholar]
- Vadas O, Burke JE, Zhang X, Berndt A, Williams RL. Structural basis for activation and inhibition of class I phosphoinositide 3-kinases. Sci Signal. 2011;4:re2. doi: 10.1126/scisignal.2002165. [DOI] [PubMed] [Google Scholar]
- Kong D, Yamori T. ZSTK474 is an ATP-competitive inhibitor of class I phosphatidylinositol 3 kinase isoforms. Cancer Sci. 2007;98:1638–42. doi: 10.1111/j.1349-7006.2007.00580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Western blot analysis of phosphorylated AKT and acetyl-histones in PC3 cells treated with FK-A5 or FK-A21.
Fig. S2. The effect of FK-A11 on inhibition of mTOR activity.
Doc. S1 Detailed materials and methods.